

Abstract
Ringfused bicyclic 2-pyridones exhibit interesting biological properties against pili assembly in uropathogenic Escherichia coli (Pinkner, J. S. et al. Proc. Natl. Acad. Sci. U. S. A.2006, 103, 17897–17902; Åberg, V. et al. Org. Biomol. Chem.2007, 5, 1827–1834) as well as curli formation (Cegelski, L. et al. Nat. Chem. Biol.2009, 5, 913–919). In the search for new ring-fused central fragments, highly selective synthetic routes to the 2-furanone or 2-pyrone containing tricyclic scaffolds 1 and 2 have been developed.
Introduction
2-Pyridones are commonly found in biologically active natural products(3) (e.g., champtothecin), and they have also attracted researchers for the design and synthesis of peptide backbone mimetics.(4) We have earlier described the synthesis of highly substituted bicyclic dihydro thiazolo ring-fused 2-pyridones 3–6 shown in Figure 1.5−8 These compounds, as their corresponding carboxylic acids, were originally designed to inhibit the formation of pili in uropathogenic Escherichia coli by interfering with the chaperone usher pathway.(1) Interestingly, by varying the substitution pattern on these central fragments, compounds with amyloid inhibiting properties could also be synthesized. From this collection, compounds named curlicides were identified that inhibit the formation of bacterial fibers, curli, which are functional amyloid structures.(2)
Figure 1.

Several methods to selectively introduce substituents onto dihydro thiazolo ring-fused 2-pyridones.
Since there is a tremendous need for new central fragments in drug discovery,(9) we have, in parallel with the development of antibacterial compounds, taken a more general approach to synthesize different rigidified tricyclic peptidomimetic scaffolds based on the 2-pyridone core10,11 (e.g., 7 and 8, Figure 2).
Figure 2.

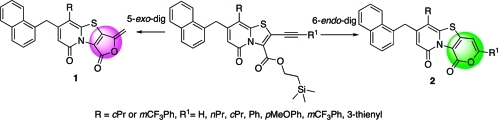
Previously developed tricyclic peptidomimetic scaffolds 7 and 8, together with the new 2-furanone (1) and 2-pyrone (2) containing tricyclic scaffolds described in this article. The peptide backbone connectivity is highlighted in red.
2-Pyrones and 2-furanones are interesting compounds with widespread biological characteristics and are common in many natural products, for instance Terreulactone A(12) and (−)-Tetrodecamycin.(13) In all ring-fused 2-pyridone derived peptidomimetics previously developed in the group, the C-terminal carboxylic ester was positioned in the ring system as a substituent with freedom to rotate (see Figure 1). However, introducing a ring-fused 2-pyrone or 2-furanone to the central fragment would result in a more rigidified scaffold but still with a peptide backbone connectivity. Herein, we present the development of methods to regioselectively synthesize either 2-furanone or 2-pyrone fused tricyclic scaffolds (1 and 2, Figure 2). These are two new central fragments, which can be substituted with a variety of substituents, thus having great potential in the future design and synthesis of peptidomimetic compounds.
Results and Discussion
It is known from the literature that 2-pyrones and 2-furanones can be synthesized via a cyclization of a carboxylic acid ester onto an alkyne.(14) In some cases the selectivity between 5-exo-dig and 6-endo-dig cyclization is poor, and for the reaction to proceed well, high concentrations of acid and/or high temperatures are demanded.(15) Previously published results show that Pd-catalyzed cross couplings such as the Suzuki–Miyaura(8) and Heck(5) couplings are possible in the 2-position of the ring-fused bicyclic 2-pyridones (Figure 3). On the basis of these results, we envisioned a retrosynthetic strategy starting with an oxidation/bromination reaction, followed by a Sonogashira coupling, and finally an acid induced cyclization onto the acetylene (Figure 3).
Figure 3.

Reterosynthetic strategy to the 2-pyrone or 2-furanone containing tricyclic scaffolds.
Synthesis of the acetylene functionalized bicyclic 2-pyridone was straightforward and started from the known methylester 9(16) (Scheme 1). The oxidation/bromination reaction previously reported(8) followed by the Pd(PPh3)2Cl2/CuI catalyzed Sonogashira coupling with trimethylsilylacetylene (TMSA) gave 11 in 73% yield. However, the following cyclization to give compounds 12 and 13 did not proceed as expected. Even when using TFA as solvent for 18 h at room temperature, only starting material remained.
Scheme 1. Synthesis of the Acetylene Functionalized Ring-Fused Bicyclic 2-Pyridone and Attempted Cyclization.

We anticipated that the corresponding carboxylic acid would ring-close under milder conditions, hence changing the methyl ester to a more acid labile ester that could withstand the conditions used in the synthetic sequence; our choice was the trimethylsilylethyl ester (TMSE).
The TMSE esters (15 and 16) were synthesized via the corresponding acid chlorides to give the racemic TMSE esters 15 and 16 in excellent yields (Scheme 2). The following oxidation/bromination with excess NaH and BrCCl3 in MeCN, previously developed,(8) was slightly modified by changing MeOH for 2-(trimethylsilyl)ethanol (TMSE–OH) because of problems with transesterification. The reaction was yielding surprisingly well, considering that TMSE esters have been reported labile toward NaH in some cases.(17) The bromides 17 and 18 were exposed to a Pd(PPh3)2Cl2/CuI catalyzed Sonogashira coupling with TMSA followed by deprotection of the TMS group with K2CO3 in MeOH/THF 4:1 to yield the TMSE protected key intermediates 19 and 20 in 61 and 62% yield, respectively (Scheme 2).
Scheme 2. Synthesis of the TMSE Protected Key Intermediates.

To our delight, this time the following ring closure worked, and the 6-endo-dig (12) and 5-exo-dig (13) products were obtained in a 1:1 ratio. Since the first conditions (20% TFA in DCM at room temperature) did not give any selectivety, a screen with different catalysts and conditions was performed (Table 1). Although most of the conditions tested resulted in a preference for the 5-exo-dig (13) product with almost complete selectivity with Pd(OAc)2 and 1,3-bis(diphenylphosphino)propane (DPPP) (Table 1, entry 3). Catalysts based upon N-heterocyclic carbenes (NHC) gave rewarding selectivity in favor of the 6-endo-dig product 12 (Table 1, entries 4 and 5).
Table 1. Catalyst Screen to Obtain Selectivity in the Cyclization Reactiona.

| entry | catalyst (5 mol %) | ligand (5 mol %) | reaction time (h) | ratio 12:13 | conversion (isolated yield) (%) |
|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | 2 | 20:80 | 100b | |
| 2 | Pd(OAc)2 | DPPFc | 2 | 5:95 | 100b |
| 3 | Pd(OAc)2 | DPPP | 1 | >99% of 13 | 100 (88) |
| 4 | Pd-IPr-NHC | 2.5 | 90:10 | 100 (88) | |
| 5 | Pd-SIPr-NHC | 2.5 | 95:5 | 100b | |
| 6 | PdCl2 | 2.5 | 50:50 | 80b | |
| 7 | Pd(PPh3)2Cl2 | 2.5 | complex mixture | ||
| 8 | Pd(PPh3)4 | 2 | complex mixture | ||
| 9 | Pd2(dba)3*CHCl3d | 1.5 | 20:80 | 100b | |
| 10 | PPh3AuCl/AgSbF6 | 3 | 5:95 | 100b | |
| 11 | AuCl3 | 3 | 50:50 | 100b |

The reactions were performed in a 10 mg/mL concentration.
