

Abstract
Potentiation of neuronal nicotinic acetylcholine receptors by exogenous ligands is a promising strategy for treatment of neurological disorders including Alzheimer's disease and schizophrenia. To gain insight into molecular mechanisms underlying potentiation, we examined ACh-induced single-channel currents through the human neuronal α7 acetylcholine receptor in the presence of the α7-specific potentiator PNU-120596 (PNU). Compared to the unusually brief single-channel opening episodes elicited by agonist alone, channel opening episodes in the presence of agonist and PNU are dramatically prolonged. Dwell time analysis reveals that PNU introduces two novel components into open time histograms, indicating at least two degrees of PNU-induced potentiation. Openings of the longest potentiated class coalesce into clusters whose frequency and duration change over a narrow range of PNU concentration. At PNU concentrations approaching saturation, these clusters last up to several minutes, prolonging the submillisecond α7 opening episodes by several orders of magnitude. Mutations known to reduce PNU potentiation at the whole-cell level still give rise to multisecond-long single-channel clusters. However mutation of five residues lining a cavity within each subunit's transmembrane domain abolishes PNU potentiation, defining minimal structural determinants of PNU potentiation.
Introduction
Neuronal α7 nicotinic acetylcholine receptors belong to the Cys-loop superfamily of pentameric ligand-gated ion channels and mediate synaptic transmission in many brain regions. High levels of α7 expression in both hippocampus and cerebral cortex suggest contributions to higher brain functions such as memory and learning (Dani and Bertrand, 2007). Furthermore, impaired α7 signaling has been implicated in cognitive deficits associated with Alzheimer's disease and schizophrenia (Freedman et al., 1995, 1997; Qi et al., 2007). As a result, α7 is a potential therapeutic target for offsetting the effects of cognitive decline (Leiser et al., 2009; Taly et al., 2009; Hajós and Rogers, 2010; Haydar and Dunlop, 2010; Williams et al., 2011).
Because coordination of synaptic events is crucial in neuronal communication, an ideal therapeutic approach would augment the strength of an existing synapse without disrupting temporal control provided by an endogenous neurotransmitter. At the single ion channel level, this would require a drug that does not activate the channel by itself but instead modifies the agonist-induced responses (Maelicke and Albuquerque, 1996; Hogg et al., 2005). Compounds that potentiate agonist responses have been called positive allosteric modulators (PAMs), while those that attenuate responses have been called negative allosteric modulators (Bertrand and Gopalakrishnan, 2007).
Several α7 PAMs, ranging in potency and specificity, have been identified (Faghih et al., 2008; Lightfoot et al., 2008; Arias, 2010; Williams et al., 2011). Of these, PNU-120596 (PNU) has garnered significant attention, largely because in vivo studies demonstrate that systemic administration of PNU alleviates cognitive impairment in rats (Hurst et al., 2005). Electrophysiological measurements of macroscopic currents show that whereas PNU alone fails to trigger α7 current, application of PNU with agonist dramatically increases the magnitude and duration of agonist-induced currents (Hurst et al., 2005). Furthermore, PNU is specific for α7 in that it does not potentiate other members of the Cys-loop receptor superfamily. Studies exploiting this specificity used chimeric receptors to identify regions within the transmembrane (TM) domain of α7 that are important for PNU potentiation (Bertrand et al., 2008; Young et al., 2008). Those observations, combined with docking simulations, suggest PNU binds to an intrasubunit cavity within the TM domain of α7 (Young et al., 2008).
While the effects of PNU on α7 have been studied at the macroscopic/whole-cell level, little is known about the effects at the microscopic/single-channel level. Initial studies showed that PNU did not increase single-channel conductance (Hurst et al., 2005). Instead, agonists elicited longer channel openings when applied in the presence of PNU (Hurst et al., 2005; Gusev and Uteshev, 2010). To gain more definitive insight, we here examine the effects of PNU on single-channel currents through human α7 acetylcholine receptors expressed in HEK 293 cells. We find that mutations known to reduce PNU potentiation of whole-cell currents still potentiate single-channel currents. By exploiting the sensitivity of single-channel recording, we identify a quintuple mutant for which PNU potentiation is essentially abolished. This mutant includes novel mutations that together with previously characterized mutations define structural determinants of PNU potentiation.
Materials and Methods
Materials.
1-(5-Chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea, known as PNU-120596, was from Tocris Biosciences. 125I-α-Bungarotoxin (α-BTX) was from PerkinElmer. All other chemicals, including acetylcholine chloride (ACh), were from Sigma-Aldrich.
Site-directed mutagenesis and human α7 expression.
Mutant α7 cDNAs were constructed using the QuikChange Site-Directed Mutagenesis kit (Stratagene) and then confirmed by sequencing the entire coding region. BOSC 23 cells, modified HEK 293 cells (Pear et al., 1993), were transfected by calcium phosphate precipitation as described previously (Sine, 1993; Bouzat et al., 1994; Sine et al., 1994). Furthermore, expression of human α7 (GenBank accession number X70297) in mammalian cells lines requires cotransfection with the intracellular chaperone Ric-3 (Williams et al., 2005). Ric-3 and α7 DNA were cotransfected at a ratio of 12:1 (w/w), with the total α7 DNA ranging from ∼0.02 to 1 μg for a 35 mm culture dish. Transfections in DMEM with 10% fetal bovine serum were carried out between 3 and 12 h and terminated by exchanging the medium. Because some conditions benefitted from a large number of channels in the patch (i.e., no PNU), and others from relatively few channels (i.e., ≥1 μm PNU), the expression level of α7 was manipulated by adjusting the transfection time and DNA concentration during transfection, as well as time after transfection. Cells were used for either whole-cell or single-channel recordings 0.5–7 d posttransfection. To facilitate identification of transfected cells, a separate plasmid encoding green fluorescent protein was included in all transfections.
Patch-clamp recordings.
Macroscopic currents were recorded in the whole-cell configuration at a holding potential of −50 mV, filtered at 5 kHz, digitized at 20 kHz, and analyzed using the IgorPro software (WaveMetrics). The pipette solution contained 134 mm KCl, 5 mm EGTA, 1 mm MgCl2, and 10 mm HEPES, pH 7.3. The extracellular solution (ECS) contained the following (in mm): 150 NaCl, 1.8 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4. The perfusion system consisted of solution reservoirs, manual switching valves, a four-channel valve controller (ALA Scientific Instruments), a solenoid-driven pinch valve, and two tubes (inner diameter, 0.3 mm), orientated at 90°, inserted into the culture dish (Liu and Dilger, 1991; Bouzat et al., 2008). For control responses, one tube contained ECS without agonist and the other contained ECS with ACh. The solution exchange time was estimated by applying ECS diluted 1:2 with water and varied between 0.1 and 1 ms (Bouzat et al., 2008; Corradi et al., 2009). Each current trace presented is the ensemble average of 3–5 individual traces obtained from the same cell that were aligned at the point where the current reached 50% of the peak current. Macroscopic responses were recorded using the following protocol of PNU application. First, the cell was exposed to ECS, followed by a 1.5 s pulse of ECS containing 100 μm ACh (first control). The same cell was then preincubated for 1 min with ECS containing 3 μm PNU before applying a 1.5 s pulse of ECS containing both 100 μm ACh and 3 μm PNU. The stability of the seal and the reversibility of the PNU effect was confirmed by washing the cell for 1 min with ECS and then applying another 1.5 s pulse of ECS containing 100 μm ACh (second control).
