

Abstract
Model quality assessment (MQA) is an integral part of protein structure prediction methods that typically generate multiple candidate models. The challenge lies in ranking and selecting the best models using a variety of physical, knowledge-based, and geometric consensus (GC)-based scoring functions. In particular, 3D-Jury and related GC methods assume that well-predicted (sub-)structures are more likely to occur frequently in a population of candidate models, compared to incorrectly folded fragments. While this approach is very successful in the context of diversified sets of models, identifying similar substructures is computationally expensive since all pairs of models need to be superimposed using MaxSub or related heuristics for structure-to-structure alignment. Here, we consider a fast alternative, in which structural similarity is assessed using 1D profiles, e.g., consisting of relative solvent accessibilities and secondary structures of equivalent amino acid residues in the respective models. We show that the new approach, dubbed 1D-Jury, allows to implicitly compare and rank N models in O(N) time, as opposed to quadratic complexity of 3D-Jury and related clustering-based methods. In addition, 1D-Jury avoids computationally expensive 3D superposition of pairs of models. At the same time, structural similarity scores based on 1D profiles are shown to correlate strongly with those obtained using MaxSub. In terms of the ability to select the best models as top candidates 1D-Jury performs on par with other GC methods. Other potential applications of the new approach, including fast clustering of large numbers of intermediate structures generated by folding simulations, are discussed as well.
Key words: 1D-Jury, 3D-Jury, clustering, geometric consensus, model quality assessment, protein structure prediction, solvent accessibility, structural similarity
1. Introduction
Protein structure prediction methods typically generate large sets of alternative models by sampling alternative plausible conformations, e.g., using alternative structural templates or fragment libraries (Zhang, 2008; Lee, 2009; Simons et al., 1997; Xia et al., 2000; Wu et al., 2007). Therefore, scoring functions and methods for model quality assessment (MQA) that can be used to select the best models from a set of candidate structures are essential for progress in the field (Cozzetto et al., 2009; Moult et al., 2007, 2009). MQA methods use a variety of physical, knowledge-based and geometric measures to rank available models and discriminate between native-like and incorrect structures (Lüthym et al., 1992; Gatchell et al., 2000; Ginalski et al., 2003; Eramian et al., 2006; Swanson et al., 2009). For example, inter-residue or inter-atomic pairwise potentials are commonly used as measures of model quality (Samudrala and Moult, 1998; Lu and Skolnick, 2001; Zhang et al., 2004), often in combination with other signals, such as the agreement between predicted (from sequence) and observed secondary structures (Pettitt et al., 2005; Benkert et al., 2008). These effective combinations of physical and knowledge-based measures can be optimized using machine learning approaches, involving extrapolation from a set of properly stratified and representative examples (Lundström et al., 2001; Qiu et al., 2008).
Methods that can be used to assess individual models independently are referred to as “true” MQA methods. On the other hand, clustering or structural consensus-based methods require a set of models to identify well-predicted (sub-)structures by assessing consistency between the models (McGuffin, 2007; Kaján and Rychlewski, 2007; Cozzetto et al., 2009). An example of the latter approach, which will be referred to as geometric consensus (GC), is the 3D-Jury method (Ginalski et al., 2003). 3D-Jury uses MaxSub heuristic (Siew et al., 2000) to identify similar (and frequently occurring) substructures as a basis for ranking. Other GC methods use very similar overall strategy. For example, top performing in CASP8 MQA evaluation Pcons (Larsson et al., 2009) and ModFoldClust (McGuffin, 2008) methods are very similar to 3D-Jury, although they employ different structure similarity measures: LGscore and TM-score, respectively (Cristobal et al., 2001; Zhang and Skolnick, 2004).
Even though 3D-Jury has originally been devised to combine a relatively small number of high-quality models from well-benchmarked methods, GC-based methods have subsequently been shown to perform very well in the context of the Critical Assessment of Techniques for Protein Structure Prediction (CASP) evaluations, outperforming “true” MQA methods (Cozzetto et al., 2009) and stimulating further development of such methods. In order to address some of the limitations of GC-based methods, i.e., the dependence on a set of (at least partially correct) models, some methods successfully derive measures of structural consistency using template and fragment-based approaches or combine structural consensus evaluation with physical and knowledge-based features to enhance the performance (Zhou and Skolnick, 2008; Qiu et al., 2008; Benkert et al., 2008; Larsson et al., 2009).
However, the computational cost of performing structure-to-structure alignment for all pairs of models, which is required to identify frequently occurring substructures, limits applications of 3D-Jury and related GC approaches to relatively small data sets. At the same time, protein structure prediction methods are increasingly capable of generating very large numbers of models (Moult et al., 2005, 2009; Lee, 2009). Assessing GC could not only improve ranking and selection of best models in this context, but also help monitoring the quality of sampling and convergence of the simulations. Molecular dynamics, computational studies on kinetics of folding, and other types of protein simulations also increasingly generate large numbers of intermediate structures (of the same protein) that are often assigned to distinct states based on clustering of their conformations (Laboulais et al., 2002; Elmer et al., 2005; Elber and West, 2010).
In this work, a fast alternative to 3D-Jury and related approaches is introduced and evaluated. The new method is dubbed 1D-Jury as it relies on simple 1D structural profiles consisting of secondary structures and solvent accessibilities of individual amino acid residues. Such profiles have been used before, e.g., to enhance fold recognition and threading methods (Zhou and Zhou, 2005; Fischer, 2003), and are also at the core of some “true” MQA methods, such as Verify-3D (Lüthym et al., 1992). In addition, various ways of reducing 3D information for fast structure-to-structure alignment have been proposed in the past, e.g., by projecting backbone distance matrices into feature profiles (Choi et al., 2003), or representing overall structure as frequency profile over a library of fragments (Budowski-Tal et al., 2010). However, a general case of structure comparison for arbitrary structures is not considered here. In MQA multiple models of the same protein are given, which simplifies the problem. Our goal is to exploit that special structure further in order to provide a basis for efficient and accurate clustering-based approach for model quality assessment.
