

Abstract
S100A4, a member of the S100 family of Ca2+-binding proteins, displays elevated expression in malignant human tumors compared with benign tumors, and increased expression correlates strongly with poor patient survival. S100A4 has a direct role in metastatic progression, likely due to the modulation of actomyosin cytoskeletal dynamics, which results in increased cellular motility. We developed a fluorescent biosensor (Mero-S100A4) that reports on the Ca2+-bound, activated form of S100A4. Direct attachment of a novel solvatochromatic reporter dye to S100A4 results in a sensor that, upon activation, undergoes a 3-fold enhancement in fluorescence, thus providing a sensitive assay for use in vitro and in vivo. In cells, localized activation of S100A4 at the cell periphery is observed during random migration and following stimulation with lysophosphatidic acid, a known activator of cell motility and proliferation. Additionally, a screen against a library of FDA-approved drugs with the biosensor identified an array of phenothiazines as inhibitors of myosin-II associated S100A4 function. These data demonstrate the utility of the new biosensor both for drug discovery and for probing the cellular dynamics controlled by the S100A4 metastasis factor.
S100A4, a member of the S100 family of proteins, is directly involved in driving the progression of metastatic disease. S100A4 protein levels are elevated in malignant forms of breast, gallbladder, pancreatic, prostate, esophageal, gastric, lung and thyroid carcinomas (1). Furthermore, nonmetastatic cancer cells engineered to overexpress S100A4 produce a high incidence of tumors with a shorter latency period and higher frequency of metastasis (2, 3). However, the most compelling evidence has emerged from animal studies using S100A4 overexpression or genetic deletion of S100A4 in models of osteosarcoma, breast and lung cancer, which conclusively demonstrate that S100A4 plays a direct role in tumor metastasis (4–7). These in vivo studies support an important but poorly understood role for S100A4 in mediating the metastatic process.
More recent efforts have focused on the prognostic significance of S100A4 expression and its use as a marker for metastasis and poor patient outcome (8, 9). In a study of 349 invasive human breast cancer specimens, those that stained positively for S100A4 were highly correlated with patient death (8, 10). Additionally, S100A4 is a prognostic marker in other cancers, including prostate, bladder, non-small lung, pancreatic, esophageal-squamous, primary gastric and malignant melanomas (1). These studies not only illustrate the potential use of S100A4 as a marker but also suggest S100A4 as a target for novel therapeutic drugs.
S100 proteins are typically homodimers that contain N-and C-terminal EF-hands connected by a loop or hinge region (1). Ca2+-binding to the C-terminal EF-hand induces a significant change in the angle between helices 3 and 4 that flank the C-terminal Ca2+-binding loop, and exposes a hydrophobic cleft that constitutes a binding surface for target proteins (1). S100A4 specifically binds to the C-terminal end of the coiled-coil of myosin-IIA in a Ca2+-dependent manner (11). Our previous studies demonstrate that Ca2+-activated S100A4 promotes the monomeric, unassembled state of myosin-IIA by depolymerizing preassembled filaments and inhibiting the assembly of myosin-IIA monomers into filaments (11).
Myosin-IIA localizes to the lamellae of migrating cells (12, 13). Localization studies of S100A4 show that it is diffuse throughout the cytoplasm and enriched at sites of protrusion along the leading edge (14, 15). Recent studies demonstrate that S100A4-expressing cells become highly polarized upon chemotactic stimulation by extending enhanced forward protrusions and suppressing side protrusions (15). Furthermore, the ability of S100A4 to promote directional motility is an immediate consequence of its interaction with myosin-IIA (15). These observations suggest that S100A4 expression may promote a metastatic phenotype via the regulation of myosin-IIA assembly and cell motility.
Here we describe the development of a biosensor that reports on the Ca2+-induced activation of S100A4. This biosensor has been used to examine the spatial activation of S100A4 in living cells. Additionally we describe the use of this biosensor as a screening tool to initiate a search for inhibitory compounds, thus providing the first step toward the development of novel S100A4-based therapies.
EXPERIMENTAL PROCEDURES
Protein Preparation
Wild-type human S100A4 was expressed and purified as described previously (11). S100A4 mutants C3R/C86S and C81S/C86S were cloned into the NdeI/HindIII sites of pET23a (Invitrogen, Carlsbad, CA). Cysteine mutants were expressed in BL21(DE3)* without IPTG induction and purified as described for wild-type S100A4. The S100A4 protein concentration was determined using the Bradford protein assay (Bio-Rad, Hercules, CA) and a S100A4 standard of known concentration. The concentration of the S100A4 standard was determined by amino acid analysis (Keck Biotechnology Resource Laboratory at Yale University, New Haven, CT).
Human myosin-IIA rods were purified as described previously (11). The concentration of the myosin-IIA rods was determined by absorbance at 280 nm.
A peptide, FITC-DAMNREVSSLKNKLRR-OH,1 derived from the human myosin-IIA (residues 1908–1923), was synthesized using solid-phase peptide synthesis (Biosynthesis Inc., Lewisville, TX). A 5 mM stock solution of the FITC-MIIA1908–1923 was prepared in 20 mM Tris pH 7.5, 150 mM KCl, 1 mM DTT and 0.02% NaN3, aliquoted and stored at −80 °C. The purity of the peptide (>95%) was determined using HPLC and mass spectrometry.
Fluorescent Labeling of S100A4
Cys81 on S100A4 was labeled in a reaction containing 100 μM C3R/C86S S100A4 in phosphate buffer (50 mM NaPO4 pH 7.5, 20 mM NaCl, 0.02% NaN3 and 0.25 mM TCEP) containing 10% acetonitrile and a 4.8-fold excess of I-SO-iodoacetamide (sodium3-{(2Z)-5-[(iodoacetyl)amino]-2-[(2E,4Z)-4-(1,1-dioxido-3-oxo-1-benzothien-2(3H)-ylidene)but-2-enylidene]-3,3-dimethyl-2,3-dihydro-1H-indol-1-yl}propane-1-sulfonate). The I-SO-iodoacetamide was synthesized as described previously (16). The labeling was quenched by the addition of 1 μL of 100% β-mercapotethanol (14.3 M). Free dye was removed by chromatography on G25 Sephadex gel filtration media in phosphate buffer containing 0.5 mM β-mercapotethanol instead of TCEP. The dye to protein ratio was calculated by determining the protein concentration using the modified Lowry assay and the dye concentration by measuring the absorbance at 600 nm (ε = 138,000). This method consistently resulted in greater than 90% labeling efficiency. The labeling was confirmed by ESI-MS, which detected a major peak with a mass of 12,189.0 Da. This is within 1 Da of the mass of an S100A4 monomer with a single dye attached (calculated mass of labeled monomer = 12,188.5 Da; mass of unlabeled monomer = 11,634.5 Da).
To prepare the Fluo-S100A4, the C81S/C86S S100A4 was labeled with fluorescein-5-maleimide (Invitrogen) on Cys3 as described above.
Characterization of Mero-S100A4
Fluorescence spectra were acquired on a PTI fluorimeter (Photon Technology International). Reactions contained 5 μM Mero-S100A4 or Fluo-S100A4 dimer in 20 mM Tris pH 7.5, 150 mM KCl, 0.02% NaN3, 1 mM DTT and either 0. 3 mM CaCl2 or 2 mM EGTA. For Mero-S100A4, excitation scans were acquired from 500 to 630 nm (em: 635 nm) and emission scans acquired from 610 to 700 nm (ex: 600 nm) using a 4 nm slit width. For Fluo-S100A4, fluorescence excitation scans were acquired from 400 to 510 nm (em: 516 nm) and emission scans acquired from 500 to 590 nm (ex: 494 nm) using a 2 nm slit width.
For cell lysate experiments, NIH-3T3 fibroblasts at 80–90% confluency were washed two times with TBS followed by the addition of 75–100 μL of lysis buffer (50 mM Tris pH 7.1, 20 mM NaCl, 1 mM DTT and 5 μg/μL each of chymostatin, leupeptin, pepstatin A). After incubating on ice for 15 min, the cells were collected and sonicated at the lowest setting (three 4 s pulses). After sonication the salt concentration was adjusted to 150 mM NaCl and the lysates were clarified by centrifugation at 13,000 rpm in a tabletop microcentrifuge for 15 min at 4 °C. The total protein concentration was determined using the Bradford Assay (BioRad). The lysate was added to 1.25 μM Mero-S100A4 dimer in lysis buffer with either no addition, 1 mM CaCl2 or 5 mM EGTA. Fluorescence emission spectra were obtained as described above.