Single-channel recordings were obtained in the cell-attached patch configuration (Hamill et al., 1981) at a membrane potential of −70 mV and at 20°C, essentially as described previously (Rayes et al., 2005; Bouzat et al., 2008). The bath solution (BS) contained the following (in mm):142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, and 10 HEPES, pH 7.4. All pipette solutions were made directly from BS by adding 1000× concentrated stock solutions of ACh in BS and PNU in DMSO. Pipette solutions not containing PNU had the equivalent volume of DMSO added to ensure that all pipette solutions contained a final concentration of 0.1% (v/v) DMSO. Pipette solutions were aliquoted and stored at −80°C until immediately before each experiment. Patch pipettes were pulled from glass capillary tubes (No.7052, Garner Glass) and coated with Sylgard (Dow Corning). Single-channel currents were recorded using an Axopatch 200B patch-clamp amplifier (Molecular Devices) and digitized at 50 kHz with an InstruTECH ITC-16 interface (Heka Elektronik). Open and closed times were determined using the program TAC 4.2.0 (Bruxton), which digitally filters the data (Gaussian response, final effective bandwidth, 10 kHz), interpolates the digitized points using a cubic spline function and detects events using the half-amplitude threshold criterion as described (Colquhoun and Sigworth, 1983). Dwell time histograms were plotted using a logarithmic abscissa and a square root ordinate (Sigworth and Sine, 1987) with a uniformly imposed dead time of 18 μs and fitted by the sum of exponentials by maximum likelihood using the program TACFit 4.2.0 (Bruxton Corporation). Clusters of openings were identified as a series of closely spaced openings preceded and followed by closed intervals longer than a specified critical duration (τcrit). This duration was taken as the point of intersection between consecutive components in the closed time histogram and ranged between 1 and 10 ms. Thus, to determine the dwell time of each cluster, the total time of a series of opening events was combined with that of the intervening closing events briefer than τcrit.
For the PNU adding experiments (see Figs. 1b,c, 6a), cells were bathed in 0.7 ml of BS, and a GΩ seal was formed in the cell-attached patch configuration. The patch pipette contained 100 μm ACh in BS. After 10 min of recording, 0.3 ml of 10 μm PNU in BS was added dropwise to the external solution surrounding the patch ([PNU]final = 3 μm). The single-channel traces before addition of PNU are representative of steady-state activity, whereas traces after the addition of PNU are taken immediately following PNU addition (i.e., not at steady state).
Figure 1.

Macroscopic versus microscopic potentiation of wild-type (wt) α7 by PNU. a, Whole-cell currents elicited by separate applications of 100 μm ACh to a single HEK 293 cell expressing wild-type human α7 before (left) and after (right) a 60 s incubation with 3 μm PNU. b, Addition of 3 μm PNU to the extracellular solution surrounding a cell-attached patch of wild-type human α7 results in a marked increase in current in the continued presence of 100 μm ACh in the patch pipette. c, Expanded view of the boxed region in b showing that the rise in current results from a dramatic PNU-induced prolongation of channel openings. Note that in b the trace was filtered at 0.1 kHz, while in c the same trace was filtered at 5 kHz to better resolve brief openings.
Figure 6.

Characterization of the quintuple TSLMF mutant. a, Addition of 3 μm PNU to the extracellular solution surrounding a cell-attached patch containing the TSLMF mutant, activated by 100 μm ACh, shows no PNU-induced change in single-channel activity. b, Whole-cell currents elicited by separate applications of 100 μm ACh for a single HEK 293 cell expressing the TSLMF mutant before (left) and after (right) a 60 s incubation with 3 μm PNU. c, PNU dose–response relationships for wild-type (black circles) and TSLMF mutant α7 (white triangles) determined from single-channel recordings (n = 2 or 3 recordings for each data point; see Materials and Methods). d, Ability of PNU to allosterically modulate agonist affinity. ACh binding was measured by its competition against the initial rate of α-bungarotoxin binding for both wild-type (circles) and TSLMF mutant (triangles) receptors in the presence (black circles, white upward triangles) and absence (gray circles and downward triangles) of 3 μm PNU (n = 3 for each data point). Error bars in c and d represent ± standard deviation of the means.
PNU dose–response curves (Fig. 6c) were generated from dwell time analysis of single-channel recordings. Cell-attached recordings of wild-type and quintuple (TSLMF) mutant α7 receptors with 100 μm ACh and 0, 0.1, 0.3, 1.0, 3.0, or 10.0 μm PNU in the patch pipette were collected and analyzed. Comparison of open time histograms, in the presence and absence of PNU, revealed additional components attributable to PNU potentiation. For wild-type α7, two additional components of longer duration were evident in open time histograms (Fig. 2). For the quintuple mutant in which open time histograms contained three components in the absence of PNU, a single longer/potentiated component was only apparent in recordings with 3 μm or greater PNU (see Fig. 5). For both wild-type and mutant receptors, the fraction (between 0 and 1) of potentiated openings was plotted as a function of PNU concentration (Fig. 6c). Each data point is the mean potentiation from two to three individual recordings, where the error bars represent ± standard deviation of the means. Fits of the Hill equation to the data were obtained using GraphPad Prism 5.0 software.
Figure 2.
