

Abstract
Cerebral β-amyloidosis and associated pathologies can be exogenously induced by the intracerebral injection of small amounts of pathogenic Aβ-containing brain extract into young β-amyloid precursor protein (APP) transgenic mice. The probable β-amyloid-inducing factor in the brain extract has been identified as a species of aggregated Aβ that is generated in its most effective conformation or composition in vivo. Here we report that Aβ in the brain extract is more proteinase K (PK) resistant than is synthetic fibrillar Aβ, and that this PK-resistant fraction of the brain extract retains the capacity to induce β-amyloid deposition upon intracerebral injection in young, pre-depositing APP23 transgenic mice. After ultracentrifugation of the brain extract, <0.05% of the Aβ remained in the supernatant fraction, and these soluble Aβ species were largely PK sensitive. However, upon intracerebral injection, this soluble fraction accounted for up to 30% of the β-amyloid induction observed with the unfractionated extract. Fragmentation of the Aβ seeds by extended sonication increased the seeding capacity of the brain extract. In summary, these results suggest that multiple Aβ assemblies, with various PK sensitivities, are capable of inducing β-amyloid aggregation in vivo. The finding that small and soluble Aβ seeds are potent inducers of cerebral β-amyloidosis raises the possibility that such seeds may mediate the spread of β-amyloidosis in the brain. If they can be identified in vivo, soluble Aβ seeds in bodily fluids also could serve as early biomarkers for cerebral β-amyloidogenesis and eventually Alzheimer's disease.
Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disorder and the most common form of senile dementia. Characteristic pathological hallmarks are proteinaceous deposits of amyloid-β (Aβ) (senile plaques) and hyperphosphorylated tau (neurofibrillary tangles), accompanied by an inflammatory response and damage to neurons and synapses (Duyckaerts et al., 2009; Holtzman et al., 2011). According to the amyloid cascade hypothesis, the misfolding and aggregation of Aβ is a fundamental event in the pathogenesis of AD (Hardy and Selkoe, 2002). While a wealth of studies have characterized Aβ aggregation in vitro (Harper and Lansbury, 1997; Roychaudhuri et al., 2009; Du et al., 2011), much less is known about the in vivo trigger for cerebral β-amyloidosis.
We and others have shown that intracerebral injections of minute amounts of Aβ-containing extract from AD brains or aged APP-transgenic (tg) mouse brains can induce β-amyloidosis and associated pathologies (activation of microglia and astrocytes; dystrophic neurites) in the brains of young APP-tg mice (Kane et al., 2000; Meyer-Luehmann et al., 2006; Eisele et al., 2009; Watts et al., 2011). β-Amyloid deposition also can be induced in APP-tg mice by intraperitoneal infusion of Aβ-containing brain extracts (Eisele et al., 2010), indicating that the seeds can translocate from the periphery to the brain. The probable β-amyloid-inducing factor in the brain extract has been identified as a species of aggregated Aβ because (1) brain extracts from wild-type mice or pre-depositing APP-tg mice were ineffective in seeding β-amyloid; and (2) Aβ-immunodepletion or formic acid denaturation of the extract abolished the induction of β-amyloidosis (Meyer-Luehmann et al., 2006). The finding that synthetic Aβ preparations fail to show robust β-amyloid-inducing activity indicates that the misfolding of Aβ in vivo results in a different conformation and/or that brain-specific cofactors are needed for effective seeding (Meyer-Luehmann et al., 2006).
The exogenous induction of cerebral β-amyloidosis, either by intracerebral or intraperitoneal application of Aβ seeds, is reminiscent of the induction of prion disease in mice (Aguzzi and Rajendran, 2009; Colby and Prusiner, 2011). Prions consist of PrPSc, an abnormally folded isoform of the cellular prion protein (PrPc). Upon inoculation of mice, PrPSc can induce the templated misfolding of PrPc and thereby cause fatal neurodegeneration (Wadsworth et al., 2010). Infectious prions are multimers of PrPSc, and their size, solubility, resistance to proteinase K (PK) treatment, and ease of fragmentation govern their infectivity and neurotoxicity (Aguzzi et al., 2007; Collinge and Clarke, 2007).
The objective of the present study was to further characterize the β-amyloid-inducing factor in the Aβ-containing extract from aged APP-tg mouse brains. Guided by the prion findings, we have tested the PK resistance, solubility, and fragmentation of the amyloidogenic seeds. Our results bolster the assumption that the β-amyloid-inducing factor is Aβ, and indicate that, rather than consisting of a single type of multimer, β-amyloid seeds comprise a range of Aβ aggregates of various sizes and PK-sensitivities.
Materials and Methods
Mice.
For all experiments, 3- to 4 month-old male or female APP23 transgenic mice were used (Sturchler-Pierrat et al., 1997). The mice have been backcrossed with C57BL/6J mice for 20 generations (C57BL/6J-Tg(Thy1-APPK670N;M671L)23). APP23 mice currently bred at the Hertie Institute for Clinical Brain Research first develop individual β-amyloid plaques in the neocortex at 7 months of age. In the hippocampus, β-amyloid deposition starts at 8–10 months of age (females 8–9 months; males 9–10 months). All mice were kept under specific pathogen-free conditions. The experimental procedures were undertaken in accordance with the veterinary office regulations of Baden-Württemberg (Germany) and approved by the local Animal Care and Use Committees.
Brain tissue extracts.