Consequently, rather than using 1D profiles to assess individual structures independently, as for example in Verify-3D, the score of a model (and individual residues within this model) is derived using a set of models, in analogy to 3D-Jury. Structural consistency between models is evaluated efficiently by comparing secondary structure (SS) and solvent accessibility (SA) states of equivalent residues (that can be found by aligning each model to the target sequence using sequence alignment methods). Thus, computationally expensive superposition of 3D structures for pairs of models is avoided. Moreover, as shown in this contribution, formulating the problem in terms of SS/SA (and related) profiles allows one to implicitly compare and rank N models in O(N) time, as opposed to quadratic complexity of 3D-Jury and related GC-based methods.
While the linear time complexity of 1D-Jury for a set of models reduces dramatically running times compared to current GC methods, approximating structural similarity in terms of 1D projection of 3D coordinates could lead to significant loss in accuracy. As demonstrated using multiple data sets, however, this is not the case in the context of MQA. First of all, the resulting profile-based similarity measure is shown to correlate strongly with MaxSub scores derived using superposition of 3D structures. For example, correlation coefficient of 0.9 (or more) between the two measures is obtained on the CASP7 and CASP8 sets of protein models. Moreover, applying 1D-Jury to data sets of models derived using both fragment and template-based protein structure prediction methods, the accuracy of 1D-Jury in terms of its ability to select the best models is shown to be on par with 3D-Jury and other top performing GC methods.
2. Methods
2.1. 1D-Jury scoring function
The 3D-Jury scoring function (Ginalski et al., 2003) simply counts residues within similar substructures found for each pair of models by way of superimposing these models using the MaxSub program (Siew et al., 2000), and it can be written as follows:
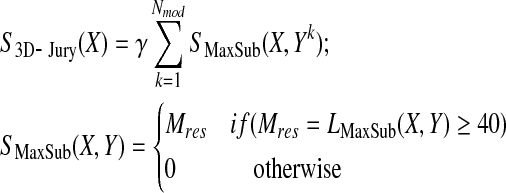 |
Mres in the above formula is defined as the number of residues in MaxSub alignment of models X and Y, i.e., the number of equivalent residues such that their distance is smaller than certain threshold (typically chosen to be 3.5 or 5 Ang, here we use the latter since it yields somewhat better results in the context of MQA) once X and Y are superimposed using the MaxSub heuristic. Models that do not share similar substructures of length of at least 40 do not contribute to the score. The normalization constant, γ, which does not affect the relative ranking of models (but could affect transferability of scores between different sets of models), was originally chosen to be one over the number of models and we used the same normalization here. Thus, 3D-Jury relies on the consensus among models to identify well-predicted (sub-)structures.
The 1D-Jury scoring function takes a similar form. However, consensus between models is assessed without the computationally expensive 3D superposition. Rather, simple structural profiles, consisting of relative solvent accessibilities (RSA) and secondary structures (SS), are used to compare pairs of models in conjunction with sequence alignments (to the target sequence) to establish equivalent positions. Namely, equivalent residues contribute to the score if they are assigned to the same (or sufficiently similar, as indicated by ≅ relationship) secondary structure and solvent accessibility states (it should be noted that this can be generalized to include other per residue measures of local environment, e.g., the number of neighbors, backbone torsion angles, contact potentials, etc.):
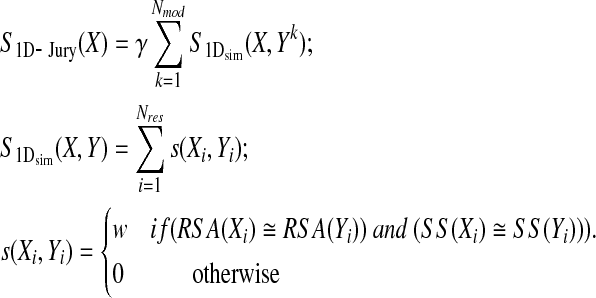 |
where Nres is the number of residues in X, w is a positive number that may be different for different joint (RSA, SS) states, and the normalization constant is defined as previously. The relative solvent accessibility of residue i, RSA(Xi), is a real number between 0 and 1, which is defined as the surface exposed area (computed here using the DSSP program (Kabsch and Sander, 1983)) relative to the maximum surface exposed area (these values are the same as used in Adamczak et al., 2004). Real-valued RSAs are projected here onto 10 classes using bins [0, 0.1), [0.1, 0.2), … , [0.9,1.0], represented by integers from 0 to 9. Since such projection introduces some noise (e.g., two residues may be considered to have different RSA states, although their real RSAs are very close), we also considered an alternative definition in which two residues are regarded as having the same solvent accessibility state if the difference between their real-valued RSAs is smaller than 0.1 (the results of 1D-Jury are largely insensitive to the particular choice of this threshold). However, improvements with respect to discrete approximation are insignificant, yielding 0.01–0.02 higher correlation coefficients between predicted and true ranks on CASP6 data. On the other hand, projection into discrete classes allows one to implement 1D-Jury for a set of models in linear time with respect to the number of models, as discussed later.