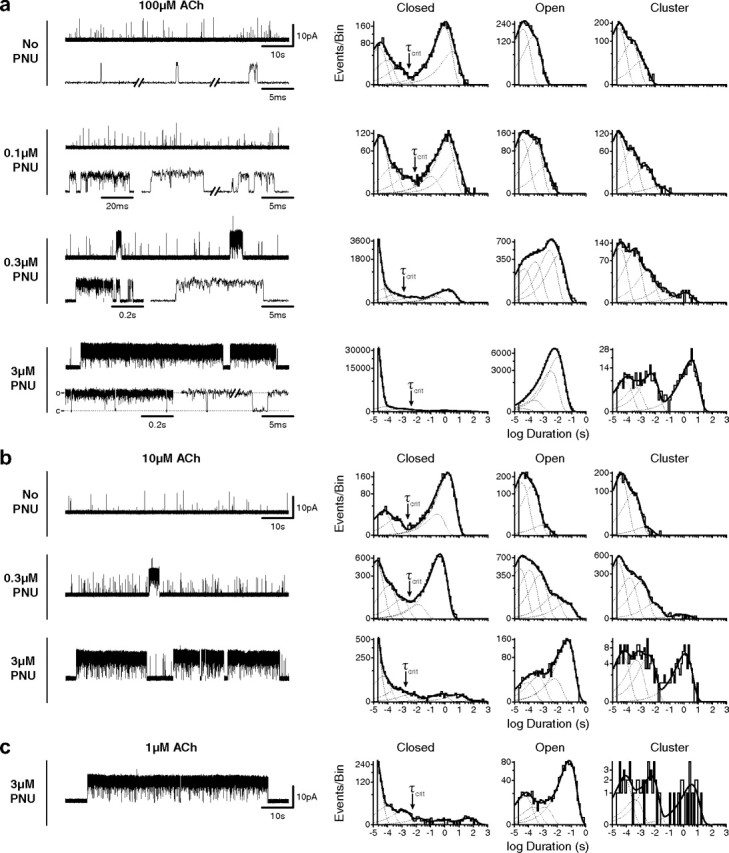
Single-channel traces and dwell time histograms for wild-type α7 in the presence of increasing concentrations of PNU in the pipette solution. Recordings were made in the cell-attached patch configuration at an applied potential of −70 mV. Currents activated by 100 μm (a), 10 μm (b), and 1 μm (c) ACh are all shown at a bandwidth of 5 kHz, with channel openings as upward deflections. Corresponding histograms of dwell times are shown on logarithmic time axes with overlaid fits to the sum of exponentials (solid line, fits; dotted lines, individual components). Cluster duration histograms (far right) are open time histograms generated by summing open times with flanking closings briefer than a critical time (τcrit, indicated by arrows) determined from corresponding closed time histograms (see Materials and Methods).
Figure 5.
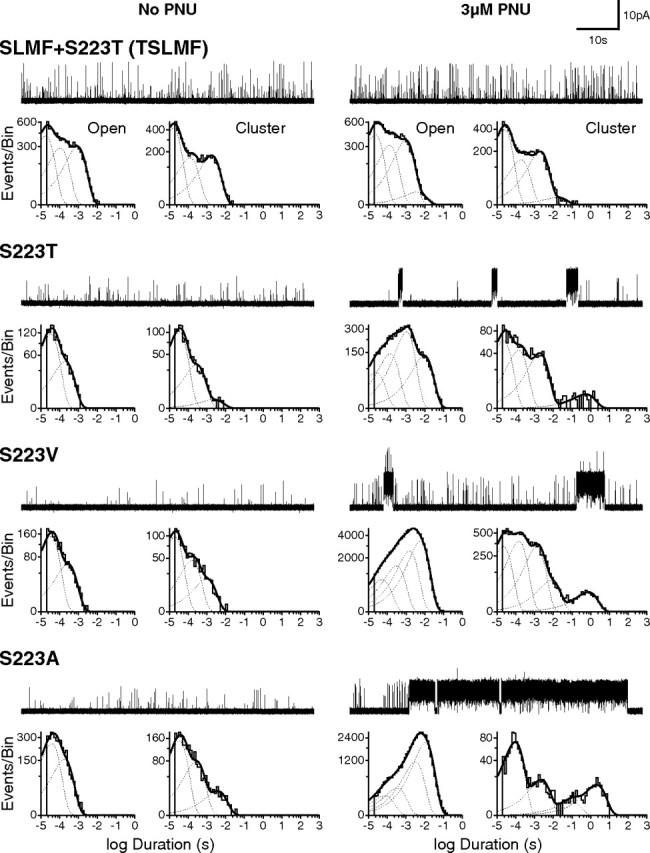
Quintuple mutant eliminates potentiation by 3 μm PNU. Single-channel current traces and dwell time histograms from quintuple (SLMF+S223T) and single (S223T; S223V; S223A) mutant α7 receptors in the absence (left) and presence (right) of 3 μm PNU in the pipette solution. Openings were elicited by 100 μm ACh, and all recordings were made in the cell-attached patch configuration with an applied voltage of −70 mV. Openings are upward deflections, and all traces are shown at a bandwidth of 5 kHz.
Agonist binding.
Agonist binding was determined by competition of specified concentrations of ACh against the initial rate of α-BTX binding as described previously (Sine et al., 1995). Briefly, transfected cells were harvested by gentle agitation in phosphate-buffered saline, centrifuged at 1000 × g for 1 min, and resuspended in potassium Ringer's solution (140 mm KCl, 5.4 mm NaCl, 1.8 mm CaCl2, 1.7 mm MgCl2, 25 mm HEPES, and 30 mg/L bovine serum albumin, adjusted to pH 7.4) with or without 3 μm PNU. For experiments with PNU, the concentration of PNU was maintained at 3 μm throughout. The total number of binding sites was determined by incubating cells with 25 nm α-BTX for 1 h and subtracting a blank determined in the presence of 1 mm d-tubocurarine. After computing fractional occupancy from the initial rates of toxin binding (Sine and Taylor, 1979), the following equation was fitted to the data: 1 − Fractional Occupancy = 1 − [L]nH/([L]nH + KdnH), where [L] is ACh concentration, Kd is the apparent dissociation constant, and nH is the Hill coefficient.
Results
PNU potentiation at the macroscopic and single-channel levels
Application of 100 μm ACh to human α7 ACh receptors (AChRs) expressed in HEK 293 cells elicits small and rapidly decaying macroscopic currents (Fig. 1a). Both the low amplitude and rapid decay have been explained by termination of the α7 openings by unusually rapid desensitization (Bouzat et al., 2008). When the same cell is preincubated with 3 μm PNU, ACh elicits a much greater peak current that decays only partially over the time of agonist application. These results, obtained with HEK 293 cells, agree well with those previously reported for α7 AChRs expressed in Xenopus oocytes and isolated neurons (Hurst et al., 2005; Grønlien et al., 2007; Ng et al., 2007; Bertrand et al., 2008; Young et al., 2008; López-Hernández et al., 2009; Papke et al., 2009; Sattelle et al., 2009).
While previous studies suggest PNU alters mean channel open time (Hurst et al., 2005), the macroscopic effects of PNU could result from an increased frequency of channel-opening events. To distinguish between changes in opening frequency and changes in open time, we examined ACh-evoked single-channel currents recorded from the same patch before and after adding PNU. Application of 100 μm ACh in the cell-attached patch configuration triggers brief and infrequent single-channel openings (Fig. 1c). Subsequent application of PNU (3 μm) to the extracellular solution elicits, within minutes, an increase in brief openings succeeded by multisecond-long clusters of single-channel openings (Fig. 1b,c). This simple graphic depiction of PNU potentiation at the microscopic/single-channel level demonstrates that increased channel open duration underlies the macroscopic manifestations of PNU.