Ca2+-Binding Measurements
Ca2+-binding affinities were determined using a 5,5′Br2-BAPTA competition assay as described previously (17). Calcium was titrated into a solution containing 25 μM 5,5′Br2-BAPTA and 25 μM wild-type S100A4 or Mero-S100A4 monomer at 25 °C in 20 mM Tris pH 7.5, 150 mM KCl, 1 mM DTT, 0.02% NaN3, and the decrease in absorbance monitored at 263 nm. Total Ca2+ in the protein preparation was less than 0.1 mol of Ca2+/mol of protein. Using the program Caligator, averaged data from 4 independent experiments were fit to two Ca2+ binding sites using a stepwise macroscopic binding equation in the presence of chelator (18). Models for single-site binding or four independent binding sites with different affinities produced poor fits to the experimental data. Additionally, since the low affinity site (EF1) does not compete well with 5,5′Br2-BAPTA, lower limits for this value were obtained by monitoring the χ2 value as a function of the low affinity constant. This approach was verified with experiments using S100B for which we obtained similar constants to those reported previously (19).
Fluorescence Ca2+ titration experiments were carried out in a buffer containing 20 mM Tris pH 7.5, 150 mM KCl, 1 mM DTT, 0.02% NaN3, using an EGTA/Ca2+ buffering system (20). 5 μL aliquots from a 1 mM Ca2+ stock were added to a solution containing 10 μM Mero-S100A4 and 25 μM EGTA with a 2 min mixing interval at 25 °C. The free Ca2+ concentration at each point was calculated using WebMaxChelator standard constants (21). The fluorescence emission at 634 nm for each titration point was plotted as a function of the free Ca2+. Data from four independent experiments was fit using both the Hill equation and a stepwise macroscopic binding equation for two sites as described previously (22).
Circular Dichroism Spectroscopy
S100A4 (wild-type, C3R/C86S and Mero) was dialyzed into 20 mM Tris pH 7.5, 20 mM KCl, 0.25 mM TCEP, 0.02% NaN3. Protein concentrations were determined by the Bradford protein assay (BioRad) for wild-type and C3R/C86S S100A4 and by the modified Lowry assay (Pierce, Rockford, IL) for the Mero-S100A4 using a S100A4 standard of known concentration. Spectra were collected using 12.5 μM S100A4 dimer in a 125 μL circular quartz cuvette on a Jasco J-700 circular dichroism spectrophotometer. The data shown represents the average of six scans for each protein. The raw CD data was transformed into molar ellipticity using the Jasco software package and the known concentration of protein in the sample.
Myosin-IIA Binding Assays
Sedimentation assays were performed as described previously (11), using 1.5 μM myosin-IIA rods and 0.1–35 μM wild-type S100A4 dimer or 0.1–18 μM Mero-S100A4 dimer. Binding constants were determined by fitting the data to the equation b = Bmax[S]/(Kd + [S]).
Inhibition of assembly and promotion of disassembly assays comparing wild-type S100A4 and Mero-S100A4 were carried out as described by Li et al. (11). For promotion of disassembly assays performed in the presence of FDA-approved drugs, 100 μM compound or 10% DMSO was added to the reaction.
Fluorescence anisotropy measurements were performed at 22 °C using a Fluoromax-3-spectrofluorometer (Jobin Yvon Inc.). Individual reactions (200 μL) contained 100 nM FITC-MIIA1908–1923 and 0–30 μM S100A4 dimer in 20 mM Tris pH 7.5, 150 mM KCl, 1 mM DTT, 0.02% NaN3, and 0.5 mM CaCl2. Anisotropy was measured with excitation at 494 nm and emission at 516 nm. Measurements were acquired at the magic angle corresponding to the angle of 55° between the vectors of polarization of the excitation and emission light using a G factor of 0.634 previously determined for FITC on this instrument. Data from three independent experiments was plotted using Graphpad Prism v4 (GraphPad Software, Inc., San Diego, CA), and the dissociation constant was calculated by fitting to a single site saturation binding equation allowing for a floating Ymin value.
Competition assays with lead compounds contained 15 μM S100A4 dimer, 100 nM FITC-MIIA1908–1923, 0.3 mM CaCl2, and 0–600 μM compound in 20 mM Tris pH 7.5, 150 mM KCl, 1 mM DTT, 0.02% NaN3, and 0.5 mM CaCl2, 2% DMF. Controls included experiments representing maximum anisotropy (15 μM S100A4, 100 nM peptide and calcium) and minimum anisotropy (100 nM peptide and calcium). Experiments were performed in triplicate and fit to a sigmoidal dose response equation with a variable slope to obtain the EC50. Using this value the inhibition constant for the compounds was obtained using the equation Ki = [KdP•EC50]/[S], where EC50 is the concentration at half-maximal binding of the competing peptide determined from fitting the raw data, KdP is the dissociation constant for the binding of FITC-MIIA1908–1923 to S100A4 and [S] represents the concentration of S100A4 dimer.
Molecular Modeling of the Mero-S100A4
The three-dimensional structure of the dye was built and energy minimized using the BUILDER module from Insight II (Accelrys Software Inc., San Diego, CA). The merocyanine-S100A4 complexes were modeled using COOT, and the structures of the apo-S100A4 (PDB: 1M31) and the Ca2+-bound S100A4 (A.R.B. unpublished results) (23). The dye molecule was covalently attached to Cys81 through the α carbon of the iodoacetamide reactant. The conformation of the dye in both complexes was modeled to maximize surface complementarity and hydrophobic interfaces between the protein and the dye molecule. No energy minimization or molecular dynamics simulations were performed, or protein–dye interaction restraints derived from alternative methods were taken into account.
Small Molecule Screen
A library of nearly 400 FDA-approved drugs (see Supporting Information, Table 1) was screened in a 96-well plate format using 5 μM Mero-S100A4 dimer, 0.3 mM CaCl2 and 150 μM compound in 50 mM Tris pH 7.5, 150 mM NaCl, 0.02% NaN3, and 1 mM DTT (screening buffer). Controls included Mero-S100A4 in the absence of any added Ca2+ and Mero-S100A4 in the presence 0.3 mM CaCl2 or 0.3 mM CaCl2 and 2 mM EGTA. The fluorescence intensity was measured using a Molecular Probes SpectraMax M5 plate reader (ex, 600 nm; em, 634 nm).
IC50 values were calculated from reactions containing 5 μM Mero-S100A4, 1–300 μM compound and 0.3 mM CaCl2. Data were plotted using GraphPad Prism v4 and IC50 values were determined using a three parameter logistic equation with a standard slope (24).
LPA Stimulation: Microscopy, Image Acquisition and Analysis
NIH-3T3 fibroblasts were maintained in DMEM supplemented with 10% fetal bovine serum and grown at 37 °C in a humidified atmosphere of 5% CO2. Prior to microinjection, cells were starved in phenol red free DMEM containing 50 mM HEPES for 2 h. Cells were injected with a 1:2 ratio of Fluo-S100A4:Mero-S100A4 (60 μM total protein) in phosphate buffered saline pH 7.1 containing 0.2 mM DTT using an Eppendorf semiautomatic microinjection system and a 37 °C heated stage. Following injection, cells were allowed to recover for 25 min and then either fixed in 3.7% formaldehyde (control) or stimulated with 1 μg/mL lysophosphatidic acid (LPA) for 1 or 5 min and then fixed. Dishes were mounted with a coverslip using Pro-long Anti-Fade (Invitrogen/Molecular Probes).
