

Abstract
We measured the inhibition of infectivity of HIV-1 isolates and derivative clones by combinations of neutralizing antibodies (NAbs) and other entry inhibitors in a single-cycle-replication assay. Synergy was analyzed both by the current linear and a new nonlinear method. The new method reduced spurious indications of synergy and antagonism. Synergy between NAbs was overall weaker than between other entry inhibitors, and no stronger where one ligand is known to enhance the binding of another. However, synergy was stronger for a genetically heterogeneous HIV-1 R5 isolate than for its derivative clones. Enhanced cooperativity in inhibition by combinations, compared with individual inhibitors, correlated with increased synergy at higher levels of inhibition, while being less variable. Again, cooperativity enhancement was stronger for isolates than clones. We hypothesize that genetic, post-translational or conformational heterogeneity of the Env protein and of other targets for inhibitors can yield apparent synergy and increased cooperativity between inhibitors.
INTRODUCTION
The functional human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein complex (Env) is a trimer of hetero-dimers that each consists of the outer gp120 subunit attached non-covalently to the transmembrane glycoprotein, gp41. The docking of gp120 onto the primary receptor, CD4, triggers conformational changes that allow interactions with the CCR5 or CXCR4 co-receptor; these interactions in turn activate a refolding of the Env complex that unleashes the fusogenic potential of gp41, ultimately allowing the viral core to enter the cytoplasm of the target cell (Doms and Peiper, 1997; Pantophlet and Burton, 2006). Neutralizing antibodies (NAbs) interfere with this process at different stages by binding to different epitopes; some recognize gp120 and impede receptor interactions; others interact with gp41 and interfere with later stages of entry (Klasse and Sattentau, 2002; Ugolini et al., 1997; Zwick and Burton, 2007). The induction of broadly active and potent NAbs is a crucial but elusive requirement for an effective vaccine to prevent HIV-1 infection. The access to epitopes on the native Env complex is restricted, as is the immunogenicity of the few epitopes that bind broadly active NAbs (Burton et al., 2004; Karlsson Hedestam et al., 2008; Klasse et al., 2011; Pantophlet and Burton, 2006; Poignard et al., 1996b; Poignard et al., 2001; Zwick and Burton, 2007). Env-vaccine design aims to induce protective levels of NAbs against these neutralization epitopes. But how well do NAbs act in combination? Do they have stronger or weaker effect when combined than when acting individually?
Various small organic molecules, as well as peptides and proteins, can also inhibit HIV-1 entry, again by acting at different stages of the entry process. Some such compounds are used in therapy or may become components of microbicides or oral prevention regimens to block sexual transmission (Grant et al., 2010; Klasse et al., 2008; Lederman et al., 2006; Veazey et al., 2005). The use of inhibitor combinations has long been standard for treating HIV-1 infection, but it may also be advantageous for prevention. For example, more than one inhibitor may be needed in a microbicide to counteract HIV-1 sequence diversity, while any enhanced potency of a combination may allow lower amounts of each drug to be used, reducing cost and improving safety (Grant et al., 2008; Ketas et al., 2007b). The quantitative analysis of combinatorial effects is therefore important both in prevention and therapy.
Synergy is a special case of combined effects (Berenbaum, 1977; Greco et al., 1995; Loewe, 1953). Its potential occurrence between NAbs and other entry inhibitors merits a rigorous, quantitative investigation. Synergy can be defined as a greater potency of combined inhibitors than would be predicted from their individual effects (Loewe, 1953); weaker than predicted potency is called antagonism; when the combined potency is neither enhanced nor reduced, it is categorized as additivity. The method most commonly used to quantify synergy in the inhibition of HIV-1 replication analyzes the inhibitor-concentration dependence after a linear transformation of the data (Chou and Talalay, 1981, 1984). Here, we compare that method with a new, non-linear approach.
What types of infectivity-inhibition assays are suitable for synergy analyses? Valid synergy assessments require proportionality between the infectious dose and the resulting propagation of the virus, which can only be guaranteed in certain titration zones of single-cycle replication assays; the distortions inherent in multi-cycle replication can create artifactual, or obliterate authentic, synergy (Ferguson et al., 2001). Moreover, PBMC assays based on production of the HIV-1 p24 Gag antigen lack precision (Choudhry et al., 2006; Heredia et al., 2007a; Heredia et al., 2007b; Ketas et al., 2007). Despite that problem, PBMC or T-cell-line assays with a p24 read-out have been used extensively in studies of synergy involving NAbs and other inhibitors (Dorr et al., 2005; Eron et al., 1992; Gantlett et al., 2007; Johnson et al., 1989; Johnson et al., 1990; Johnson et al., 1992; Kennedy et al., 1991; Laal et al., 1994; Li et al., 1997; Mascola et al., 1997; McKeating et al., 1992; Nakata et al., 2008; Strizki et al., 2005; Tremblay et al., 1999; Tremblay et al., 2005a; Tremblay et al., 2005b; Tremblay et al., 2002; Tremblay et al., 2000; Vermeire et al., 2004; Xu et al., 2001; Zwick et al., 2001). Here, we explored whether data obtained from a PBMC assay are amenable to synergy analysis.
Cooperativity differs from synergy in that it can occur for each individual ligand; it typically arises when ligands interact with multimeric proteins (Hill, 1913; Koshland et al., 1966; Monod et al., 1965). At least some NAbs may show negative coperativity in binding to the trimeric HIV-1 Env complex (Gustchina et al., 2010). However, cooperativity as measured by slope coefficients is subject to other molecular influences, and slope differences complicate synergy determinations (Chou and Talalay, 1981; Greco et al., 1995; Hoffman and Goldberg, 1994; Loewe, 1953; Weiss, 1997). We have therefore also investigated the relationship between synergy and changes in cooperativity.
Overall, we found that synergy between NAbs was weaker than between certain other entry inhibitors, and that both synergy and cooperativity enhancement were stronger for HIV-1 isolates than derivative clones. We suggest that heterogeneity among the target molecules, both viral and cellular, is a general source of apparent synergy and enhanced cooperativity between entry inhibitors. Although the molecular basis of these phenomena may differ from what has previously been assumed, the resulting effects may nevertheless be biologically important. We propose changes in both in the methods and the interpretations of combinatorial analyses of viral inhibitors. We emphasize that both the synergy and cooperativity phenomena observed can be regarded as apparent, i.e. as merely fulfilling the criteria of their respective mathematical definitions. We also try to keep the two problems we have addressed strictly separate: first, under what conditions these quantities can be measured correctly; and second, how certified observations of synergy and changes in cooperativity should be interpreted in molecular terms.
RESULTS
Comparison of linear and non-linear methods for determining inhibitory concentrations
The commonly used method for evaluating synergy is based on linear transformation of infectivity-inhibition data (Chou and Talalay, 1981, 1984). Our new one uses non-linear regression to fit a non-linear function, based on the Law of Mass Action, to untransformed data: I=(C/Ki)η/(1+(C/Ki)η)), where I = relative inhibition of infectivity; Ki is the half-maximal inhibitory concentration by analogy with the dissociation constant, Kd, in the Law of Mass Action; C = concentration of an inhibitor; η = slope coefficient or cooperativity factor, corresponding to the Hill constant of binding. One advantage of this non-linear function is that it can be fitted without assuming any reduction in the plateau of maximum inhibition (which is beyond the resolution of our current data); it also avoids the problem that data transformed by the other method, so as to become linear, are often far from linear. In addition, unlike the linear function, the non-linear one does not require values that lie just above 100% (or below 0%) inhibition to be censored, whereby loss of many high-quality data points is avoided. Here we did not, however, include such data in the non-linear analysis; we opted for a comparison of how the two methods perform based on exactly same data. In a previous informal comparison of the linear method with a different non-linear one, only 38 out of 136 published data sets yielded conclusions about synergy that were in close agreement (Greco et al., 1995). As laid out in what follows, it should be noted that there are other differences between the standard approach and ours than how inhibitory concentrations are calculated (see also Supplement).
To generate data for our own comparative synergy analyses, we titrated four NAbs, other entry inhibitors and one non-nucleoside reverse-transcriptase inhibitor (NNRTI), both alone and in selected combinations, against HIV-1 isolates CC1/85 and IIIB, as well as against clones derived from these viruses (9-6, 9-7, and 9-8 for CC1/85; NL4.3 and LAI for IIIB). The residual infectivity was quantified on Tzm-bl cells. The linear and non-linear methods were used for calculating synergy indices for data from 316 such inhibition experiments, including all six possible pair-wise combinations of the four NAbs.
Some examples of how the two methods can generate different synergy or antagonism outcomes are illustrated in Fig. 1 and Table 1. For each combination, the left-hand panel in Fig. 1 shows the median-effect plot of the Chou-Talalay method. The linear transformation involves calculating the ratio between the “fraction affected”, fa (i.e. the fraction of virus prevented from replicating) and the “fraction unaffected”, fu (i.e. the residual infectivity). The logarithm of that ratio is plotted as a function of the logarithm of the concentration of each inhibitor or their combination. Although this transformation is the standard basis for calculating synergy, it is hypersensitive to errors in the upper and lower zones of inhibition. Thus, a change from 90% to 99.999% inhibition involves a 1000-fold greater change in the fa/fu ratio than a change from 90% to 99%, although the difference between 99% and 99.999% is well within the margin of error for the assay. The plots in each right-hand panel show the fits by non-linear regression of the non-linear function described above to untransformed fractional data. The degree of inhibition is plotted as a function of inhibitor concentration on a log scale (Fig. 1).
Figure 1. Synergy analyses of HIV-1 inhibitor combinations by linear and nonlinear methods.