To quantify the action of PNU at the single-channel level, we examined dwell time histograms generated from recordings obtained in the presence of ACh and increasing concentrations of PNU (Fig. 2). In the presence of ACh alone, open time histograms are described by the sum of two exponential components, the briefest of which approaches the resolution limit of the patch-clamp apparatus (Fig. 2a). As a result, openings briefer than the dead time are not fully resolved and thus appear with submaximal current amplitudes. Inspection of the current traces reveals an apparent third component of openings, corresponding to several openings separated by brief closings. These infrequent opening episodes are unmasked by constructing cluster duration histograms in which successive openings are combined with closings briefer than a defined critical time, τcrit, to form clusters (Materials and Methods) and yielded three exponential components of clusters.
In the presence of 100 μm ACh and 0.1 μm PNU, open duration and cluster duration histograms shift toward longer durations and are described by the sum of three and four components, respectively (Fig. 2a), demonstrating that even at a concentration well below its EC50 (see Fig. 6c), PNU prolongs α7 openings. At 0.3 μm PNU (∼EC50), more profound potentiation becomes obvious from the appearance of multisecond long clusters. Briefer 10–100 ms and subsecond clusters are also apparent, as are brief unpotentiated events. The appearance of longer potentiated openings further expands the open duration histogram, which is described by the sum of four exponential components. The component with longest mean duration originates from openings that occur exclusively within clusters, giving rise to a fifth component in the cluster duration histogram. Raising the PNU concentration to 3 μm, a concentration that maximally potentiates (see Fig. 6c), increases both the frequency and duration of clusters, which coalesce into superclusters lasting from ten to several hundred seconds. The appearance of superclusters coincides with an increase in the relative area of the corresponding openings, eclipsing that of unpotentiated events, as well as an increase in the relative area of the longest component of clusters.
At the level of macroscopic currents, PAMs have been reported to increase agonist potency (Hogg et al., 2005; Hurst et al., 2005; Bertrand and Gopalakrishnan, 2007). To determine whether PNU increases agonist potency at the single-channel level, we examined the relationship between agonist concentration and PNU potentiation. Comparing open time histograms obtained at either 10 or 100 μm ACh in the presence of PNU (0.3 or 3 μm PNU) reveals longer openings at the lower agonist concentration, suggesting PNU potentiation depends on agonist concentration (Fig. 2a,b). However, longer openings at 10 μm ACh likely arise from reduced open channel block by ACh, accounting for the increased open time and the decreased relative area of brief closings. Cluster duration histograms are insensitive to these blocking events because by design, closings briefer than a defined τcrit are combined with the flanking openings. Accordingly, cluster duration histograms obtained at the different ACh concentrations show essentially no difference in the mean durations or relative areas of potentiated and unpotentiated events, which was also apparent from inspection of the single-channel traces. In fact, 1 μm ACh, a concentration that triggers only rare openings in the absence or presence of PNU, still elicits multisecond-long clusters in the presence of 3 μm PNU (Fig. 2c). Despite a low number of total events at 1 μm ACh, cluster duration histograms show that the relative proportion of potentiated and unpotentiated events is essentially the same as that at 10 and 100 μm ACh. Thus, agonist concentration does not affect the relative proportion of potentiated and unpotentiated events, and at the single-channel level PNU potentiation does not depend on ACh concentration.
Structural determinants of PNU potentiation
Previous studies identified several residues within the transmembrane domain of rat α7 crucial for PNU potentiation (Young et al., 2008). Two mutations in particular, A225D and M253L (A226 and M254 in human α7), reduced PNU potentiation of macroscopic currents by ∼90%. We therefore examined how these mutations affected PNU potentiation at the microscopic/single-channel level. When installed in human α7, the A226D mutation abolished surface expression in HEK 293 cells, but the mutant A226S expressed well, as did M254L. To our surprise, in the presence of 3 μm PNU, receptors with either of these substitutions still give rise to unmistakable multisecond-long clusters (Fig. 3), as do receptors containing both mutations (A226S+M254L; SL). Both open duration and cluster duration histograms show that compared to wild-type α7, the ratio of potentiated to unpotentiated events is lower in each of the three mutants. Thus A226S and M254L do not abolish clusters, but instead reduce their frequency of occurrence relative to unpotentiated events.
Figure 3.

Mutations that markedly reduce PNU potentiation at the whole-cell level exhibit potentiation at the single-channel level. Current traces with associated open time and cluster duration histograms from mutant α7 receptors (A226S; M254L; A226S+M254L double mutant), both in the absence (left) and presence (right) of 3 μm PNU in the pipette solution, are shown. Openings were elicited by 100 μm ACh; all recordings were made in the cell-attached patch configuration with an applied voltage of −70 mV. Single-channel traces are shown at a bandwidth of 5 kHz, and openings are upward deflections.
Given that A226 and M254 are not the sole determinants of PNU potentiation, we hypothesized that nearby residues also contribute to the putative PNU site. Thus, we introduced additional substitutions into the double mutant background and examined potentiation by PNU. Of the many triple mutants initially screened (Table 1), two further decreased potentiation (SL+I281M and SL+V288F), although both I281M and V288F by themselves only marginally reduced potentiation (Fig. 4). However, in the presence of 100 μm ACh and 3 μm PNU, both triple mutants still exhibited multisecond-long clusters. Thus we combined I281M and V288F into the double mutant to form the quadruple mutant (SLMF). The SLMF mutant reduced potentiation to the point where clusters were even briefer and less frequent (Fig. 4). Based on open duration and cluster duration histograms, 3 μm PNU potentiated the SLMF mutant comparable to 0.3 μm PNU applied to wild-type α7, indicating roughly a 10-fold decrease in PNU potency.
Table 1.
List of mutations
| Background | Mutated residue | Substitutions | Abbreviation |
|---|---|---|---|
| Single mutants | |||
| wt(α7) | C219 | A, D, T | |
| wt(α7) | S223 | A, T, V | |
| wt(α7) | A226 | D, K, L, N, S | |
| wt(α7) | M254 | L | |
| wt(α7) | I281 | D, F, M | |
| wt(α7) | S285 | A, D, I | |
| wt(α7) | V288 | F, I, S | |
| wt(α7) | G463 | F, V, W | |
| Double mutant | |||
| A226S+M254L | SL | ||
| Triple mutants | |||
| A226S+M254L | C219 | A, D, T | |
| A226S+M254L | I281 | D, F, M | SLM |
| A226S+M254L | S285 | A, D, I | |
| A226S+M254L | V288 | F, I, S | SLF |
| A226S+M254L | G463 | F, V, W | |
| Quadruple mutants | |||
| A226S+M254L+V288F | I281 | F, M | SLMF |
| Quintuple mutants | |||
| A226S+M254L+I281M+V288F | S223 | A, M, T, V | TSLMF |
| A226S+M254L+I281M+V288F | L250 | I, M, Y | |
| A226S+M254L+I281M+V288F | V257 | I, M | |
| A226S+M254L+I281M+V288F | F456 | A | |
| A226S+M254L+I281M+V288F | I459 | F, M | |
| A226S+M254L+I281M+V288F | C460 | Y |
Bold indicates individual mutations combined to make the quintuple TSLMF mutant. The abbreviation wt is wild type.