The secondary structures are also computed using DSSP, and are compared in terms of 3-state projection of the original 8 states (G, I, H - > H; B - > E; C otherwise). Consistency of secondary structure elements is expected to play an important role when assessing model quality. Therefore, we tested a version of the 1D-Jury scoring function in which residues assigned to different secondary structure states contribute differently to the overall score. Namely, if equivalent residues with the same RSA state are in the same secondary structure (i.e., they are both either in H or E state), they contribute twice as much as residues that are both in C state (w = 2 for (H,H) and (E,E) pairs, as opposed to w = 1 for (C,C) pairs). In addition, in order to avoid penalizing edge effects too strongly, pairs of equivalent residues with the same RSA that match C state with H or E, contribute w = 0.5 to the overall score. Such weighting of different secondary structure states resulted in somewhat improved performance (increasing correlation coefficients between true and predicted rankings by about 0.04 on CASP6 data), compared to the baseline method, in which all matching pairs contribute a score of one. Residues with missing coordinates are assigned to an arbitrary GAP state, so that they do not contribute to the score. It should be noted that further regularization and slight improvements in the overall accuracy can be achieved by introducing, in analogy to 3D-Jury, a threshold for the number of equivalent positions with the same SS and similar RSA in a model for that model to contribute to the score. However, this increases the computational complexity as discussed below, and we do not use such a threshold in the implementation presented here.
2.2. Computational complexity of 1D-Jury
Discretizing RSAs, and avoiding the requirement of contiguous fragments of certain length to satisfy similarity criteria to contribute to the score, allows one to implement the problem of computing scores for all Nmod models with 1D-Jury without a loop over pairs of models. This can be shown by considering the following implementation. In the first step, sequences of all models are aligned to the reference target sequence (in linear time in the number of models) to establish equivalent positions by way of constructing pseudo-multiple alignment of all model sequences, and to account for incomplete models and models with gaps. In our current implementation, this step is not optimized, involving 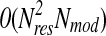 complexity since the Smith-Waterman algorithm, as implemented in LOOPP (Meller and Elber, 2001), is used to compute alignments. In the next step, the DSSP program is used to compute RSAs and SSs for each residue (independently for each model). Initial Nmod alignments to the target sequence, and subsequent Nmod DSSP runs, provide all information required to compute 1D-Jury score, i.e., equivalent positions and their joint state assignments, e.g., (RSA = 3, SS = H).
complexity since the Smith-Waterman algorithm, as implemented in LOOPP (Meller and Elber, 2001), is used to compute alignments. In the next step, the DSSP program is used to compute RSAs and SSs for each residue (independently for each model). Initial Nmod alignments to the target sequence, and subsequent Nmod DSSP runs, provide all information required to compute 1D-Jury score, i.e., equivalent positions and their joint state assignments, e.g., (RSA = 3, SS = H).
The contribution of each position is independent in 1D-Jury, adding to the score whenever both SS and RSA states of equivalent residues are the same (or regarded as sufficiently similar). Therefore, by pre-computing the number of residues in each joint (RSA, SS) state in each column of pseudo-multiple alignment (an operation which is again linear in terms of the number of models, i.e., rows in the multiple alignment), one can obtain a score at that position in any given model by a simple look-up operation. Namely, the score due to a residue assigned to one of the joint (RSA, SS) states is given by the total number of residues in that state (or states regarded as similar) observed for equivalent positions in this residue's column of the multiple alignment. This is illustrated in Figure 1 and can be better understood in terms of exchange of summation indices in the formula for the 1D-Jury score:
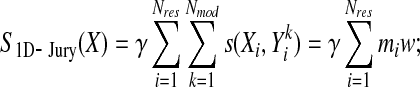 |
FIG. 1.

Schematic illustration of an efficient 1D-Jury implementation with linear time complexity in the number of models. The total number of residues in each 1D state (here represented by black and white boxes for a simplified two state problem) at each position is pre-computed first, allowing one to compute the 1D-Jury score for a model without pairwise comparison with other models. For example, the score of 25 for the model M2 is computed as the sum of the numbers of the black states in columns 1–4 and 6–8, and the numbers of the white states in columns 5 and 9–10, in which the corresponding residues in M2 are also in the black or white state, respectively.
where mi is the total number of residues 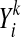 in column i that are the assigned to the same state as that of Xi.
in column i that are the assigned to the same state as that of Xi.
Thus, since each of the steps requires 0(Nmod) operations, the assignment of scores for all models in the set can be achieved in linear time. 3D-Jury, on the other hand, requires 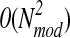 operations to score all models: for each model all other models need to be superimposed with it using MaxSub heuristic. In addition, the use of MaxSub (or similar programs) implies a significant (and length dependent) cost of structure-to-structure alignment, which makes the use of 3D-Jury prohibitive for large sets of models. It should be also noted that even in quadratic implementations of 1D-Jury that allow for further refinements of the scoring scheme as discussed above, the cost of pairwise comparison in 1D-Jury is reduced to a simple evaluation of 1D profiles, resulting in significant decreases in the running time compared to 3D-Jury (compare 1D-Jury(Q) and 1D-Jury in the Results section). This kind of implementation could be considered, e.g., in application to studies on kinetics of folding that require explicit structure clustering of intermediate states.
operations to score all models: for each model all other models need to be superimposed with it using MaxSub heuristic. In addition, the use of MaxSub (or similar programs) implies a significant (and length dependent) cost of structure-to-structure alignment, which makes the use of 3D-Jury prohibitive for large sets of models. It should be also noted that even in quadratic implementations of 1D-Jury that allow for further refinements of the scoring scheme as discussed above, the cost of pairwise comparison in 1D-Jury is reduced to a simple evaluation of 1D profiles, resulting in significant decreases in the running time compared to 3D-Jury (compare 1D-Jury(Q) and 1D-Jury in the Results section). This kind of implementation could be considered, e.g., in application to studies on kinetics of folding that require explicit structure clustering of intermediate states.