Brain extracts were derived from aged (22–28 months old) β-amyloid-depositing male or female APP23 transgenic mice and from age-matched nontransgenic mice. After removal of the cerebellum and lower brainstem, the forebrain was immediately fresh frozen on dry ice and stored at −80°C until use. Tissue was then homogenized (Ultra Turrax T8, IKA-Werke) at 10% (w/v) in sterile PBS (Lonza), vortexed, sonicated three times for 5 s (LabSonic, B. Braun Biotech International; 0.5-mm-diameter sonotrode, cycle 1, amplitude 80%), and centrifuged at 3000 × g for 5 min as previously described (Meyer-Luehmann et al., 2006; Eisele et al., 2009). The supernatant was aliquoted and immediately frozen. For all experiments, the 10% (w/v) extract was used unless otherwise stated.
Synthetic Aβ preparations.
Aβ1–40 and Aβ1–42 (American Peptide) were dissolved at equimolar concentrations in PBS to reach a final concentration of 100 μm total Aβ. To generate fibrils, the peptides were incubated on a rotator for 5 d at 37°C as previously described (Meyer-Luehmann et al., 2006). Fibrils were stored at −80°C until use.
Proteinase K treatment of the brain extract and synthetic Aβ.
Brain extracts and synthetic Aβ preparations were thawed on ice and subsequently treated with 50 μg/ml PK (Roche Diagnostics) for up to 2 h at 37°C. After 30 min, 1 h, or 2 h, samples were boiled for 5 min at 95°C to heat-inactivate the PK.
Ultracentrifugation of the brain extract.
Brain extracts were thawed on ice and centrifuged in a Beckman Centrifuge at 100,000 × g for 1 h at 4°C in Protein LoBind Tubes (Eppendorf). Supernatant was transferred to a new tube and the pellet was resuspended in sterile PBS to obtain the same volume as the supernatant. Samples were stored on ice until use.
Fragmentation of the brain extract.
Brain extracts [10% (w/v) in PBS; 3000 × g supernatant] were thawed on ice and sonicated with a LabSonic (B. Braun Biotech International) three times for 20 s (0.5-mm-diameter sonotrode, cycle 1, amplitude 80%).
Aβ quantification of the extract by electrochemiluminescence-linked immunoassay.
Aβ levels (Aβx–38, Aβx–40, and Aβx–42) in extracts were determined with an electrochemiluminescence-linked immunoassay using the MSD 96-well MULTI-SPOT Human (6E10) Aβ Triplex Assay (Meso Scale Discovery). To this end, the extracts were first treated with formic acid (final concentration: 70%) (Sigma). Samples were sonicated for 30 s on ice and centrifuged at 25,000 × g for 1 h at 4°C. Supernatants were equilibrated in neutralization buffer (1 m Tris base, 0.5 m Na2HPO4, 0.05% NaN3). The 100,000 × g supernatant was measured directly. Aβ detection was further conducted according to the manufacturer's instructions. In brief, 96-well plates prespotted with capture antibodies against Aβx–38, Aβx–40, Aβx–42, and bovine serum albumin were blocked for 1 h with 1% Blocker A solution and then washed three times with 1× Tris buffer. In a second step, samples were diluted 1:100 in 1% Blocker A solution, except supernatant of ultracentrifugation, and coincubated with the SULFO-TAG 6E10 detection antibody solution on the plate for 2 h. After washing, MSD Read Buffer T was added and the plate was read immediately on a Sector Imager 6000. Data analysis used MSD DISCOVERY WORKBENCH software 2.0. For total Aβ, Aβx–38, Aβx–40, and Aβx–42 were combined.
Total protein quantification.
Total protein of the brain extract was quantified with a microplate bicinchoninic acid (BCA) assay (Pierce Biotechnology). Samples were measured on an ELISA plate reader (Mithras, Berthold Technologies).
SDS-PAGE and silver staining of proteins.
Brain extracts and synthetic Aβ preparations were analyzed on NuPage Bis-Tris mini gels using NuPage LDS sample buffer and MES running buffer (Invitrogen). To stain for total protein, silver staining was performed according to a previously established protocol (Nesterenko et al., 1994) with the following modifications. Gels were fixed with a solution of 50% acetone, 1% trichloroacetic acid (TCA) and 0.015% formaldehyde (HCHO) for 5 min, washed three times with double-distilled water (ddH2O), incubated again with ddH2O for 5 min and washed again thrice. For pretreatment, gels were incubated in pure acetone for 5 min and afterward 1 min in 1.4 mm dithionite solution. After washing with ddH2O, the staining solution (15.7 mm silver nitrate in ddH2O with 0.18% HCOH) was added and incubated for 8 min. This step was followed by incubation in a developing solution (70 mm soda-decahydrate, 0.015% HCHO, and 0.004% sodium thiosulfate) and the staining was stopped afterward with 3% glacial acetic acid. After washing with ddH2O, the gel was incubated in a gel-drying solution (5% glycerol and 30% methanol in ddH2O) for 30 min. Staining of the gel was documented with a scanner, and afterward the gel was dried with a Gel Dryer (Bio-Rad).
Western blot analysis.
Brain extracts and synthetic Aβ preparations were analyzed on NuPage Bis-Tris mini gels using NuPage LDS sample buffer and MES running buffer (Invitrogen). For Western blotting, samples were wet-blotted onto a nitrocellulose membrane, probed with 6E10 antibody (Covance Research Products) and visualized with chemiluminescence using SuperSignal West Pico (Thermo Scientific). Densitometric values of band intensities were analyzed using the public domain software ImageJ, version 1.34 (http://imagej.nih.gov/ij/).
Stereotactic injection of brain extracts.