Phase, fluorescein and merocyanine images were acquired using IPLab Spectrum software (Scanalytics, Inc., Rockville, MD) with a CoolSNAP HQ interline 12-bit, cooled CCD camera (Roper Scientific, Tucson, AZ) mounted on an Olympus IX70 microscope with a PlanApo 60× 1.4NA oil immersion objective (Olympus, Melville, NY). For emission ratio imaging, fluorescein (HQ470/40, HQ525/50) and merocyanine (HQ580/30, HQ630/40) filter sets were used with a custom manufactured dichroic mirror (Chroma Technology, Rockingham, VT). Cells were illuminated through a 0.6 neutral density filter using exposure times of 0.3–0.6 s and 2 × 2 binning. All images were analyzed using ImageJ v1.37 (NIH). Images were background subtracted. The fluorescein image, which has the strongest signal, was thresholded to generate a binary mask with all values within the cell equal to 1 and all values outside the cell equal to 0. The fluorescein and merocyanine images were multiplied by this binary mask to eliminate background. The merocyanine image was then divided by the fluorescein image. After ratioing, the images were registered after adjusting for proper pixel alignment to minimize artifacts. Cells were traced in phase, and the outline of the cell was overlaid onto the respective ratio image. Areas outside of the cell were set to zero. The ratio image was pseudocolored, creating a map of S100A4 activation, with blue representing low and red representing high activation.
Live Cell Imaging and Analysis
Mouse embryonic fibroblasts (MEF/3T3, Clontech Inc., Mountain View, CA) were used in live cell imaging. To prevent nonspecific binding of Mero-S100A4 to fibronectin-coated coverslips during microinjection, coverslips were treated with 4% octadecyl-trichlorosilane (OTS; Sigma Aldrich, St. Louis, MO) in anhydrous chloroform, followed by 3 successive washes in clean chloroform, and allowed to air-dry. Excess OTS was removed by gentle polishing, and the coverslips were coated with 10 μg/mL fibronectin (Sigma Aldrich). 4 × 104 cells were seeded per coverslip and allowed to adhere and spread for 2 h. Medium was exchanged with standard culture medium supplemented with 10 mM HEPES and 5% w/v fatty acid free bovine serum albumin (Sigma Aldrich), and incubated for an additional 2 h prior to microinjection. Cells were microinjected with a mixture containing a 2:1 ratio of Mero-S100A4:Fluo-S100A4, at a final concentration of 73 μM in buffer containing 10 mM KH2PO4, 150 mM KCl, 0.8 mM DTT. Injected cells were allowed to recover for 30 min and imaged in Ham’s F-12K medium without phenol red (Invitrogen, Carlsbad, CA), with 2% fetal bovine serum, 10 mM HEPES and 10 μg/mL Oxy-Fluor supplemented with 5 mM DL-lactate (Oxyrase Inc., Mansfield, OH), in a heated closed chamber (20/20 Technologies, Wilmington, NC). Images were obtained using an Olympus 40× 1.3NA UIS2 UPlan FL N DIC lens on an Olympus IX71 inverted microscope (Olympus, Center Valley, PA), a CoolsnapES CCD camera (Roper Scientific, Tucson, AZ), and Metamorph software (Molecular Devices, Downington, PA) using the filter sets described above. Cells were illuminated through a 22% neutral density filter using exposure times of 1200 ms and 300 ms for the ISO and FITC images respectively, and 2 × 2 binning. Cells were imaged at 5 min time intervals for 2.5 h.
Metamorph ver 7.1.3 (Molecular Devices) was used to perform the image processing and data analysis. All images were flat-field corrected and background subtracted. ISO and FITC images were carefully aligned automatically (25) to ascertain correct whole and subpixel registration. The ISO image, because it had the largest signal-to-noise ratio and therefore provided the best distinction between the cell and the background, was intensity thresholded to generate a binary mask with a value of zero outside the cell and a value of one inside the cell. ISO and FITC images were multiplied by this binary mask to set areas outside the cell uniformly to zero to minimize noise and other artifacts. The masked ISO image was divided by the masked FITC image to yield a ratio, reflecting Mero-S100A4 activation throughout the cell. A linear pseudocolor lookup table was applied and the ratio values were normalized to the lower scale value. In every data set, the FITC and ISO images were inspected to verify that all portions used to create the ratio image had a high enough signal/noise ratio. This was especially important in thin parts of the cell where fluorescence was low. In time-lapse experiments, ISO and FITC bleached at different rates. The ratio was corrected for bleaching using published methods (26).
RESULTS
Development of a Biosensor That Reports on the Calcium-Induced Activation of S100A4
S100A4 undergoes a large conformational change upon binding Ca2+ at the C-terminal EF-hand, which is necessary for interactions with protein targets and to generate a biological effect (27–30). We took advantage of this Ca2+-induced conformational change and created a fluorescent S100A4 biosensor by exploiting dyes designed for reporting protein activity in living cells. The merocyanine dyes exhibit bright fluorescence at long wavelengths, are photostable, and are sensitive to both solvent polarity and hydrogen bonding, which enables them to report changes in their protein environment that occur due to conformational changes or protein–protein interactions (Figure 1A) (16, 31, 32). Using our NMR structure of the apo-S100A4 and the structures of the Ca2+-bound and Ca2+-target peptide complex of S100B (30, 33, 34), a structure-based alignment suggested several potentially useful sites for dye attachment, including Cys81 and Cys86. Site-specific labeling was achieved by making single serine substitutions at either Cys81 or Cys86 and replacing Cys3 with an Arg, which is found in the rodent homologues. Cys76 is buried at the dimer interface and is resistant to modification. The merocyanine dye exhibits the greatest response at position 81 (Figure 1B) with a 3-fold Ca2+-induced increase in fluorescence intensity. By contrast, target binding in the presence of Ca2+ (e.g., myosin-IIA) does not affect fluorescence intensity, producing only a small red shift. In the presence of myosin-IIA and EGTA, Mero-S100A4 displays the same fluorescence intensity as observed with EGTA alone (data not shown). This dye yields an unusually sensitive reagent for both in vitro screening and live cell studies as merocyanine dyes in hydrophobic environments have quantum yields >0.7 and extinction coefficients greater than 150,-000 cm−1 M−1 (16).
Figure 1.

Mero-S100A4 reports activation by Ca2+. (A) Structure of the I-SO merocyanine dye. (B) Fluorescence excitation and emission spectra of 5 μM Mero-S100A4 dimer. Dashed line: Mero-S100A4 in the presence of EGTA. Solid line: Mero-S100A4 in the presence of Ca2+. Dotted line: Mero-S100A4 in the presence of Ca2+ and myosin-IIA. Mero-S100A4 exhibits a 3-fold increase in fluorescence upon Ca2+ addition. The addition of Ca2+ and a 10-fold molar excess of myosin-IIA results in a slight red shift, but no additional increase in fluorescence intensity. (C) Mero-S100A4 was added to a fibroblast lysate in the presence of EGTA or calcium. The data represent the average peak intensity at 634 nm for three independent experiments and the standard deviation.
The biosensor can also be used to assay S100A4 activation in cell lysates (Figure 1C). In the absence of exogenous Ca2+, purified Mero-S100A4 and Mero-S100A4 in a fibroblast lysate exhibit comparably low fluorescence intensities. In the presence of Ca2+-supplemented lysate Mero-S100A4 displays a similar 3-fold increase in fluorescence intensity as observed with the purified biosensor. These data indicate that other potential S100A4 targets do not affect the Ca2+-induced fluorescence increase of Mero-S100A4.
The merocyanines are solvatochromatic dyes that display sensitivity to both solvent polarity and hydrogen bonding, thus small changes in protein conformation can strongly influence the fluorescence properties of the dyes (16, 31, 32). Our NMR structure of the apo-S100A4 shows that helices 3 and 4 have a roughly parallel orientation with respect to one another (35). Molecular modeling of the merocyanine in the apo-S100A4 suggests that the fluorophore is solvent accessible. The structural rearrangement of helices 3 and 4 that occurs upon Ca2+-binding should afford the merocyanine access to the hydrophobic pocket that comprises the target binding site of S100A4 (Figure 2). This interhelical separation likely decreases the average polarity of the fluorophore’s environment, resulting in an increase in the fluorescence intensity, and thus allowing the biosensor to report on the conformational rearrangements that are associated with Ca2+-binding in S100A4. Although the placement of the merocyanine is not expected to interfere with Ca2+-binding, biochemical studies were necessary to examine potential effects of the fluorescent moiety on the Ca2+ and myosin-IIA binding activities of S100A4.
Figure 2.