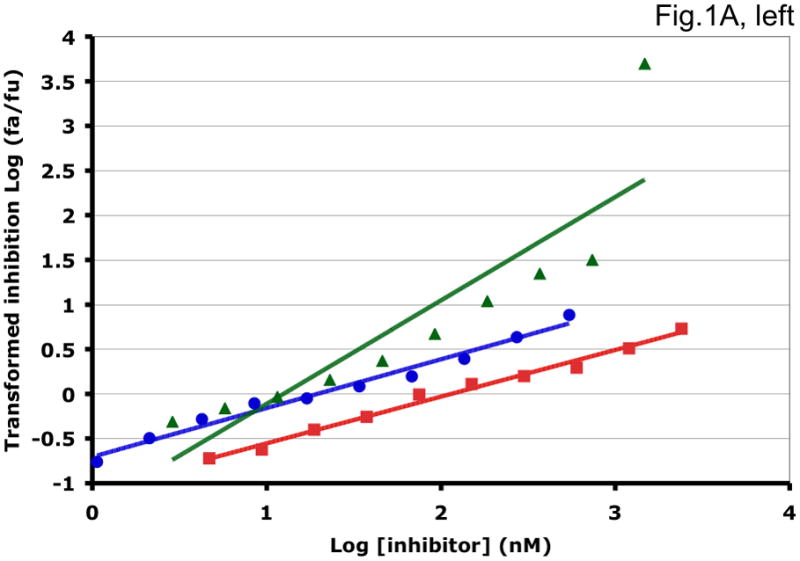
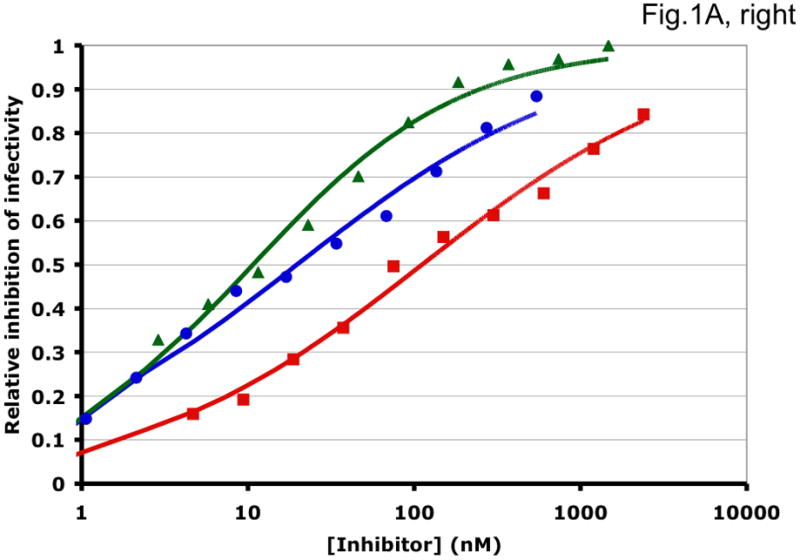
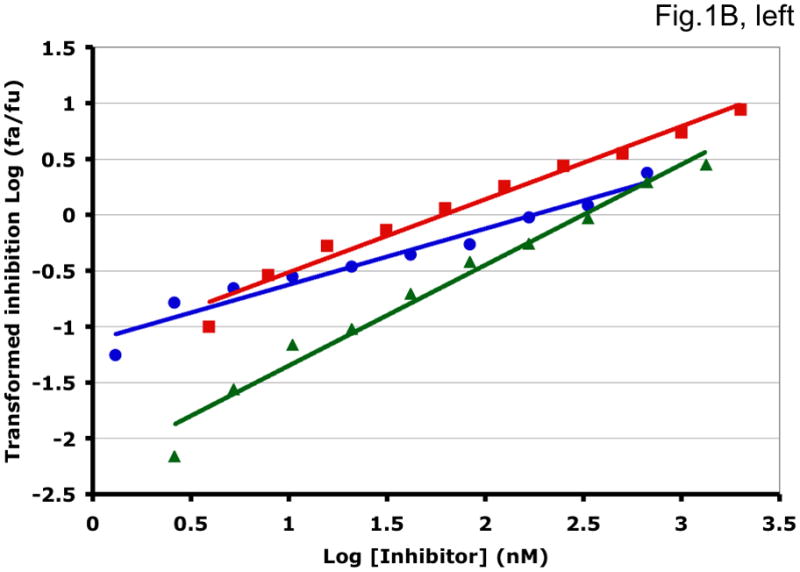
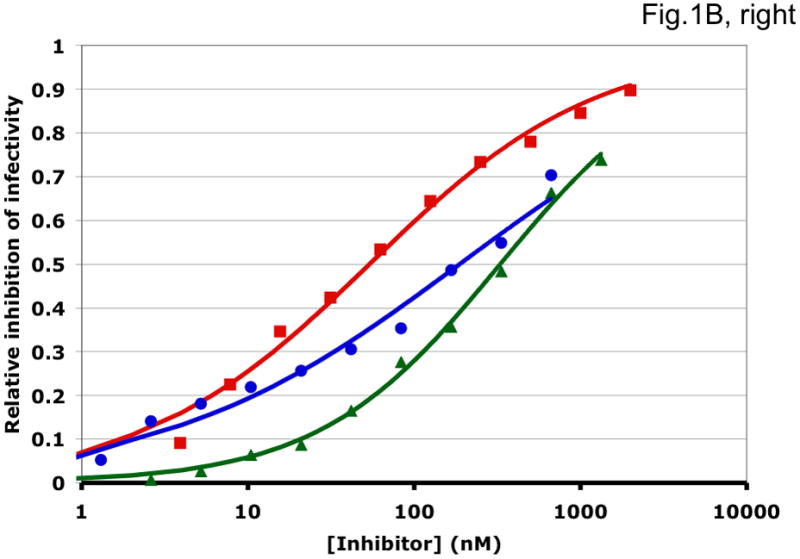
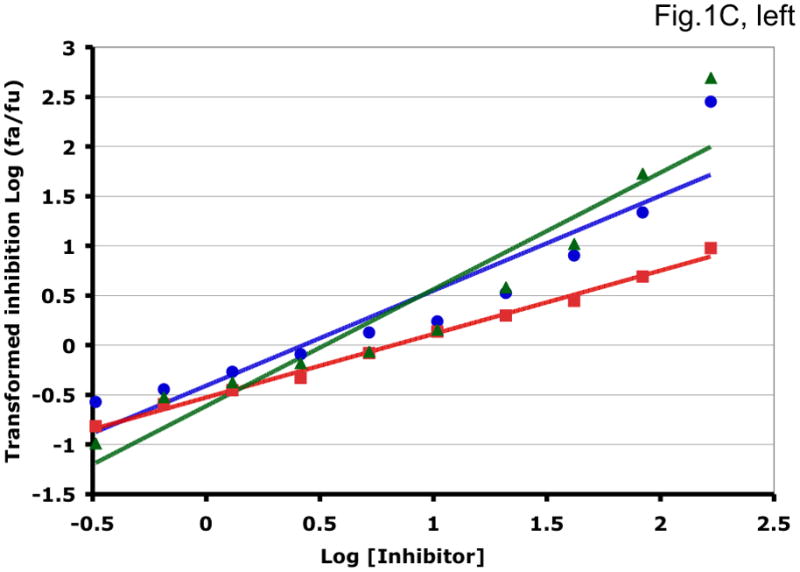
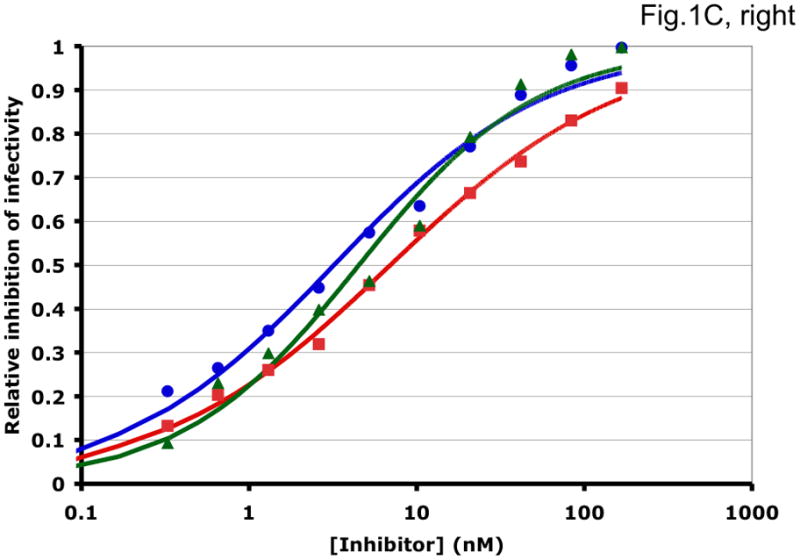
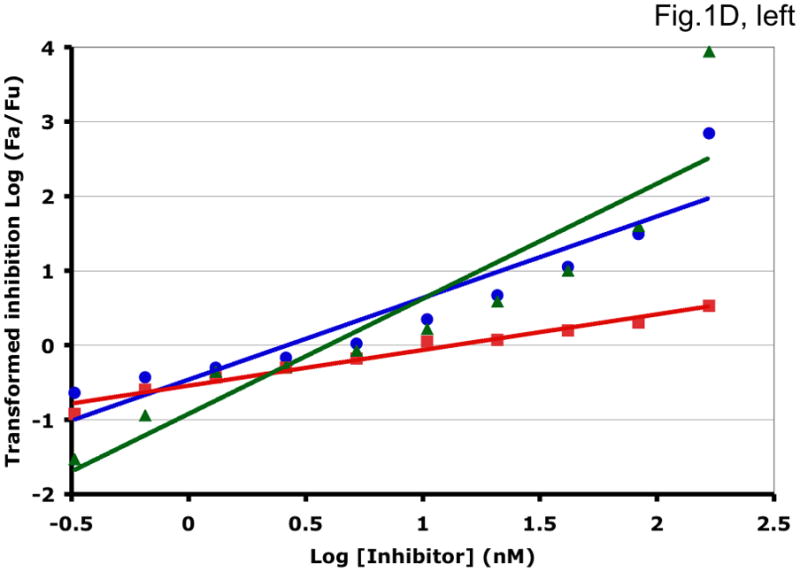
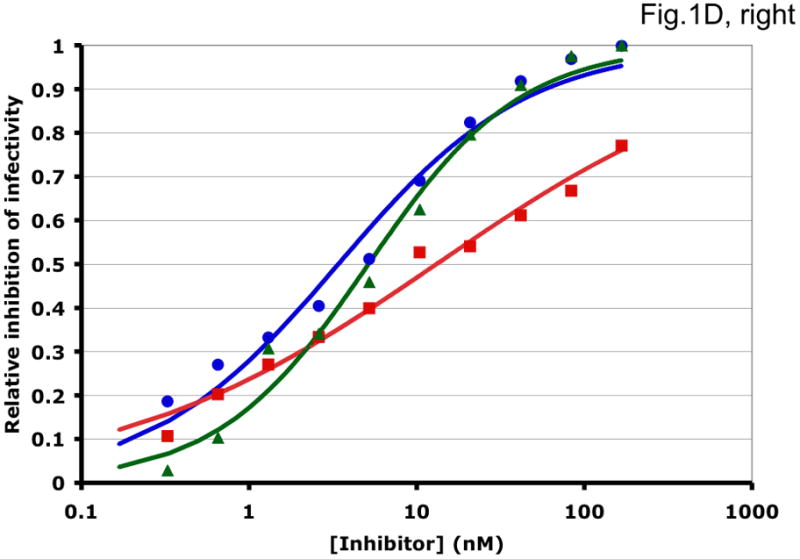
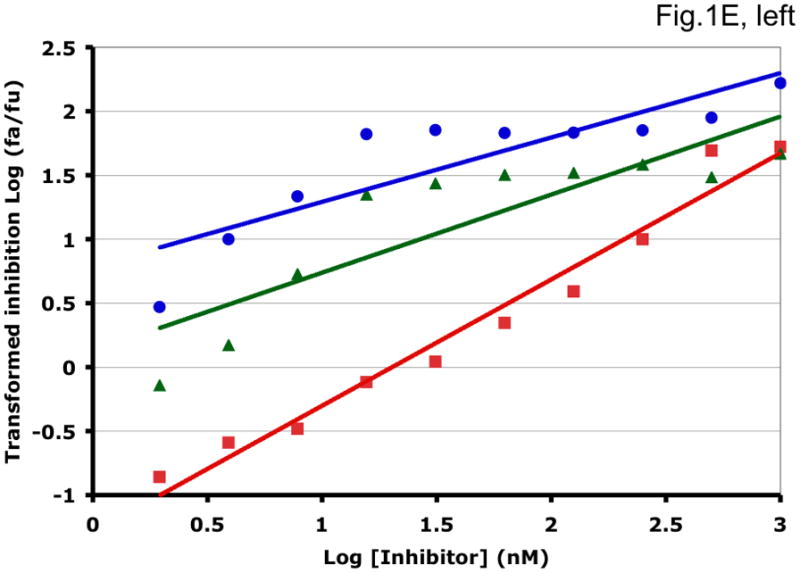
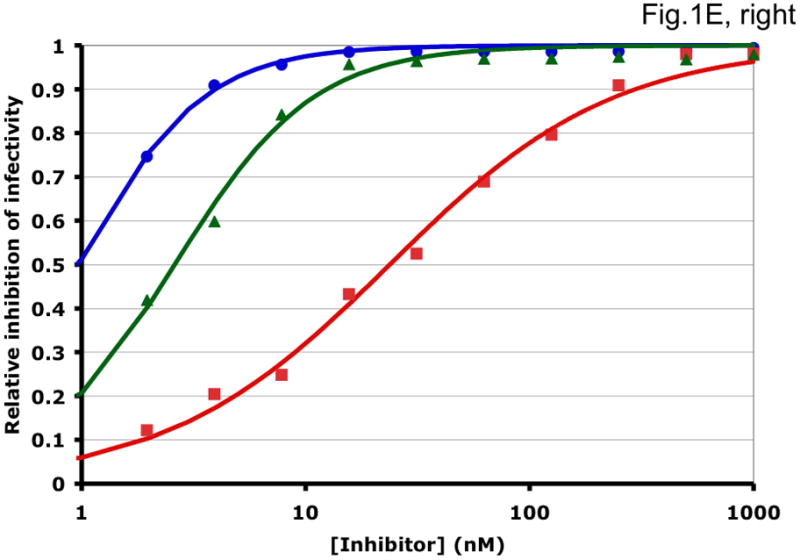
Left-hand panels show median-effect plots of the linearly transformed data. Y-axis: the log10 of the ratio of the fraction of inhibited infectivity over the fraction remaining. X-axis: the log10 of the concentration of each inhibitor, or their sum in combinations. Right-hand panels show the non-linear function fitted to the same data; relative inhibition of infectivity is plotted on the y-axis as a function of the concentration of individual inhibitors, or their sum, on a log scale on the x-axis. A. Inhibition of CC1/85 by VCV (blue, circles), T-20 (red, squares), and their combination at a molar ratio of 17/75 (green, triangles). The shift to the left of the green curve indicates strong synergy. B. Inhibition of CC1/85 by NAb 2F5 (blue, circles), T-20 (red, squares), and their combination at a ratio of 120/27 (green, triangles). The shift to the right of the green curve indicates antagonism. C. Neutralization of CC1/85 by NAbs b12 (blue, circles), 2G12 (red, squares), and their combination at a molar ratio of 1/1 (green, triangles). The intermediate position of the green curve indicates approximate additivity. D. Neutralization of the X4 isolate IIIB by NAbs b12 (blue, circles), 2G12 (red, squares), and their combination at a molar ratio of 1/1 (green, triangles). The intermediate position of the green curve indicates approximate additivity. E. Inhibition of IIIB by AMD3100 (blue, circles), Nevirapine (red, squares), and their combination at a molar ratio of 1/1 (green, triangles). The poor fit of the linear function results in a spurious indication of antagonism, whereas the non-linear method yields an outcome that is close to additivity (Table 1). The graphs are simplified for the purpose of illustration: only three curves are shown in each, the inhibition by the combined inhibitors being expressed as a function of the sum of their concentrations. The computations of the synergy indices are performed on values obtained by fitting the function to four separate data series, each having individual inhibitor concentrations on the x axis.
Table 1.
Comparison of linear and non-linear methods of synergy assessment
| Virus | Inhibitor | Lin/Non-lina | Lin/Non-lina | Lin/Non-lina | Lin/Non-lina | |
|---|---|---|---|---|---|---|
| Half-max. inhib. conc. (Dm/Ki)b | Half-max. inhib. conc. c.i. (fold)c | R2/R2d | CI/σ e | |||
| CC1/85 | VCV | 20/20 | 5.0/1.5 | 0.98/0.99 | 0.79/0.99 | 0.21/0.18 |
| T-20 | 110/110 | 5.5/1.4 | 0.99/0.99 | |||
| CC1/85 | 2F5 | 180/190 | 29/1.8 | 0.95/0.98 | 0.98/1.0 | 4.3/5.0 |
| T-20 | 61/54 | 11/1.4 | 0.98/0.99 | |||
| CC1/85 | b12 | 2.7/3.2 | 13/1.6 | 0.88/0.99 | 0.90/0.98 | 0.87/1.0 |
| 2G12 | 6.7/6.9 | 1.8/1.2 | 0.99/1.0 | |||
| IIIB | b12 | 2.6/3.4 | 14/1.6 | 0.87/0.98 | 0.84/0.99 | 0.89/0.95 |
| 2G12 | 14/13 | 4.4/1.5 | 0.97/0.99 | |||
| IIIB | AMD3100 | 0.027/0.97 | 104/1.4 | 0.76/1.0 | 0.74/0.99 | 11/1.4 |
| Nevirapine | 20/24 | 8.3/1.3 | 0.97/0.99 | |||
The values obtained by the linear and non-linear methods are expressed as the numerators and the denominators in ratios, respectively.