APP23 host mice were anesthetized with a mixture of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight) in saline. Bilateral stereotactic injections of 2.5 μl brain extract were made with a Hamilton syringe into the hippocampus (AP −2.5 mm, L ±2.0 mm, DV −1.8 mm). Injection speed was 1.0 μl/min and the needle was kept in place for an additional 2 min before it was slowly withdrawn. The surgical area was cleaned with sterile saline, the incision was sutured, and the mice were monitored until recovery from anesthesia.
Histology and immunohistochemistry.
Brains were removed and immersion fixed for 48 h in 4% paraformaldehyde in PBS, then cryoprotected in 30% sucrose in PBS for an additional 2 d. After freezing, serial, 25-μm-thick coronal sections were cut through the brains using a freezing-sliding microtome. The sections were collected in 0.1 m Tris-buffered saline, pH 7.4, and stained immunohistochemically according to previously published protocols (Stalder et al., 2005). Polyclonal antibody CN3 (Eisele et al., 2010) was used for immunostaining of Aβ. Adjacent sections were stained with Congo red according to standard protocols and viewed under cross-polarized light. The following additional antibodies were used: rabbit polyclonal antibody against cow glial fibrillary acidic protein (GFAP) (Dako); rabbit polyclonal antibody against ionized calcium binding adapter molecule 1 (Iba1) (Wako); and rabbit polyclonal antibody A4CT against APP (Calhoun et al., 1999) (gift from K. Beyreuther, Netzwerk Alternsforschung, Ruprecht-Karls-Universität, Heidelberg, Germany).
Quantification of total Aβ load and Congo red-positive Aβ deposits.
Aβ load was quantified on an Aβ-immunostained set of every 12th systematically sampled, serial, coronal section throughout the entire hippocampus. Researchers who were blinded to the inoculation groups performed the analysis. Stereological analysis of CN3-positive staining was performed using a microscope equipped with a motorized x–y–z stage coupled to a video-microscopy system and the Stereo Investigator software (MicroBrightField) as previously described (Bondolfi et al., 2002). The β-amyloid load (percentage) was determined by calculating the areal fraction occupied by CN3-positive immunostaining in two-dimensional sectors at a single focal plane (20×/0.45 objective). The percentage of Aβ deposits that were also Congo red positive was determined manually on an adjacent set of systematically sampled sections that were double stained by the anti-Aβ antibody and Congo red. Quantification was performed using a 20× objective and a Zeiss Axioskop 2 microscope (Zeiss).
Results
The β-amyloid-inducing factor is partially PK resistant
Brain extracts from aged APP23 transgenic mice (Tg extract) and preparations of synthetic Aβ fibrils in PBS or spiked into brain extract from aged nontransgenic (wild-type) mice (Wt extract) were incubated with 50 μg of PK/ml for up to 2 h. Subsequent Aβ immunoblotting revealed that Aβ in the Tg extract was to a high degree PK resistant (80% remaining after 30 min PK treatment; 70% remaining after 2 h PK treatment), whereas synthetic Aβ fibrils were almost completely digested after 30 min (Fig. 1A–C). The spiking of the synthetic Aβ fibrils into Wt extract made the fibrils more PK resistant (50% remaining after 30 min PK treatment; 35% remaining after 2 h PK treatment), although they were still significantly less resistant than were the Aβ aggregates in the Tg extract (Fig. 1A–C).
Figure 1.

The β-amyloid-inducing activity is partly proteinase K-resistant. Brain extracts from an aged APP23 tg mouse (Tg extract), an aged non-tg mouse (Wt extract) spiked with synthetic Aβ fibrils (syn. Aβ fibrils in Wt extract; 10 μm Aβ), and synthetic Aβ fibrils in PBS (syn. Aβ fibrils in PBS; 10 μm) were treated with 50 μg/ml PK for 0 min, 30 min, 1 h, or 2 h at 37°C. A, Silver staining of a 12% Bis-Tris NuPage gel for total protein reveals the digestion of the majority of proteins in the brain extracts and of synthetic Aβ fibrils already after 30 min PK treatment. B, Immunoblot analysis with an antibody specific to human Aβ (6E10) shows that Aβ in the Tg extract is largely PK resistant for up to 2 h. Aβ in the spiked Wt extract revealed some, but significantly less PK resistance, whereas synthetic Aβ fibrils in PBS were almost completely digested by the PK treatment. C, Densitometric quantification of Aβ-immunoblots after PK treatment of five Tg extracts (each from a different animal) and three independent synthetic Aβ fibril preparations are shown. Each preparation was tested at least three times and the mean was taken. Aβ concentration at time point 0 was designated as 100%. Indicated is the mean ± SEM. Repeated-measures two-way ANOVA revealed a significant difference between the groups (F(2,34) = 112.2). Multiple Bonferroni post hoc tests showed significances after PK digestion for all group comparisons, p < 0.001. D–F, In vivo seeding activity of brain extracts with or without PK treatment (50 μg/ml PK for 30 min) was tested by intrahippocampal injections into young, pre-depositing 4-month-old APP23 mice. Brains were immunohistochemically analyzed for Aβ deposition 5 months later. Shown is the dentate gyrus of the hippocampus, which revealed robust β-amyloid induction by the Tg extract (D) and the PK-treated Tg extract (E), but not with Wt extract (F). Note that extracts were boiled before injection to deactivate residual PK activity. The small insert shows double staining for Aβ immunoreactivity and Congo red binding of the induced β-amyloid in an adjacent section. Scale bars: 200 μm and 20 μm. G, Stereological quantification of Aβ load in the hippocampus. Although PK treatment reduced the β-amyloid induction, the PK-resistant Tg material still showed potent seeding activity compared to the Wt extract (n = 5–6 mice per group; mean ± SEM: ANOVA followed by Bonferroni post hoc tests revealed a significant difference between the extracts (F(2,14) = 18.95, ***p < 0.001; *p < 0.05). Note that neither PK-treated nor untreated Wt extract induced any amyloid, and therefore these groups were combined.