No byproducts detected by HPLC.
DPPF = 1,1′-Bis(diphenylphosphino)ferrocene.
dba = dibenzylideneacetone.
In an attempt to further increase the selectivity between 5-exo-dig and 6-endo-dig cyclization in the Pd-NHC catalyzed reactions, a set of Lewis acid additives were screened, and here BF3*Et2O was by far the most effective Lewis acid (Table 2, entry 7). Indeed, complete selectivity for the 6-endo-dig product 12 was obtained. As a control, the reaction was also performed without Pd-catalyst, and under these conditions, fast deprotection of the TMSE ester was observed but no cyclization. Even when the BF3*Et2O amount was increased to 20%, only TMSE ester deprotection was observed.
Table 2. Lewis Acid Screena.

| entry | Lewis acid (10 eq) | ratio 12:13 | reaction time (h) | comment |
|---|---|---|---|---|
| 1 | ZrCl4 | 18 | complex mixture | |
| 2 | TiCl4 | 18 | only ester deprotection | |
| 3 | SnCl4 | 18 | complex mixture | |
| 4 | TiCl3 | 18 | only ester deprotection | |
| 5 | InCl3 | 80:20 | 18 | 70% starting material |
| 6 | SbF3 | 18 | no reaction | |
| 7 | BF3*Et2O | 100% of 12 | 3 | 100% conversion |
| 8 | SnCl2 | 18 | no reaction |
The reactions were performed in a 10 mg/mL concentration.
Changing the cPr substituent on the 2-pyridone scaffold (19) for the more electron withdrawing mCF3Ph substituent (20) had a surprisingly large impact on the cyclization rate, even though it is located far away from the reaction center. Under noncatalyzed conditions, the reaction only reached 20% conversion after 18 h reaction at room temperature with DCM/TFA 80:20 as solvent. However, the catalyzed reactions were efficient and carried out with excellent regioselectivity between the 5-exo-dig (21) and 6-endo-dig (22) in 1 and 3 h, respectively (Scheme 3).
Scheme 3. Selective Cyclizations of the mCF3Ph Substituted Bicyclic 2-Pyridone.

The mechanism of the selective Pd-IPr-NHC catalyzed 6-endo-dig cyclization in DCM/BF3*Et2O 95:5 is still not known. The selective Pd(OAc)2/DPPP catalyzed 5-exo-dig cyclization is believed to proceed via the Pd(II) catalytic cycle shown in Scheme 4.(18) The Pd(OAc)2/DPPP catalyzed reaction was also performed with 5% TFA in DCM as solvent, but these conditions gave less selectivity, with about 5% of the 6-endo-dig product 22 formed.
Scheme 4. Tentative Mechanism of the Pd(OAc)2/DPPP Catalyzed Selective 5-exo-dig Cyclization.

The great results obtained with the terminal acetylenes, regarding regioselectivity and yields in the ring-forming reaction, encouraged further studies but with substituted acetylenes. Hence, a set of different internal acetylenes was synthesized in high yields from the bromo intermediate 17 (Table 3).
Table 3. Synthesis of the Internal Acetylenes.

| compound | R | yield (%) |
|---|---|---|
| 23a | nPr | 86 |
| 23b | cPr | 84 |
| 23c | Ph | 95 |
| 23d | pMeOPh | 89 |
| 23e | mCF3Ph | 86 |
| 23f | 3-thienyl | 95 |
Next, the internal acetylenes 23a–f were exposed to the ring closing procedure under the optimized conditions (Table 4). Surprisingly, the reactions only gave the 6-endo-dig products 24a–f regardless of which conditions were used; without catalyst the reaction rates were much slower (Table 4, entries 1, 7, 10, 13, and 16). The faster ring-closure of the cPr substituted acetylene under noncatalyzed conditions (Table 4, entry 4) is attributed to the cation stabilizing properties of the “sp2-like” hybridization of the cPr carbons.(19)
Table 4. Cyclization of the Internal Acetylenes.

| entry | compound | R | catalyst (5 mol %) | ligand (5 mol %) | solvent | time (h) | conversion (isolated yield) (%) |
|---|---|---|---|---|---|---|---|
| 1 | 24a | nPr | I | 28 | 100 (99) | ||
| 2 | 24a | nPr | Pd(OAc)2 | DPPP | II | 1 | 100a |
| 3 | 24a | nPr | Pd-IPr-NHC | III | 2 | 100a | |
| 4 | 24b | cPr | I | 1 | 100 (99) | ||
| 5 | 24b | cPr | Pd(OAc)2 | DPPP | II | 1 | 100a |
| 6 | 24b | cPr | Pd-IPr-NHC | III | 4 | 100a | |
| 7 | 24c | Ph | I | 18 | 100 (98) | ||
| 8 | 24c | Ph | Pd(OAc)2 | DPPP | II | 1 | 100a |
| 9 | 24c | Ph | Pd-IPr-NHC | III | 18 | 100a | |
| 10 | 24d | pMeOPh | I | 1 | 100 (99) | ||
| 11 | 24d | pMeOPh | Pd(OAc)2 | DPPP | II | 1 | 100a |
| 12 | 24d | pMeOPh | Pd-IPr-NHC | III | 2.5 | 100a | |
| 13 | 24e | mCF3Ph | I | 96 | 90 (88) | ||
| 14 | 24e | mCF3Ph | Pd(OAc)2 | DPPP | II | 2 | 100a |
| 15 | 24e | mCF3Ph | Pd-IPr-NHC | III | 2 | 100a | |
| 16 | 24f | 3-thienyl | I | 20 | 100 (99) | ||
| 17 | 24f | 3-thienyl | Pd(OAc)2 | DPPP | II | 1 | 100a |
| 18 | 24f | 3-thienyl | Pd-IPr-NHC | III | 1.5 | 100a |
No byproducts detected by HPLC.
Conclusions
In conclusion, we have shown that the 2-furanone or the 2-pyrone containing tricyclic scaffolds (12, 13, 21, 22) can be synthesized with excellent selectivity in good yields from the TMSE protected terminal acetylenes 19 and 20 under Pd-catalysis. In the latter case, it was discovered that exchanging TFA for BF3*Et2O as an acidic additive gave complete 6-endo-dig selectivity in the Pd-NHC catalyzed reaction. The substituted 2-pyrone tricyclic scaffolds 24a–f were synthesized in quantitative yields from the corresponding internal acetylenes 23a–f. This was proven to work excellently for alkyl, cycloalkyl, aryl, and heteroaryl substituted acetylenes. Hence, two new rigid tricyclic central fragments, which both have a peptide backbone connectivity, onto which it is easy to introduce substituents and fuctional groups, have been constructed. These central fragments are interesting in the synthesis of future peptidomimetics.
Experimental Section
General Methods
Unless stated otherwise, all reagents and solvents were used as received from commercial suppliers. DMF was distilled under a vacuum and stored over 3 Å molecular sieves. MeCN and MeOH were dried with 3 Å molecular sieves for at least 20 h before use. CuI was purified by refluxing with DCM for 20 h in a Soxhlet apparatus and stored under dark and dry conditions. All HPLC was performed on a C18 reversed-phase column with H2O/MeCN mixtures as eluent. Microwave heated reactions were performed in a microwave reactor; temperatures were monitored with an IR-probe. TLC was performed on silica gel detected with UV light. Column chromatography was employed on normal phase silica gel (eluents given in brackets). Optical rotation was measured with a polarimeter at 20 °C and 589 nm. IR was recorded on a spectrometer equipped with an ATR device. 1H and 13C NMR spectra were recorded on a 400 or 500 MHz spectrometer at 298 K and calibrated using the residual peak of solvent as internal standard [CDCl3 (CHCl3 δH 7.26 ppm, CDCl3 δC 77.16 ppm), DMSO-d6 (DMSO-d5 δH 2.49 ppm, DMSO-d6 δC 39.5 ppm), DCM-d2 (DCM-d1 δH 5.32 ppm, DCM-d2 δC 53.84 ppm)]. HRMS was performed using a mass spectrometer with electrospray ionization (ES+); sodiumformate was used as calibration chemical.