2.3. Model sets
We used several sets of models and decoys to test the performance of 1D-Jury, and compare it with the 3D-Jury and other GC approaches. These sets include CASP6-8 models (Moult et al., 2005, 2007, 2009), MOULDER (Eramian et al., 2006), and LOOPP (Vallat et al., 2008, 2009) sets of decoys of varying quality generated using fold recognition and homology modeling, as well as the TASSER set obtained by de novo (fragment-based) folding approach (Wu et al., 2007; Zhou et al., 2007).
The latter set represents a case in which a single method with specific biases is used to generate all the models. As a result, TASSER models tend to cluster more tightly compared to CASP models, which is indicated by the mean standard deviation of the mean (per target) MaxSub score (Table 1, last column). This allows one to probe limits of the applicability of clustering-based MQA approaches, which generally work well when sufficiently stratified sets of models are available. Further details regarding sets of models used here are described in the Supplementary Material (available at www.liebertonline.com/cmb).
Table 1.
Summary of Data Sets Used in the Study
| Data set | Targets | Models per target | Mean Top5 | Mean MaxSub per target | Mean SD of MaxSub per targ. |
|---|---|---|---|---|---|
| CASP7 | 94 | 407 ± 28 | 0.69 ± 0.21 | 0.49 ± 0.25 | 0.15 ± 0.06 |
| 0.65 ± 0.20* | |||||
| TASSER | 56 | 1868 ± 598 | 0.81 ± 0.11 | 0.63 ± 0.19 | 0.06 ± 0.05 |
| MOULDER | 20 | 320 ± 39 | 0.82 ± 0.06 | 0.42 ± 0.06 | 0.22 ± 0.04 |
| LOOPP | 91 | 231 ± 114 | 0.63 ± 0.21 | 0.17 ± 0.09 | 0.15 ± 0.08 |
For each set, the number of targets, average number of models per target, averaged per target mean MaxSub (or GDT_TS, if denoted by *) score for five best models (Mean Top5), which is the prediction limit for Top5 measure, averaged per target mean MaxSub score for all models, and averaged per target standard deviation of the mean MaxSub score for all models, which reflects diversity of models, are given in the respective columns.
3. Results and Discussion
Using the CASP7 data set, we first compared MaxSub and the new 1Dsim profile-based measures of structural similarity, which are then used to detect similar substructures, and as a basis of model ranking in 3D- and 1D-Jury, respectively. In order to enable comparison across different targets, both of these scores were normalized to the [0, 1] interval by dividing the raw scores by the respective maximum scores, i.e., by the total length of the target sequence in the case of the MaxSub score, and by the length of secondary structure elements in the native state with appropriate weights (w = 2 for H and E, and w = 1 for C) for the 1Dsim score. If no additional weights are used for 1D-Jury, the maximum score is again just the length of the target sequence.
As can be seen from Figure 2, there is a very clear correlation between MaxSub and 1Dsim similarity measures. An overall Pearson correlation coefficient (CC(P)) of 0.91, and Spearman rank CC (CC(S)) of 0.90 were obtained when considering similarity scores between (ZT,YkT) pairs of CASP7 models, YkT, and their respective target native structures, ZT, over all targets indexed by T (which corresponds to data included in Fig. 2). Very similar correlations (CC(P) of 0.9) were also observed between MaxSub and 1Dsim scores computed for pairs of models, (YkT, YlT), even though matching two models (rather than a model and its target native structure) could, in principle, result in more inconsistencies between MaxSub and 1Dsim scores. When these correlations were assessed for each target separately, a mean per target CC(P) of 0.81 was obtained, which is somewhat lower primarily due to several relatively short targets for which many low-quality models obtain zero MaxSub scores.
FIG. 2.

Comparison of normalized MaxSub (x-axis) and 1Dsim (y-axis) structural similarity scores for CASP7 models and their respective native (target) structures. 1Dsim and MaxSub measures correlate with the Pearson correlation coefficient of 0.90 and Spearman rank correlation of 0.91 in this case. Note that MaxSub scores for models with less than 20 residues aligned to the native structure are set to zero, resulting in the observed gap near the y-axis and affecting correlation between 1Dsim and MaxSub.
Despite these strong correlations, some off-diagonal density can be observed in Figure 2, mainly with low 1DSim scores and high MaxSub scores. In order to understand better the limitations of the proposed approach, we analyzed the sources of such discrepancies. These include the above mentioned cutoff for the length of similar substructures, used by MaxSub and 3D-Jury, as well as loss of information due to projection of 3D coordinates into 1D (SS, RSA) profiles. Regarding the latter, it should be noted that in addition to backbone only models that were excluded, a fraction of about 3.5% of models had incomplete coordinates for at least some side chains. Such residues, if exposed, are assigned to incorrect RSA states, adding to the noise. Excluding models that contain such residues increases the observed correlations by about 0.01 on CASP7 set. Nevertheless, we decided to include such models in our analysis to test the performance on a possibly large subset of CASP models.
Other types of discrepancies between 1Dsim and MaxSub are related to models that although very similar in terms of 3D superposition, are characterized by distorted secondary structures, resulting in low 1Dsim scores. Examples of such models are shown in the Supplementary Material. While some of these limitations could potentially be addressed, e.g., by using backbone torsion angles to define 1D profiles, one can conclude that the overall and per target correlations are already very strong, especially for relatively high-quality models, which bodes well for the use of much more efficient 1Dsim measure in the context of MQA.
In order to assess the performance of the new approach, the results of 1D-Jury were first compared to those obtained using 3D-Jury on the data sets from Table 1. 3D-Jury constitutes a useful reference for evaluation of 1D-Jury not only because of their conceptual similarity, but also because 3D-Jury was found to perform on par with the best GC methods in the context of CASP (McGuffin, 2007). Additional support for this conclusion is included in Supplementary Material (and below), based on the comparison of 3D-Jury with other state-of-the-art GC-based methods using CASP7 and CASP8 data.