To test whether PK-treated Tg extracts harbor β-amyloid-inducing activity in vivo, Tg extracts with and without PK treatment were injected into the hippocampus of 4-month-old pre-depositing APP23 mice and compared with the injection of Wt extracts (Fig. 1D–G). Inactivation of PK was achieved by boiling the samples (including the controls) before injection. Boiling has been found to reduce, but not abolish, the β-amyloid-inducing activity of the brain extracts (Meyer-Luehmann et al., 2006). Therefore, to optimize the seeding signal, an incubation time of 5 months was chosen. Immunohistochemical analysis revealed robust β-amyloid deposition in the hippocampus of mice receiving either the Tg extract or the PK-treated Tg extract, whereas β-amyloid was not induced by the Wt extracts (Fig. 1D–F); i.e., the effect of the Wt extract was not different from that of the PBS injection (Aβ load: 0.06 ± 0.03% vs 0.03 ± 0.01%; n = 5–6 per group, mean ± SEM, t(9) = 1.893; p > 0.05). β amyloid induced by the PK-treated Tg extract was both congophilic and diffuse (51.1 ± 8.1% of the Aβ deposits had a detectable Congo red-positive core). All congophilic β-amyloid plaques were surrounded by intensely stained hypertrophic GFAP-positive astrocytes, activated Iba1-positive microglia, as well as APP-positive dystrophic neurites, as previously described (Meyer-Luehmann et al., 2006). Quantification of the induced β-amyloid load indicated, however, that the PK-treated Tg extracts yielded only 55% of the β-amyloid-inducing activity of the Tg extract without PK treatment (Fig. 1G), suggesting a partial digestion of the β-amyloid-inducing activity by PK. Histological analysis of the brains 1 d after injection of the PK-treated Tg extract revealed no Aβ deposits, ruling out the possibility that the induced Aβ deposition represents the injected material itself (n = 3; data not shown), consistent with previous studies (Meyer-Luehmann et al., 2006; Eisele et al., 2009).
The β-amyloid-inducing factor is partially soluble
Tg extract was ultracentrifuged (100,000 × g for 1 h at 4°C) and the supernatant and pellet fractions were subsequently assessed for total protein content and Aβ levels. Measurements of total protein concentration (by BCA assay) revealed 3.6 ± 0.2 μg/μl and 2.8 ± 0.3 μg/μl protein in the supernatant and in the pellet fraction, corresponding to ∼60% and 40% of total protein of the original extract, respectively. This is consistent with the silver staining of acrylamide gels (Fig. 2A). In contrast, Aβ immunoblotting and Aβ-ELISA revealed that the great majority (>99.9%) of Aβ was in the pellet fraction (Fig. 2A, Table 1).
Figure 2.
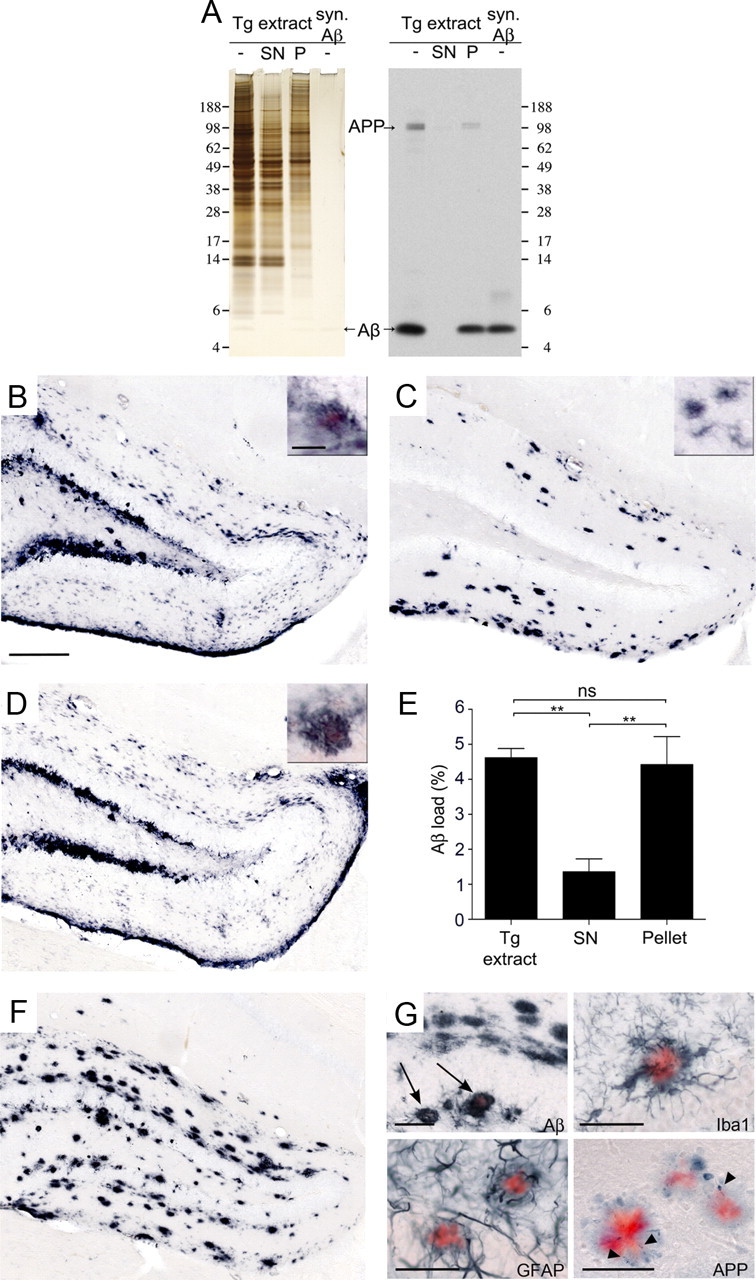
The β-amyloid-inducing activity is partly soluble. A, Brain extracts (10%, w/v) from aged APP23 mice (Tg extract) were subjected to 100,000 × g ultracentrifugation (1 h, 4°C). Left panel, Silver staining of a 12% Bis-Tris gel shows somewhat less protein in the pellet fraction (P) compared to the supernatant (SN) fraction (this was confirmed by total protein measurements revealing ∼60% of the protein in the supernatant and 40% in the pellet, see results). Right panel, Immunoblotting for human Aβ (antibody 6E10) using 12% Bis-Tris gels reveals that most Aβ is retained in the pellet fraction. Aβ in the supernatant was below the level of detection by this immunoblot, but was estimated to be <0.05% of total Aβ concentration in the original Tg extract using ELISA (see Table 1). Loading control: synthetic Aβ, 15 ng/lane. B–D, In vivo seeding activity of the supernatant and pellet fractions was tested by intrahippocampal injections into young, pre-depositing 4-month-old APP23 mice. Brains were immunohistochemically analyzed for Aβ deposition 4 months after injection. Shown is the dentate gyrus of the