Methyl 8-Cyclopropyl-2-ethynyl-7-(naphthalen-1-ylmethyl)-5-oxo-5H-thiazolo[3,2-a]pyridine-3-carboxylate (11)
Compound 10 (1.73 mmol, 0.81 g), CuI (0.17 mmol, 32 mg), Pd(PPh3)3Cl2 (0.09 mmol, 63 mg), and TEA (3.46 mmol, 0.48 mL) were dissolved in dry DMF (15 mL). Ethynyltrimethylsilane (5.19 mmol, 0.73 mL) dissolved in dry DMF (0.5 mL) was added, and the reaction was heated in the microwave oven at 110 °C for 10 min. The reaction mixture was diluted with saturated NaHCO3 (aq) and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The crude product was dissolved in MeOH/THF 4:1 (15 mL), and K2CO3 (1.73 mmol, 239 mg) was added; the reaction was stirred at rt for 15 min. The reaction mixture was diluted with saturated NaHCO3 and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The crude reaction mixture was purified by column chromatography on silica gel (heptane/EtOAc 90:10 → 70:30) to give the desired compound (0.52 g, 73%) as a yellow foam: 1H NMR (400 MHz, DMSO-d6) δ 8.00–7.92 (m, 1H), 7.92–7.83 (m, 2H), 7.57–7.43 (m, 3H), 7.39–7.31 (m, 1H), 5.56 (s, 1H), 5.15 (s, 1H), 4.55 (s, 2H), 3.82 (s, 3H), 1.89–1.79 (m, 1H), 1.02–0.94 (m, 2H), 0.77–0.70 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 159.5, 157.1, 155.1, 144.9, 134.3, 133.8, 133.4, 131.4, 128.6, 127.5, 127.4, 126.3, 125.8, 125.6, 123.9, 112.0, 110.1, 108.9, 92.9, 70.5, 53.2, 35.4, 10.6, 7.4 (2C); IR λ 1739, 1655, 1567, 1468, 1430; HRMS (ES) calcd [M + Na] for C25H19NNaO3S 436.0984, obsd 436.0972.
(3RS)-2-(Trimethylsilyl)ethyl 8-Cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylate (15)
Compound 9 (2.55 mmol, 1 g) was dissolved in THF (20 mL), and 1 M LiOH (5.1 mmol, 5.1 mL) was added; the reaction was stirred at rt for 20 min. The reaction mixture was quenched with acidic water (pH ∼ 1, set with 1 M HCl) and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The solid material was suspended in DCM (30 mL), and oxalylchloride (2.81 mmol, 0.25 mL), together with DMF (5 drops), was added; the reaction was stirred at rt for 10 min. 2-(Trimethylsilyl)ethanol (7.65 mmol, 1.1 mL) was added, and the reaction was stirred at rt for additional 20 min. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated. The crude material was purified by column chromatography on silica gel (heptane/EtOAc 90:10 → 60:40) to give the desired compound (1.15 g, 94%) as a colorless foam: 1H NMR (400 MHz, CDCl3) δ 7.92–7.87 (m, 1H), 7.85–7.78 (m, 2H), 7.52–7.40 (m, 3H), 7.32–7.29 (m, 1H), 5.78 (s, 1H), 5.55 (dd, J = 2.2, 8.6 Hz, 1H), 4.54–4.35 (m, 2H), 4.35–4.24 (m, 2H), 3.67 (dd, J = 8.6, 11.7 Hz, 1H), 3.51 (dd, J = 2.2, 11.7 Hz, 1H), 1.71–1.62 (m, 1H), 1.08–0.88 m, 4H), 0.80–0.71 (m, 2H), 0.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 168.4, 161.4, 156.7, 147.1, 134.1, 134.0, 132.0, 128.9, 127.7, 127.6, 126.3, 125.8, 125.6, 123.9, 115.5, 113.6, 65.0, 63.0, 36.3, 31.8, 17.3, 11.3, 7.8, 7.5, −1.5 (3C); IR λ 1734, 1653, 1577, 1483, 1425; [α]D = 0 (c0.5, CHCl3); HRMS (ES) calcd [M + Na] for C27H31NNaO3SSi 500.1692, obsd 500.1694.
(3RS)-2-(Trimethylsilyl)ethyl 7-(Naphthalen-1-ylmethyl)-5-oxo-8-(3-(trifluoromethyl)phenyl)-3,5-dihydro-2H-thiazolo[3,2-a]pyridine-3-carboxylate (16)
Compound 14 (2.02 mmol, 1 g) was dissolved in THF (20 mL), and 1 M LiOH (4.0 mmol, 4 mL) was added; the reaction was stirred at rt for 15 min. The reaction mixture was quenched with acidic water (pH ∼ 1, set with 1 M HCl) and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The solid material was suspended in DCM (30 mL), and oxalylchloride (2.22 mmol, 0.19 mL), together with DMF (5 drops), was added; the reaction was stirred at rt for 10 min. 2-(Trimethylsilyl)ethanol (6.1 mmol, 0.87 mL) was added, and the reaction was stirred at rt for additional 15 min. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated. The crude material was purified by column chromatography on silica gel (heptane/EtOAc 90:10 → 60:40) to give the desired compound (1.06 g, 90%) as a colorless foam: 1H NMR (400 MHz, CDCl3) δ 7.83–7.80 (m, 1H), 7.75–7.71 (m, 1H), 7.61–7.53 (m, 3H), 7.51–7.32 (m, 5H), 7.21–7.16 (m, 1H), 5.99–5.92 (m, 1H), 5.60 (dd, J = 2.3, 8.6 Hz, 1H), 4.39–4.24 (m, 2H), 4.04–3.87 (m, 2H), 3.72–3.63 (m, 1H), 3.51–3.43 (m, 1H), 1.08–1.00 (m, 2H), 0.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 168.0, 161.3, 153.9, 147.3, 137.2, 133.9, 133.6 (d, J = 41 Hz, 1C), 133.4, 131.7, 131.4 (splitted q, J = 32 Hz, 1C), 129.6, 128.9, 127.9, 127.8, 127.0 (splitted d, J = 31 Hz, 1C), 126.2, 125.7, 125.4, 125.2, 123.9 (q, J = 270 Hz, 1C), 123.5, 115.8, 114.6, 65.3, 63.9, 36.9, 31.9, 17.4, −1.4 (3C); IR λ 1740, 1653, 1579, 1482, 1436, 1331; [α]D = 0 (c0.5, CHCl3); HRMS (ES) calcd [M + Na] for C31H30F3NNaO3SSi 604.1566, obsd 604.1566.