Figure 4.
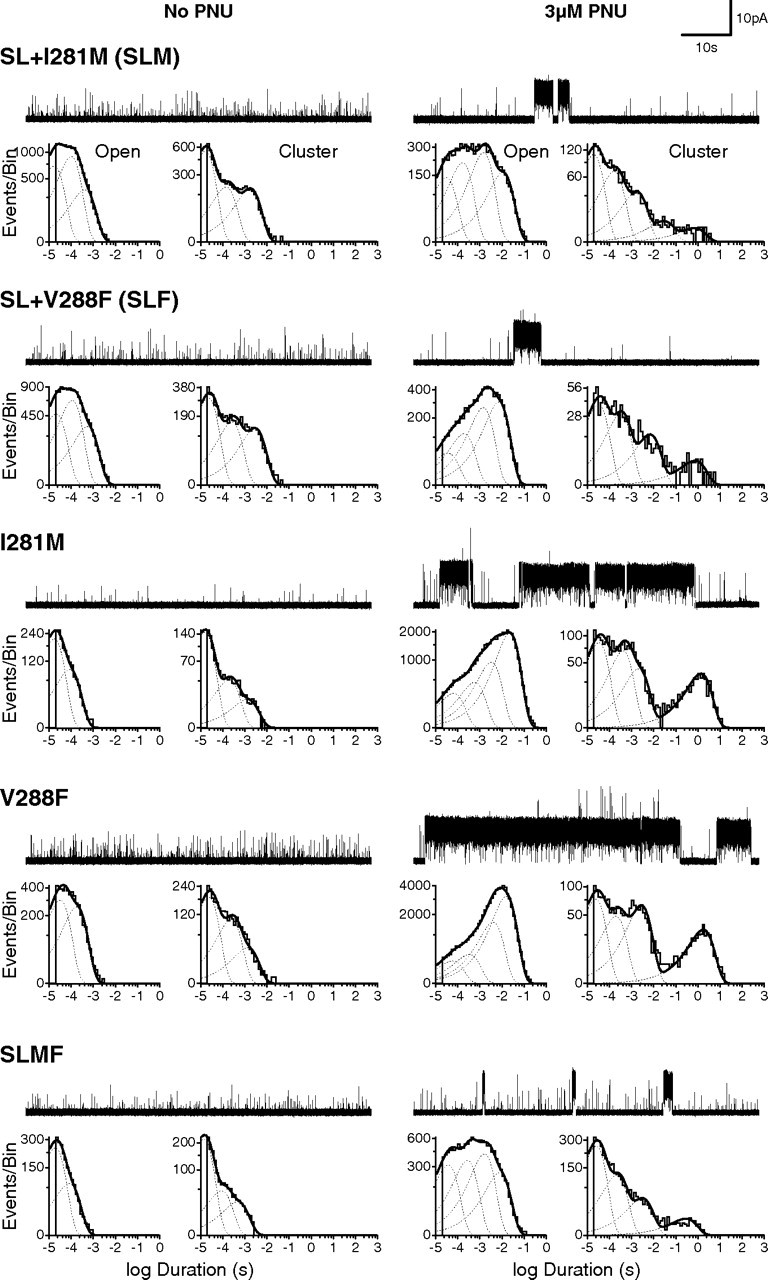
Combining individual mutations progressively reduces PNU potentiation. Single-channel traces and dwell time histograms from triple (SL+I281M; SL+V288F), single (I281M; V288F) and quadruple (SL+I281M+V288F) mutant α7 receptors in the absence (left) and presence (right) of 3 μm PNU in the pipette solution. All recordings were made in the cell-attached configuration at an applied voltage of −70 mV, with 100 μm ACh in the patch pipette. Single-channel openings are upward deflections, and all traces are shown at a bandwidth of 5 kHz.
Buoyed by the further reduction in cluster duration and frequency of the quadruple mutant, we screened additional mutations on the SLMF mutant background (Table 1). Combining the mutation S223T to form the quintuple mutant (TSLMF) abolished clusters at 3 μm PNU (Fig. 5). Despite the structural similarity between the Ser and Thr side chains, the S223T mutant by itself reduced potentiation to a similar extent as the quadruple (SLMF) mutant. Given the marked reduction of potentiation by S223T, we tested different side chains at this position. Substitution of larger and bulkier residues (S223T and S223V) reduced PNU potentiation, whereas substitution of the smaller alanine (S223A) had little effect (Fig. 5). Thus, side chain volume at position 223 appears important for PNU potentiation.
To further illustrate the profound reduction in PNU potentiation by the quintuple mutant (TSLMF), we performed a PNU adding experiment akin to that in Figure 1 for wild-type α7. Whereas addition of 3 μm PNU to an active cell-attached patch of wild-type receptors results in multiple superimposed clusters, addition of PNU to a comparable patch of quintuple mutant receptors has no noticeable effect (Fig. 6a). This insensitivity to PNU was confirmed by whole-cell recordings in which preincubation with 3 μm PNU had little effect on the magnitude and duration of the agonist-evoked response (Fig. 6b). To quantify the decrease in potentiation by the TSLMF mutant, we constructed a single-channel PNU dose–response relationship, plotting the ratio of the number of potentiated to total events based on fitted exponential components in open time histograms as a function of PNU concentration (Fig. 6c). For wild-type α7, the EC50 was 0.28 μm (95% confidence interval, 0.22–0.35 μm), in accord with previous measurements (Hurst et al., 2005), whereas for the quintuple mutant the EC50 was ∼17 μm (95% confidence interval, 12.5–23.1 μm). Thus, the TSLMF mutant is not potentiated by concentrations of PNU up to 3 μm and is only marginally potentiated at 10 μm PNU, a concentration approaching the solubility limit of PNU in our buffer solution.
We also examined the ability of PNU to allosterically alter agonist binding affinity through measurements of ACh competition against the initial rate of radio-labeled α-BTX binding (Fig. 6d). In the case of wild-type α7, 3 μm PNU increases apparent affinity for ACh, whereas the quintuple TSLMF mutant is unaffected. Thus, our overall data show that 3 μm PNU, a concentration that maximally potentiates wild-type α7 receptors (see Fig. 6c), does not potentiate the quintuple TSLMF mutant.