Table 2 summarizes this comparison using several measures commonly applied to evaluate MQA methods. In particular, mean Pearson and Spearman rank correlation coefficients between 3D- and 1D-Jury scores/rankings on the one hand, and true scores/ranks on the other hand, are reported. These mean values were obtained by computing correlation coefficients for individual targets, and then computing averages over all targets (which also provides standard deviations of the mean CCs as a measure of variability for individual targets).
Table 2.
Comparison of Results for 1D-Jury and 3D-Jury Approaches
| Data set | 1D-Jury: Mean Top5 | 3D-Jury: Mean Top5 | 1D-Jury: Mean CC(P) | 3D-Jury: Mean CC(P) | 1D-Jury: Mean CC(S) | 3D-Jury: Mean CC(S) |
|---|---|---|---|---|---|---|
| CASP7 (GDT_TS) | 0.59 ± 0.22 | 0.60 ± 0.26 | 0.78 ± 0.16 | 0.83 ± 0.25 | 0.75 ± 0.13 | 0.77 ± 0.23 |
| CASP7 (MaxSub) | 0.62 ± 0.25 | 0.64 ± 0.26 | 0.75 ± 0.16 | 0.83 ± 0.25 | 0.71 ± 0.13 | 0.77 ± 0.23 |
| TASSER (MaxSub) | 0.66 ± 0.19 | 0.66 ± 0.19 | 0.35 ± 0.19 | 0.50 ± 0.30 | 0.20 ± 0.17 | 0.32 ± 0.28 |
| MOULDER (MaxSub) | 0.71 ± 0.10 | 0.71 ± 0.10 | 0.91 ± 0.04 | 0.93 ± 0.04 | 0.91 ± 0.05 | 0.94 ± 0.04 |
| LOOPP (MaxSub) | 0.54 ± 0.23 | 0.51 ± 0.28 | 0.63 ± 0.22 | 0.71 ± 0.25 | 0.61 ± 0.20 | 0.64 ± 0.27 |
Mean Top5 (mean MaxSub score for top 5 models for each target averaged over all targets), mean Pearson (CC(P)) and Spearman rank (CC(S)) correlation coefficients (per target CC averaged over all targets), and their standard deviations are given for both 1D- and 3D-Jury, respectively. None of the observed differences between the means is significant at 95% confidence level, as measured by the t-test.
The “true” scores are defined using either MaxSub or GDT_TS (for CASP models) measures of structural similarity. Similarly, mean Top5 measure is defined as the per target average of mean MaxSub (or GDT_TS) scores of the top 5 ranking models for each target. The MaxSub scores are computed using the MaxSub program, whereas GDT_TS scores were downloaded from the CASP Prediction Center for comparison with methods that were evaluated primarily using this measure. It should be noted that the Pearson correlation coefficient between MaxSub and GDT_TS measures on CASP7 is equal to 0.97, leading to very similar results irrespective of the measure used. Similar trends are expected for other data sets, for which only MaxSub measure is used.
In terms of the ability to select high-quality models among predicted top models, 1D-Jury performed marginally worse on the CASP7 set, marginally better on the LOOPP set, whereas identical mean Top5 scores for both 1D-and 3D- Jury methods on the TASSER and MOULDER sets were obtained. For example, per target mean GDT_TS1 (Top1) of 0.60 and 0.61, and mean GDT_TS5 (Top5) of 0.59 and 0.60, were obtained on the CASP7 set for 1D-Jury and 3D-Jury, respectively. On the other hand, 1D-Jury yielded consistently lower correlations between predicted and true (MaxSub or GDT_TS) MQA scores. Per target mean CC(S) of 0.77 was obtained for 3D-Jury, as opposed to mean CC(S) of 0.75 or 0.71 for 1D-Jury, when using GDT_TS or MaxSub score, respectively. However, due to significant variations between targets, which are in fact higher for 3D-Jury compared to 1D-Jury, as indicated by standard deviations of mean GDT_TS (and other) scores, these differences are not statistically significant at the level of per target means.
Very good results were obtained for both 1D-Jury and 3D-Jury on the MOULDER set (mean CC(P) of 0.91 and 0.93, respectively), for which Sali and colleagues reported a mean CC(P) of 0.87 using their SVMod (true MQA method) and RMSD as the measure of structural (dis-)similarity (Eramian et al., 2006). Both 1D-Jury and 3D-Jury yielded mean Top5 score of 0.71 while the best possible score was 0.82 in this case. On the other hand, both 3D-Jury and 1D-Jury performed significantly worse on the TASSER set, yielding mean Top5 score of 0.66 with the theoretical limit of 0.81. The TASSER benchmark provides a direct illustration of some of the pitfalls of clustering-based model assessment when the set of available models in not sufficiently stratified, e.g., because it was generated using an approach with specific biases that increase the likelihood of obtaining models with similar characteristics.
1D-Jury, 3D-Jury and other representative GC methods, namely Pcons (Pcons_Pcons), ModFoldClust, and QmeanClust, were further compared using CASP8 data. It should be noted that Pcons and ModFoldClust were ranked as the best performing methods in CASP8 evaluation (Cozzetto et al., 2009), with QmeanClust ranked just below. As can be seen from Table 3, all these methods (including 1D-Jury) performed on par in terms of the selection of best models: ModFoldClust, Pcons, 3D-Jury, and 1D-Jury achieved essentially identical Top5 (GDT_TS5) scores of 0.64, while QMEANClust yielded marginally lower GDT_TS5 of 0.63. On the other hand, 1D-Jury performed somewhat worse in terms of measures of correlation between predicted and true MQA scores, yielding per target mean CC(S) of 0.75 compared to 0.84, 0.80 for ModFoldClust, 3D-Jury, respectively.
Table 3.