2-(Trimethylsilyl)ethyl 2-Bromo-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-5H-thiazolo[3,2-a]pyridine-3-carboxylate (17)
Compound 15 (2.76 mmol, 1.32 g) was dissolved in dry MeCN (30 mL, dried over 3 Å MS), NaH (8.3 mmol, 200 mg), BrCCl3 (8.3 mmol, 0.82 mL), and 2-(trimethylsilyl)ethanol (5.52 mmol, 0.79 mL); the reaction was stirred at rt for 8 h. MeCN was evaporated, and the crude material was quenched with saturated NaHCO3 (aq) and extracted with EtOAc. The organic phase was dried (Na2SO4), filtered, and concentrated. The crude material was purified by column chromatography on silica gel (heptane/EtOAc 95:5 → 85:15) to give the desired compound (1.1 g, 72%) as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.89–7.84 (m, 1H), 7.81–7.75 (m, 2H), 7.50–7.43 (m, 2H), 7.43–7.36 (m, 1H), 7.24–7.19 (m, 1H), 5.89 (s, 1H), 4.50 (s, 2H), 4.52–4.44 (m, 2H), 1.81–1.71 (m, 1H), 1.20–1.12 (m, 2H), 1.07–1.00 (m, 2H), 0.78–0.71 (m, 2H), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 159.8, 158.1, 153.9, 146.8, 134.1, 134.0, 132.0, 131.2, 129.0, 127.9, 127.5, 126.4, 125.9, 125.6, 123.7, 112.2, 112.1, 103.1, 65.7, 36.4, 17.3, 11.0, 7.9 (2C), −1.4 (3C); IR λ 1723, 1654, 1567, 1469; HRMS (ES) calcd [M + Na] for C27H28 BrNNaO3SSi 576.0640, obsd 576.0635.
2-(Trimethylsilyl)ethyl 2-Bromo-7-(naphthalen-1-ylmethyl)-5-oxo-8-(3-(trifluoromethyl)phenyl)-5H-thiazolo[3,2-a]pyridine-3-carboxylate (18)
Compound 16 (1.01 mmol, 0.59 g) was dissolved in dry MeCN (15 mL), NaH (3.03 mmol, 73 mg), BrCCl3 (3.03 mmol, 0.3 mL) and 2-(trimethylsilyl)ethanol (2.02 mmol, 0.29 mL); the reaction was stirred at rt for 10 h. MeCN was evaporated, and the crude material was quenched with saturated NaHCO3 (aq) and extracted with EtOAc. The organic phase was dried (Na2SO4), filtered, and concentrated. The crude material was purified by column chromatography on silica gel (heptane/EtOAc 95:5 → 85:15) to give the desired compound (0.55 g, 83%) as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.83–7.78 (m, 1H), 7.74–7.70 (m, 1H), 7.65–7.61 (m, 1H), 7.58–7.50 (m, 3H), 7.47–7.30 (m, 4H), 7.16–7.11 (m, 1H), 6.10 (s, 1H), 4.57–4.47 (m, 2H), 4.06 (s, 2H), 1.25–1.15 (m, 2H), 0.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 159.4, 158.0, 151.3, 147.1, 136.1, 133.9, 133.4, 133.3, 132.1 (q, J = 33 Hz, 1C), 131.5, 131.3, 130.3, 128.9, 128.0, 127.8, 126.7 (q, J = 3 Hz, 1C), 126.3, 125.9 (q, J = 3 Hz, 1C), 125.8, 125.4, 123.7 (q, J = 271 Hz, 1C), 123.4, 113.3, 112.3, 102.9, 65.9, 36.8, 17.3, −1.4 (3C); IR λ 1734, 1661, 1570, 1467, 1436; HRMS (ES) calcd [M + Na] for C31H27 BrF3NNaO3SSi 680.0514, obsd 680.0528.
2-(Trimethylsilyl)ethyl 8-Cyclopropyl-2-ethynyl-7-(naphthalen-1-ylmethyl)-5-oxo-5H-thiazolo[3,2-a]pyridine-3-carboxylate (19)
Compound 17 (1.8 mmol, 1 g), CuI (0.18 mmol, 34 mg), Pd(PPh3)3Cl2 (0.09 mmol, 63 mg) and TEA (3.6 mmol, 0.5 mL) were dissolved in dry DMF (15 mL). Ethynyltrimethylsilane (5.4 mmol, 0.76 mL) dissolved in dry DMF (1 mL) was added, and the reaction was heated in the microwave oven at 110 °C for 10 min. The reaction mixture was diluted with saturated NaHCO3 (aq) and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The crude product was dissolved in MeOH/THF 4:1 (15 mL), and K2CO3 (1.8 mmol, 0.25 g) was added; the reaction was stirred at rt for 10 min. The reaction mixture was diluted with saturated NaHCO3 and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The crude reaction mixture was purified by column chromatography on silica gel (heptane/EtOAc 95:5 → 85:15) to give the desired compound (0.55 g, 61%) as a yellow foam: 1H NMR (400 MHz, DMSO-d6) δ 7.98–7.92 (m, 1H), 7.90–7.83 (m, 2H), 7.54–7,43 (m, 3H), 7.36–7.31 (m, 1H), 5.53 (s, 1H), 5.11 (s, 1H), 4.54 (s, 2H), 4.36–4.28 (m, 2H), 1.88–1.79 (m, 2H), 1.04–0.93 (m, 4H), 0.76–0.69 (m, 2H), −0.02 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 159.0, 157.1, 155.1, 144.9, 134.3 (splitted, 2C), 133.5, 131.5, 128.6, 127.6, 127.4, 126.4, 125.8, 125.7, 123.9, 112.0, 110.1, 108.5, 92.8, 70.6, 64.7, 35.5, 16.6, 10.6, 7.4 (2C), −1.6 (3C); IR λ 1732, 1654, 1568, 1466, 1247; HRMS (ES) calcd [M + Na] for C29H29 NNaO3SSi 522.1535, obsd 522.1530.
2-(Trimethylsilyl)ethyl 2-Ethynyl-7-(naphthalen-1-ylmethyl)-5-oxo-8-(3-(trifluoromethyl)phenyl)-5H-thiazolo[3,2-a]pyridine-3-carboxylate (20)
Compound 18 (0.8 mmol, 525 mg), CuI (0.08 mmol, 15 mg), Pd(PPh3)3Cl2 (0.04 mmol, 28 mg), and TEA (1.6 mmol, 0.22 mL) were dissolved in dry DMF (10 mL). Ethynyltrimethylsilane (2.4 mmol, 0.34 mL) dissolved in dry DMF (1 mL) was added, and the reaction was heated in the microwave oven at 110 °C for 10 min. The reaction mixture was diluted with saturated NaHCO3 (aq) and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The crude product was dissolved in MeOH/THF 4:1 (10 mL), and K2CO3 (0.8 mmol, 0.11 g) was added; the reaction was stirred at rt for 10 min. The reaction mixture was diluted with saturated NaHCO3 and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The crude reaction mixture was purified by column chromatography on silica gel (heptane/EtOAc 95:5 → 85:15) to give the desired compound (0.3 g, 62%) as a yellow foam: 1H NMR (400 MHz, DMSO-d6) δ 7.91–7.85 (m, 1H), 7.82–7.63 (m, 6H), 7.49–7.34 (m, 3H), 7.26–7.20 (m, 1H), 5.89 (s, 1H), 5.11 (s, 1H), 4.42–4.32 (m, 2H), 4.14 (s, 2H), 1.09–1.01 (m, 2H), 0.02 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 158.8, 157.1, 152.1, 145.5, 136.0, 134.4, 134.1, 133.8, 133.3, 131.1, 130.6, 130.1 (q, J = 31 Hz, 1C), 128.5, 127.5, 127.4, 126.6 (q, J = 3 Hz, 1C), 126.2, 125.7, 125.6 (q, J = 3 Hz, 1C), 125.4, 123.8 (q, J = 272 Hz, 1C), 123.6, 113.1, 110.5, 108.3, 93.1, 70.2, 64.9, 35.8, 16.6, −1.6 (3C); IR λ 1734, 1662, 1570, 1471, 1329; HRMS (ES) calcd [M + Na] for C33H28F3NNaO3SSi 626.1409, obsd 626.1412.