The half-maximal inhibitory concentrations (nM) in the linear function (log (fa/fu) = m log(D/Dm)) is Dm, and in the non-linear function (I=(Ki/D)η/(1+(Ki/D)η)), it is Ki.
The 95% confidence interval (c.i.) of the half-maximal inhibitory concentrations obtained by the respective fits are described by their fold variation from the lower to the upper limit.
The R2 values describe the closeness of the fit: R2=1 represents the ideal fit. The values in the lower two boxes to the left pertain to the individual inhibitors, the values in the taller box to the right, to their combination.
The combination index (CI) of the linear, and the synergy indicator σ of the nonlinear method, indicate synergy when <1, additivity when =1, and antagonism when >1.
Synergistic inhibition of the HIV-1 R5 isolate CC1/85 by the CCR5 small molecule ligand vicriviroc (VCV) and the gp41 HR2 peptide T-20 is suggested by the displacement of the combination curve (green) to the left of the individual curves (red and blue) (Fig. 1A) Conversely, the gp41-directed NAb 2F5 and the HR2 peptide T-20 were antagonistic, and the combination curve is shifted to the right of both individual curves (Fig. 1B). The latter outcome was expected because T-20 contains the 2F5 epitope; the peptide binds the NAb to form an inert complex. Subtler synergy or additivity arose when isolates CC1/85 and IIIB were neutralized by NAbs b12 and 2G12 in combination (Figs. 1C and 1D). Here, the curves for the combinations fell mostly between those for the individual NAbs. Finally, an experiment with the CXCR4 ligand AMD3100 combined with the NNRTI Nevirapine generated several data points that represented nearly complete inhibition (Fig. 1E). Such data are less amenable to analysis by the linear than the non-linear method. Thus, the linear transformation (Fig. 1E, left-hand panel) gave a poorer curve fit than the non-linear one (right-hand panel) (Table 1). Table 1 shows that poor fits can substantially distort the outcome of the analyses.
It should be noted that the slopes of the curves in Fig. 1 differ and deviate from 1. Such complicating factors make synergy or antagonism hard to detect by inspecting the data. For example, when the slopes for the individual inhibitors differ, the combined curve will theoretically be bent, with different slopes at high and low inhibitor concentrations. A linear curve may therefore fit poorly even to theoretically perfect data. Greater complications arise with predictions of the combined fractional effect than when the inhibitory concentrations are compared for individual and combined inhibitors (Ferguson et al., 2001; Greco et al., 1995). Calculating a synergy index using Loewe’s formula is the best way to make the latter comparison (Greco et al., 1995; Loewe, 1953). We use this formula throughout, whereas Chou and Talalay apply it only to what is deemed to be mutually exclusive inhibition.
For clarity, we designate the synergy indices calculated by the linear method as CI (combination index; (Chou and Talalay, 1981, 1984)) and those by the non-linear one as σ (sigma) (Table 1). CI and σ both indicate synergy when <1, additivity when =1 and antagonism when >1. The non-linear method gave narrower confidence intervals of the half-maximal inhibitory concentrations (Dm and Ki) and higher R2 values (i.e. better fits), both for individual inhibitors and for combinations. In individual experiments, the nonlinear method indicated marginally stronger synergy than the linear for the combination of VCV with T-20, and somewhat stronger antagonism for 2F5 combined with T-20 (Table 1). In the two examples of NAbs b12 combined with 2G12 against isolates CC1/85 and IIIB, the linear method indicated modest synergy, the non-linear method mere additivity (Table 1). The displayed data for the AMD3100 and Nevirapine combination were dealt with conspicuously less well by the linear method, which works poorly when inhibition approaches 100% (Figure 1E). Thus, R2 was low, the imprecision of the Dm value was substantial, and the CI value of 11 indicated strong antagonism (Table 1). In contrast, the non-linear method generated excellent fits to the same data and indicated only modest antagonism (σ50% = 1.4), which was close to the mean from four replicate experiments (σ50% = 1.6 ± 0.27). Overall, these various examples illustrate analytic artifacts that the linear method is more prone to than is the non-linear one.
To compare the validity of the two methods further, we combined each inhibitor with itself in all experiments by adding it twice, each time at half the total concentration. A synergy analysis of such controls should theoretically give exact additivity (i.e. CI = σ = 1). We therefore refer to these titrations as additivity controls. The deviation of the additivity controls from ideality, as analyzed by the two methods, is shown in Supplementary Table 1. When we analyzed a total of 632 such titrations (two additivity controls from each of 316 experiments), the variability, expressed as the fold-spread from minimum to maximum, was greater for the linear method. However, both methods frequently indicated spurious synergy as well as antagonism, particularly at the higher levels of inhibition. The fold-spread CI or σ at 50% inhibition was 11-fold narrower for the non-linear than the linear method; at 75% inhibition it was 45-fold narrower; at 90% 180-fold (Supplementary Table 1). Further comparisons of the variability of the results obtained by the two methods are given in the Supplement.
We conclude that although neither method is acceptably accurate or precise at the higher levels of inhibition, the non-linear one yields more valid results. Hence, from here on, we only cite results obtained with the non-linear method and focus on the mid-range zone of inhibition. Furthermore, we avoid the standard classification of combinations as mutually exclusive and non-exclusive, as well as the concomitant modifications of the formula for calculating combination indices (Chou and Talalay, 1981). Both theoretical and pragmatic reasons for this choice are given in the Supplement.
Single- vs. multi-cycle replication
Multi-cycle replication of viruses can distort synergy analyses (Ferguson et al., 2001). It was therefore imperative to determine how many replication cycles can occur in the Tzm-bl assay under the conditions we used; although the input virus is not defective, the limited culture period (40 h) might prevent any progeny virus from initiating new cycles. Accordingly, we compared the infectivity of the 9-7 clone in the Tzm-bl cell and PBMC assays, both in the presence and absence of a protease inhibitor, Atazanavir, that prevents the formation of infectious progeny viruses. The luciferase signal generated in the Tzm-bl assay was proportional to the virus input, whereas the amount of p24 antigen produced in the PBMC assay varied exponentially with the input (Supplementary Figure 1). Furthermore, Atazanavir had a negligible effect in the Tzm-bl assay, confirming that progeny virus did not contribute to the endpoint. In contrast, the protease inhibitor eliminated p24 production in the PBMC assay, suggesting that the first replication cycle contributes only marginally. Both lines of evidence thus strongly indicate that the Tzm-bl assay records single-cycle replication while the PBMC assay involves multiple cycles.
We then explored whether multi-cycle replication would allow meaningful synergy assessments. The most accurate synergy measurement we identified, σ50% (Table 1, Supplementary Table 1), varied drastically with the time of sampling, and as much for the additivity controls as for the NAb b12-2G12 combination (Figure 2). The variation in σ50% among three repeat experiments was also extensive (Figure 2). Hence, the results from the PBMC assay were unsuitable for synergy analyses. Multiple cycles of replication complicate synergy calculations based on fractional effects (Ferguson et al., 2001). However, the large errors we found in concentration-based synergy calculations by Loewe’s formula can be attributed to the imprecision of the PBMC assay.
Figure 2. Lability of synergy measurements in a multi-cycle replication assay.
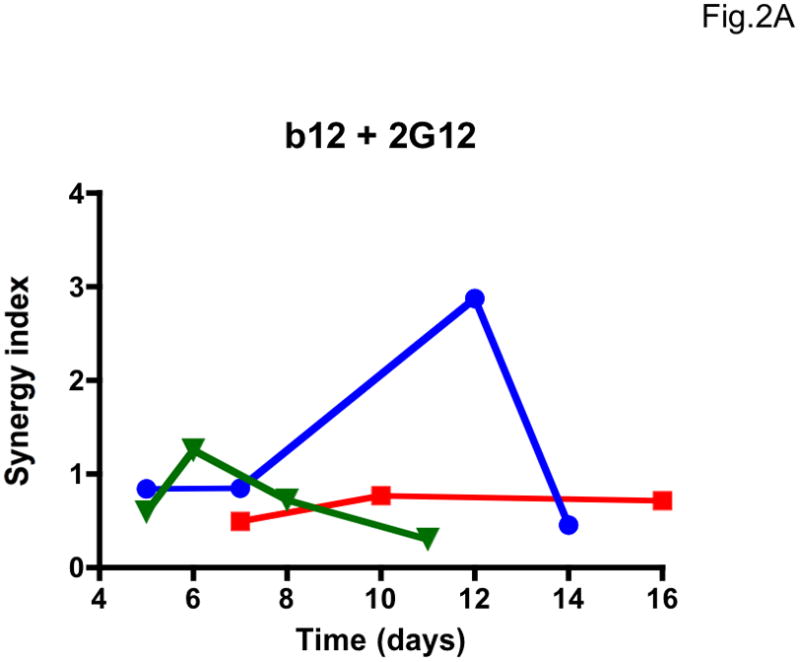
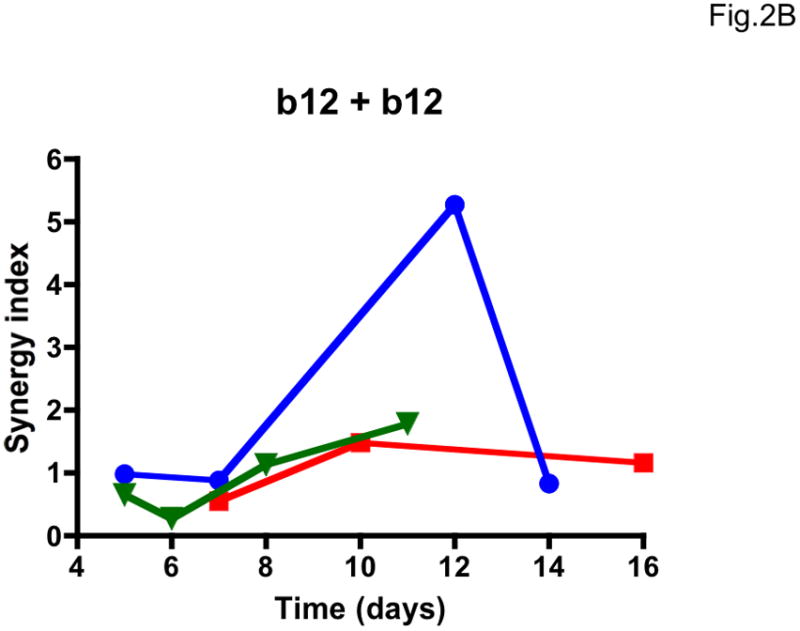
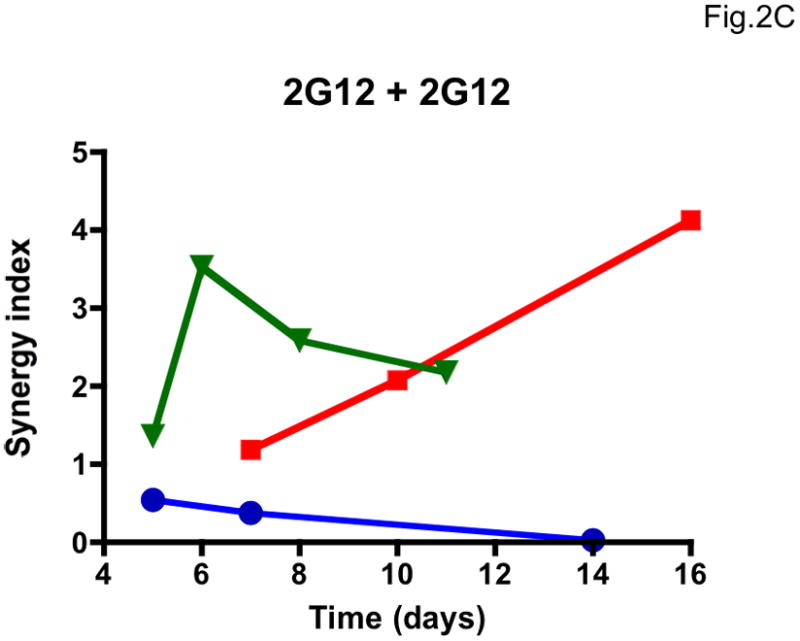
The dependence of apparent synergy on the duration of PBMC cultures, and hence the number of replication cycles, was investigated by taking samples for p24 quantification from days 5 to 16 after infection with the clonal virus 9-7. Each diagram shows the results from three experiments (Exp 1, blue circles; Exp 2, red squares; Exp 3, green triangles; symbols for each experiment are connected by lines). A. Synergy index at 50% inhibition, σ50%, for the combination b12 + 2G12 used at a constant ratio close to their IC50 ratio; B. σ50% for the additivity control b12 + b12. C. σ50% for the additivity control 2G12 + 2G12. The results varied over time from considerable synergy to antagonism for the b12 + 2G12 combination. However, the additivity controls for both NAbs gave even more divergent results over time, and since any deviation from 1 is an error in B and C, it is likely to be so in A as well.