Comparison of Geometric Consensus Methods on CASP8 Data
| Method | Mean Top5 | Mean CC(S) |
|---|---|---|
| ModFoldClust | 0.64 ± 0.20 | 0.84 ± 0.14 |
| Pcons_Pcons | 0.64 ± 0.19 | 0.83 ± 0.14 |
| QMEANclust | 0.63 ± 0.20 | 0.82 ± 0.15 |
| 3D-jury | 0.64 ± 0.19 | 0.80 ± 0.15 |
| 1D-jury | 0.64 ± 0.19 | 0.75 ± 0.14 |
Mean Top5 (mean GTD_TS score for top 5 models for each target averaged over all targets, with theoretical limit of 0.67 ± 0.18) and mean Spearman rank correlation coefficients, as well as their standard deviations are given for each method.
Although the differences between per target mean correlation coefficients for 1D-Jury and 3D-Jury were not statistically significant, as assessed by the two-sided T-test without assuming equal variances, correlations for individual targets were also analyzed to capture better trends that may be hidden by large target to target variation. Using Fisher transformation (z-statistic) to test if correlation coefficients for different methods observed for individual targets were statistically significant, we found that 3D-Jury resulted in significantly better (rank) correlation for 96 targets and 1D-Jury for 5 targets, with no significant differences for the remaining 14 targets at the 95% confidence level. For comparison, when contrasting results of 3D-Jury with Pcons, the former was significantly better for 22 targets, and the latter for 55 targets (with no significant differences for the remaining 38 targets).
The differences between 1D-Jury and other methods in terms of correlation with true rankings become less significant when removing poor models, such as those with very short correctly predicted fragments and models with partially missing side-chain coordinates. This is consistent with trends observed on other data sets considered here. In fact, differences between 1D- and 3D-jury are somewhat larger for sets with significant fraction of low-quality models, as indicated by the differences between the results on LOOPP and MOULDER sets. Thus, 1Dsim and the resulting 1D-Jury scores are less consistent with MaxSub measure and 3D-Jury for non-native-like models, for which the notion of a relative distance from the native structure (or dis-similarity score) may not be well defined (Swanson et al., 2009). Overall, and most importantly from the point of view of the selection of best models, these results support the conclusion that 1D-Jury performs on par with 3D-Jury and other top performing GC (or clustering-based) methods in the field.
At the same time, as shown in the Methods section, the computational complexity of 1D-Jury is linear with respect to the number of models, resulting in dramatic reduction of running times compared to 3D-Jury, especially when the number of models becomes large. Dependence of the computation time on the number of models is shown for a particular CASP7 target of length 75 in Figure 3. As expected, based on the quadratic complexity of 3D-Jury, the running time for 3D-Jury can be approximated very well using quadratic polynomial fit (225, 1112, and 2261 CPU sec for 180, 360, and 530 models, respectively), whereas 1D-Jury follows linear scaling (0.6, 1.0, and 1.4 CPU sec for 180, 360, and 530 models, respectively). On average, 1D-Jury takes (on a single 3-GHz Xeon processor) 3.3 CPU sec per CASP7 target to compute scores and rank all models for that target (and the maximum CPU time of 12.2 CPU sec), with similar times observed for CASP8.
FIG. 3.

Running times versus the number of models for 3D-Jury, quadratic (1D-Jury(Q)), and linear implementation of 1D-Jury, respectively.
The fact that comparison of all pairs of models can be performed implicitly in 1D-Jury by pre-computing the number of joint (RSA,SS) states at each position is clearly advantageous in applications to model quality assessment. At the same time, however, pairwise similarity scores (or distances between models) are not computed explicitly, which may be a problem in other potential applications. For example, pairwise distances are required for K-means, hierarchical and other classical clustering algorithms that can be used for partitioning of state space in folding simulations. Therefore, we included in the analysis of running times a suboptimal implementation of 1D-Jury, referred to as 1D-Jury(Q), which performs explicit comparison of pairs of models in terms of their 1D-profiles and computes pairwise similarity scores in addition to overall quality scores. As indicated in the Methods section, 1D-Jury(Q) also offers some additional flexibility, e.g., allowing one to introduce cutoffs for the minimum length of similar substructures that contribute to the score, as used in MaxSub and 3D-Jury, although the gains in accuracy due to a more flexible model are limited.
As can be seen from Figure 3, 1D-Jury(Q) is still significantly faster compared to 3D-Jury, even though both methods involve a loop over all pairs of models. This is primarily achieved by avoiding 3D superposition with MaxSub heuristic, illustrating the relative gains due to linear versus quadratic complexity, and due to 1D versus 3D structure alignment (which depend on the number of models, but also on their length). These speed-ups bode well for applications to explicit model clustering. On the other hand, increased computational complexity of 1D-Jury(Q) makes it significantly slower than linear 1D-Jury in the context of MQA, with mean per target running time of 170 CPU sec on CASP7 data (compared with 3.3 CPU sec for 1D-Jury).
4. Conclusion
In this contribution, we showed that similarity between protein models can be effectively assessed in terms of consistency of their 1D profiles consisting of secondary structures and solvent accessibilities of individual amino acid residues. In particular, such defined simple profile-based similarity measure can be used to identify similar substructures in a set of models, with the goal of ranking and selecting high-quality models based on the presence of frequently shared (and thus likely well-predicted) substructures. The resulting 1D-Jury approach can be viewed as a simple alternative to much more involved computationally 3D-Jury and related GC-based methods for model quality assessment.