6-Cyclopropyl-7-(naphthalen-1-ylmethyl)-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (12)
Compound 19 (0.04 mmol, 20 mg) and Pd-NHC (0.002 mmol, 1.4 mg) were dissolved in DCM/BF3*Et2O 95:5 (2 mL), and the reaction was stirred at rt for 3 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4) filtered and concentrated. The crude material was purified by HPLC and freeze-dried to give the desired compound (14 mg, 88%) as a yellow solid: 1H NMR (400 MHz, CDCl3) δ 7.90–7.85 (m, 1H), 7.82–7.76 (m, 2H), 7.56 (d, J = 8 Hz, 1H), 7.51–7.43 (m, 2H), 7.43–7.37 (m, 1H), 7.27–7.22 (m, 1H), 6.57 (d, J = 8 Hz, 1H), 6.04 (s, 1H), 4.51 (s, 2H), 1.82–1.73 (m, 1H), 1.10–1.03 (m, 2H), 0.80–0.73 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 159.2, 153.2, 150.3, 149.4, 143.6, 142.5, 134.1, 134.0, 132.0, 129.0, 127.9, 127.6, 126.4, 125.9, 125.6, 123.7, 122.8, 115.8, 112.7, 100.7, 36.2, 11.2, 8.2 (2C); IR λ 1735, 1655, 1602, 1477; HRMS (ES) calcd [M + Na] for C24H17NNaO3S 422.0827, obsd 422.0818.
5-Cyclopropyl-3-methylidene-6-(naphthalen-1-ylmethyl)-8H-furo[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,8(3H)-dione (13)
Compound 19 (0.04 mmol, 20 mg), Pd(OAc)2 (0.002 mmol, 0.4 mg), and DPPP (0.002 mmol, 0.8 mg) were dissolved in DCM/TFA 85:15 (2 mL), and the reaction was stirred at rt for 1 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4) filtered and concentrated. The crude material was purified by HPLC and freeze-dried to give the desired compound (14 mg, 88%) as a yellow solid: 1H NMR (400 MHz, CDCl3) δ 7.91–7.85 (m, 1H), 7.83–7.74 (m, 2H), 7.52–7.38 (m, 3H), 7.27–7.21 (m, 1H), 6.01 (s, 1H), 5.34 (d, J = 3.4 Hz, 1H), 5.03 (d, J = 3.4 Hz, 1H), 4.52 (s, 2H), 1.82–1.72 (m, 1H), 1.14–1.05 (m, 2H), 0.84–0.76 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 158.6, 154.7, 153.3, 149.8, 145.8, 141.1, 134.2, 133.7, 133.3, 131.9, 129.1, 128.1, 127.6, 126.5, 126.0, 125.7, 123.7, 115.4, 114.0, 96.8, 36.5, 11.8, 8.2 (2C); IR λ 1782, 1664, 1569, 1464, 1134; HRMS (ES) calcd [M + Na] for C24H17NNaO3S 422.0827, obsd 422.0815.
3-Methylidene-6-(naphthalen-1-ylmethyl)-5-[3-(trifluoromethyl)phenyl]-8H-furo[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,8(3H)-dione (21)
Compound 20 (0.07 mmol, 40 mg), Pd(OAc)2 (0.004 mmol, 0.8 mg), and DPPP (0.004 mmol, 1.6 mg) were dissolved in DCM/TFA 85:15 (4 mL), and the reaction was stirred at rt for 1 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated. The crude material was purified by HPLC and freeze-dried to give the desired compound (30 mg, 85%) as a yellow solid: 1H NMR (400 MHz, DMSO-d6) δ 7.92–7.87 (m, 1H), 7.83–7.68 (m, 6H), 7.50–7.37 (m, 3H), 7.25–7.20 (m, 1H), 5.96 (s, 1H), 5.64 (d, J = 4 Hz, 1H), 5.43 (d, J = 4 Hz, 1H), 4.15 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 157.2, 152.8, 151.6, 151.3, 144.7, 140.7, 135.6, 134.3, 133.7, 133.2, 132.3, 131.1, 130.6, 130.1 (q,J = 30 Hz, 1C), 128.4, 127.4, 127.3, 126.6 (splitted 1C), 126.1, 125.6 (2C), 125.3, 123.7 (q, J = 271 Hz, 1C), 123.5, 114.2, 113.0, 98.8, 35.7; IR λ 1798, 1680, 1583, 1479, 1332; HRMS (ES) calcd [M + Na] for C28H16F3NNaO3S 526.0701, obsd 526.0698.
7-(Naphthalen-1-ylmethyl)-6-[3-(trifluoromethyl)phenyl]-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (22)
Compound 20 (0.07 mmol, 40 mg) and Pd-NHC (0.004 mmol, 2.7 mg) were dissolved in DCM/BF3*Et2O 95:5 (4 mL), and the reaction was stirred at rt for 3 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated. The crude material was purified by HPLC and freeze-dried to give the desired compound (32 mg, 91%) as a yellow solid: 1H NMR (400 MHz, CDCl3) δ 7.85–7.78 (m, 1H), 7.76–7.70 (m, 1H), 7.67–7.61 (m, 1H), 7.60–7.49 (m, 3H), 7.57 (d, J = 4 Hz, 1H), 7.48–7.29 (m, 4H), 7.15–7.09 (m, 1H), 6.46 (d, J = 4 Hz, 1H), 6.28 (s, 1H), 4.08 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 158.9, 150.7, 150.6, 149.2, 143.8, 142.2, 136.3, 134.0, 133.5, 133.3, 132.2 (q, J = 33 Hz, 1C), 131.6, 130.4, 129.0, 128.1, 127.9, 126.9 (splitted 1C), 126.4, 126.0 (splitted 1C), 125.8, 125.4, 123.7 (q, J = 272 Hz, 1C), 123.4, 122.9, 115.9, 114.2, 100.5, 36.9; IR λ 1748, 1660, 1603, 1482, 1328; HRMS (ES) calcd [M + Na] for C28H16F3NNaO3S 526.0701, obsd 526.0705.
Typical Procedure for Synthesis of Internal Acetylenes 23
2-(Trimethylsilyl)ethyl 8-Cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2-(pent-1-ynyl)-5H-thiazolo[3,2-a]pyridine-3-carboxylate (23a)
Compound 17 (0.47 mmol, 0.26 g), CuI (0.047 mmol, 9 mg), Pd(PPh3)3Cl2 (0.023 mmol, 16 mg), and TEA (0.94 mmol, 0.13 mL) were dissolved in dry DMF (4.5 mL). 1-Pentyne (1.41 mmol, 0.14 mL) dissolved in dry DMF (0.5 mL) was added, and the reaction was heated in the microwave oven at 110 °C for 10 min. The reaction mixture was diluted with saturated NaHCO3 (aq) and extracted with EtOAc; the organic phase was dried (Na2SO4), filtered, and concentrated. The crude reaction mixture was purified by column chromatography on silica gel (heptane/EtOAc 95:5 → 85:15) to give the desired compound (0.22 g, 86%) as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.88–7.83 (m, 1H), 7.82–7.74 (m, 2H), 7.50–7.42 (m, 2H), 7.42–7.36 (m, 1H), 7.24–7.19 (m, 1H) 5.87 (s, 1H), 4.50 (s, 2H), 4.49–4.42 (m, 2H), 2.43 (t, J = 8 Hz, 2H) 1.79–1.70 (m, 1H), 1.63 (m, 2H), 1.18–1.11 (m, 2H), 1.04–0.97 (m, 2H) 1.03 (t, J = 4 Hz, 3H), 0.76–0.71 (m, 2H), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 160.4, 158.4, 154.0, 145.7, 134.2, 134.0, 133.2, 132.0, 128.9, 127.7, 127.5, 126.3, 125.8, 125.6, 123.8, 111.8 (2C), 111.1, 102.7, 68.4, 65.2, 36.3, 21.9, 21.7, 17.3, 13.5, 11.0, 8.0 (2C), −1.4 (3C); IR λ 1734, 1662, 1474; HRMS (ES) calcd [M + Na] for C32H35NNaO3SSi 564.2005, obsd 564.2016.