Comparing isolates and clones for synergy in entry inhibition
The combination of the CCR5 ligands VCV (small molecule) and PRO 140 (MAb) was synergistic against the CC1/85 R5 isolate and the derivative clones 9-6 and 9-7, with a σ50% value of 0.33–0.35. Synergy was weaker (σ50% = 0.58) for this combination against the other CC1/85 clone, 9-8 (Table 2). The strongest synergy recorded (σ50% = 0.22) was for VCV combined with T-20 against CC1/85, but it was weaker (σ50% = 0.68–0.84) for the same combination against the three R5 clones. In contrast, the corresponding combination of the CXCR4 ligand AMD3100 with T-20 was not synergistic for any of the X4 isolates or clones (σ50% = 0.90–1.2). VCV combined with the NNRTI Nevirapine was moderately synergistic against the CC1/85 isolate (σ50% = 0.58), weakly synergistic or additive against the 9-6, 9-7 and 9-8 clones (σ50% = 0.85–0.93). There was a tendency towards antagonism between AMD3100 and Nevirapine for the X4 viruses, particularly for IIIB (σ50% = 1.6).
Table 2.
Synergy (σ50%) in blocking the infection of Tzm-bl cells by R5 and X4 HIV-1 isolates and clones
| Combination | Virus | ||||||
|---|---|---|---|---|---|---|---|
| R5 | X4 | ||||||
| CC1/85 | 9-6 | 9-7 | 9-8 | IIIB | LAI | NL4.3 | |
| VCV + PRO 140 | 0.35a±0.075 (n=5) | 0.33±0.035 (n=7) | 0.35±0.045 (n=4) | 0.58±0.27 (n=4) | - | - | - |
| VCV/AMD3100b + T-20 | 0.22±0.023 (n=4) | 0.68±0.066 (n=8) | 0.84±0.062 (n=5) | 0.84±0.075 (n=6) | 1.2±0.23 (n=8) | 0.90±0.141 (n=6) | 1.1±0.094 (n=7) |
| VCV/AMD3100b + Nevirapinec | 0.58±0.099 (n=3) | 0.93±0.10 (n=3) | 0.87±0.058 (n=3) | 0.85±0.036 (n=3) | 1.6±0.27 (n=4) | 1.3±0.30 (n=3) | 1.1±0.14 (n=3) |
| CD4-IgG2 + 17b | 1.1±0.23 (n=4) | 0.62±0.16 (n=4) | 0.78±0.16 (n=6) | 0.73±0.12 (n=5) | 1.1±0.14 (n=4) | 1.2±0.18 (n=3) | 0.79±0.099 (n=5) |
| b12 + 2G12 | 0.88±0.12 (n=4) | 0.72±0.066 (n=3) | 0.60±0.038 (n=5) | 0.75±0.051 (n=4) | 0.77±0.13 (n=3) | 0.66±0.033 (n=3) | 0.85±0.081 (n=4) |
| b12 + 2F5 | 0.73±0.16 (n=3) | 1.1±0.050 (n=3) | 1.6±0.39 (n=3) | 0.71±0.12 (n=4) | 0.95±0.065 (n=3) | 0.75±0.014 (n=3) | 1.2±0.17 (n=3) |
| b12 + 17b | 0.68±0.15 (n=3) | 1.5±0.12 (n=4) | 1.2±0.289 (n=4) | 1.9±0.28 (n=3) | 1.2±0.21 (n=4) | 0.93±0.066 (n=3) | 0.69±0.10 (n=3) |
| 2G12 + 2F5 | 0.50±0.094 (n=3) | 1.6±0.78 (n=3) | 0.69±0.141 (n=3) | 0.60±0.055 (n=3) | 0.67±0.16 (n=3) | 0.60±0.17 (n=3) | 1.00±0.105 (n=3) |
| 2G12 + 17b | 0.64±0.049 (n=8) | 1.2±0.13 (n=7) | 0.78±0.097 (n=6) | 0.80±0.063 (n=9) | 0.53±0.12 (n=7) | 0.72±0.11 (n=7) | 1.2±0.16 (n=8) |
| 2F5 + 17b | 1.4±0.50 (n=6) | 1.6±0.47 (n=6) | 1.1±0.28 (n=5) | 0.83±0.20 (n=6) | 0.65±0.19 (n=4) | 0.50±0.050 (n=4) | 0.98±0.125 (n=4) |
The values are the mean values of σ50%from n (3 ≤ n ≤ 9) replicate experiments± SEM. σ50% denotes synergy when <1, additivity when = 1, and antagonism when >1. Mean σ50% values − 2 SEM >1 are shown in underlined italics; mean σ50% values + 2 SEM < 1 are in bold. Values marked thus indicate synergy or antagonism ascertained with 95% confidence.
VCV was used against R5 viruses, AMD3100 against X4 viruses.
Nevirapine is an NNRTI and thus the only inhibitor tested that blocks a post-entry step.
The receptor mimic CD4-IgG2, a tetravalent soluble CD4 construct, enhances the binding of the 17b antibody to monomeric gp120. Conversely, 17b inhibits soluble CD4 binding to gp120 (Moore and Sodroski, 1996). In line with this lack of mutually enhanced binding, CD4-IgG2 was not consistently synergistic together with 17b (Table 2). For this pair, synergy was no stronger or more consistent than for some of the NAb-NAb combinations (see below). For example, the combination of b12 with 2G12 gave σ50% values < 1 for all viruses (Table 2).
Moderate synergy was detected sporadically for the six NAb-NAb combinations against the seven test viruses (Table 2). Overall, the virus-NAb combinations gave σ50% + 2 SEM values that sometimes fell below 1, but only twice were the values of σ50% >2 SEM above 1 (for b12 + 17b against clones 9-6 and 9-8). The inconsistent outcome may reflect the considerable variability inherent to even the most precise assessment of synergy (i.e. σ50%). Although we performed 3–9 replicates of each experiment, it was still difficult to identify, or exclude, genuine moderate synergy in a matrix containing as many as 67 virus-inhibitor combinations, given the experimental variation (Supplementary Table 1): the mass-significance problem and the magnification of the experimental variation by the synergy calculations together exacerbate the difficulties of interpretation. Against that background it can be noted that with 95% confidence intervals, three σ50% values out of 67 deviating significantly from 1 would be expected by chance, although that could be in either direction as for the additivity controls (Supplementary Table 1). In contrast to that expected noise, 32 σ50% values were significantly below 1; three were above. In other words, it is beyond doubt that synergy occurs with combinations of these inhibitors. Nevertheless, we conclude that multiple replicates in assays with high precision, as well as focusing on half-maximal effect where σ varies least, are necessary steps to render synergy analyses of viral inhibition meaningful.
The strongest synergy among the NAb combinations was observed for 2G12 + 2F5 against CC1/85 (σ50%= 0.50), 2G12 + 17b against IIIB (σ50%= 0.53), and 2F5 + 17b against LAI (σ50%= 0.50). Although the other entry inhibitors were generally more synergistic than the NAbs, the above NAb-NAb combinations showed stronger synergy than did VCV + PRO 140 against 9-8 (σ50% = 0.58) or VCV + T-20 against the three R5 clones (σ50% = 0.68–0.84). The b12 + 17b combination yielded a particularly stark contrast between the R5 isolate (σ50% = 0.68) and the three R5 clones (σ50% 1.2–1.9).
More robust patterns were revealed when data were pooled across all ten inhibitor combinations. When this was done, synergy measurements were shown to differ between viral isolates and clones. Thus, overall, σ50% values tended to be lower for isolates than clones (p = 0.051). However, this outcome was restricted to the R5 viruses, for which the difference was significant (p = 0.0061; NS for X4 virus, p = 0.45).
Optimizing the sensitivity of synergy detection
We explored whether the ratio at which two inhibitors were mixed affected the detection of synergy. In summary, we found that using two inhibitors at a ratio as close as possible to that of their respective half-maximal inhibitory concentrations, Ki, modestly enhanced the sensitivity of synergy detection. The σ50% values correlated weakly but highly significantly with the deviation from the intra-experimental Ki ratio (Supplementary Figure 2, Spearman rank correlation coefficient, r=0.22, p=0.0001). In other words, the more the actual concentration ratio for the two inhibitors deviated (in either direction) from the ratio of their Ki values, as measured in the same assay, the higher were the values of σ50% (i.e., the weaker the indication of synergy). The theoretical background for this effect on the sensitivity of synergy detection is given in the Supplement.
Cooperativity and its relationship to synergy
We favor restricting synergy measurement to half-maximal inhibition, which is more precise (Table 1, Supplementary Table 1) and also slope-independent; the slope, an indicator of cooperativity, provides complementary information. It is common for ligands to bind cooperatively to multiple sites on oligomeric proteins. Cooperativity differs from synergy in that a single agent can cooperate with itself, as in the classic case of the binding of oxygen to tetrameric hemoglobin (Hill, 1913; Koshland et al., 1966; Monod et al., 1965), whereas two different agents are required for synergy. In our non-linear function, the quantity η (eta) corresponds to the Hill coefficient and therefore may partly reflect classic cooperativity but can be influenced by other factors too. Henceforth we refer to the apparent cooperativity, measured by the slope coefficient, merely as cooperativity, regardless of the underlying molecular mechanism.
We investigated whether individual inhibitors and their combinations show cooperativity, and how changes in cooperativity relate to synergy. First, we examined how accurately and precisely we could measure a change in cooperativity, ηrel, which we calculate as ((ηAB/ηA) + (ηAB/ηB))/2. In that formula, ηAB is the cooperativity factor for inhibition by the combination, while ηA and ηB are the corresponding factors for inhibition by the individual components. Again, we used additivity controls, for which ((ηAB/ηA) + (ηAB/ηB))/2 should equal 1. Thus, although the individual inhibitors have different intrinsic cooperativities, these values should not change when the inhibitors are combined with themselves. We were encouraged to find that these additivity controls did indeed vary little. Thus, the range of change in η among the 632 controls was only 4.4-fold, whereas for σ50% it was 79-fold (for minima and maxima, see Supplementary Tables 1 and 2). In conclusion, the ηrel values were measured more accurately than σ50% (which in turn was more accurate than σ75%, σ90% or linearly derived CI values, as shown in Table 1 and Supplementary Table 1).
Combining two inhibitors increased cooperativity to differing extents (Table 3). The effect was particularly strong and consistent for the VCV + PRO 140 and 2G12 + 17b combinations against R5 viruses. We observed no example of a decrease in η of more than 2 SEM below the mean; overall, ηrel ranged from ~1 (i.e. no change) to 2.
Table 3.
Change in cooperativity, ηrel
| Combination | Virus | ||||||
|---|---|---|---|---|---|---|---|
| R5 | X4 | ||||||
| CC1/85 | 9-6 | 9-7 | 9-8 | IIIB | LAI | NL4.3 | |
| VCV + PRO 140 | 1.8±0.11 | 1.5±0.060 | 2.0±0.19 | 1.4±0.15 | - | - | - |
| VCV/AMD3100 + T-20 | 1.5±0.15 | 1.4±0.045 | 1.4±0.089 | 1.2±0.092 | 1.2±0.13 | 1.2±0.033 | 1.0±0.079 |
| VCV/AMD3100 + Nevirapine | 1.2±0.034 | 1.1±0.072 | 1.0±0.017 | 1.1±0.038 | 1.2±0.064 | 1.0±0.20 | 1.0±0.050 |
| CD4-IgG2 + 17b | 1.3±0.16 | 1.0±0.14 | 1.2±0.068 | 1.6±0.27 | 1.4±0.16 | 1.3±0.083 | 1.1±0.047 |
| b12 + 2G12 | 1.2±0.048 | 1.1±0.030 | 1.1±0.042 | 1.2±0.11 | 1.6±0.060 | 1.2±0.077 | 1.1±0.046 |
| b12 + 2F5 | 1.1±0.130 | 1.4±0.28 | 1.1±0.019 | 0.93±0.057 | 1.2±0.096 | 1.0±0.048 | 1.1±0.052 |
| b12 + 17b | 1.0±0.30 | 1.0±0.078 | 1.0±0.043 | 1.1±0.075 | 1.3±0.132 | 1.1±0.048 | 1.2±0.039 |
| 2G12 + 2F5 | 1.6±0.30 | 1.6±0.34 | 1.4±0.15 | 1.6±0.32 | 1.7±0.191 | 1.5±0.0081 | 1.4±0.23 |
| 2G12 + 17b | 1.6±0.17 | 1.8±0.16 | 1.3±0.077 | 1.8±0.077 | 1.4±0.065 | 1.5±0.35 | 1.3±0.099 |
| 2F5 + 17b | 1.3±0.19 | 1.2±0.11 | 1.1±0.13 | 1.2±0.118 | 1.3±0.091 | 0.9±0.12 | 1.3±0.058 |
The values are the mean values of fold change in η (calculated as ηrel = ((ηAB/ηA) + (ηAB/ηB))/2) from n (3 ≤ n ≤ 9) replicate experiments ± SEM. Mean values of ηrel + 2 SEM > 1 are given in bold. There were no mean values of ηrel + 2 SEM < 1. Note that n for each combination of inhibitors and virus is the same as in Table 2 and is given only there.