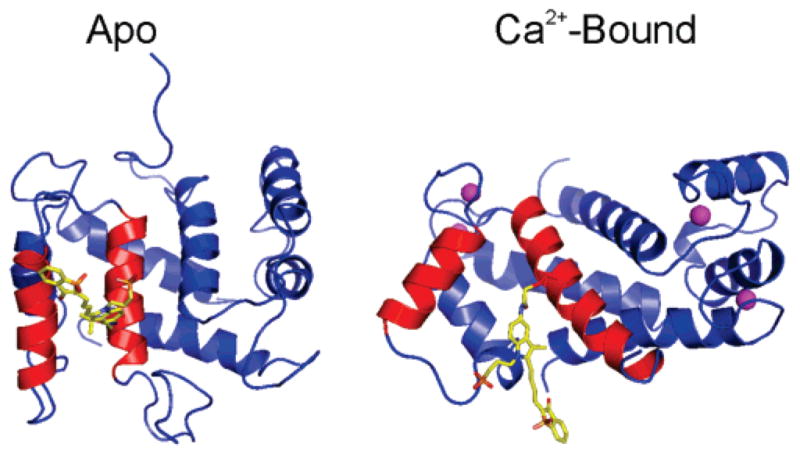
Model of Mero-S100A4. Model of the apo and Ca2+-bound Mero-S100A4. Helices 3 and 4 of one monomer are shown in red, and the merocyanine dye attached to Cys81 is shown in yellow. Merocyanine attached to only one monomer is shown for simplicity. The bound calcium ions are depicted as pink spheres.
Ca2+-Binding Properties of Wild-Type and Mero-S100A4
The Ca2+ binding capabilities of the wild-type S100A4 and biosensor were examined in a competition assay with the chromophoric Ca2+ chelator 5,5′Br2-BAPTA (17). For the wild-type and Mero-S100A4, the dissociation constants (Kd) for the high-affinity EF-hands (EF2) are 2.6 ± 1 μM and 0.4 ± 0.3 μM, respectively (Table 1, Figures 3A and 3B). For the low-affinity EF-hands (EF1) Kd’s of ≥574 μM and ≥447 μM were obtained for the wild-type and Mero-S100A4 (Table 1, Figures 3A and 3B). 5,5′Br2-BAPTA has a relatively high affinity for Ca2+ (2.3 μM), thus only limiting values could be estimated for the low affinity EF-hands (EF1) as they do not effectively compete for Ca2+ binding in this assay (17). These data show that Mero-S100A4 binds Ca2+ with slightly higher affinity at the EF2 site than wild-type S100A4 yet maintains the same affinity at the EF1 site. Previously, Ca2+-binding constants for S100B were reported as ≥350 μM and 56 ± 9 μM for EF1 and EF2 respectively (36). Using the same 5,5′Br2-BAPTA assay and fitting procedure as for S100A4, we obtained comparable Kd’s of 209 ± 60 μM and 17.5 ± 40 μM for Ca2+-binding to EF1 and EF2 of S100B (Supporting Information, Figure 1).
Table 1.
Dissociation Constants for Ca2+-Binding to S100A4
| dissociation constant (μM)
|
||
|---|---|---|
| EF1 | EF2 | |
| wild-type S100A4 | ≥574a | 2.6 ± 1.0a |
| Mero-S100A4 | ≥447a | 0.4 ± 0.3a |
| ≥830b | ≤0.5b | |
The dissociation constants were determined using competition studies of Ca2+ with the chromophoric chelator 5,5′Br2-BAPTA.
The dissociation constants were determined by monitoring the fluorescence of the Mero-S100A4 during titration with Ca2+. The data from both assays were fit to a stepwise macroscopic binding equation for two sites (22).
Figure 3.

Wild-type and Mero-S100A4 bind Ca2+ with the same affinities. Competition assay with the chelator 5,5′Br2-BAPTA was used to examine the affinity of Mero-S100A4 for Ca2+. The decrease in absorbance was monitored at 263 nm for a mixture containing 25 μM 5,5′Br2-BAPTA and either 25 μM wild-type S100A4 (A) or Mero-S100A4 (B). The data represent 6–8 titrations from 4 independent experiments. The insets show the saturation curve representation for the best fit in Caligator. (C) Ca2+ titration experiments were carried out using a Ca2+/EGTA buffering system and 5 μM Mero-S100A4 dimer. The data represent four independent experiments.
We examined the response of Mero-S100A4 as a function of Ca2+ concentration in the absence of myosin-IIA (Figure 3C). A cooperative binding model provides the best fit to the titration data, and the data were fit using both the Hill equation and a stepwise two-site binding equation. Macroscopic binding constants for Ca2+-binding to EF1 and EF2 are ≥830 μM and ≤0.5 μM in the absence of myosin-IIA with a Hill coefficient of nh = 2.0, which suggests strong positive cooperativity for Ca2+-binding. These are in good agreement with the values obtained from the 5,5′Br2-BAPTA competition assay and show the ability of the biosensor to report activation at Ca2+ levels attainable by the cell.
Interaction of Mero-S100A4 with Myosin-IIA
Our previous studies demonstrate that Ca2+-activated S100A4 promotes the monomeric, unassembled state of myosin-IIA by depolymerizing preassembled filaments and inhibiting the assembly of myosin-IIA monomers into filaments (11). Wild-type and Mero-S100A4 bind to myosin-IIA filaments with similar affinities (Kd’s of 2.1 ± 0.3 μM and 4.3 ± 1.1 μM, respectively) (Supporting Information, Figure 2), indicating that dye attachment does not prevent binding to myosin-IIA. However, Mero-S100A4 does not regulate the monomer–polymer equilibrium of myosin-IIA. At a ratio of one S100A4 dimer per myosin-IIA rod, wild-type S100A4 maximally disassembles pre-existing myosin-IIA filaments and completely inhibits the assembly of myosin-IIA monomers into filaments. At a ratio of five dimers per myosin-IIA rod, Mero-S100A4 does not destabilize pre-existing myosin-IIA filaments nor does it prevent the assembly of myosin-IIA monomers (Figure 4A and B). Circular dichroism spectroscopy indicates that the C3R/C86S mutant and Mero-S100A4 exhibit comparable secondary structures to the wild-type protein (Figure 5), therefore the inability of Mero-S100A4 to modulate the monomer–polymer equilibrium of myosin-IIA does not result from significant structural alterations in the protein.
Figure 4.
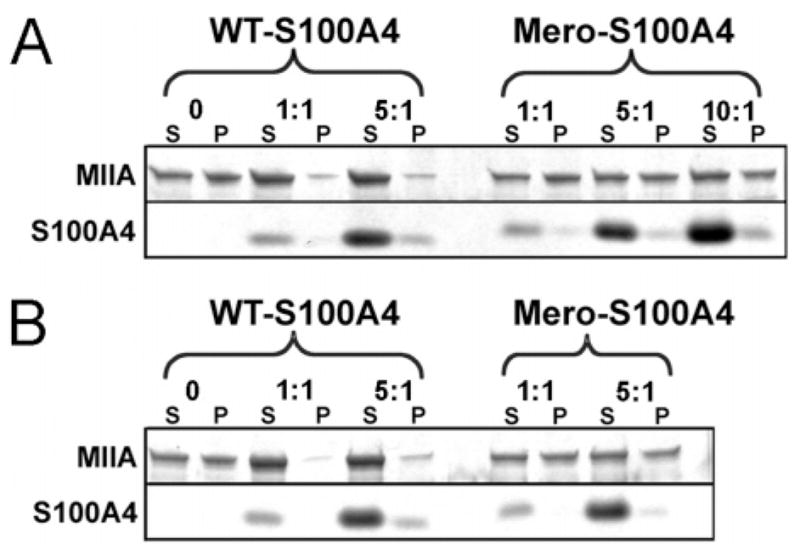
Mero-S100A4 does not regulate the monomer–polymer equilibrium of myosin-IIA filaments. (A) Promotion of myosin-IIA filament disassembly by wild-type and Mero-S100A4. At a ratio of one wild-type S100A4 dimer per myosin-IIA rod, most of the myosin-IIA rods are present in the supernatant. A ratio of five Mero-S100A4 dimers per myosin-IIA rod has no effect on the myosin-IIA filaments. (B) Inhibition of myosin-IIA filament assembly by wild-type and Mero-S100A4. At a ratio of one S100A4 dimer per myosin-IIA rod, wild-type S100A4 inhibits the assembly of myosin-IIA monomers into filaments. Mero-S100A4 does not inhibit the assembly of the myosin-IIA rods at a ratio of five Mero-S100A4 dimers per myosin-IIA rod.
Figure 5.