Discussion
Our single-channel recordings from neuronal α7 AChRs show that PNU potentiates agonist-induced responses by prolonging individual channel openings and, further, by condensing these openings into clusters of many openings in quick succession. Two kinetic classes of prolonged openings and clusters are observed, with the first and briefer class predominating at low and intermediate PNU concentrations and the second and longer class predominating at high concentrations. The two brief kinetic classes of α7 openings without PNU are prolonged ∼3000- to 30,000-fold by high PNU concentrations. Thus, rather than increasing the frequency of individual activation episodes, PNU increases the mean duration and number of openings per activation episode. By combining single-channel recording with mutagenesis, we provide evidence that a transmembrane region within each α7 subunit is required for PNU potentiation, and we identify novel determinants within this region.
At the single-channel level, α7 AChRs activate predominantly as single brief openings flanked by long closed periods. The mean duration of these openings coincides with the time constant for desensitization determined by rapid application of agonist to out-side-out patches (Bouzat et al., 2008). Thus channel openings of the α7 AChR are unusual in that they terminate by desensitization rather than by return to a closed, activatable state. Termination via desensitization explains why, at high agonist concentrations, openings of the α7 AChR do not form clusters of many openings in quick succession and, further, why the kinetics are independent of agonist concentration. Our single-channel studies show that the most pronounced effect of PNU is to increase the number and duration of openings within clusters, accounting for the slowing of desensitization observed with macroscopic currents.
Previous studies concluded that PNU activates desensitized α7 AChRs (Hurst et al., 2005; Grønlien et al., 2007; Ng et al., 2007; Barron et al., 2009). This conclusion is based on recordings of whole-cell macroscopic current in which addition of PNU to agonist-desensitized receptors stimulates new current. Our single-channel experiment in Figure 1 was done under similar conditions in which a cell-attached patch was exposed to a high agonist concentration for several minutes in the absence of PNU. Under these conditions, α7 AChRs spend most of their time desensitized, yet openings nonetheless appear at a low frequency owing to recovery of individual AChRs from desensitization followed by activation. At the level of macroscopic currents, these brief and infrequent openings would result in negligible current, making them essentially undetectable. Following application of PNU, however, the apparent “activation” of desensitized receptors may instead result from the ability of PNU to dramatically prolong these infrequent basal openings by slowing their entry to the desensitized state, resulting in detectable macroscopic current (Fig. 1). Furthermore, by stabilizing the open state, PNU may draw additional receptors from the desensitized to the activatable pool, further increasing current. In an alternative scenario, even if PNU increased the rate of recovery from desensitization 100-fold, the current from an increased frequency of brief openings would pale compared to the current resulting from prolongation of openings by slowed desensitization.
We find that PNU produces two kinetic classes of prolonged channel openings in a concentration-dependent manner, suggesting multiple PNU binding sites. Because the site required for PNU potentiation is present in each subunit of the homopentamer, the two classes of prolonged openings may arise from variable site occupancy. While it is premature to speculate on the number of sites required for each type of potentiation, it is reasonable to expect that clusters of tens to hundreds of milliseconds originate from suboptimal occupancy of PNU sites, while the appearance of multisecond-long clusters arises from occupancy of a greater number of sites.
Although a kinetic mechanism for α7 activation remains to be determined, we suggest the following framework consistent with the overall single-channel dwell times in the absence and presence of PNU. In the presence of agonist alone we can draw the fully occupied closed state (Cn), the open state (On), and the desensitized state (Dn), linked by reversible equilibria, where n is the optimal number of bound agonists (Fig. 7a). After the channel opens, transition from On to Dn predominates as opposed to the transition back to Cn, indicating that Dn corresponds to a low energy basin. Because optimal agonist occupancy promotes essentially complete desensitization, the energy basin for Dn is lower than that for On and Cn. Similarly, following optimal occupancy of PNU sites, we draw the agonist-occupied closed state with PNU bound (ppCn), open state (ppOn), and desensitized state (ppDn) linked by reversible equilibria. After the channel opens, transition from ppOn to ppDn is dramatically suppressed, indicating that ppOn corresponds to a deep energy basin and that a large energy barrier separates ppOn from ppDn. Additionally, brief closings between successive openings may arise from transitions back and forth between ppOn and ppCn, suggesting that the two states are separated by a low energy barrier. Because our patches contain multiple channels, we cannot determine the absolute probabilities of ppOn and ppDn, but in the presence of PNU our recordings show that clusters of openings and intervening closings last on the order of minutes, corresponding to two deep energy basins, each flanked by large energy barriers.
Figure 7.
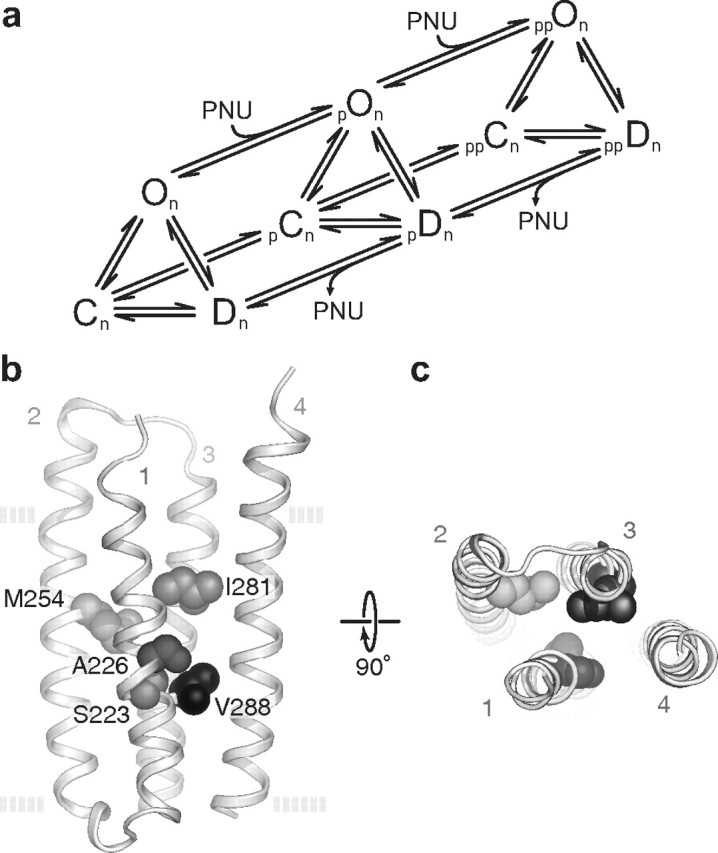
a, A possible mechanism for PNU potentiation. The scheme does not include agonist binding steps and assumes closed (C), open (O), and desensitized (D) states are bound with an optimal number of n agonists. The scheme also includes a minimum of two classes of PNU potentiation (p, pp) thought to relate to different levels of PNU occupancy. b, c, Structural determinants of PNU potentiation. Homology model (Cheng et al., 2006) based on Protein Data Bank identifier 2BG9 (Unwin, 2005) of the isolated transmembrane region of a single α7 subunit as viewed from the plane of the membrane (b) and the synaptic space (c). The side chains of the five amino acids mutated in the quintuple (TSLMF) mutant are shown as space-filling spheres. The four transmembrane helices (TM1-4) are also labeled. The dashed lines in b represent the approximate location of the plasma membrane.