As with synergy, the cooperativity change for inhibitor combinations differed between isolates and clones. Thus the ηrel values were significantly greater for isolates than clones (p=0.0031), more so among the X4 than the R5 viruses (IIIB vs. LAI and NL4.3, p=0.0005; CC1/85 vs. 9-6, 9-7 and 9-8, p=0.095, NS).
Indirectly, the limited change in cooperativity for the additivity controls (Supplementary Table 2) also validates the determination of intrinsic η for all the inhibitors we used (Table 4). Most of the intrinsic η values were <1. It should be noted that negative cooperativity does not necessarily confer values <1, although it does reduce the slope value below the maximum value that is set by the number of interacting subunits (Weiss, 1997). In contrast, heterogeneity, i.e. the width of inhibitory effects over a population of target molecules, often reduce the slope values to <1 (Greco et al., 1995; Hoffman and Goldberg, 1994). The R5 isolate had lower values of η than its derivative clones (p < 0.0001), and the X4 isolate and clones had significantly higher values of η than the R5 viruses (p < 0.0001). These differences are interpreted in the Discussion.
Table 4.
Intrinsic cooperativity, η, of inhibitors of HIV-1 infection
| Inhibitor | Virus | ||||||
|---|---|---|---|---|---|---|---|
| R5 | X4 | ||||||
| CC1/85 | 9-6 | 9-7 | 9-8 | IIIB | LAI | NL4.3 | |
| PRO 140 | 0.80a±0.039 (n=12) | 0.90±0.027 (n=14) | 1.0±0.055 (n=8) | 1.0±0.050 (n=8) | - | - | - |
| VCV | 0.64±0.030 (n=25) | 0.86±0.035 (n=36) | 0.94±0.024 (n=24) | 0.93±0.015 (n=26) | - | - | - |
| AMD3100 | - | - | - | - | 1.2±0.065 (n=24) | 1.0±0.038 (n=18) | 1.2±0.081 (n=20) |
| T-20 | 0.59a±0.027 (n=16) | 0.80±0.022 (n=22) | 1.1±0.054 (n=12) | 0.98±0.98 (n=18) | 1.2±0.064 (n=22) | 1.4±0.12 (n=16) | 1.0±0.12 (n=24) |
| Nevirapine | 0.68±0.080 (n=6) | 0.84±0.018 (n=6) | 0.83±0.050 (n= 6) | 0.83±0.028 (n= 6) | 0.92±0.031 (n=8) | 0.71±0.034 (n=6) | 0.86±0.017 (n= 6) |
| CD4-IgG2 | 0.74±0.027 (n= 8) | 0.65±0.030 (n= 8) | 0.84±0.048 (n=12) | 0.88±0.042 (n=10) | 1.1±0.053 (n=8) | 0.88±0.047 (n=6) | 0.81±0.044 (n=10) |
| 17b | 0.84±0.054 (n= 42) | 0.60±0.031 (n=42) | 0.70±0.023 (n=42) | 0.53±0.026 (n=46) | 0.74±0.091 (n=38) | 1.0±0.14 (n=34) | 0.66±0.023 (n=40) |
| b12 | 0.79±0.030 (n= 20) | 0.71±0.018 (n=20) | 0.80±0.015 (n=24) | 0.80±0.016 (n=24) | 0.88±0.014 (n=20) | 1.0±0.023 (n=18) | 0.88±0.027 (n=20) |
| 2G12 | 0.54±0.036 (n=30) | 0.95±0.031 (n=26) | 0.74±0.023 (n=28) | 0.87±0.036 (n=32) | 0.76±0.025 (n=26) | 1.1±0.038 (n=26) | 0.79±0.028 (n=30) |
| 2F5 | 0.57±0.035 (n=32) | 0.48±0.021 (n=28) | 0.69±0.021 (n=24) | 0.73±0.037 (n=32) | 0.72±0.032 (n=24) | 1.3±0.061 (n=24) | 0.69±0.035 (n=30) |
The values are the means of intrinsic cooperativity factors, η, obtained by fitting the non-linear function (I=(C/Ki)η/(1+(C/Ki)η)) to data from n (6 ≤ n ≤ 46) replicate titrations ± SEM. Since most inhibitors were included in different combinations, the results titrations of individual inhibitors, which were pooled here, had higher n values and gave greater precision than those listed in Tables 2 and 3. Mean values of η + 2 SEM < 1 are given in bold; mean values of η − 2 SEM > 1 are given in underlined italics. The factors that raise and lower η (number of subunits of oligomeric targets, positive and negative cooperativity, and heterogeneity among target molecules) are outlined in the Discussion.
We have given several reasons for believing that σ50% and ηrel measure distinct and partially independent biological quantities. Indeed, σ50% and ηrel did not correlate (Figure 3A, Spearman r=0.021, p=0.72). Instead, the change in cooperativity correlated well with the ratio of σ50%/σ90% (Figure 3B, Spearman r=0.80, p<0.0001). Different degrees of synergy at different levels of inhibition are commonly reported (Greco et al., 1995). However, as we show here, synergy measurements are subject to extreme error at the higher levels of inhibition, as well as being theoretically unjustified (Loewe, 1953). The more accurate determination of change in cooperativity therefore provides a valuable complement to the slope-independent synergy measurement at 50% inhibition. In theory, synergy can occur as a perfectly parallel displacement of the inhibition curves. This is exemplified by values of σ50% <1 in Table 2 that correspond to values of ηrel ~ 1 in Table 3. Conversely, cooperativity can increase markedly when the same combination is merely additive at 50% inhibition. These discrepancies account for the lack of correlation between σ50% and ηrel. The two effects can also occur together, however, as illustrated by the VCV + PRO 140 combination (Tables 2 and 3), and both are worth evaluating. We suggest that non-linearly determined σ50% and ηrel values together capture biologically important and partly independent combinatorial effects with optimal accuracy and precision.
Figure 3. Variable relationship between synergy and enhanced cooperativity.
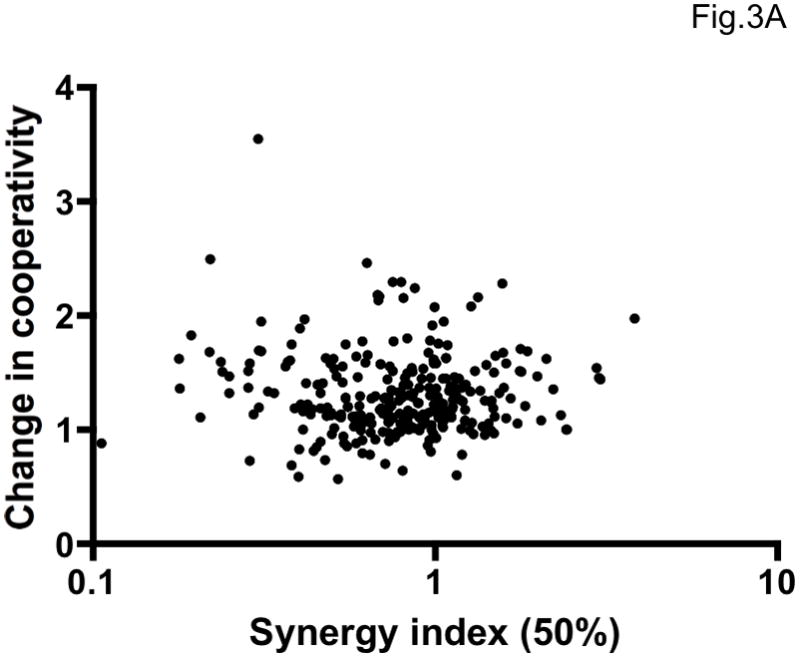
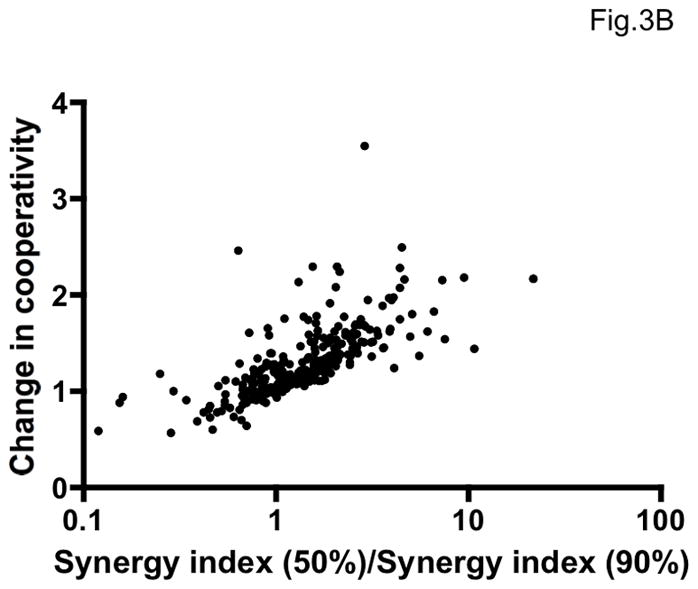
A. The average increase in cooperativity (fold change in η, ηrel) is plotted on the y-axis as a function of synergy as measured by the non-linear method at 50% inhibition (σ50%) on the x-axis. The individual results from 297 experiments (antagonistic combination excluded) are shown as dots. The two variables were uncorrelated (Spearman r = 0.021, p = 0.72), indicating at least partial independence. B. The average fold change in cooperativity, ηrel, is plotted on the y-axis against the ratio of synergy indices at 50% and 90% inhibition, σ50%/σ90%, for all combinations against all viruses (antagonistic combination excluded, n=297). The correlation was strong and highly significant (Spearman rank correlation, r = 0.80, p < 0.0001).
A summary listing all the differences between the standard approach to synergy in the inhibition of HIV-1 infectivity and our proposed framework for combinatorial analysis can be found at the end of the Supplement.
DISCUSSION
Multiple studies have addressed whether NAbs synergize in blocking HIV-1 infection, the outcomes ranging from strong synergy to mere additivity or even antagonism (Allaway et al., 1993; Buchbinder et al., 1992; Cavacini et al., 1993; Hrin et al., 2008; Kennedy et al., 1991; Laal et al., 1994; Li et al., 1997; Li et al., 1998; Mascola et al., 1997; McKeating et al., 1992; Montefiori et al., 1993; Potts et al., 1993; Thali et al., 1992; Tilley et al., 1992; Verrier et al., 2001; Vijh-Warrier et al., 1996; Xu et al., 2001; Zwick et al., 2001). Those studies used both T-cell line-adapted (TCLA) viruses and primary isolates in infectivity assays that allow multi-cycle replication to different extents. One study compared multi-cycle replication with single-cycle pseudovirus infection: in a multi-cycle PBMC assay the triple-NAb combination b12, 2F5, and 2G12 showed synergy at both 50% and 90% inhibition of the JR-CSF primary isolate (CI=0.6–0.8); in a single-cycle assay with a JR-CSF Env-pseudotyped virus, weak antagonism occurred at 50% inhibition (CI=1.2) but strong synergy at 90% (CI=0.4) (Zwick et al., 2001). Hence, single- and multi-cycle-replication assays can clearly give different results. We found that widely different degrees of synergy or antagonism arose in a multi-cycle experiment at different times of sampling. Different synergy indices at middle and high levels of inhibition are also problematic; in our analyses, CI values were more error-prone at 90% inhibition than at 50%. Furthermore, most of the cited studies used the linear transformation method, which, as illustrated here and previously explained, can contribute to errors (Suhnel, 1998). For example, the median-effect linearization of combination data will often result in curves that are actually non-linear, and the commonly found CI values ≫1 in the region close to zero inhibition are calculation artifacts (Greco et al., 1995).
It is not surprising that the above sources of error together can generate the highly divergent results that are found in the literature on HIV-1 NAb-NAb synergy. We conclude, nevertheless, that synergy can sometimes occur between different NAbs as measured by the more precise non-linear method in the most reliable zone, i.e. at 50% inhibition. The synergy we observed was, however, only moderate and sporadic (Table 2); it was markedly less pronounced and consistent than when a small-molecule CCR5 inhibitor was combined with a CCR5 MAb. We will now discuss possible mechanisms for these different synergies, and then suggest the best approach for analyzing the divergent combinatorial effects that frequently arise at different levels of inhibition.
Because an IgG molecule is bulky, similar in size to a gp120-gp41 hetero-dimer, the binding of a single NAb could be sufficient to inactivate the function of that protomer sterically; indeed, models of neutralization stoichiometry suggest that a single molecule of certain NAbs can inactivate an entire Env trimer (Burton et al., 2001; Klasse, 2007; Magnus and Regoes, 2010; Yang et al., 2006). What, then, would the binding of a second NAb to a different (or even the same) Env protomer in the trimer accomplish? A similar argument can be applied to all the obligate components of an entry complex, including CD4 and CCR5 or CXCR4. When one necessary step of the fusion process is blocked, impeding a second would conceivably be redundant. But does this reasoning erroneously assume all-or-nothing, static equilibrium effects? Perhaps models of these inhibitory processes should instead be both kinetic and dynamic, allowing for ligand association and dissociation over time, as well as for binding competition among Env, NAbs, and receptors. The binding of two NAbs to the same Env protomer could significantly increase the proportion of time that the antigen is occupied and thereby non-functional. In addition, the binding of some NAbs irreversibly inactivates a protomer by inducing the dissociation of gp120 from gp41 (Poignard et al., 1996a; Ruprecht et al., 2011), a process that might be accelerated for combinations of NAbs. Whether any such dynamic or kinetic effects could yield synergy is an unsolved problem.