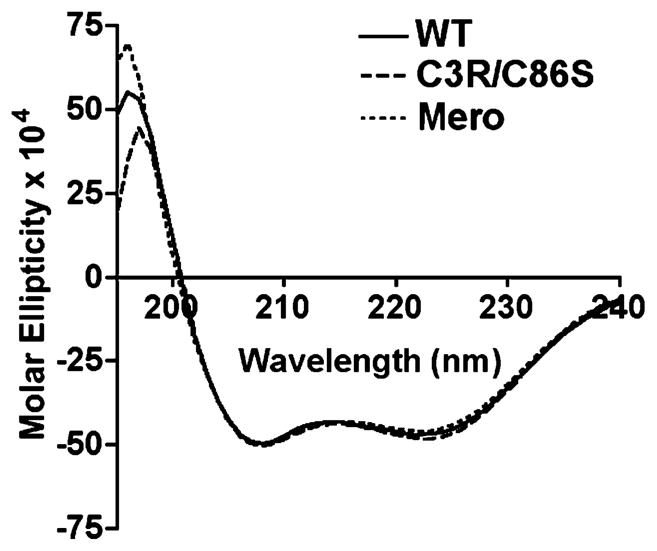
Far-UV circular dichroism spectra of wild-type, C3R/C86S and Mero-S100A4. The spectra show wild-type (solid line), C3R/C86S (dashed line) and Mero-S100A4 (dotted line) at a protein concentration of 12.5 μM dimer.
Identification of Inhibitors of S100A4 Function
A screen against a library of FDA-approved drugs identified twelve compounds that inhibit the Ca2+-induced fluorescence increase of the biosensor (Figure 6A). There was no shift in the emission maxima of Mero-S100A4 in the presence of hit compounds that would suggest inhibition caused by direct interaction with the merocyanine. Half of these compounds are members of the phenothiazine class of drugs, and include perphenazine, prochlorperazine, trifluoperazine, fluphenazine, chlorprothixene, and flupenthixol. The phenothiazines and other compounds identified in the screen display IC50 values in the low to mid micromolar range for inhibition of the Ca2+-induced fluorescence increase of the biosensor (Figure 6B). To eliminate the possibility that these compounds quench the fluorescence of the merocyanine dye and to evaluate their ability to inhibit the biochemical activity of S100A4, we examined their efficiency in blocking S100A4-mediated disassembly of myosin-IIA filaments (Figure 7A). Trifluoperazine completely abolishes the ability of S100A4 to depolymerize myosin-IIA filaments. Prochlorperazine, fluphenazine and bepridil also inhibit S100A4’s depolymerizing activity, albeit to a lesser extent than trifluoperazine. By contrast, loperamide has no effect on the ability of S100A4 to promote myosin-IIA filament disassembly. None of the compounds affect the assembly state of myosin-IIA in the absence of S100A4. To examine whether these compounds compete with myosin-IIA for binding to S100A4, we developed a fluorescence anisotropy assay using a fluorescein-tagged myosin-IIA peptide that comprises the S100A4 binding site (FITC-MIIA1908–1923). The FITC-MIIA1908–1923 exhibited Ca2+-dependent binding to S100A4 with a Kd of 1.7 ± 0.2 μM (Figure 7B). Titration of the S100A4-FITC-MIIA1908–1923 complex with lead compounds demonstrates that trifluoperazine, prochlorperazine and fluphenazine compete for myosin-IIA binding, whereas loperamide does not (Figures 7C and D). A modest increase in the anisotropy signal was observed in assays using high concentrations of the phenothiazines as compared to controls due to the intrinsic fluorescence of the compounds at the wavelengths used to monitor the fluorescein signal. Assays with bepridil could not be performed due to insolubility at the high compound concentrations needed for effective competition. These results suggest that the phenothiazines bind to the target-binding cleft of S100A4 in a manner that inhibits myosin-IIA binding. Although loperamide inhibits the Ca2+-induced fluorescence increase of the biosensor, these biochemical assays demonstrate that it does not compete with myosin-IIA for binding S100A4 nor does it promote myosin-IIA filament disassembly. These observations suggest that loperamide may interact with a region of the target binding cleft that affects the fluorescence of the merocyanine, but does not affect the interaction of S100A4 with myosin-IIA, or it may be a false positive due to direct interaction with the merocyanine.
Figure 6.

Identification of small molecule inhibitors using the Mero-S100A4. (A) Hit compounds identified in a screen of a library of FDA-approved drugs. Compounds that inhibited the Ca2+-induced fluorescence increase of the Mero-S100A4 to a similar extent as EGTA were considered to be hits. (B) IC50 values for inhibition of the Ca2+-induced fluorescence increase of the bio-sensor. The data represents the best fit to a three parameter logistic equation from two independent experiments.
Figure 7.

Hit compounds disrupt the interaction of S100A4 and myosin-IIA. (A) A sedimentation assay was used to examine the ability of hit compounds to attenuate the destabilization of myosin-IIA filaments by S100A4. At a molar ratio of one wild-type S100A4 dimer per myosin-IIA rod, ~90% of the myosin-IIA rods are recovered in the supernatant. Values represent the mean and the standard error of the mean from at least four independent experiments. 1, trifluoperazine; 2, prochlorperazine; 3, loperamide; 4, bepridil, and 5, fluphenazine. (B) Fluorescence anisotropy measurements of S100A4 binding to FITC-MIIA1908–1923. Values represent the mean and the standard deviation from three independent experiments. A Kd of 1.7 ± 0.2 μM was determined from the fit to a single site saturation binding curve. (C) Titration experiment with trifluoperazine showing a loss of anisotropy as TFP competes off the bound FITC-MIIA1908–1923. The values represent the mean and the standard deviation from three independent experiments. The data were fit to a three parameter logistic equation to obtain the EC50. (D) Bar graph representation of anisotropy competition assays with lead compounds. Values represent the mean and the standard deviation for the endpoint of the titration (600 μM compound) from three independent experiments. The numbers above the bar indicate the Ki in units of μM.
In Vivo Imaging of the Mero-S100A4
Cell-based studies examining S100A4 activation were performed in fibroblasts since they express high levels of S100A4. Stimulation with lysophosphatidic acid (LPA), a lipid mediator that signals through a specific G-protein-coupled receptor, triggers a phospholipase C-mediated rise in intracellular Ca2+. We used biosensor imaging to examine where cell surface stimulation leads to localized S100A4 activation. For in vivo experiments, ratiometric imaging is required to correct for variations in cell thickness, uneven illumination, and other variables that may affect the local dye intensity (37). Therefore, we created a second fluorescent analogue of S100A4 by attaching a fluorescein moiety to Cys3 (Fluo-S100A4). This analogue does not show any change in fluorescence upon Ca2+ or myosin-IIA binding (Supporting Information, Figure 3). To determine the level of S100A4 activation in specific subcellular compartments following LPA stimulation, the merocyanine image is divided by the corresponding fluorescein image to normalize any alterations in dye intensity that do not result from S100A4 activation. Prior to stimulation, little to no activated S100A4 is detected (Figure 8A), consistent with the low intracellular Ca2+ observed in quiescent cells. Upon stimulation with LPA, the biosensor reveals localized S100A4 activation at the cell periphery and in cell extensions, at 1 min poststimulation. Importantly, activation occurs concurrently with the rapid rise in intracellular Ca2+ that peaks within 1–2 min of LPA stimulation (38–40). By 5 min poststimulation, cells show reduced S100A4 activation, consistent with the corresponding drop in intracellular Ca2+ levels. These data suggest that the observed localization of S100A4 activity is due to the structural rearrangements associated with Ca2+-binding, and is supported by control experiments demonstrating that cells coinjected with Mero-S100A4 and EGTA show no appreciable activation poststimulation (Figure 8B).
Figure 8.

S100A4 activity in LPA-stimulated fibroblasts. (A) Cells were injected with a 1:2 ratio of Fluo-S100A4:Mero-S100A4, stimulated with LPA for the indicated times and fixed. A representative cell for each time point is shown with purple to red signifying low to high activation. At 1 min poststimulation, S100A4 activation is restricted to cell extensions with no activated S100A4 detected in the cell body. This pattern of activation was very similar in the 6 cells examined at 1 min and the 8 cells examined at 5 min. (B) Cells were injected with a 1:2 ratio of Fluo-S100A4:Mero-S100A4 in a buffer containing 1 mM EGTA, stimulated with LPA for the indicated times and fixed. A representative cell for each time point is shown with purple to red signifying low to high activation. No activation of S100A4 is detected in cells 1 min poststimulation (10 cells were examined). Scale bar = 20 μm.