hippocampus. While the Tg extract (B) induced robust Aβ deposition, the SN fraction (C) induced largely diffuse Congo red-negative Aβ deposits. Aβ deposition induced by the pellet fraction (D) is similar to that induced by the Tg extract. The small insert shows double staining of Aβ and Congo red in seeded β-amyloid in an adjacent section. Note that the β-amyloid-induction by the SN was largely Congo red negative after this 4 month incubation period, but was partly Congo red positive after a 6 month incubation (see G below). E, Stereological quantification of Aβ load in the hippocampus 4 months after injection (n = 5 mice per group; mean ± SEM; ANOVA followed by Bonferroni post hoc test (F(2,13) = 11.93; **p < 0.01; n.s., not significant). Note that an additive effect of the β-amyloid induction by the SN and P fractions cannot be assumed (Meyer-Luehmann et al., 2006). F, β-Amyloid induction by the SN fraction 6 months after injection. G, The majority of the SN fraction-induced β-amyloid-deposits at 6 months after injection were still Congo red negative; however, a subset of the induced deposits were congophilic (arrow) and surrounded by activated, darkly stained Iba-1-positive microglia, hypertrophic GFAP-positive astrocytes, and APP-positive dystrophic boutons and neurites (arrowheads). Shown are sections adjacent to F. Scale bars: 200 μm, 20 μm (B–D, F), and 50 μm (G).
Table 1.
Ultracentrifugation of the Tg extract
| Total Aβa [ng/μl] (relative amount to Tg extract) | β-amyloid loadb [%] (relative amyloid load to Tg extract) | |
|---|---|---|
| Tg extract | 14.838 ± 0.308 (1) | 4.62 ± 0.26 (1) |
| Supernatant | 0.005 ± 0.003 (0.004) | 1.35 ± 0.37 (0.29) |
| Pellet | 14.885 ± 1.181 (1) | 4.42 ± 0.80 (0.95) |
aELISA measurements were done with three different Tg extracts. Mean ± SEM is indicated. Note that 2.5 μl of each fraction were injected.
bStereological quantification of induced β-amyloid. Mean ± SEM is indicated. Data are taken from Fig. 2E. Note that β-amyloid induction is not linear to the Aβ concentration in the extract (see Meyer-Luehmann et al., 2006). Thus, an additive effect of β-amyloid induction of the two fractions cannot be assumed.
To investigate the β-amyloid-inducing activity of these fractions, 3- to 4-month-old, pre-depositing APP23 mice were given intracerebral injections of the material. Histological analysis was performed 4 months later (Fig. 2B–D). Both the supernatant and pellet fractions induced β-amyloid deposition. The β-amyloid induction by the pellet fraction achieved 95% of the activity observed with unfractionated, total Tg extract. Strikingly, the supernatant induced almost 30% of the β-amyloid load observed with the total Tg extract (Fig. 2E). Given that this fraction harbors <0.05% of the Aβ in the total Tg extract, these observations indicate a very high seeding activity of the 100,000 × g soluble Aβ assemblies in the brain extract (Table 1). The β-amyloid deposits induced by the pellet fraction were morphologically similar to those in the original Tg extract, while the β-amyloid induced by the supernatant was mostly diffuse and Congo red-negative (only 2.4 ± 1.0% of the compact Aβ deposits had a Congo red-positive core in comparison to 39.8 ± 6.5% with the unfractionated, total extract) after the 4 month incubation period (Fig. 2C). After a 6 month incubation period, however, the compact deposits induced by the supernatant fraction also were increasingly Congo red positive (29.8 ± 1.2%) (Fig. 2F). Again, all of the Congo red-positive Aβ deposits were accompanied by cytopathologic changes, i.e., activation of microglia and astrocytes, as well as dystrophic neurites (Fig. 2G), all identical in appearance to those induced by the total extract and in normal aged APP23 mice (Meyer-Luehmann et al., 2006).