HIV-1 entry into susceptible cells is a non-linear process, involving complex consecutive molecular interactions. The virions may attach to ancillary receptors, then dock onto a requisite number of CD4 molecules, and thereafter recruit several chemokine receptor molecules (Klasse et al., 1999; Kuhmann et al., 2000; Platt et al., 2005; Sougrat et al., 2007). These interactions ultimately trigger the fusion reaction in which an unknown number of Env trimers participate in forming a fusion pore through which the viral core can enter the cytoplasm. The inhibition of this process may have critical thresholds at the levels of both the virion and the Env trimer (Herrera et al., 2006; Klasse, 2007; Klasse and Moore, 1996; Magnus and Regoes, 2010; Magnus et al., 2009; Schonning et al., 1999; Yang et al., 2005). Because of this complexity, inhibition may not increase with binding in a simple proportional, incremental manner. Therefore, when the kinetics, dynamics, and threshold effects of inhibition are taken into account, synergy in biological activity might result without synergistic binding of NAbs or other inhibitors to components of the entry machinery.
MAbs and soluble CD4 (sCD4) do, however, affect each other’s binding to the gp120 monomer. For example, CD4-binding-site (CD4bs) MAbs enhance the binding of some V3-directed MAbs, and sCD4 greatly increases the binding of MAbs to the CD4i epitopes in the bridging sheet (Moore and Sodroski, 1996). But not a single case of mutually enhanced binding is known: when two ligands are added to gp120, the binding of one ligand is enhanced while that of the other is unaffected or reduced, or else there is mutual inhibition (Moore and Sodroski, 1996). These effects fall short of the potentiated binding of both ligands at a linked equilibrium, which would provide a straightforward molecular basis for synergy through interactions in the ternary complex. Indeed, we did not observe any more consistent synergy when CD4-IgG2 was combined with the CD4i MAb 17b than when 17b was combined with the CD4bs MAb b12 or with the mannose-epitope-specific MAb 2G12 (Table 2). Yet CD4-IgG2 uni-directionally enhances 17b binding to gp120, whereas the binding of 17b and b12 is mutually antagonistic, and 17b and 2G12 have no effect on each other’s binding (Moore and Sodroski, 1996). It should be noted, however, that these bi- or uni-directional effects of MAb binding could differ between the gp120 monomer and the functional Env trimer. Synergy in NAb binding to trimers also does not correlate with synergistic virus neutralization, although the binding assays have involved uncleaved Env precursors that are irrelevant to HIV-1 entry and its inhibition (Thali et al., 1992; Zwick et al., 2001). No assay preferentially detects NAb binding to functional Env, and no soluble Env trimer presents all conserved neutralization epitopes (Burton and Poignard, 2009).
Some, but not all, NAbs to the V3 region of gp120 reportedly synergize with CD4bs NAbs and recombinant CD4 constructs (Allaway et al., 1993; Laal et al., 1994; Potts et al., 1993; Thali et al., 1992; Tilley et al., 1992; Verrier et al., 2001; Vijh-Warrier et al., 1996). Even different NAbs against overlapping epitopes within the same CD4bs cluster on Env have been reported to synergize (Laal et al., 1994). In one study, synergy was detected between sCD4 and a V3 NAb, but for no combination of Env-directed MAbs (Verrier et al., 2001). As outlined above, it is difficult to explain such differential findings if the requirement for neutralization were simply a certain degree of sCD4 or NAb occupancy on indistinguishable Env protomers (Burnet et al., 1937; Burton, 2001; Klasse and Sattentau, 2002). We found cases of synergy for all NAb combinations as well as for CD4-IgG2 combined with 17b. Although the detection of moderate synergy is to some extent subject to chance, its sporadic occurrence across the NAb panel argues against a role for special molecular interactions within the ternary complex consisting of two NAbs and one Env spike or protomer; that some combinations gave more synergy than others can have other explanations than mutually enhanced binding.
We propose instead that antigenic differences in Env within each virus population may contribute to the occasionally synergistic effects. The only discernable overall pattern in our own data was that synergy was stronger for the R5 isolate than for its corresponding clones. A similar difference among X4 isolates and clones has been reported previously (Vijh-Warrier et al., 1996). Genetic variation within the viral quasispecies is one obvious source of Env heterogeneity, but we suggest that other contributors include variable carbohydrate processing (with direct and indirect effects on NAb recognition), and the conformational flexibility of the Env complex (Harris et al., 2011; Kwong et al., 2002; Kwong et al., 1998; Pantophlet and Burton, 2006; Wyatt and Sodroski, 1998). Functional and non-functional Env complexes (not only trimeric) on the surface of virions have been described (Moore et al., 2006; Pancera and Wyatt, 2005; Poignard et al., 2003), but here we are postulating heterogeneity also within the functional subset. Overall, any difference that affects the antigenicity of functional Env trimers could explain why NAbs with distinct specificities can apparently synergize when combined. Indeed, antigenic heterogeneity could even account for synergy between two NAbs directed to the same epitope cluster, e.g. the CD4bs (Laal et al., 1994).
We also tested combinations of inhibitors of HIV-1 entry other than NAbs. Thus, we found that T-20 synergized with the CCR5 ligand VCV but not with the CXCR4 ligand AMD3100 (Table 2). In earlier reports, fusion-blocking gp41-derived heptad-repeat 2 (HR2) peptides such as T-20 have shown variable degrees of synergy with co-receptor ligands against some HIV-1 isolates but mere additivity against others (Nakata et al., 2008; Tremblay et al., 2005a; Tremblay et al., 2005b; Tremblay et al., 2002; Tremblay et al., 2000; Veazey et al., 2005). Interactions between Env and co-receptors elicit the gp41 pre-hairpin-intermediate, which is the target for T-20, but co-receptors are also involved in promoting later, T-20-insensitive steps of the fusion reaction (Matthews et al., 2004; Mkrtchyan et al., 2005; Nagashima et al., 2001). The lower the CCR5 expression levels on the target cells, the more potently T-20 acts against R5 viruses, an explanation being that the fewer the co-receptor molecules available the more time T-20 has to intercede (Platt et al., 2005; Reeves et al., 2002). Similarly, CCR5 inhibitors may potentiate the action of T-20, but other observations of synergy in entry inhibition are less readily explained by such an effect: CD4-IgG2, which prevents binding to cellular CD4 but induces the co-receptor binding site on Env, also synergizes with T-20 (Nagashima et al., 2001); sulfated polyanions that block both HIV-1 attachment and co-receptor interactions can synergize with an HR2 peptide, as well as with a CCR5 ligand (Gantlett et al., 2007).
The sequence of events in entry, which is common to most strains of HIV-1, does not provide a comprehensive explanation for the variable observations of synergy among different inhibitors and viral strains. That synergy between HR2 peptides has only been observed with certain HIV-1 strains might be related to amino-acid variation in the V3 region of gp120, which strongly influences the sensitivity to inhibitors of both classes (Anastassopoulou et al., 2011; Berro et al., 2009; Derdeyn et al., 2000; Derdeyn et al., 2001; Kuhmann et al., 2004; Ogert et al., 2010). Therefore, heterogeneity among Env and co-receptor molecules could conceivably contribute to explaining these diverse phenomena. In this regard, we note that T-20 and VCV synergized more strongly against the R5 isolate CC1/85 than against all three clones derived from it, whereas no synergy was observed for AMD3100 combined with T-20 against X4 viruses. On the heterogeneity-based interpretation, our results would thus suggest that cell-surface CCR5 is more heterogeneous than CXCR4, particularly in how it interacts with Env. Although there is evidence that both co-receptors are heterogeneous, the relative extent is unknown (Anastassopoulou et al., 2009; Baribaud et al., 2001; Berro et al., 2011; Lee et al., 1999). It is also possible that, compared with X4 gp41, R5 gp41 varies more on each virion in how it interacts with T-20 and other HR2 peptides: the lower sensitivity of R5 viruses to such peptides is strongly influenced by the conformationally flexible and sequence-variable V3 region, as is their fusogenicity in general (Derdeyn et al., 2000; Derdeyn et al., 2001). Finally, mutants of HR2 peptides and a 5-helix-bundle construct, with reduced affinity for each other, can synergize strongly in blocking infection (Kahle et al., 2009). Heterogeneity might contribute to that example of synergy as well, in that the pre-hairpin intermediate of gp41 assumes different conformations during the fusion process, the HR1 region becoming more exposed, the HR2 region less so (Abrahamyan et al., 2005; Kahle et al., 2009). Since the multiple co-receptor interactions of Env trimers on a single virion would be asynchronous, the greater activity of HR1 peptides against some trimers would be complemented by the greater activity of HR2 peptides against others, creating synergy.
A related but simpler case is the strong synergy observed here and previously between a MAb and a small molecule that both bind to CCR5 (Ji et al., 2007; Latinovic et al., 2011a; Murga et al., 2006). Although the small CCR5 ligand may enhance the binding of the MAb (Latinovic et al., 2011b), the binding of the two ligands can also be antagonistic (Murga et al., 2006). Furthermore, the MAb and the small molecule can be active in different (Safarian et al., 2006) or the same (Ji et al., 2007) time windows and still synergize. The best explanation for their synergy might therefore be that they have distinct affinities for different forms of CCR5, which the virus can use (Anastassopoulou et al., 2009; Berro et al., 2011; Lee et al., 1999). Thus, combining two co-receptor ligands may potentiate both, one ligand compensating for when the other binds weakly to a subset of targets and vice versa. We note the smaller difference in synergy between the R5 isolate and its clones for the CCR5-ligand combination than for VCV + T-20 (Table 2). Hence Env heterogeneity may have a greater impact when the target molecules consist of both CCR5 and Env (i.e., VCV + T-20) rather than only CCR5 (i.e., VCV + PRO 140).
Many studies also report synergy between agents that block distinct steps in HIV-1 replication, e.g., among inhibitors of entry, the reverse transcriptase and the protease (Johnson et al., 1989; Johnson et al., 1990; Johnson et al., 1992; Johnson et al., 1991; Nakata et al., 2008; Strizki et al., 2005; Tremblay et al., 2005a; Tremblay et al., 2005b; Vermeire et al., 2004). We observed moderate synergy between the NNRTI Nevirapine and the CCR5 ligand VCV against the CC1/85 R5 isolate and to a smaller extent against the R5 clones; in contrast, Nevirapine and the CXCR4 ligand AMD3100 tended to be weakly antagonistic against the X4 viruses (Table 2). The weak antagonism may be due to fortuitous binding between the two molecules (although we have no evidence for this), but what could explain synergy when separate replicative steps are targeted? Since each blocked event is necessary for HIV-1 replication, why is blocking a second one not merely redundant? Again, viral heterogeneity could be the explanation, one inhibitor acting against variants that are relatively resistant to the other, and vice versa. In some cases, the viral quasispecies may be sufficiently pan-sensitive to exclude such effects, particularly when ligands are directed to the more invariable non-viral sites (e.g., CD4), or to relatively conserved sites on viral proteins other than Env. In the case of the combination of a co-receptor blocker and an NNRTI, differential sensitivity among Env trimers to inhibition by, e.g., VCV could be complemented by any heterogeneity in the reverse transcriptase that affects the action of, e.g., Nevirapine. The only requirement for this mechanism of synergy is that the maxima in sensitivity to the two inhibitors do not exactly coincide in the virion population.
In biochemistry or pharmacology, synergy is generally observed, and readily explained, when two parallel, mutually redundant pathways are blocked (Yeh and Kishony, 2007). Such situations differ from the consecutive steps of the HIV-1 replication cycle, with one informative exception. Thus, when both CCR5 and CXCR4 are expressed on the same target cells, a mixture of R5 and X4 HIV-1 isolates can be completely blocked only by a combination of ligands against both co-receptors. In that black-and-white situation, clear synergy was indeed observed when CCR5 and CXCR4 ligands were combined in a multi-cycle infectivity assay (Nakata et al., 2008). The heterogeneity that we postulate among Env, CCR5, and other target molecules would have more shades of grey. However, the basic principle of tightening the blockade by combining inhibitors would still apply. It can be shown by a schematic example when the outcome would be synergy rather than additivity. Suppose the inhibitor A has a Kd = 1 nM for half the target molecules and a Kd = 100 nM for the other half. Inhibitor B has the converse affinities. At 100 nM, each inhibitor alone therefore achieves ~ 75% occupancy on the total population of target molecules. When they are combined, however, 75% occupancy is achieved at 3 nM of each. Suppose that is the minimal inhibitory occupancy. Then the σ50% for the combination at equal concentrations will be ~ 0.06, which indicates strong synergy.
We investigated the slope of the inhibition curves. The fitted quantity η in our formula corresponds to the cooperativity coefficient in Hill’s equation for ligand-receptor binding (Hill, 1913). It is notable that η was < 1 for all individual inhibitors against all viruses, with the exceptions of each of AMD3100 and T-20 against all X4 viruses. Among R5 viruses, the values of η were markedly lower for the isolate than the clones, perhaps because the greater genetic heterogeneity widens the spectrum of sensitivities, which would reduce the slope coefficient (Greco et al., 1995; Hoffman and Goldberg, 1994). The η-values were also lower overall for R5 (primary-isolate-derived) than X4 (TCLA) viruses, possibly because there are fewer functional Env trimers on TCLA virions (Moore and Klasse, 1992; Moore et al., 1992). The difference between the minimum number of Env trimers required for entry and the total number on each virion is the number that is spare (Klasse and Moore, 1996). The larger the proportion of spare trimers the greater the potential number on each virion that are heterogeneously sensitive to inhibition, and the greater the heterogeneity the less steep the inhibition curve would be. By analogy, pseudoviruses, which have fewer Env spikes than natural viruses but were not included in this study, could be expected to show higher intrinsic values of η (Table 5).