Previous macroscopic current measurements showed an increase in agonist potency in the presence of PNU (Hurst et al., 2005). However, we find that for a given concentration of PNU, distributions of single-channel cluster durations are independent of the agonist concentration. In whole-cell measurements, noninstantaneous agonist perfusion, combined with rapid desensitization, underestimates the true EC50 for activation (Papke and Thinschmidt, 1998). Because PNU markedly slows desensitization, a limited agonist perfusion rate does not affect the measured EC50 in the presence of PNU, potentially accounting for the apparent increase in agonist potency.
Previous whole-cell experiments identified multiple transmembrane residues important for PNU potentiation (Young et al., 2008). These residues line an internal cavity between the four transmembrane helices (TM1-4) of a single subunit. Our single-channel data show that mutation of any of these residues individually does not eliminate PNU potentiation. Instead combination of multiple mutations progressively reduces potentiation. We identified five substitutions that cluster in and around this cavity (Figure 7) that, when combined, almost completely abolish potentiation at a high concentration of PNU. Of all the individual substitutions we made, the relatively conservative mutation S223T had the largest effect. This same substitution is found in the Caenorhabditis elegans homolog of human α7 called ACR-16, which despite having 47% sequence identity is not potentiated by PNU (Sattelle et al., 2009). In fact, ACR-16 has substitutions at all five positions mutated in our quintuple mutant, three of which are identical to the substitutions we arrived at independently in our screening (S223T, M254L, and V288F).
While our findings are consistent with an intrahelical PNU binding site, we emphasize two points of caution. First, we selected our mutations by assuming that residues lining the interhelical cavity contribute to PNU binding, and thus our data cannot be considered de novo evidence supporting a PNU binding site. Second, mutagenesis experiments cannot rule out indirect allosteric effects. Thus, unequivocal localization of the PNU binding site awaits direct evidence from structural or chemical-labeling studies. However, mounting evidence suggests that this intrahelical cavity may be a general modulatory site within the Cys-loop receptor superfamily. Anesthetics, alcohols, and neurosteroids are thought to bind to a similar location in GABA and glycine receptors (Belelli et al., 1997; Mihic et al., 1997; Hosie et al., 2006), while photoactivatable hydrophobic probes label nearby residues in the Torpedo AChR (Arevalo et al., 2005; Garcia et al., 2007; Hamouda et al., 2011). Furthermore, x-ray structures of a prokaryotic homolog show general anesthetics interacting with residues lining the cavity walls (Nury et al., 2011). The presence of an intrahelical binding site within a subunit contrasts with the intersubunit site for the PAM ivermectin revealed in the crystal structure of a glutamate-activated Cys-loop receptor (Hibbs and Gouaux, 2011).
If this intrahelical cavity is the PNU binding site, it immediately suggests a physical mechanism by which PNU alters desensitization kinetics. Channel opening and closing result from structural rearrangements of the transmembrane helices, with particular emphasis on TM2, which lines the channel pore. In the process of knocking down potentiation, we found that several substitutions altered mean channel open times in the absence of PNU. While these changes are small compared to the effects of PNU, they show that the chemical identity of residues lining the interhelical cavity affect open channel lifetime, and thus the rate of α7 desensitization. Spacing and interactions between transmembrane helices likely determine energy barriers controlling their movements, and by occupying a space that is normally vacant, PNU may present a steric barrier to helical movements required for desensitization. Analogously, substitutions that introduce bulkier side chains, effectively filling the interhelical cavity, may have a similar effect while at the same time occluding PNU binding. Thus, our findings provide a mechanistic and structural framework to understand channel opening, desensitization, and potentiation of neuronal α7 AChRs at the single-molecule level.
Footnotes
This work was supported by National Institutes of Health Grants NS053521 (S.M.S., C.B.) and NS031744 (S.M.S.), as well as a Canadian Institutes of Health Research Fellowship (C.J.B.D.). We thank Dr. Hai-Long Wang for providing the α7 homology model and Dr. Nuriya Mukhtasimova for contributions in the initial development of this work.
The authors declare no competing financial interests.
References
- Arevalo E, Chiara DC, Forman SA, Cohen JB, Miller KW. Gating-enhanced accessibility of hydrophobic sites within the transmembrane region of the nicotinic acetylcholine receptor's δ-subunit. A time-resolved photolabeling study. J Biol Chem. 2005;280:13631–13640. doi: 10.1074/jbc.M413911200. [DOI] [PubMed] [Google Scholar]
- Arias HR. Positive and negative modulation of nicotinic receptors. Adv Protein Chem Struct Biol. 2010;80:153–203. doi: 10.1016/B978-0-12-381264-3.00005-9. [DOI] [PubMed] [Google Scholar]
- Barron SC, McLaughlin JT, See JA, Richards VL, Rosenberg RL. An allosteric modulator of alpha7 nicotinic receptors, N- (5-Chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea (PNU-120596), causes conformational changes in the extracellular ligand binding domain similar to those caused by acetylcholine. Mol Pharmacol. 2009;76:253–263. doi: 10.1124/mol.109.056226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci U S A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;74:1155–1163. doi: 10.1016/j.bcp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Bertrand S, Cassar S, Gubbins E, Li J, Gopalakrishnan M. Positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2–M3 segment. Mol Pharmacol. 2008;74:1407–1416. doi: 10.1124/mol.107.042820. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Bren N, Sine SM. Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron. 1994;13:1395–1402. doi: 10.1016/0896-6273(94)90424-3. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Bartos M, Corradi J, Sine SM. The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J Neurosci. 2008;28:7808–7819. doi: 10.1523/JNEUROSCI.0448-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Wang H, Grant B, Sine SM, McCammon JA. Targeted molecular dynamics study of C-loop closure and channel gating in nicotinic receptors. PLoS Comput Biol. 2006;2:e134. doi: 10.1371/journal.pcbi.0020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single channel records. In: Sakmann B, Neher E, editors. Single channel recording. New York: Plenum; 1983. pp. 191–264. [Google Scholar]