The soluble β-amyloid-inducing factor is largely PK sensitive
Next we assessed the PK-sensitivity of the soluble β-amyloid-inducing fraction (Fig. 3). Results demonstrated that <5% of the Aβ in the 100,000 × g supernatant fraction was resistant to 30 min PK treatment, while the original Tg extract again revealed 80% PK resistance (Fig. 3A,B). The pellet fraction resulting from ultracentrifugation showed PK resistance similar to that of the untreated Tg extract (data not shown). The in vivo β-amyloid-inducing activity of the PK-digested supernatant fraction was then evaluated by intracerebral injections into 3-month-old pre-depositing APP23 mice, and compared with the activity of the undigested supernatant fraction 6 months after injection. Inactivation of PK was again achieved by boiling the samples (including the controls) before injection. Results revealed the expected β-amyloid induction by the supernatant fraction, whereas PK treatment almost completely abolished the β-amyloid-inducing activity of this fraction (Fig. 3C–E), suggesting that the potent, β-amyloid-inducing soluble Aβ assemblies are PK sensitive.
Figure 3.

The soluble β-amyloid-inducing activity is mostly proteinase K sensitive. A, Brain extracts (10%, w/v) from aged APP23 mice (Tg extract) and the 100,000 × g supernatant (SN) of the Tg extract (ultracentrifugation, 1 h, 4°C) were treated with 50 μg PK/ml for 30 min at 37°C. Immunoblotting for human Aβ (antibody 6E10) using 12% Bis-Tris gels reveals the expected PK resistance of the Tg extract. In contrast, only a minor fraction of the SN was not cleaved by PK (see long exposure). Note that the Tg extract was diluted 10-fold, and that 10-fold more of the SN fraction was loaded compared to that in Figures 1B and 2A, respectively. Also note that there is an empty lane between the two Tg extract lanes and the two lanes with the SN. B, Densitometric analysis of Aβ immunoblots after PK treatment [4 Tg extracts (each from a different animal) and the corresponding 100,000 × g supernatant were used; each preparation was tested at least 2 times and the mean was taken]. Aβ concentration at time point 0 was defined as 100%. Indicated is the mean ± SEM. C, D, In vivo seeding activity of brain extracts with or without PK treatment was tested by intrahippocampal injections into young, pre-depositing 3 month-old APP23 mice. Brains were immunohistochemically analyzed for Aβ-deposition 6 months later. Shown is the dentate gyrus of the hippocampus. SN of Tg brain extract was boiled for 5 min at 95°C (C). The small inset shows double labeling of Aβ immunoreactivity and Congo red binding of the induced β-amyloid in an adjacent section. D, PK-treated (30 min) and boiled SN of Tg brain extract. Scale bars: 200 μm and 20 μm. E, Stereological quantification of Aβ load in the hippocampus. PK treatment of the 100,000 × g supernatant of Tg brain extract diminished the seeding activity. t test revealed a significant difference between the two groups (n = 4–5 mice per group, mean ± SEM, t(7) = 4.713; **p < 0.01).
Extended sonication increases β-amyloid-inducing activity
To determine whether fragmentation of aggregates can augment the seeding capacity of brain extracts, the impact of extended sonication on the β-amyloid-inducing activity of the Tg extracts was assessed. Immunoblot analysis of the original (briefly sonicated) Tg extract and Tg extract with extended sonication revealed no change in total Aβ concentration (Fig. 4A). However, ultracentrifugation of the extracts revealed a higher amount of soluble Aβ in the supernatant of the Tg extract that had undergone extended sonication, suggesting the breakage of insoluble Aβ assemblies into smaller, 100,000 × g soluble units (Fig. 4A). The additional Aβ released into the supernatant fraction after extended sonication showed PK sensitivity similar to that of the Aβ in the supernatant of the untreated Tg extract (data not shown).
Figure 4.

Extended sonication increases β-amyloid-inducing activity. Brain extracts [10% (w/v)] from aged APP23 mice (Tg extract) were subjected to additional sonication (3 × 20 s). A, Immunoblot analysis with an antibody specific to human Aβ reveals no change in total Aβ between the original Tg extract (−) and the extract with extended sonication (+). However, more Aβ was found in the supernatant (SN) of the extra-sonicated extract after ultracentrifugation (100,000 × g; 1 h) (long exposure). B, C, In vivo seeding activity of the original and extra-sonicated extracts was tested by intrahippocampal injections into young, pre-depositing APP23 mice. Brains were immunohistochemically analyzed for Aβ deposition 4 months after injection. Shown is the dentate gyrus of the hippocampus. Injection of original Tg extract (B) induced robust congophilic Aβ deposition with a filamentous and dense pattern, while Aβ induction with the extra-sonicated Tg extract generated more punctate and small deposits (C). The inset shows double labeling of Aβ immunoreactivity and Congo red binding of the induced β-amyloid. Scale bars: 200 μm and 20 μm. D, Stereological quantification of Aβ load in the hippocampus revealed a significant increase in β-amyloid induction by extended sonication (n = 5–6 mice per group; mean ± SEM, t(9) = 3.188; *p < 0.05).
To test the β-amyloid-inducing activity of the Tg extract with and without extended sonication, extracts were injected intracerebrally into 4-month-old APP23 mice, and the brains of the injected animals were analyzed 4 months later. Aβ immunostaining revealed increased induction of β-amyloid deposits in the mice injected with the extra-sonicated brain extract compared to the mice injected with the original seeding extract (Fig. 4B–D). Morphologically, the original seeding extract induced a more filamentous and dense pattern of β-amyloid aggregation, whereas the deposits induced by the extra-sonicated seeding extract appeared smaller and more punctate. In both groups, a subset of the induced Aβ deposits were Congo red positive (Fig. 4B,C).