Table 5.
Summary of relationships between synergy and cooperativity in the inhibition of HIV-1 entry
| Synergy index, σ50%, for combinations | Change in cooperativity, ηrel, for combinations | Intrinsic cooperativity for individual inhibitors, η | |
|---|---|---|---|
| Range of values | ~0.20 – ~1.9a | ~1.0 – ~2.0a | ~0.50 – ~1.5a |
| Isolates/clonesb | Low (R5) | High (mainly X4) | Low (R5) |
| R5 (PI)/X4 (TCLA)b | NSc | NSc | Low |
| Correlates | Weakly with deviation of used ratio from Ki ratio | Strongly with σ50%/σ90%, synergy skewness | Inversely with SD/EC50d |
| Moderate Emax differences as confounding factor | Weake | Strong | Strong |
| Plausible influence of heterogeneity in Env and of trimer number per virion | Yes | Yes | Yes |
The ratios of the values obtained for isolates and clones or R5 and X4 viruses are described.
NS = non-significant deviation from 1.
The ratio of the standard deviation of the half-maximal effective concentration over the mean of the latter among a hypothetical population of heterogeneous receptors was shown to have an inverse relationship to the slope coefficient or cooperativity factor (Hoffman and Goldberg, 1994).
Large Emax differences among individual inhibitors and combinations will
invalidate regular concentration-based or fractional synergy analyses.
When the cooperativity factor for inhibitors other than NAbs was analyzed by the Chou-Talalay median-effect principle in an X4 pseudovirus-PBMC assay with a 4-log dynamic range, the slopes of the viral inhibition curves were inhibitor-class-specific (Shen et al., 2008; Zhang et al., 2004). NRTI and integrase inhibitors had slopes of ~1, NNRTIs and fusion-blocking peptides of ~1.7, while protease inhibitors had the highest slopes of 1.8–4.5. Furthermore, the differences in slope affect how HIV-1 escapes the different classes of ARV. Thus, escape from drugs with low slope values takes the form of increased IC50 values, whereas escape from inhibitors with high slope values is manifested by a reduction of the slope (Sampah et al., 2011). We obtained comparable slope values >1 for T-20 with two of three X4 viruses (Table 4), but the corresponding values for the R5 viruses (not studied in (Shen et al., 2008)) were lower (0.59–1.1). The slope (η) influences the instantaneous inhibitory potential (the log reduction in single-round infectivity at clinical drug concentrations), which correlates better with in vivo efficacy than does IC50 or the inhibitory quotient (the ratio of plasma drug concentration over IC50) (Shen et al., 2008). Although many other factors than instantaneous inhibitory potential and inhibitory quotient influence clinical outcome during ART, both correlate with long-term viral suppression (Henrich et al., 2010; Shen and Siliciano, 2010). It may therefore be worth considering how efficacy is affected by synergy and changes in cooperativity at inhibitor concentrations close to and above IC50. Suppose the combination A+B is synergistic in the mid-zone (σ50%=0.5), whereas C+D is not (σ50%=1.0). A+B involves no slope change (ηrel=1), but C+D shows enhanced cooperativity (ηrel=2). For simplicity, all single-inhibitor Ki values are the same and all intrinsic η values = 1. Then at an inhibitory quotient of 1, C+D reduces the remaining infectivity 1.5-fold less than A+B; but at inhibitory quotients of 10 and 100, C+D reduces the remaining infectivity, respectively, 5- and 50-fold more than A+B. The example shows that at high concentrations, which may be medically relevant, changes in cooperativity can give more drastic combinatorial effects than synergy that is manifested merely as a parallel curve shift.
It can be argued that a high slope of inhibition would be crucial for vaccine-induced NAbs. For their protective capability may be determined not by their mid-range potency but by how much they can reduce the infectivity of virus deposited in the mucosae. The values of η we measured here for the NAbs were low, <1 or ~1, with one exception (2F5 against LAI, η=1.3). Hence it should be investigated whether other, newly discovered, broadly acting and potent NAbs (Walker et al., 2011; Walker et al., 2009; Zhou et al., 2010) perform better in this regard. Overall, the relationship between the in vitro potency and the in vivo protective capacity of NAbs remains to be fully elucidated (Hessell et al., 2009).
We extended the slope analysis to combinations of inhibitors and found near-uniform elevations of η (i.e. ηrel >1) (Table 3). This quantity, change in slope upon combination, may be useful in dealing with the problem of inconsistent synergy over different degrees of inhibition. Such skewness in synergy - usually manifested as lower CI90% than CI50% values - is a common finding (Allaway et al., 1993; Buchbinder et al., 1992; Gantlett et al., 2007; Hrin et al., 2008; Ji et al., 2007; Mascola et al., 1997; Murga et al., 2006; Nakata et al., 2008; Strizki et al., 2005; Tilley et al., 1992; Tremblay et al., 2002; Tremblay et al., 2000; Xu et al., 2001; Yoshimura et al., 2006; Zwick et al., 2001). The CI90% or σ90% value is theoretically often invalid and is, in any case, practically impossible to determine with acceptable precision (Supplementary Table 1). However, ηrel correlates strongly with the σ50%/σ90% ratio (Figure 3B) and is more readily determined with precision than σ90% (Supplementary Table 2). The ηrel quantity is therefore appropriate for characterizing combinatorial effects at higher levels of inhibition, as a complement to the slope-independent, σ50% value (Table 5). However, adding the measurement of η and ηrel to the standard arsenal of inhibition analyses, as we advocate, raises the question of how these quantities should be interpreted biologically.
Originally, the Hill coefficient, n, was suggested to reflect the number of ligands interacting with each receptor complex, but the value of n is generally lower than that stoichiometric number and only approaches it under conditions of extreme positive cooperativity (Hill, 1913; Weiss, 1997). In some binding schemes, 10 sites for the ligand on the receptor can give n = 2 without any cooperativity; negative cooperativity reduces n further below its theoretical maximum, but not below 1 (Weiss, 1997). In contrast, heterogeneity in the population of target molecules, whether Env, co-receptor or reverse transcriptase, can bring the value of the slope coefficient below 1. The greater the heterogeneity in target molecule sensitivity, expressed as SD/EC50 (the standard deviation of the half-maximal effective concentration divided by the mean of the latter) the smaller will be the slope coefficient, n (Hoffman and Goldberg, 1994), or in our case η (Table 5). This basis of reduced η-values for individual inhibitors also explains why ηrel becomes >1 for combinations. Thus, the virions that are least sensitive to one inhibitor will statistically have a more ordinary sensitivity to another; the resulting reduction in the heterogeneity in sensitivity to the combined action of the two inhibitors raises the value of η. In this regard, it is notable that the broad and potent neutralizing capacity of sera from some HIV-1-infected persons has been attributed to the combined action of NAbs directed to several different Env epitopes (Scheid et al., 2009), although in most subjects only one or two specificities may be involved (Walker et al., 2011; Walker et al., 2010). Hypotheses now worth testing are whether the values of η are higher for polyclonal sera than for their constituent specificities, and whether ηrel for pooled NAbs > 1. Since it was the breadth of the neutralizing capacity rather than its potency that could be reconstituted by the pooling of NAb clones (Scheid et al., 2009), a generalized version of the heterogeneity hypothesis might have further test implications: individual NAbs would be predicted to give lower values of η for a mixture of different isolates than for individual ones, and the greater the diversity of the virus mix, the higher the predicted ηrel values for cross-neutralizing pools of NAbs.
In summary, the synergy in neutralization that we observed was modest, sporadic, and interspersed with some cases of antagonism. In contrast, the positive change in cooperativity upon combination was nearly ubiquitous and frequently strong (Table 5). In particular, combining 2G12 with 2F5 or 17b gave consistently high values of ηrel (1.3–1.8) across the panel of viruses (Table 3). Heterogeneity among target molecules may provide a mechanistic basis for both synergy and enhanced cooperativity. Genetic heterogeneity in env may contribute to synergy among NAbs in a way that the data for R5 viruses support; heterogeneity in the glycan shield can both stem from genetically determined differences at glycosylation sites and be affected by non-uniform carbohydrate processing. The involvement of the NAb 2G12, which is directed to a mannose-dependent epitope (Sanders et al., 2002; Scanlan et al., 2002), in some of the strongest synergy, as well as in enhanced cooperativity, suggests a role for glycan-shield heterogeneity. Conformational heterogeneity may instead particularly affect the antigenicity of the 17b and 2F5 epitopes (Dimitrov et al., 2007; Finnegan et al., 2001, 2002; Harris et al., 2011; Sattentau et al., 1995; Shen et al., 2010). Each of these NAbs also gave many instances of marked synergy and slope increases when combined with 2G12; they synergized considerably less together, suggesting that two different kinds of heterogeneity together are particularly conducive to synergy and enhanced cooperativity.
New assays for quantifying the inhibition of HIV-1 infectivity over a wide dynamic range, and the concomitant focus on slope analysis, constitute significant advances (Sampah et al., 2011; Shen et al., 2008; Zhang et al., 2004). Such experimental systems may also allow rigorous determinations of the maximal extent of inhibition, Emax, which is currently beyond the scope of our data. Determining that additional quantity could improve our analyses, since real differences in Emax may masquerade as variation in η (Table 5). Since these in vitro measurements might predict the efficacy of NAbs and viral inhibitors in vivo, they seem worth pursuing in the quest for better preventive and therapeutic strategies.
MATERIALS AND METHODS
Cells and viruses
Tzm-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program (ARRRP). They express the luciferase gene under control of the HIV-1 LTR (Wei et al., 2002), which is activated by viral Tat upon infection. The 293T cell line was obtained from the American Type Culture Collection (ATCC; Manassas, VA). Tzm-bl and 293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Invitrogen Inc., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 100 U/ml penicillin + 100 μg/ml streptomycin (1× PenStrep; HyClone, Logan, UT). All cells were incubated at 37 °C in a 5% CO2 atmosphere.
The primary R5 HIV-1 isolate CC1/85 and its env-derivative chimeric clonal viruses NL4.3/9-6, NL4.3/9-7, and NL4.3/9-8 have been described (Kuhmann et al., 2004; Trkola et al., 2002). These clones are hereafter described as 9-6, 9-7, and 9-8. The HIV-1 isolate IIIB and the derivative clonal viruses LAI and NL4.3 were obtained from the ARRRP. Stocks of infectious clones were prepared by transfecting the full-length proviral plasmids into 293T cells by Lipofectamine 2000 (Invitrogen).
Antibodies and reagents
NAbs 2G12 to a mannose-dependent epitope on the outer domain of gp120, b12 to the CD4bs, 17b to a CD4-induced epitope overlapping the co-receptor binding site, and 2F5 to a membrane-proximal epitope on gp41 were all provided by the IAVI Neutralizing Antibody Consortium Repository. The humanized anti-CCR5 MAb PRO 140 was obtained from Bill Olson (Progenics Pharmaceuticals), and the small molecule CCR5 inhibitor vicriviroc (VCV) from Julie Strizki (Schering-Plough; now Merck). Michael Greenberg (Trimeris) supplied the gp41-derived HR2 fusion-blocking peptide T-20. AMD3100, a small-molecule CXCR4 inhibitor, was obtained from Gary Bridger (AnorMed; now Genzyme). Nevirapine, a non-nucleoside reverse transcriptase inhibitor, was purchased from Boehringer Ingelheim.
Infectivity assays
Neutralization and entry inhibition experiments were carried out using Tzm-bl cells (Wei et al., 2003). One day before infection, 1 × 104 Tzm-bl cells in 200 μl DMEM-FBS were seeded into each well of 96-well plates. NAbs were serially diluted in 2-fold steps in DMEM. A 50 μl aliquot of each dilution was then mixed with 50 μl of virus suspension (pre-titrated to yield a luminescence of ~5 × 105 counts per second) per well, and incubated at 37ºC for 1 h. The mixture (100 μl per well) was then added to the cells, which were incubated at 37ºC for 40 h. Each NAb concentration was tested in quadruplicate. The other inhibitors (T-20, VCV, AMD3100, Neviparine, PRO 140) were titrated similarly, except that the 50 μl aliquots from each dilution step were incubated with the Tzm-bl cells for 1 h at 37ºC before addition of 50 μl of virus for a further 40 h. The cells were then washed with PBS, lysed and freeze-thawed. The luciferase activity in the lysate was measured by the Bright-Glo method (Promega) on a Victor3 1420 plate-reading luminometer (Perkin Elmer, Wellesley, MA). Background luminescence values from control wells with cells and no virus were subtracted. Inhibitory concentrations were calculated by non-linear regression in Prism (Graphpad, see Modeling section below).
Inhibitors were then combined in proportions approximately corresponding to the ratios of their IC50 (Ki) values derived from the single-inhibitor titrations. As controls, each inhibitor was combined with itself at equimolar concentrations. All such combinations were titrated at fixed ratios in octuplicate in parallel with single inhibitors in quadruplicate.