We furthermore demonstrated that implicit comparison of all pairs of models and the computation of 1D-Jury scores for a set of models can be achieved in linear time with respect to the number of models, as opposed to quadratic complexity of 3D-Jury. Moreover, the computationally expensive superposition of 3D structures, using MaxSub or related heuristic methods for structure-to-structure alignment, is avoided. Instead, each model is aligned to the target sequence using sequence alignment methods, and their similarity is measured in terms of consistency of joint SS/RSA states for equivalent residues. As a result, dramatic reductions in running times are achieved compared to 3D-Jury. For example, 530 models for a CASP7 target of length 75 residues can be ranked in 1.4 seconds CPU time (on a 3-GHz Xeon processor) with 1D-Jury, as opposed to about 2,200 sec with MaxSub-based 3D-Jury implementation. Increasing the number of models, e.g., 100-fold increases these times to about 140 sec for 1D-Jury and to about 22 mln sec for 3D-Jury, respectively.
At the same time, using CASP and other data sets, we found that structural similarity between models and the respective native structures, as well as between pairs of models, can be measured without significant loss of accuracy using simple 1D profiles, yielding correlation coefficient of about 0.9 between 1Dsim and MaxSub scores. We also showed that when applied to the problem of model selection and ranking, the new approach performs on par with 3D-Jury and related GC-based methods in terms of the ability to select the best models, with somewhat lower overall correlations between the true and predicted ranking.
The advantage of geometric clustering methods, such as 3D-Jury is that essentially no parameter optimization and no training set are required. However, the results are dependent on the population of models (and methods used to generate these models), which may require a carefully optimized (e.g., to account for specific training set biases) combination of true MQAs with clustering-based methods to obtain more universally applicable and transferable scores. As a fast alternative, 1D-Jury can facilitate repeated evaluation and parameter optimization with the goal of achieving such robust combination of various physical, knowledge-based, and structural consensus-based features. The ability to quickly evaluate structural similarity and the degree of stratification within large set of models by generating distribution of 1D-Jury scores is also expected to provide valuable guidance and on-the-fly assessment of the convergence and quality of sampling for protein structure prediction methods. Other potential applications of 1D profile based assessment of structural similarity include fast clustering of large number of intermediate structures generated in studies on folding kinetics and other folding simulations.
A web server that enables scoring and ranking large sets of models using 1D-Jury is available at http://sift.chmcc.org. In addition, 1D-Jury is also available as part of the stand-alone version of the SIFT package, which combines several clustering-based, as well as true MQA approaches. Importantly, due to reduced computational complexity, the on-line version of 1D-Jury is available to the users essentially without limitations on the number of models (except for those implied by the size of files that can be submitted).
Supplementary Material
Acknowledgments
We are grateful to the authors of decoy sets, MQA programs, and CASP organizers, for making their data sets and assessment results available. This work was supported in part by the National Institutes of Health (grants GM067823, A1055649, and P30-ES006096). Computational resources were made available by Cincinnati Children's Hospital Research Foundation and University of Cincinnati College of Medicine.
Disclosure Statement
No competing financial interests exist.
References
- Adamczak R. Porollo A. Meller J. Accurate prediction of solvent accessibility using neural networks-based regression. Proteins. 2004;56:753–767. doi: 10.1002/prot.20176. [DOI] [PubMed] [Google Scholar]
- Benkert P. Tosatto S.C. Schomburg D. QMEAN: a comprehensive scoring function for model quality assessment. Proteins. 2008;71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- Benkert P. Schwede T. Tosatto S.C. QMEANclust: estimation of protein model quality by combining a composite scoring function with structural density information. BMC Struct. Biol. 2009;9:35. doi: 10.1186/1472-6807-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budowski-Tal I. Nov Y. Kolodny R. FragBag, an accurate representation of protein structure, retrieves structural neighbors from the entire PDB quickly and accurately. Proc. Natl. Acad. Sci. USA. 2010;107:3481–3486. doi: 10.1073/pnas.0914097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai I. Kwon J. Kim S.-H. Local feature frequency profile: a method to measure structural similarity in proteins. Proc. Natl. Acad. Sci. USA. 2004;101:3797–3802. doi: 10.1073/pnas.0308656100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristobal S. Zemla A. Fischer D., et al. A study of quality measures for protein threading models. BMC Bioinformatics. 2001;2:5. doi: 10.1186/1471-2105-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzetto D. Kryshtafovych A. Tramontano A. Evaluation of CASP8 model quality predictions. Proteins. 2009;77(Suppl 9):157–166. doi: 10.1002/prot.22534. [DOI] [PubMed] [Google Scholar]
- Elber R. West A. Atomically detailed simulation of the recovery stroke in myosin by Milestoning. Proc. Natl. Acad. Sci. USA. 2010;107:5001–5005. doi: 10.1073/pnas.0909636107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer S. Park S. Pande V.S. Foldamer dynamics expressed via Markov state models. II. State space decomposition. J. Chem. Phys. 2005;123:114903. doi: 10.1063/1.2008230. [DOI] [PubMed] [Google Scholar]
- Eramian D. Shen M. Devos D., et al. A composite score for predicting errors in protein structure models. Protein Sci. 2006;15:1653–1666. doi: 10.1110/ps.062095806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D. 3D-SHOTGUN: a novel, cooperative, fold-recognition meta-predictor. Proteins. 2003;51:434–441. doi: 10.1002/prot.10357. [DOI] [PubMed] [Google Scholar]