- Corradi J, Gumilar F, Bouzat C. Single-channel kinetic analysis for activation and desensitization of homomeric 5-HT(3)A receptors. Biophys J. 2009;97:1335–1345. doi: 10.1016/j.bpj.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Faghih R, Gopalakrishnan M, Briggs CA. Allosteric modulators of the alpha7 nicotinic acetylcholine receptor. J Med Chem. 2008;51:701–712. doi: 10.1021/jm070256g. [DOI] [PubMed] [Google Scholar]
- Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry. 1995;38:22–33. doi: 10.1016/0006-3223(94)00252-X. [DOI] [PubMed] [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci U S A. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G, 3rd, Chiara DC, Nirthanan S, Hamouda AK, Stewart DS, Cohen JB. [3H]Benzophenone photolabeling identifies state-dependent changes in nicotinic acetylcholine receptor structure. Biochemistry. 2007;46:10296–10307. doi: 10.1021/bi7008163. [DOI] [PubMed] [Google Scholar]
- Grønlien JH, Håkerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, Malysz J. Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. 2007;72:715–724. doi: 10.1124/mol.107.035410. [DOI] [PubMed] [Google Scholar]
- Gusev AG, Uteshev VV. Physiological concentrations of choline activate native alpha7-containing nicotinic acetylcholine receptors in the presence of PNU-120596 [1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)-urea] J Pharmacol Exp Ther. 2010;332:588–598. doi: 10.1124/jpet.109.162099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajós M, Rogers BN. Targeting alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia. Curr Pharm Des. 2010;16:538–554. doi: 10.2174/138161210790361434. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hamouda AK, Stewart DS, Husain SS, Cohen JB. Multiple transmembrane binding sites for p-trifluoromethyldiazirinyl-etomidate, a photoreactive Torpedo nicotinic acetylcholine receptor allosteric inhibitor. J Biol Chem. 2011;286:20466–20477. doi: 10.1074/jbc.M111.219071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar SN, Dunlop J. Neuronal nicotinic acetylcholine receptors - targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer's disease. Curr Top Med Chem. 2010;10:144–152. doi: 10.2174/156802610790410983. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg RC, Buisson B, Bertrand D. Allosteric modulation of ligand-gated ion channels. Biochem Pharmacol. 2005;70:1267–1276. doi: 10.1016/j.bcp.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Hajós M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, Rutherford-Root KL, Berkenpas MB, Hoffmann WE, Piotrowski DW, Groppi VE, Allaman G, Ogier R, Bertrand S, Bertrand D, Arneric SP. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser SC, Bowlby MR, Comery TA, Dunlop J. A cog in cognition: how the alpha 7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol Ther. 2009;122:302–311. doi: 10.1016/j.pharmthera.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Lightfoot AP, Kew JN, Skidmore J. Alpha7 nicotinic acetylcholine receptor agonists and positive allosteric modulators. Prog Med Chem. 2008;46:131–171. doi: 10.1016/S0079-6468(07)00003-3. [DOI] [PubMed] [Google Scholar]
- Liu Y, Dilger JP. Opening rate of acetylcholine receptor channels. Biophys J. 1991;60:424–432. doi: 10.1016/S0006-3495(91)82068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Hernández GY, Thinschmidt JS, Morain P, Trocme-Thibierge C, Kem WR, Soti F, Papke RL. Positive modulation of alpha7 nAChR responses in rat hippocampal interneurons to full agonists and the alpha7-selective partial agonists, 4OH-GTS-21 and S 24795. Neuropharmacology. 2009;56:821–830. doi: 10.1016/j.neuropharm.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelicke A, Albuquerque EX. New approach to drug therapy in Alzheimer's dementia. Drug Discov Today. 1996;1:53–59. [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Ng HJ, Whittemore ER, Tran MB, Hogenkamp DJ, Broide RS, Johnstone TB, Zheng L, Stevens KE, Gee KW. Nootropic alpha7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc Natl Acad Sci U S A. 2007;104:8059–8064. doi: 10.1073/pnas.0701321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Papke RL, Thinschmidt JS. The correction of alpha7 nicotinic acetylcholine receptor concentration-response relationships in Xenopus oocytes. Neurosci Lett. 1998;256:163–166. doi: 10.1016/s0304-3940(98)00786-1. [DOI] [PubMed] [Google Scholar]
- Papke RL, Kem WR, Soti F, López-Hernández GY, Horenstein NA. Activation and desensitization of nicotinic alpha7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther. 2009;329:791–807. doi: 10.1124/jpet.108.150151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi XL, Nordberg A, Xiu J, Guan ZZ. The consequences of reducing expression of the alpha7 nicotinic receptor by RNA interference and of stimulating its activity with an alpha7 agonist in SH-SY5Y cells indicate that this receptor plays a neuroprotective role in connection with the pathogenesis of Alzheimer's disease. Neurochem Int. 2007;51:377–383. doi: 10.1016/j.neuint.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Rayes D, Spitzmaul G, Sine SM, Bouzat C. Single-channel kinetic analysis of chimeric alpha7–5HT3A receptors. Mol Pharmacol. 2005;68:1475–1483. doi: 10.1124/mol.105.015438. [DOI] [PubMed] [Google Scholar]
- Sattelle DB, Buckingham SD, Akamatsu M, Matsuda K, Pienaar IS, Pienaar I, Jones AK, Sattelle BM, Almond A, Blundell CD. Comparative pharmacology and computational modelling yield insights into allosteric modulation of human alpha7 nicotinic acetylcholine receptors. Biochem Pharmacol. 2009;78:836–843. doi: 10.1016/j.bcp.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine S, Taylor P. Functional consequences of agonist-mediated state transitions in the cholinergic receptor. Studies in cultured muscle cells. J Biol Chem. 1979;254:3315–3325. [PubMed] [Google Scholar]
- Sine SM. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of residues that determine curare selectivity. Proc Natl Acad Sci U S A. 1993;90:9436–9440. doi: 10.1073/pnas.90.20.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the alpha subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem. 1994;269:8808–8816. [PubMed] [Google Scholar]
- Sine SM, Kreienkamp HJ, Bren N, Maeda R, Taylor P. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of determinants of alpha-conotoxin M1 selectivity. Neuron. 1995;15:205–211. doi: 10.1016/0896-6273(95)90077-2. [DOI] [PubMed] [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Williams DK, Wang J, Papke RL. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem Pharmacol. 2011 doi: 10.1016/j.bcp.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Burton B, Urrutia A, Shcherbatko A, Chavez-Noriega LE, Cohen CJ, Aiyar J. Ric-3 promotes functional expression of the nicotinic acetylcholine receptor alpha7 subunit in mammalian cells. J Biol Chem. 2005;280:1257–1263. doi: 10.1074/jbc.M410039200. [DOI] [PubMed] [Google Scholar]
- Young GT, Zwart R, Walker AS, Sher E, Millar NS. Potentiation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci U S A. 2008;105:14686–14691. doi: 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]