Time-lapse imaging of Mero-S100A4 in MEFs revealed dynamic populations of activated S100A4 present in cell edges undergoing remodeling. During random migration, S100A4 activation was observed specifically in cellular regions undergoing retraction (Figure 9). This area of elevated S100A4 activation persists even after the start of a new protrusion in the same area of the cell (Figure 9, arrowhead). These observations suggest a possible role for S100A4 in modulating edge retraction events.
Figure 9.

S100A4 is activated during cell retraction. The panels show different time points (hours:minutes) during random cell movement over fibronectin. Upper panels: DIC. Lower panels: ratio of Mero over FITC signal. An area of high activation is prominent during retraction. The area of elevated activation persists after the start of a new protrusion in the same area (arrowhead). Elevated Mero-S100A4 ratios were observed in cellular retractions in 5 cells examined. Scale bar = 10 μm. The ratio image was scaled by setting the lower and upper threshold levels to exclude the lower 7% and upper 3%, respectively, and a linear pseudocolor lookup table was applied where the upper threshold limit value was divided by the lower threshold limit value to show the dynamic range of ratio signal.
DISCUSSION
The direct covalent derivatization of S100A4 with a novel reporter dye has resulted in the development of the first biosensor for the S100 family of proteins. The merocyanines are solvatochromatic dyes that display sensitivity to both solvent polarity and hydrogen bonding, thus small changes in protein conformation can strongly influence the fluorescence properties of the dyes (16, 31, 32). For S100A4, conjugation with the merocyanine dye afforded a biosensor (Mero-S100A4) that reports on Ca2+-induced activation and the associated structural rearrangements that result in the exposure of the hydrophobic target binding cleft. This new reagent provides a convenient and sensitive readout to screen for inhibitors in vitro, and to begin examining the spatial and temporal dynamics of S100A4 activation in living cells.
Biochemical characterization demonstrated that Mero-S100A4 closely mimics the wild-type protein although slight differences are observed. Ca2+-binding measurements indicate that EF2 of Mero-S100A4 binds calcium ~6-fold tighter than the wild-type protein; however, this difference is unlikely to be significant in vivo due to the submicromolar (0.1–0.2 μM) concentrations of calcium in quiescent cells (41, 42). In addition, although Mero-S100A4 binds myosin-IIA, it does not disassemble filaments, thus the merocyanine moiety is associated with the target binding cleft in a manner that does not block the formation of the binary complex (S100A4–myosin-IIA). Interestingly, the phenothiazines interfere with both myosin-IIA binding and depolymerization. These observations suggest that there are distinct regions of the binding pocket that are involved in target recognition and S100A4’s depolymerizing activity. More importantly, our biochemical findings suggest that the Mero-S100A4 will function as a quiescent indicator of the activated state in vivo since it should not perturb the monomer–polymer equilibrium of myosin-IIA.
Previous studies examining Ca2+-binding to wild-type S100A4 used different methodologies from that reported here. Competition assays with the fluorescent Ca2+ indicator Fluo-3, which binds calcium with a 6-fold tighter affinity than 5,5′Br2-BAPTA, reported a Kd of 2.6 μM for two identical sites (43). This value likely reflects Ca2+-binding to only the high affinity EF2 site since Fluo-3 will not compete effectively with Ca2+ for the low affinity sites. Moreover, this value is consistent with our measurements for the EF2 site. In studies using flow dialysis to measure Ca2+-binding, Kd’s of 100 μM and 222 μM were reported for EF1 and EF2, respectively (44, 45). Several factors may account for the observed differences between these studies and those reported here. Given the difficulties associated with preparing Ca2+-free protein solutions, the contaminating calcium may be sufficiently high to partially occupy the high affinity sites, resulting in titration of only the weak sites, and is consistent with the poor fitting of the data at low calcium concentrations (44). Second, increased turbidity of the protein solution was reported during titration, which further biases the measurements toward lower affinities (44). Furthermore, although our experiments were performed at physiological salt, the geometric mean constant (~36 μM) is the same as that obtained by Gibbs and colleagues at low ionic strength (34–38 μM) (45). However, they reported a 5-fold lower affinity at physiological salt that, if true, would make it impossible for S100A4 to bind calcium in vivo even though its target interactions are calcium dependent (1, 11). This trend of reduced affinity with increasing ionic strength could be an artifact due to the documented effects on adsorption and equilibration of the membrane during dialysis (46). Most importantly, we were able to validate our methodology by measuring Ca2+-binding constants for S100B that were similar to those previously obtained.
Importantly, this biosensor has allowed for the identification of the first compounds of any kind that block S100A4 function. The use of a library of FDA-approved drugs as an initial screen to identify biological effectors of S100A4 has the following advantages: (1) the library members constitute an array of extraordinary structural diversity; (2) for nearly every library member there exists a vast number of structurally related analogues that can be examined for activity; and (3) the majority of library members are approved for human use. Therefore the identification of an effective S100A4 inhibitor has the potential of finding rapid clinical utility.
Based on the properties of the merocyanine dye and the design of the biosensor, we anticipated that our screen would identify compounds that bind within the target binding cleft of S100A4. Compounds that bind the hydrophobic cleft should prevent movement of the merocyanine into the target binding site of S100A4, resulting in an inhibition of the Ca2+-induced fluorescence increase of the biosensor. Our biochemical studies of TFP binding to S100A4 are consistent with this idea. TFP binding does not inhibit Ca2+-binding, rather TFP binds to the target binding cleft that is exposed due to the conformational rearrangements that occur upon Ca2+-binding. Our identification of phenothiazines supports previous reports indicating interactions between S100a, S100b, S100a0 and the phenothiazines chlorpromazine and trifluoperazine (47, 48). However, these studies only report binding of these compounds with no evidence of inhibiting or competing with target interactions. Our screen also identified several other compounds that are structurally unrelated to the phenothiazines.
Selectivity and potency are always major considerations in any drug development process. Accordingly, these issues need to be fully addressed prior to the use of phenothiazines in the treatment or prevention of metastatic disease. Phenothiazines are antipsychotics that not only are anxiolytics but also exhibit antiemetic and hypotensive attributes. These compounds may prove useful as adjuvants for cancer chemotherapy since they interfere with multidrug resistance in tumor cells (49). However, to our knowledge, these agents have not been examined as antimetastatics. At a minimum, these studies provide proof of principle that the biosensor can be used to identify bioactive compounds that block S100A4 function.
Biosensors based on direct covalent attachment of reporter dyes to the targeted protein are used rarely in living cells, and the Mero-S100A4 represents one of the first in vivo applications of these new merocyanine dyes (31, 50). Given the causal role of S100A4 in promoting tumor metastasis (1), the biosensor affords a unique opportunity to examine the spatiotemporal dynamics of S100A4 activation during chemotactic motility of carcinoma cells, and will provide new information on previously inaccessible biology of a major metastasis factor. For example, pathways leading to localized activation of S100A4 can now be studied in living cells using the sensitive readout provided with this new biosensor. To test the validity of the biosensor in vivo we used LPA stimulation of fibroblasts as a well-characterized system to create a condition under which cells would experience a transient and globally high Ca2+ signal. LPA primarily promotes the release of intracellular calcium from the endoplasmic reticulum; resulting in high perinuclear calcium levels (39, 51). Our observation that S100A4 activation is highest at the cell periphery indicates that the biosensor is not merely a calcium indicator, but rather reports on the specific and localized activation of S100A4.
Previous immunofluorescence studies examining total S100A4 demonstrated localization to the leading edge and perinuclear region (14, 15). Importantly, our live cell experiments on randomly migrating cells indicate that activated S100A4 localizes to regions of the cell perimeter undergoing dynamic remodeling. In addition, the biosensor reveals that activation is transient and is associated with specific cellular activities, thus although the overall distribution of S100A4 remains relatively unchanged, activation occurs in specific subcellular locations. These experiments highlight the utility of the biosensor for deciphering the spatial and temporal dynamics of S100A4 activation, and for understanding how S100A4 regulates cell migration.