Discussion
Converging experimental data implicate corruptive protein templating in the induction and spread of proteinaceous lesions in a number of neurodegenerative diseases (Walker et al., 2006; Aguzzi and Rajendran, 2009; Colby and Prusiner, 2011). Pathologic similarities with prion disease suggest that these other disorders, though not known to be infectious, could involve the prion-like propagation of proteopathic seeds (Jucker and Walker, 2011). While it is now evident that the aggregation of Aβ can be exogenously induced in vivo by brain extracts containing aggregated Aβ (Kane et al., 2000; Meyer-Luehmann et al., 2006; Eisele et al., 2009, 2010; Watts et al., 2011), the seeds themselves have not been rigorously characterized. The present findings, together with previous work (Meyer-Luehmann et al., 2006), indicate that the β-amyloid-inducing factor in brain extracts from aged, APP-tg mice consists of a range of aggregated Aβ species of different sizes and of various sensitivities to PK digestion.
We previously found that synthetic Aβ fibrils—in PBS or mixed with wild-type brain extract—do not effectively seed Aβ deposition in vivo (Meyer-Luehmann et al., 2006). Our present results reveal that aggregated Aβ from β-amyloid-laden brains is more PK resistant than are synthetic Aβ fibrils. Because the amino acid sequence is the same, this finding indicates that Aβ fibrils in vivo either assume a multidimensional conformation that confers PK resistance compared to that of synthetic Aβ fibrils, or that posttranslational modifications and/or auxiliary factors in the brain inhibit the interaction of proteinase K with Aβ. Conformation has been shown to influence the resistance of Aβ to proteases, presumably by determining the accessibility of putative cleavage sites (Nordstedt et al., 1994), and differences in the structural properties of brain-derived Aβ fibrils and synthetic Aβ fibrils have been reported (Paravastu et al., 2009). In addition to these conformational differences, cofactors in the brain may protect Aβ fibrils from protease cleavage (Tennent et al., 1995; Söderberg et al., 2005), which may explain our finding of increased PK resistance of synthetic Aβ fibrils that are mixed with wild-type brain extract. Alternatively, the increased PK resistance could be an indirect effect of the increase in total protein when synthetic Aβ is mixed with wild-type brain extract.
Misfolded proteins with increased protease resistance may escape proteolytic degradation and become proteopathic seeds (Lansbury, 1997; Powers et al., 2009). However, PK resistant Aβ seeds are not the sole inductive species in our assay. PK-treated extracts induced β-amyloid deposition at a magnitude of only ∼55% of that of untreated extract (Fig. 1), arguing that PK-sensitive seeds must exist, and that they are highly efficient β-amyloid-inducing agents. This conclusion is supported by our ultracentrifugation experiments. Although <0.05% of the Aβ was in the 100,000 × g supernatant fraction, this soluble fraction was responsible for up to 30% of the seeding activity and was PK sensitive (Figs. 2, 3). Similarly, PK resistance was initially thought to be an important feature of infectious prions (McKinley et al., 1983), but more recent studies reveal that PK-sensitive PrP aggregates also are pathogenic, suggesting a range of infectious PrP species (Safar et al., 1998; Pastrana et al., 2006; Gambetti et al., 2008; Colby et al., 2010).
Small Aβ aggregates provide relatively more molecular interfaces for templated misfolding and efficient propagation compared to larger aggregates or fibrils. Thus, a higher fibrillogenic activity of small, soluble Aβ species compared to larger, insoluble assemblies would be predicted (Lee et al., 2007; Shankar and Walsh, 2009; Keshet et al., 2010). This view is supported by the present finding that extended sonication of Aβ-containing brain extract, i.e., fragmentation of larger β-amyloid assemblies into numerous smaller seeds, increases the seeding activity of the extract, in line with in vitro studies of amyloidogenic proteins (Soto et al., 2002; Knowles et al., 2009; Xue et al., 2009). Similarly, for prions, nonfibrillar small particles were found to be the most infectious agent, and sonication has been reported to increase prion infectivity (Silveira et al., 2005).
In AD research, a variety of pathogenic Aβ species have been described, ranging from Aβ dimers to larger oligomers, protofibrils, and fibrils (for review, see Haass and Selkoe, 2007; Glabe, 2008). In most studies of AD postmortem brain samples, the soluble pools consisting of small Aβ aggregates were found to correlate highly with AD severity and progression (Lue et al., 1999; McLean et al., 1999; Wang et al., 1999; Tomic et al., 2009; Mc Donald et al., 2010). In the present study, the morphological appearance of the induced β-amyloid deposits differed between the soluble and insoluble fractions. The β-amyloid deposits induced by the insoluble and PK-treated brain extracts were rather large and frequently congophilic aggregates that appeared to be nonuniformly distributed throughout the injected area. In contrast, the soluble Aβ species from brain extracts induced smaller, more often Congo red-negative patches of Aβ aggregates with a more homogeneous distribution throughout the injected region. Similarly, fragmentation of seeds by extended sonication induced smaller and more punctate β-amyloid deposits compared to the original, briefly sonicated extract. While these findings imply that the size of the seeds influences the nature of the induced deposits, the distributional and morphological differences of the induced lesions may also represent a difference in diffusion/transport of the larger, bulkier, insoluble Aβ seeds versus the smaller soluble seeds. In all cases, and independent of the morphological appearance, the congophilic aggregates were similarly surrounded by activated microglia, hypertrophic astrocytes, and dystrophic neurites, as previously described (Meyer-Luehmann et al., 2006). The relatively delayed emergence of Congo red-positive plaques in mice receiving the soluble material suggests either a different time course of initiation following seeding, and/or that these lesions eventually develop from the diffuse plaques.