2-(Trimethylsilyl)ethyl 8-Cyclopropyl-2-(cyclopropylethynyl)-7-(naphthalen-1-ylmethyl)-5-oxo-5H-thiazolo[3,2-a]pyridine-3-carboxylate (23b)
Following the procedure for 23a, the title compound (82 mg, 84%) was isolated as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.89–7.83 (m 1H), 7.82–7.74 (m, 2H), 7.50–7.42 (m, 2H), 7.39 (t, J = 8 Hz, 1H), 7.21 (d, J = 8 Hz, 1H), 5.86 (s, 1H), 4.50 (s, 2H), 4.50–4.42 (m, 2H), 1.78–1.68 (m, 1H), 1.52–1.44 (m, 1H), 1.18–1.10 (m, 2H), 1.04–0.97 (m, 2H), 0.97–0.90 (m, 2H), 0.89–0.82 (m, 2H), 0.77–0.70 (m, 2H), 0.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 160.4, 158.4, 154.0, 145.7, 134.2, 134.0, 133.3, 132.0, 128.9, 127.7, 127.5, 126.3, 125.8, 125.6, 123.8, 111.9 (2C), 111.2, 105.9, 65.2, 63.2, 36.4, 17.3, 11.0, 9.4 (2C), 8.0 (2C), 0.7, −1.4 (3C); IR λ 1732, 1659, 1569, 1473; HRMS (ES) calcd [M + Na] for C32H33NNaO3SSi 562.1848, obsd 562.1849.
2-(Trimethylsilyl)ethyl 8-Cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2-(phenylethynyl)-5H-thiazolo[3,2-a]pyridine-3-carboxylate (23c)
Following the procedure for 23a, the title compound (147 mg, 95%) was isolated as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.90–7.85 (m, 1H), 7.84–7.76 (m, 2H), 7.54–7.45 (m, 4H), 7.44–7.34 (m, 4H), 7.24 (d, J = 4 Hz, 1H), 5.91 (s, 1H), 4.57–4.48 (m, 4H), 1.83–1.73 (m, 1H), 1.23–1.15 (m, 2H), 1.08–1.00 (m, 2H), 0.81–0.73 (m, 2H), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 160.2, 158.5, 154.2, 145.7, 134.1, 134.0, 133.9, 132.0, 131.9 (2C), 129.8, 128.9, 128.6 (2C), 127.8, 127.5, 126.3, 125.8, 125.6, 123.7, 121.4, 112.0 (2C), 110.4, 100.1, 76.7, 65.4, 36.4, 17.4, 11.0, 8.0 (2C), −1.5 (3C); IR λ 1733, 1665, 1570, 1473; HRMS (ES) calcd [M + Na] for C35H33NNaO3SSi 598.1848, obsd 598.1850.
2-(Trimethylsilyl)ethyl 8-Cyclopropyl-2-((4-methoxyphenyl)ethynyl)-7-(naphthalen-1-ylmethyl)-5-oxo-5H-thiazolo[3,2-a]pyridine-3-carboxylate (23d)
Following the procedure for 23a, the title compound (145 mg, 89%) was isolated as a yellow foam: 1H NMR (400 MHz, DMSO-d6) δ 7.98–7.91 (m, 1H), 7.91–7.82 (m, 2H), 7.55–7.42 (m, 5H), 7.34 (d, J = 8 Hz, 1H), 7.03–6.95 (m, 2H), 5.54 (s, 1H), 4.53 (s, 2H), 4.41–4.32 (m, 2H), 3.79 (s, 3H), 1.88–1.77 (m, 1H), 1.08–1.00 (m, 2H), 1.00–0.91 (m, 2H), 0.76–0.67 (m, 2H), −0.03 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 160.6, 159.2, 157.0, 154.6, 144.9, 134.3, 133.4, 133.3 (2C), 132.5, 131.4, 128.6, 127.5, 127.4, 126.3, 125.8, 125.6, 123.9, 114.6 (2C), 111.9, 111.7, 110.1, 109.8, 100.3, 75.1, 64.4, 55.4, 35.4, 16.8, 10.6, 7.4 (2C), −1.7 (3C); IR λ 1728, 1654, 1605, 1568, 1507, 1466; HRMS (ES) calcd [M + Na] for C36H35NNaO4SSi 628.1954, obsd 628.1945.
2-(Trimethylsilyl)ethyl 8-Cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2-((3-(trifluoromethyl)phenyl)ethynyl)-5H-thiazolo[3,2-a]pyridine-3-carboxylate (23e)
Following the procedure for 23a, the title compound (145 mg, 86%) was isolated as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.91–7.84 (m, 1H), 7.84–7.75 (m, 3H), 7.70–7.63 (m, 2H), 7.54–7.45 (m, 3H), 7.44–7.38 (m, 1H), 7.25 (d, J = 8 Hz, 1H), 5.91 (s, 1H), 4.57–4.49 (m, 4H), 1.84–1.72 (m, 1H), 1.23–1.14 (m, 2H), 1.10–1.01 (m, 2H), 0.82–0.74 (m, 2H), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 160.0, 158.5, 154.5, 145.6, 134.9, 134.7, 134.0 (2C), 132.0, 131.3 (q, J = 33 Hz, 1C), 129.2, 129.0, 128.6 (splitted, 1C), 127.8, 127.5, 126.4, 126.3 (splitted, 1C), 125.9, 125.6, 123.7, 123.5 (q, J = 271 Hz, 1C), 122.4, 112.1 (2C), 109.5, 98.1, 78.1, 65.6, 36.4, 17.4, 11.0, 8.0 (2C), −1.5 (3C); IR λ 1732, 1662, 1570, 1472, 1329; HRMS (ES) calcd [M + Na] for C36H32F3NNaO3SSi 666.1722, obsd 666.1719.
2-(Trimethylsilyl)ethyl 8-Cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2-(thiophen-3-ylethynyl)-5H-thiazolo[3,2-a]pyridine-3-carboxylate (23f)
Following the procedure for 23a, the title compound (150 mg, 95%) was isolated as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.84–7.79 (m, 1H), 7.79–7.70 (m, 2H), 7.57–7.53 (m, 1H), 7.45–7.39 (m, 2H), 7.39–7.33 (m, 1H), 7.29–7.24 (m, 1H), 7.21–7.17 (m, 1H), 7.14–7.10 (m, 1H), 5.86 (s, 1H), 4.51–4.44 (m, 2H), 4.46 (s, 2H), 1.75–1.66 (m, 1H), 1.18–1.10 (m, 2H), 1.01–0.93 (m, 2H), 0.73–0.66 (m, 2H), 0.00 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 160.1, 158.3, 154.1, 145.6, 134.0, 133.9, 133.6, 131.9, 130.9, 129.6, 128.8, 127.7, 127.4, 126.3, 126.0, 125.7, 125.5, 123.7, 120.4, 111.9 (2C), 110.3, 95.4, 76.2, 65.3, 36.2, 17.3, 10.9, 7.9 (2C), −1.5 (3C); IR λ 1724, 1654, 1560, 1466; HRMS (ES) calcd [M + Na] for C33H31NNaO3S2Si 604.1412, obsd 604.1412.