The S100 proteins share a high degree of sequence homology and a common three-dimensional fold and show similar conformational changes upon binding Ca2+ (35). However, most S100 family members display a high degree of target specificity, suggesting that individual S100 proteins regulate specific cellular processes (52). Moreover, alterations in S100 function are associated with a number of human diseases, including cancer, inflammatory disorders, cardiomyopathies and neurodegeneration (53–55). The successful use of the merocyanine dyes for the creation of an S100A4 biosensor provides an important proof of principle and suggests that this approach can be extended to other S100 family members.
Supplementary Material
Acknowledgments
We thank Dr. Michael Brenowitz for help with the analysis of the Ca2+-binding data, Dr. Vladimir Malashkevich for the molecular models of the Mero-S100A4, and Ms. Natalie Turner for assistance with IC50 measurements for hit compounds. We are grateful to Dr. Jonathan Backer for the use of his needle-puller and to Dr. Steven Almo for critical reading of the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants GM069945 (A.R.B.), CA095019 (D.S.L.) and GM057464 (K.M.H.).
Abbreviations: MIIA, myosin-IIA; FITC, fluorescein; LPA, lysophosphatidic acid; Mero, merocyanine; TFP, trifluoperazine.
SUPPORTING INFORMATION AVAILABLE
Three figures, depicting Ca2+-binding to S100B, binding of wild-type and Mero-S100A4 to myosin-IIA and excitation/emission spectra of Fluo-S100A4, and a table listing the compounds in the library of FDA-approved drugs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Garrett SC, Varney KM, Weber DJ, Bresnick AR. S100A4, a mediator of metastasis. J Biol Chem. 2006;281:677–680. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 2.Davies BR, Davies MP, Gibbs FE, Barraclough R, Rudland PS. Induction of the metastatic phenotype by transfection of a benign rat mammary epithelial cell line with the gene for p9Ka, a rat calcium-binding protein, but not with the oncogene EJ-ras-1. Oncogene. 1993;8:999–1008. [PubMed] [Google Scholar]
- 3.Grigorian M, Ambartsumian N, Lykkesfeldt AE, Bastholm L, Elling F, Georgiev G, Lukanidin E. Effect of mts1 (S100A4) expression on the progression of human breast cancer cells. Int J Cancer. 1996;67:831–841. doi: 10.1002/(SICI)1097-0215(19960917)67:6<831::AID-IJC13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Davies MP, Rudland PS, Robertson L, Parry EW, Jolicoeur P, Barraclough R. Expression of the calcium-binding protein S100A4 (p9Ka) in MMTV-neu transgenic mice induces metastasis of mammary tumours. Oncogene. 1996;13:1631–1637. [PubMed] [Google Scholar]
- 5.Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res. 2003;63:3386–3394. [PubMed] [Google Scholar]
- 6.Maelandsmo GM, Hovig E, Skrede M, Engebraaten O, Florenes VA, Myklebost O, Grigorian M, Lukanidin E, Scanlon KJ, Fodstad O. Reversal of the in vivo metastatic phenotype of human tumor cells by an anti-CAPL (mts1) ribozyme. Cancer Res. 1996;56:5490–5498. [PubMed] [Google Scholar]
- 7.Takenaga K, Nakamura Y, Sakiyama S. Expression of antisense RNA to S100A4 gene encoding an S100-related calcium-binding protein suppresses metastatic potential of high-metastatic Lewis lung carcinoma cells. Oncogene. 1997;14:331–337. doi: 10.1038/sj.onc.1200820. [DOI] [PubMed] [Google Scholar]
- 8.Rudland PS, Platt-Higgins A, Renshaw C, West CR, Winstanley JH, Robertson L, Barraclough R. Prognostic significance of the metastasis-inducing protein S100A4 (p9Ka) in human breast cancer. Cancer Res. 2000;60:1595–1603. [PubMed] [Google Scholar]
- 9.de Silva Rudland S, Martin L, Roshanlall C, Winstanley J, Leinster S, Platt-Higgins A, Carroll J, West C, Barraclough R, Rudland P. Association of S100A4 and osteopontin with specific prognostic factors and survival of patients with minimally invasive breast cancer. Clin Cancer Res. 2006;12:1192–1200. doi: 10.1158/1078-0432.CCR-05-1580. [DOI] [PubMed] [Google Scholar]
- 10.Platt-Higgins AM, Renshaw CA, West CR, Winstanley JH, De Silva Rudland S, Barraclough R, Rudland PS. Comparison of the metastasis-inducing protein S100A4 (p9ka) with other prognostic markers in human breast cancer. Int J Cancer. 2000;89:198–208. [PubMed] [Google Scholar]
- 11.Li ZH, Spektor A, Varlamova O, Bresnick AR. Mts1 regulates the assembly of nonmuscle myosin-IIA. Biochemistry. 2003;42:14258–14266. doi: 10.1021/bi0354379. [DOI] [PubMed] [Google Scholar]
- 12.Kolega J. Cytoplasmic dynamics of myosin IIA and IIB: spatial ‘sorting’ of isoforms in locomoting cells. J Cell Sci. 1998;111(Part 15):2085–2095. doi: 10.1242/jcs.111.15.2085. [DOI] [PubMed] [Google Scholar]
- 13.Betapudi V, Licate LS, Egelhoff TT. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 2006;66:4725–4733. doi: 10.1158/0008-5472.CAN-05-4236. [DOI] [PubMed] [Google Scholar]
- 14.Kim EJ, Helfman DM. Characterization of the metastasis-associated protein, S100A4. Roles of calcium binding and dimerization in cellular localization and interaction with myosin. J Biol Chem. 2003;278:30063–30073. doi: 10.1074/jbc.M304909200. [DOI] [PubMed] [Google Scholar]
- 15.Li Z-H, Bresnick AR. S100A4 regulates cellular motility via a direct interaction with myosin-IIA. Cancer Res. 2006;66:5173–5180. doi: 10.1158/0008-5472.CAN-05-3087. [DOI] [PubMed] [Google Scholar]
- 16.Toutchkine A, Kraynov V, Hahn K. Solvent-sensitive dyes to report protein conformational changes in living cells. J Am Chem Soc. 2003;125:4132–4145. doi: 10.1021/ja0290882. [DOI] [PubMed] [Google Scholar]
- 17.Linse S. Calcium binding to proteins studied via competition with chromophoric chelators. Methods Mol Biol. 2002;173:15–24. doi: 10.1385/1-59259-184-1:015. [DOI] [PubMed] [Google Scholar]
- 18.Andre I, Linse S. Measurement of Ca2+-binding constants of proteins and presentation of the CaLigator software. Anal Biochem. 2002;305:195–205. doi: 10.1006/abio.2002.5661. [DOI] [PubMed] [Google Scholar]
- 19.Markowitz J, Rustandi RR, Varney KM, Wilder PT, Udan R, Wu SL, Horrocks WD, Weber DJ. Calcium-binding properties of wild-type and EF-hand mutants of S100B in the presence and absence of a peptide derived from the C-terminal negative regulatory domain of p53. Biochemistry. 2005;44:7305–7314. doi: 10.1021/bi050321t. [DOI] [PubMed] [Google Scholar]
- 20.Potter JD, Gergely J. The calcium and magnesium binding sites on troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J Biol Chem. 1975;250:4628–4633. [PubMed] [Google Scholar]
- 21.Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Wyman J, Gill SJ. Binding and Linkage: Functional Chemistry of Biological Macromolecules. University Science Books; Mill Valley, CA: 1990. [Google Scholar]
- 23.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Stukel T. Generalized Logistic Models. J Am Stat Assoc. 1988;83:426–431. [Google Scholar]
- 25.Shen F, Hodgson L, Hahn K. Digital autofocus methods for automated microscopy. Methods Enzymol. 2006;414:620–632. doi: 10.1016/S0076-6879(06)14032-X. [DOI] [PubMed] [Google Scholar]