In summary, the β-amyloid-inducing activity of brain extracts containing aggregated Aβ can be attributed to multiple entities with varying degrees of PK resistance or sensitivity. While insoluble pools of Aβ clearly are capable of seeding β-amyloidosis in vivo, brain extracts also contain soluble Aβ assemblies that we found to be super-proportionally active in inducing β-amyloidosis. The existence of soluble, bioactive Aβ seeds raises the possibility that these agents may mediate the spread of β-amyloidosis within the brain, and that they could serve as diagnostic biomarkers in biological fluids such as CSF or plasma. There is increasing evidence that Aβ deposition in the brain precedes the clinical symptoms of AD by many years (Holtzman et al., 2011). Sensitive and reliable identification of the earliest forms of pathogenic Aβ assemblies has the potential to expand the window for preventive interventions, and to refine current therapeutic strategies specifically targeting the initial and most pathogenic Aβ seeds.
Footnotes
This work was supported by grants from the Competence Network on Degenerative Dementias (BMBF-01GI0705), the Bundesministerium für Bildung und Forschung in the framework of ERA-Net NEURON (MIPROTRAN), and NIH Grant RR-00165. We thank Lisa Münter, Gerd Multhaup (Berlin), Heinke Schieb, Hans Klafki, Jens Wiltfang (Essen), Christoph Schall, Thilo Stehle (Tübingen), Ulrike Obermüller, Jörg Odenthal, Stephan Kaeser, and all the other members of our department for experimental help and comments on this manuscript.
References
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, Lemus A, Cohen FE, DeArmond SJ, Prusiner SB. Protease-sensitive synthetic prions. PLoS Pathog. 2010;6:e1000736. doi: 10.1371/journal.ppat.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Du D, Murray AN, Cohen E, Kim HE, Simkovsky R, Dillin A, Kelly JW. A kinetic aggregation assay allowing selective and sensitive amyloid-beta quantification in cells and tissues. Biochemistry. 2011;50:1607–1617. doi: 10.1021/bi1013744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, Roth K, Aguzzi A, Staufenbiel M, Walker LC, Jucker M. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009;106:12926–12931. doi: 10.1073/pnas.0903200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D, Belay ED, Schonberger LB, Marder K, Harris C, Burke JR, Montine T, Wisniewski T, Dickson DW, Soto C, Hulette CM, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer's disease and other neurodegenerative disorders. Ann Neurol. 2011 doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in beta-amyloid precursor protein-transgenic mice. J Neurosci. 2000;20:3606–3611. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet B, Yang IH, Good TA. Can size alone explain some of the differences in toxicity between beta-amyloid oligomers and fibrils? Biotechnol Bioeng. 2010;106:333–337. doi: 10.1002/bit.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- Lansbury PT., Jr Structural neurology: are seeds at the root of neuronal degeneration? Neuron. 1997;19:1151–1154. doi: 10.1016/s0896-6273(00)80406-7. [DOI] [PubMed] [Google Scholar]
- Lee S, Fernandez EJ, Good TA. Role of aggregation conditions in structure, stability, and toxicity of intermediates in the Abeta fibril formation pathway. Protein Sci. 2007;16:723–732. doi: 10.1110/ps.062514807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35:57–62. doi: 10.1016/0092-8674(83)90207-6. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Nesterenko MV, Tilley M, Upton SJ. A simple modification of Blum's silver stain method allows for 30 minute detection of proteins in polyacrylamide gels. J Biochem Biophys Methods. 1994;28:239–242. doi: 10.1016/0165-022x(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Nordstedt C, Näslund J, Tjernberg LO, Karlström AR, Thyberg J, Terenius L. The Alzheimer A beta peptide develops protease resistance in association with its polymerization into fibrils. J Biol Chem. 1994;269:30773–30776. [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded growth of beta-amyloid fibrils from Alzheimer's brain-derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci U S A. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, Requena JR. Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry. 2006;45:15710–15717. doi: 10.1021/bi0615442. [DOI] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg L, Dahlqvist C, Kakuyama H, Thyberg J, Ito A, Winblad B, Näslund J, Tjernberg LO. Collagenous Alzheimer amyloid plaque component assembles amyloid fibrils into protease resistant aggregates. FEBS J. 2005;272:2231–2236. doi: 10.1111/j.1742-4658.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci. 2002;25:390–394. doi: 10.1016/s0166-2236(02)02195-1. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennent GA, Lovat LB, Pepys MB. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci U S A. 1995;92:4299–4303. doi: 10.1073/pnas.92.10.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–358. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth JD, Asante EA, Collinge J. Review: contribution of transgenic models to understanding human prion disease. Neuropathol Appl Neurobiol. 2010;36:576–597. doi: 10.1111/j.1365-2990.2010.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Levine H, 3rd, Mattson MP, Jucker M. Inducible proteopathies. Trends Neurosci. 2006;29:438–443. doi: 10.1016/j.tins.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Watts JC, Giles K, Grillo SK, Lemus A, DeArmond SJ, Prusiner SB. Bioluminescence imaging of Abeta deposition in bigenic mouse models of Alzheimer's disease. Proc Natl Acad Sci U S A. 2011;108:2528–2533. doi: 10.1073/pnas.1019034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue WF, Hellewell AL, Gosal WS, Homans SW, Hewitt EW, Radford SE. Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem. 2009;284:34272–34282. doi: 10.1074/jbc.M109.049809. [DOI] [PMC free article] [PubMed] [Google Scholar]