- Gatchell D.W. Dennis S. Vajda S. Discrimination of near-native protein structures from misfolded models by empirical free energy functions. Proteins. 2000;41:518–534. [PubMed] [Google Scholar]
- Ginalski K. Elofsson A. Fischer D., et al. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kaján L. Rychlewski L. Evaluation of 3D-Jury on CASP7 models. BMC Bioinformatics. 2007;8:304. doi: 10.1186/1471-2105-8-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laboulais C. Ouali M. Le Bret M., et al. Hamming distance geometry of a protein conformational space: application to the clustering of a 4-ns molecular dynamics trajectory of the HIV-1 integrase catalytic core. Proteins. 2002;47:169–179. doi: 10.1002/prot.10081. [DOI] [PubMed] [Google Scholar]
- Larsson P. Skwark M.J. Wallner B., et al. Assessment of global and local model quality in CASP8 using Pcons and ProQ. Proteins. 2009;77(Suppl 9):167–172. doi: 10.1002/prot.22476. [DOI] [PubMed] [Google Scholar]
- Lee J. Ab initio protein structure prediction, 1-26. In: Rigden D.J., editor. From Protein Structure to Function with Bioinformatics. Springer; London: 2009. [Google Scholar]
- Lu H. Skolnick J. A distance-dependent atomic knowledge-based potential for improved protein structure selection. Proteins. 2001;44:223–232. doi: 10.1002/prot.1087. [DOI] [PubMed] [Google Scholar]
- Lundström J. Rychlewski L. Bujnicki J., et al. Pcons: a neural-network-based consensus predictor that improves fold recognition. Protein Sci. 10:2354–2362. doi: 10.1110/ps.08501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüthym R. Bowie J.U. Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- McGuffin L.J. Benchmarking consensus model quality assessment for protein fold recognition. BMC Bioinformatics. 2007;8:345. doi: 10.1186/1471-2105-8-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L.J. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics. 2008;24:586–587. doi: 10.1093/bioinformatics/btn014. [DOI] [PubMed] [Google Scholar]
- Meller J. Elber R. Linear optimization and a double statistical filter for protein threading protocols. Proteins. 2001;45:241–261. doi: 10.1002/prot.1145. [DOI] [PubMed] [Google Scholar]
- Moult J. Fidelis K. Kryshtafovych A., et al. Critical assessment of methods of protein structure prediction (CASP)–Round 6. Proteins. 2005;61(Suppl 7):3–7. doi: 10.1002/prot.20716. [DOI] [PubMed] [Google Scholar]
- Moult J. Fidelis K. Kryshtafovych A., et al. Critical assessment of methods of protein structure prediction–Round VII. Proteins. 2007;69(Suppl 7):3–9. doi: 10.1002/prot.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult J. Fidelis K. Kryshtafovych A., et al. Critical assessment of methods of protein structure prediction–Round VIII. Proteins. 2009;77(Suppl 9):1–4. doi: 10.1002/prot.22589. [DOI] [PubMed] [Google Scholar]
- Pettitt C.S. McGuffin L.J. Jones D.T. Improving sequence-based fold recognition by using 3D model quality assessment. Bioinformatics. 2005;21:3509–3515. doi: 10.1093/bioinformatics/bti540. [DOI] [PubMed] [Google Scholar]
- Porollo A.A. Adamczak R. Meller J. POLYVIEW: a flexible visualization tool for structural and functional annotations of proteins. Bioinformatics. 2004;20:2460–2462. doi: 10.1093/bioinformatics/bth248. [DOI] [PubMed] [Google Scholar]
- Qiu J. Sheffler W. Baker D., et al. Ranking predicted protein structures with support vector regression. Proteins. 2008;71:1175–1182. doi: 10.1002/prot.21809. [DOI] [PubMed] [Google Scholar]
- Samudrala R. Moult J. An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. J. Mol. Biol. 1998;275:895–916. doi: 10.1006/jmbi.1997.1479. [DOI] [PubMed] [Google Scholar]
- Siew N. Elofsson A. Rychlewski L., et al. MaxSub: an automated measure for the assessment of protein structure prediction quality. Bioinformatics. 2000;16:776–785. doi: 10.1093/bioinformatics/16.9.776. [DOI] [PubMed] [Google Scholar]
- Simons Kooperberg C. Huang E., et al. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- Swanson R. Vannucci M. Tsai J.W. Information theory provides a comprehensive framework for the evaluation of protein structure predictions. Proteins. 2009;74:701–711. doi: 10.1002/prot.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat B.K. Pillardy J. Elber R. A template finding algorithm and a comprehensive benchmark for homology modeling of proteins. Proteins. 2008;72:910–928. doi: 10.1002/prot.21976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat B.K. Pillardy J. Májek P., et al. Building and assessing atomic models of proteins from structural templates: learning and benchmarks. Proteins. 2009;76:930–945. doi: 10.1002/prot.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. Skolnick J. Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y. Huang E.S. Levitt M., et al. Ab initio construction of protein tertiary structures using a hierarchical approach. J. Mol. Biol. 2000;300:171–185. doi: 10.1006/jmbi.2000.3835. [DOI] [PubMed] [Google Scholar]
- Zemla A. Venclovas C. Moult J, et al. Processing and analysis of CASP3 protein structure predictions. Proteins. 1999;(Suppl 3):22–29. doi: 10.1002/(sici)1097-0134(1999)37:3+<22::aid-prot5>3.3.co;2-n. [DOI] [PubMed] [Google Scholar]
- Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. Liu S. Zhou Y. Accurate and efficient loop selections by the DFIRE-based all-atom statistical potential. Protein Sci. 2004;13:391–399. doi: 10.1110/ps.03411904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. Pandit S.B. Lee S.Y., et al. Analysis of TASSER-based CASP7 protein structure prediction results. Proteins. 2007;69(Suppl 8):90–97. doi: 10.1002/prot.21649. [DOI] [PubMed] [Google Scholar]
- Zhou H. Zhou Y. SPARKS 2 and SP3 servers in CASP6. Proteins. 2005;61(Suppl 7):152–156. doi: 10.1002/prot.20732. [DOI] [PubMed] [Google Scholar]
- Zhou H. Skolnick J. Protein model quality assessment prediction by combining fragment comparisons and a consensus C(alpha) contact potential. Proteins. 2008;71:1211–1218. doi: 10.1002/prot.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.