- 26.Hodgson L, Nalbant P, Shen F, Hahn K. Imaging and photobleach correction of Mero-CBD, sensor of endogenous Cdc42 activation. Methods Enzymol. 2006;406:140–156. doi: 10.1016/S0076-6879(06)06012-5. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharya S, Large E, Heizmann CW, Hemmings B, Chazin WJ. Structure of the Ca2+/S100B/NDR kinase peptide complex: insights into S100 target specificity and activation of the kinase. Biochemistry. 2003;42:14416–14426. doi: 10.1021/bi035089a. [DOI] [PubMed] [Google Scholar]
- 28.Maler L, Potts BC, Chazin WJ. High resolution solution structure of apo calcyclin and structural variations in the S100 family of calcium-binding proteins. J Biomol NMR. 1999;13:233–247. doi: 10.1023/a:1008315517955. [DOI] [PubMed] [Google Scholar]
- 29.Inman KG, Yang R, Rustandi RR, Miller KE, Baldisseri DM, Weber DJ. Solution NMR structure of S100B bound to the high-affinity target peptide TRTK-12. J Mol Biol. 2002;324:1003–1014. doi: 10.1016/s0022-2836(02)01152-x. [DOI] [PubMed] [Google Scholar]
- 30.Rustandi RR, Baldisseri DM, Weber DJ. Structure of the negative regulatory domain of p53 bound to S100B(betabeta) Nat Struct Biol. 2000;7:570–574. doi: 10.1038/76797. [DOI] [PubMed] [Google Scholar]
- 31.Nalbant P, Hodgson L, Kraynov V, Toutchkine A, Hahn KM. Activation of endogenous Cdc42 visualized in living cells. Science. 2004;305:1615–1619. doi: 10.1126/science.1100367. [DOI] [PubMed] [Google Scholar]
- 32.Pertz O, Hahn KM. Designing biosensors for Rho family proteins–deciphering the dynamics of Rho family GTPase activation in living cells. J Cell Sci. 2004;117:1313–1318. doi: 10.1242/jcs.01117. [DOI] [PubMed] [Google Scholar]
- 33.Drohat AC, Amburgey JC, Abildgaard F, Starich MR, Baldisseri D, Weber DJ. Solution structure of rat apo-S100B(beta beta) as determined by NMR spectroscopy. Biochemistry. 1996;35:11577–11588. doi: 10.1021/bi9612226. [DOI] [PubMed] [Google Scholar]
- 34.Drohat AC, Baldisseri DM, Rustandi RR, Weber DJ. Solution structure of calcium-bound rat S100B(betabeta) as determined by nuclear magnetic resonance spectroscopy. Biochemistry. 1998;37:2729–2740. doi: 10.1021/bi972635p. [DOI] [PubMed] [Google Scholar]
- 35.Vallely KM, Rustandi RR, Ellis KC, Varlamova O, Bresnick AR, Weber DJ. Solution structure of human Mts1 (S100A4) as determined by NMR spectroscopy. Biochemistry. 2002;41:12670–12680. doi: 10.1021/bi020365r. [DOI] [PubMed] [Google Scholar]
- 36.Rustandi RR, Drohat AC, Baldisseri DM, Wilder PT, Weber DJ. The Ca(2+)-dependent interaction of S100B(beta beta) with a peptide derived from p53. Biochemistry. 1998;37:1951–1960. doi: 10.1021/bi972701n. [DOI] [PubMed] [Google Scholar]
- 37.Bright GR, Fisher GW, Rogowska J, Taylor DL. Fluorescence ratio imaging microscopy. Methods Cell Biol. 1989;30:157–192. doi: 10.1016/s0091-679x(08)60979-6. [DOI] [PubMed] [Google Scholar]
- 38.Jalink K, van Corven EJ, Moolenaar WH. Lysophosphatidic acid, but not phosphatidic acid, is a potent Ca2-(+)-mobilizing stimulus for fibroblasts. Evidence for an extra-cellular site of action. J Biol Chem. 1990;265:12232–12239. [PubMed] [Google Scholar]
- 39.Lee ZW, Kweon SM, Kim BC, Leem SH, Shin I, Kim JH, Ha KS. Phosphatidic acid-induced elevation of intracellular Ca2+ is mediated by RhoA and H2O2 in Rat-2 fibroblasts. J Biol Chem. 1998;273:12710–12715. doi: 10.1074/jbc.273.21.12710. [DOI] [PubMed] [Google Scholar]
- 40.Emmert DA, Fee JA, Goeckeler ZM, Grojean JM, Wakatsuki T, Elson EL, Herring BP, Gallagher PJ, Wysolmerski RB. Rho-kinase-mediated Ca2+-independent contraction in rat embryo fibroblasts. Am J Physiol Cell Physiol. 2004;286:C8–C21. doi: 10.1152/ajpcell.00428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giovannardi S, Peres A. Nuclear and cytosolic calcium levels in NIH-3T3 fibroblasts. Exp Biol Online. 1997;2:1–9. [Google Scholar]
- 42.Hallett MB, Hodges R, Cadman M, Blanchfield H, Dewitt S, Pettit EJ, Laffafian I, Davies EV. Techniques for measuring and manipulating free Ca2+ in the cytosol and organelles of neutrophils. J Immunol Methods. 1999;232:77–88. doi: 10.1016/s0022-1759(99)00167-2. [DOI] [PubMed] [Google Scholar]
- 43.Dukhanina EA, Dukhanin AS, Lomonosov MY, Lukanidin EM, Georgiev GP. Spectral studies on the calcium-binding properties of Mts1 protein and its interaction with target protein. FEBS Lett. 1997;410:403–406. doi: 10.1016/s0014-5793(97)00576-0. [DOI] [PubMed] [Google Scholar]
- 44.Pedrocchi M, Schafer BW, Durussel I, Cox JA, Heizmann CW. Purification and characterization of the recombinant human calcium-binding S100 proteins CAPL and CACY. Biochemistry. 1994;33:6732–6738. doi: 10.1021/bi00187a045. [DOI] [PubMed] [Google Scholar]
- 45.Gibbs FE, Wilkinson MC, Rudland PS, Barraclough R. Interactions in vitro of p9Ka, the rat S-100-related, metastasis-inducing, calcium-binding protein. J Biol Chem. 1994;269:18992–18999. [PubMed] [Google Scholar]
- 46.Desoye G. Error analysis in equilibrium dialysis: evaluation of adsorption phenomena. J Biochem Biophys Methods. 1988;17:3–16. doi: 10.1016/0165-022x(88)90073-5. [DOI] [PubMed] [Google Scholar]
- 47.Donato R. Chlorpromazine inhibits the calcium-mediated effects of S-100 protein(s) on assembled brain microtubule proteins, but not those on microtubule protein assembly. Biochem Biophys Res Commun. 1984;122:983–990. doi: 10.1016/0006-291x(84)91188-4. [DOI] [PubMed] [Google Scholar]
- 48.Pingerelli PL, Mizukami H, Wagner AS, Bartnicki DE, Oliver JP. Investigation of the Ca2(+)-dependent interaction of trifluoperazine with S100a: a 19F NMR and circular dichroism study. J Protein Chem. 1990;9:169–175. doi: 10.1007/BF01025308. [DOI] [PubMed] [Google Scholar]
- 49.Tsakovska I, Pajeva I. Phenothiazines and structurally related compounds as modulators of cancer multidrug resistance. Curr Drug Targets. 2006;7:1123–1134. doi: 10.2174/138945006778226660. [DOI] [PubMed] [Google Scholar]
- 50.Hahn K, DeBiasio R, Taylor DL. Patterns of elevated free calcium and calmodulin activation in living cells. Nature. 1992;359:736–738. doi: 10.1038/359736a0. [DOI] [PubMed] [Google Scholar]
- 51.Kim JH, Johannes L, Goud B, Antony C, Lingwood CA, Daneman R, Grinstein S. Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc Natl Acad Sci USA. 1998;95:2997–3002. doi: 10.1073/pnas.95.6.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zimmer DB, Wright Sadosky P, Weber DJ. Molecular mechanisms of S100-target protein interactions. Microsc Res Tech. 2003;60:552–559. doi: 10.1002/jemt.10297. [DOI] [PubMed] [Google Scholar]
- 53.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochem Biophys Res Commun. 2004;322:1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 54.Roth J, Vogl T, Sorg C, Sunderkotter C. Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 2003;24:155–158. doi: 10.1016/s1471-4906(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 55.Most P, Bernotat J, Ehlermann P, Pleger ST, Reppel M, Borries M, Niroomand F, Pieske B, Janssen PM, Eschenhagen T, Karczewski P, Smith GL, Koch WJ, Katus HA, Remppis A. S100A1: a regulator of myocardial contractility. Proc Natl Acad Sci USA. 2001;98:13889–13894. doi: 10.1073/pnas.241393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.