6-Cyclopropyl-7-(naphthalen-1-ylmethyl)-3-propyl-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (24a)
Compound 23a (0.18 mmol, 100 mg) was dissolved in DCM/TFA 80:20 (4 mL), and the reaction was stirred at rt for 28 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated to give the desired product (79 mg, 99%) as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.89–7.83 (m, 1H), 7.83–7.73 (m, 2H), 7.50–7.35 (m, 3 h), 7.23 (d, J = 8 Hz, 1H), 6.31 (s, 1H), 6.03 (s, 1H), 4.49 (s, 2H), 2.53 (t, J = 8 Hz, 2H), 1.79–1.65 (m, 3H), 1.09–1.00 (m, 2H), 0.97 (t, J = 4 Hz, 3H), 0.78–0.70 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 165.3, 159.2, 152.9, 150.2, 144.1, 143.8, 134.0 (2C), 131.9, 129.0, 127.8, 127.5, 126.4, 125.8, 125.6, 123.7, 120.5, 115.7, 112.6, 97.1, 36.1, 35.6, 20.3, 13.5, 11.1, 8.2 (2C); IR λ 1734, 1654, 1616, 1578, 1474, 1420; HRMS (ES) calcd [M + Na] for C27H23NNaO3S 464.1296, obsd 464.1298.
3,6-Dicyclopropyl-7-(naphthalen-1-ylmethyl)-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (24b)
Compound 23b (0.19 mmol, 100 mg) was dissolved in DCM/TFA 80:20 (4 mL), and the reaction was stirred at rt for 1 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated to give the desired product (83 mg, 99%) as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 7.89–7.82 (m, 1H), 7.82–7.73 (m, 2H), 7.50–7.35 (m, 3H), 7.23 (d, J = 8 Hz, 1H), 6.37 (s, 1H), 6.01 (s, 1H), 4.49 (s, 2H), 1.87–1.69 (m, 2H), 1.20–1.12 (m, 2H), 1.09–0.96 (m, 4H), 0.78–0.69 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 166.2, 159.2, 152.7, 149.6, 144.7, 143.6, 134.0 (2C), 131.9, 128.9, 127.8, 127.5, 126.3, 125.8, 125.6, 123.7, 119.5, 115.6, 112.6, 95.5, 36.1, 14.5, 11.1, 9.1 (2C), 8.2 (2C); IR λ 1734, 1654, 1607, 1474; HRMS (ES) calcd [M + Na] for C27H21NNaO3S 462.1140, obsd 462.1135.
6-Cyclopropyl-7-(naphthalen-1-ylmethyl)-3-phenyl-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (24c)
Compound 23c (0.25 mmol, 145 mg) was dissolved in DCM/TFA 80:20 (6 mL), and the reaction was stirred at rt for 18 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated to give the desired product (117 mg, 98%) as a yellow solid: 1H NMR (400 MHz, CDCl3) δ 7.92–7.84 (m, 3H), 7.84–7.76 (m, 2H), 7.52–7.38 (m, 6H), 7.28–7.23 (m, 1H), 6.95 (s, 1H), 6.06 (s, 1H), 4.52 (s, 2H), 1.83–1.74 (m, 1H), 1.12–1.04 (m, 2H), 0.83–0.76 (m, 2H);13C NMR (125 MHz, CDCl3) δ 159.9, 159.2, 153.1, 149.2, 144.1, 143.8, 134.2, 134.1, 132.1, 131.5, 130.5, 129.3 (2C), 129.1, 128.0, 127.6, 126.5, 126.1 (2C), 125.9, 125.7, 123.8, 121.2, 116.0, 112.8, 95.2, 36.2, 11.2, 8.3 (2C); IR λ 1748, 1663, 1605, 1482; HRMS (ES) calcd [M + Na] for C30H21NNaO3S 498.1140, obsd 498.1146.
6-Cyclopropyl-3-(4-methoxyphenyl)-7-(naphthalen-1-ylmethyl)-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (24d)
Compound 23d (0.24 mmol, 145 mg) was dissolved in DCM/TFA 80:20 (6 mL), and the reaction was stirred at rt for 1 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated to give the desired product (120 mg, 99%) as a yellow solid: 1H NMR (400 MHz, CD2Cl2 + 1% TFA) δ 7.99–7.88 (m, 4H), 7.71–7.66 (m, 1H), 7.58 (s, 1H), 7.56–7.47 (m, 3H), 7.38–7.33 (m, 1H), 7.11–7.05 (m, 2H), 6.53 (s, 1H), 4.79 (s, 2H), 3.91 (s, 3H), 2.23–2.11 (m, 1H), 1.45–1.36 (m, 2H), 1.04–0.96 (m, 1H); 13C NMR (100 MHz, CD2Cl2 + 1% TFA) δ 164.8, 162.9, 161.0, 157.8, 157.0, 152.2, 148.6, 134.6, 132.3, 131.9, 129.5 (3C), 129.2, 128.8, 127.3, 126.6, 126.2, 125.5, 123.6, 120.5, 119.2, 115.7 (2C), 112.8, 97.3, 56.2, 37.0, 11.9, 9.0 (2C); IR λ 1738, 1657, 1606, 1571, 1505, 1472; HRMS (ES) calcd [M + Na] for C31H23NNaO4S 528.1245, obsd 528.1250.
6-Cyclopropyl-7-(naphthalen-1-ylmethyl)-3-[3-(trifluoromethyl)phenyl]-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (24e)
Compound 23e (0.23 mmol, 145 mg) was dissolved in DCM/TFA 80:20 (6 mL), and the reaction was stirred at rt for 120 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated to give the desired product (110 mg, 88%) as a yellow foam: 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 8.02–7.95 (m, 1H), 7.86–7.79 (m, 1H), 7.79–7.70 (m, 2H), 7.67 (d, J = 8 Hz 1H), 7.57–7.49 (m, 1H), 7.46–7.38 (m, 2H), 7.38–7.32 (m, 1H), 7.21 (d, J = 8 Hz, 1H), 7.10 (s, 1H), 6.00 (s, 1H), 4.47 (s, 2H), 1.79–1.68 (m, 1H), 1.08–0.98 (m, 2H), 0.76–0.68 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 159.0, 157.5, 153.3, 148.7, 144.0, 143.8, 133.9, 133.8, 131.8, 131.6 (q, J = 32 Hz, 1C), 131.0, 129.8, 129.0, 128.9, 127.8, 127.6 (splitted, 1C), 127.5, 126.3, 125.8, 125.5, 123.6 (q, J = 270 Hz, 1C), 123.6, 122.6 (splitted, 1C), 121.4, 115.4, 112.8, 96.6, 36.0, 11.0, 8.1 (2C); IR λ 1742, 1660, 1474, 1323; HRMS (ES) calcd [M + Na] for C31H20F3NNaO3S 566.1014, obsd 566.1023.
6-Cyclopropyl-7-(naphthalen-1-ylmethyl)-3-(thiophen-3-yl)-1H,9H-pyrano[3′,4′:4,5][1,3]thiazolo[3,2-a]pyridine-1,9-dione (24f)
Compound 23f (0.18 mmol, 105 mg) was dissolved in DCM/TFA 80:20 (4 mL), and the reaction was stirred at rt for 20 h. The reaction mixture was diluted with DCM and washed with saturated NaHCO3 (aq); the organic phase was dried (Na2SO4), filtered, and concentrated to give the desired product (86 mg, 99%) as a yellow solid: 1H NMR (400 MHz, CDCl3) δ 7.88–7.82 (m, 1H), 7.82–7.76 (m, 1H), 7.76–7.67 (m, 2H), 7.43–7.34 (m, 2H), 7.34–7.24 (m, 3H), 7.20–7.13 (m, 1H), 6.68 (s, 1H), 5.95 (s, 1H), 4.41 (s, 2H), 1.72–1.61 (m, 1H), 1.01–0.92 (m, 2H), 0.71–0.63 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 159.1, 155.9, 153.0, 149.1, 144.4, 143.7, 134.1, 134.0, 132.7, 132.0, 129.0, 127.9, 127.6 (splitted, 2C), 127.0, 126.4, 125.9, 125.6, 124.5, 123.8, 120.5, 115.7, 112.7, 95.0, 36.1, 11.1, 8.2 (2C); IR λ 1747, 1662, 1606, 1475; HRMS (ES) calcd [M + Na] for C28H19NNaO3S2 504.0704, obsd 504.0698.
Acknowledgments
We are thankful for financial support from the Swedish Research Council (621-2010-4730), the Knut and Alice Wallenberg Foundation, and the Kempe Foundation (SJCKMS).
Supporting Information Available
1H NMR and 13C NMR spectra of all new compounds. This material is available free of charge via the Internet http://pubs.acs.org.
Supplementary Material
References
- a Misra R.; Pandey R. C.; Silverton J. V. J. Am. Chem. Soc. 1982, 104, 4478–4479. [Google Scholar]; b Hsiang Y. H.; Hertzberg R.; Hecht S.; Liu L. F. J. Biol. Chem. 1985, 260, 14873–14878. [PubMed] [Google Scholar]; c Liu J. S.; Zhu Y. L.; Yu C. M.; Zhou Y. Z.; Han Y. Y.; Wu F. W.; Qi B. F. Can. J. Chem. 1986, 64, 837–839. [Google Scholar]
- a Reiner J. E.; Lim-Wilby M. S.; Brunck T. K.; Ha-Uong T.; Goldman E. A.; Abelman M. A.; Nutt R. F.; Semple J. E.; Tamura S. Y. Bioorg. Med. Chem. Lett. 1999, 9, 895–900. [DOI] [PubMed] [Google Scholar]; b Svensson A.; Larsson A.; Emtenäs H.; Hedenström M.; Fex T.; Hultgren S. J.; Pinkner J. S.; Almqvist F.; Kihlberg J. ChemBioChem 2001, 12, 915–918. [DOI] [PubMed] [Google Scholar]; c Zhang X. J.; Schmitt A. C.; Decicco C. P. Tetrahedron Lett. 2002, 43, 9663–9666. [Google Scholar]; d Dragovich P. S.; Prins T. J.; Zhou R.; Johnson T. O.; Brown E. L.; Maldonado F. C.; Fuhrman S. A.; Zalman L. S.; Patick A. K.; Matthews D. A.; Hou X.; Meador J. W.; Ferre R. A.; Worland S. T. Bioorg. Med. Chem. Lett. 2002, 12, 733–738. [DOI] [PubMed] [Google Scholar]; e Åberg V.; Norman F.; Chorell E.; Westermark A.; Olofsson A.; Sauer-Eriksson A. E.; Almqvist F. Org. Biomol. Chem. 2005, 3, 2817–2823. [DOI] [PubMed] [Google Scholar]; f Åberg V.; Sellstedt M.; Hedenström M.; Pinkner J. S.; Hultgren S. J.; Almqvist F. Bioorg. Med. Chem. 2006, 14, 7563–7581. [DOI] [PubMed] [Google Scholar]
- Chorell E.; Das P.; Almqvist F. J. Org. Chem. 2007, 72, 4917–4924. [DOI] [PubMed] [Google Scholar]
- Pemberton N.; Pinkner J. S.; Jones J. M.; Jakobsson L.; Hultgren S. J.; Almqvist F. Tetrahedron Lett. 2007, 48, 4543–4546. [Google Scholar]
- Sellstedt M.; Almqvist F. Org. Lett. 2009, 11, 5470–5472. [DOI] [PubMed] [Google Scholar]
- Chorell E.; Pinkner J. S.; Phan G.; Edvinsson S.; Buelens F.; Remaut H.; Waksman G.; Hultgren S. J.; Almqvist F. J. Med. Chem. 2010, 53, 5690–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pinkner J. S.; Remaut H.; Buelens F.; Miller E.; Åberg V.; Pemberton N.; Hedenström M.; Larsson A.; Seed P.; Waksman G.; Hultgren S. J.; Almqvist F. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 17897–17902. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Åberg V.; Almqvist F. Org. Biomol. Chem. 2007, 5, 1827–1834. [DOI] [PubMed] [Google Scholar]
- Cegelski L.; Pinkner J. S.; Hammer N. D.; Cusumano C. K.; Hung C. S.; Chorell E.; Åberg V.; Walker J. N.; Seed P. C.; Almqvist F.; Chapman M. R.; Hultgren S. J. Nat. Chem. Biol. 2009, 5, 913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt W. R.; Parry D. M.; Perry B. G.; Groom C. R. J. Med. Chem. 2009, 52, 2952–2963. [DOI] [PubMed] [Google Scholar]
- Sellstedt M.; Almqvist F. Org. Lett. 2011, 13, 5278–5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellstedt M.; Almqvist F. Org. Lett. 2008, 10, 4005–4007. [DOI] [PubMed] [Google Scholar]
- Sunazuka T.; O̅mura S. Chem. Rev. 2005, 105, 4559–4580. [DOI] [PubMed] [Google Scholar]
- Tatsuta K.; Suzuki Y.; Furuyama A.; Ikegami H. Tetrahedron Lett. 2006, 47, 3595–3598. [Google Scholar]
- Yao T.; Larock R. C. J. Org. Chem. 2003, 68, 5936–5942. [DOI] [PubMed] [Google Scholar]
- a Uchiyama M.; Ozawa H.; Takuma K.; Matsumoto Y.; Yonehara M.; Hiroya K.; Sakamoto T. Org. Lett. 2006, 8, 5517–5520. [DOI] [PubMed] [Google Scholar]; b Hellal M.; Bourguignon J.-J.; Bihel F. J.-J. Tetrahedron Lett. 2008, 49, 62–65. [Google Scholar]
- Emtenäs H.; Taflin C.; Almqvist F. Mol. Diversity 2003, 7, 165–169. [DOI] [PubMed] [Google Scholar]
- Serrano-Wu M. H.; Regueiro-Ren A.; St. Laurent D. R.; Carroll T. M.; Balasubramanian B. N. Tetrahedron Lett. 2001, 42, 8593–8595. [Google Scholar]
- Jia C.; Kitamura T.; Fujiwara Y. Acc. Chem. Res. 2001, 34, 633–639. [DOI] [PubMed] [Google Scholar]
- De Meijere A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.