To examine the influence of multi-cycle replication, we also used an infectivity assay based on p24 production by HIV-1-infected primary lymphocytes. Peripheral blood mononuclear cells (PBMC) were isolated from blood obtained at the New York Blood Center (New York, NY), then cultured in Culture Medium, which was RPMI 1640 supplemented with 10% FBS, 10 mM L-glutamine (Cellgro), and 100 U/ml IL-2 (ARRRP, contributed by Hoffman-LaRoche, Inc.). Half the PBMC culture was stimulated with 5 μg/ml of phytohemagglutinin (PHA; Sigma, St. Louis, MO), half with an anti-CD3 MAb (supernatant from the OKT3 hybridoma) in Culture Medium, directly after isolation. After 3 days, the differently stimulated cells were mixed in equal proportions, washed, and suspended in Culture Medium supplemented with 100 μg/ml of penicillin/streptomycin for the remainder of the culture. The PBMC were seeded at 1.4 × 106 cells/ml in 100 μl per well in 96-well plates. NAbs b12 and 2G12 (the only ones included in the PBMC experiment) were diluted serially in two-fold steps and incubated with virus for 1 h before adding the mixture to cells. HIV-1 replication was measured as Gag-p24 content in mixed cell-culture suspension by an in-house ELISA on different days over 2 weeks of culture (Ketas et al., 2003). The ELISA detects both p24 and Gag precursor (p55), so that antigen detection is not affected by the presence of the protease inhibitor (data not shown). The amount of residual Gag from the input virus was measured in 9 replicates per plate and subtracted from test values. Net Gag production in the test wells was compared with that in the control wells (no inhibitor in 21 replicates per plate). The yield corresponded to 0.1–3 ng p24 per ml, which was defined as 100%.
Modeling
We compared the current standard method for synergy analysis (Chou and Talalay, 1981, 1984) with a new one that we devised. The “median-effect principle” of Chou’s method is based on a linear transformation of the inhibition data. A linear function is then fitted: (log (fa/fu) = m log(D/Dm)), where fa = fraction affected (i.e., the normalized proportion of inhibited infection); fu = fraction unaffected (i.e., 1- fa, or the relative residual infectivity); m is a constant affecting the slope of the linear curve; Dm is the “median effect dose”, the equivalent of the half-maximal inhibitory concentration; D is the concentration of inhibitor yielding a degree of inhibition corresponding to fa. The combination index, CI = (D(AB)A/DA) + (D(AB)B/DB) is next calculated from the fitted values of D when inhibitors A or B are used alone (DA or DB) or as constituents of the combination (D(AB)A and D(AB)B). This formula has been suggested to apply to the case of two inhibitors with mutually exclusive actions only (Chou and Talalay, 1981, 1984). An alternative formula, CI= (D(AB)A/DA) + (D(AB)B/DB) + (D(AB)A/DA) * (D(AB)B/DB), has been proposed to apply to the case of non-exclusive inhibitors (Chou and Talalay, 1981, 1984). We only used the simpler formula here. For a justification of this choice, see the Supplementary Information. Chou’s and Talalay’s method is available in software form (CalcuSyn, Bisoft), but we fitted the published functions as polynomials of the first order in Prism (Graphpad).
We also used non-linear regression (Prism, Graphpad) to fit all data to a non-linear function that is similar in form to the law of mass action: I=(C/Ki)η/(1+(C/Ki)η)), where I = relative inhibition of infectivity; Ki is the half-maximal inhibitory concentration by analogy with the dissociation constant, Kd, in the Law of Mass Action; C = concentration of an inhibitor; η = a cooperativity factor, related to the Hill constant.
The synergy indicator was calculated according to Loewe’s formula (Loewe, 1953) as σ = (Ki(AB)A/KiA) + (Ki(AB)B/KiB), where KiA = inhibitory concentration of A when used alone; KiB = inhibitory concentration of B when used alone; Ki(AB)A = inhibitory concentration of A when used in combination with B; and Ki(AB)B = inhibitory concentration of B when used in combination with A.
Supplementary Material
Acknowledgments
We are grateful to Bill Olson, Julie Strizki, Michael Greenberg, and Gary Bridger for reagents. We thank Samson Jacob for excellent technical assistance. This work was supported primarily by NIH grant R37 AI36082, with additional support from NIH grants AI41420 and AI76982, the IAVI Neutralizing Antibody Consortium, and the Bristol Myers Squibb Foundation via an Unrestricted Infectious Disease Research grant. Michael Zwick and John Murray provided valuable comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamyan LG, Mkrtchyan SR, Binley J, Lu M, Melikyan GB, Cohen FS. The cytoplasmic tail slows the folding of human immunodeficiency virus type 1 Env from a late prebundle configuration into the six-helix bundle. J Virol. 2005;79:106–115. doi: 10.1128/JVI.79.1.106-115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaway GP, Ryder AM, Beaudry GA, Maddon PJ. Synergistic inhibition of HIV-1 envelope-mediated cell fusion by CD4- based molecules in combination with antibodies to gp120 or gp41. AIDS Res Hum Retroviruses. 1993;9:581–587. doi: 10.1089/aid.1993.9.581. [DOI] [PubMed] [Google Scholar]
- Anastassopoulou CG, Ketas TJ, Depetris RS, Thomas AM, Klasse PJ, Moore JP. Resistance of a human immunodeficiency virus type 1 isolate to a small molecule CCR5 inhibitor can involve sequence changes in both gp120 and gp41. Virology. 2011;413:47–59. doi: 10.1016/j.virol.2010.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassopoulou CG, Ketas TJ, Klasse PJ, Moore JP. Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41. Proc Natl Acad Sci U S A. 2009;106:5318–5323. doi: 10.1073/pnas.0811713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribaud F, Edwards TG, Sharron M, Brelot A, Heveker N, Price K, Mortari F, Alizon M, Tsang M, Doms RW. Antigenically distinct conformations of CXCR4. J Virol. 2001;75:8957–8967. doi: 10.1128/JVI.75.19.8957-8967.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum MC. Synergy, additivism and antagonism in immunosuppression. A critical review. Clin Exp Immunol. 1977;28:1–18. [PMC free article] [PubMed] [Google Scholar]
- Berro R, Klasse PJ, Lascano D, Flegler A, Nagashima KA, Sanders RW, Sakmar TP, Hope TJ, Moore JP. Multiple CCR5 conformations on the cell surface are used differentially by human immunodeficiency viruses resistant or sensitive to CCR5 inhibitors. J Virol. 2011 doi: 10.1128/JVI.00767-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berro R, Sanders RW, Lu M, Klasse PJ, Moore JP. Two HIV-1 variants resistant to small molecule CCR5 inhibitors differ in how they use CCR5 for entry. PLoS Pathog. 2009;5:e1000548. doi: 10.1371/journal.ppat.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchbinder A, Zolla-Pazner S, Karwowska S, Gorny MK, Burda ST. Synergy between human monoclonal antibodies to HIV extends their effective biologic activity against homologous and divergent strains. AIDS Res Hum Retroviruses. 1992;8:1395. doi: 10.1089/aid.1992.8.1395. [DOI] [PubMed] [Google Scholar]
- Burnet FM, Keogh EV, Lush D. The immunological reactions of the filterable viruses. Austral J Exp Biol Med Sci. 1937;15:227–368. [Google Scholar]
- Burton DR, editor. Antibodies in Viral Infection. Berlin: Springer; 2001. [Google Scholar]
- Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, Nabel GJ, Sodroski J, Wilson IA, Wyatt RT. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- Burton DR, Poignard P. HIV: Immune memory downloaded. Nature. 2009;458:584–585. doi: 10.1038/458584a. [DOI] [PubMed] [Google Scholar]
- Burton DR, Saphire EO, Parren PWHI. A model for neutralization of viruses based on antibody coating of the virion surface. Curr Top Microbiol Immunol. 2001;260:109–143. doi: 10.1007/978-3-662-05783-4_7. [DOI] [PubMed] [Google Scholar]
- Cavacini LA, Emes CL, Power J, Buchbinder A, Zolla-Pazner S, Posner MR. Human monoclonal antibodies to the V3 loop of HIV-1 gp120 mediate variable and distinct effects on binding and viral neutralization by a human monoclonal antibody to the CD4 binding site. J Acquir Immune Defic Syndr. 1993;6:353–358. [PubMed] [Google Scholar]
- Chou TC, Talalay P. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem. 1981;115:207–216. doi: 10.1111/j.1432-1033.1981.tb06218.x. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Choudhry V, Zhang MY, Harris I, Sidorov IA, Vu B, Dimitrov AS, Fouts T, Dimitrov DS. Increased efficacy of HIV-1 neutralization by antibodies at low CCR5 surface concentration. Biochem Biophys Res Commun. 2006;348:1107–1115. doi: 10.1016/j.bbrc.2006.07.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74:8358–8367. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Zhang Z, O’Brien WA, Ratner L, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J Virol. 2001;75:8605–8614. doi: 10.1128/JVI.75.18.8605-8614.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov AS, Jacobs A, Finnegan CM, Stiegler G, Katinger H, Blumenthal R. Exposure of the membrane-proximal external region of HIV-1 gp41 in the course of HIV-1 envelope glycoprotein-mediated fusion. Biochemistry. 2007;46:1398–1401. doi: 10.1021/bi062245f. [DOI] [PubMed] [Google Scholar]
- Doms RW, Peiper SC. Unwelcomed guests with master keys: how HIV uses chemokine receptors for cellular entry. Virology. 1997;235:179–190. doi: 10.1006/viro.1997.8703. [DOI] [PubMed] [Google Scholar]
- Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eron JJ, Jr, Johnson VA, Merrill DP, Chou TC, Hirsch MS. Synergistic inhibition of replication of human immunodeficiency virus type 1, including that of a zidovudine-resistant isolate, by zidovudine and 2′,3′-dideoxycytidine in vitro. Antimicrob Agents Chemother. 1992;36:1559–1562. doi: 10.1128/aac.36.7.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson NM, Fraser C, Anderson RM. Viral dynamics and anti-viral pharmacodynamics: rethinking in vitro measures of drug potency. Trends Pharmacol Sci. 2001;22:97–100. doi: 10.1016/s0165-6147(00)01615-1. [DOI] [PubMed] [Google Scholar]
- Finnegan CM, Berg W, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus envelope during cell-cell fusion. J Virol. 2001;75:11096–11105. doi: 10.1128/JVI.75.22.11096-11105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan CM, Berg W, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus transmembrane glycoprotein during cell-cell fusion. J Virol. 2002;76:12123–12134. doi: 10.1128/JVI.76.23.12123-12134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantlett KE, Weber JN, Sattentau QJ. Synergistic inhibition of HIV-1 infection by combinations of soluble polyanions with other potential microbicides. Antiviral Res. 2007;75:188–197. doi: 10.1016/j.antiviral.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapia M, Guanira-Carranza JV, Ramirez-Cardich ME, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47:331–385. [PubMed] [Google Scholar]
- Gustchina E, Li M, Louis JM, Anderson DE, Lloyd J, Frisch C, Bewley CA, Gustchina A, Wlodawer A, Clore GM. Structural basis of HIV-1 neutralization by affinity matured Fabs directed against the internal trimeric coiled-coil of gp41. PLoS Pathog. 2010;6:e1001182. doi: 10.1371/journal.ppat.1001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Borgnia MJ, Shi D, Bartesaghi A, He H, Pejchal R, Kang YK, Depetris R, Marozsan AJ, Sanders RW, et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1101414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich TJ, Ribaudo HJ, Kuritzkes DR. Instantaneous inhibitory potential is similar to inhibitory quotient at predicting HIV-1 response to antiretroviral therapy. Clin Infect Dis. 2010;51:93–98. doi: 10.1086/653430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia A, Gilliam B, DeVico A, Le N, Bamba D, Flinko R, Lewis G, Gallo RC, Redfield RR. CCR5 density levels on primary CD4 T cells impact the replication and Enfuvirtide susceptibility of R5 HIV-1. Aids. 2007a;21:1317–1322. doi: 10.1097/QAD.0b013e32815278ea. [DOI] [PubMed] [Google Scholar]
- Heredia A, Gilliam B, Latinovic O, Le N, Bamba D, Devico A, Melikyan GB, Gallo RC, Redfield RR. Rapamycin reduces CCR5 density levels on CD4 T cells, and this effect results in potentiation of enfuvirtide (T-20) against R5 strains of human immunodeficiency virus type 1 in vitro. Antimicrob Agents Chemother. 2007b;51:2489–2496. doi: 10.1128/AAC.01602-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera C, Klasse PJ, Kibler CW, Michael E, Moore JP, Beddows S. Dominant-negative effect of hetero-oligomerization on the function of the human immunodeficiency virus type 1 envelope glycoprotein complex. Virology. 2006 doi: 10.1016/j.virol.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, Koff WC, Watkins DI, Burton DR. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009;5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AV. The Combinations of Haemoglobin with Oxygen and with Carbon Monoxide. I. Biochem J. 1913;7:471–480. doi: 10.1042/bj0070471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A, Goldberg A. The relationship between receptor-effector unit heterogeneity and the shape of the concentration-effect profile: pharmacodynamic implications. J Pharmacokinet Biopharm. 1994;22:449–468. doi: 10.1007/BF02353789. [DOI] [PubMed] [Google Scholar]
- Hrin R, Montgomery DL, Wang F, Condra JH, An Z, Strohl WR, Bianchi E, Pessi A, Joyce JG, Wang YJ. Short communication: In vitro synergy between peptides or neutralizing antibodies targeting the N- and C-terminal heptad repeats of HIV Type 1 gp41. AIDS Res Hum Retroviruses. 2008;24:1537–1544. doi: 10.1089/aid.2008.0129. [DOI] [PubMed] [Google Scholar]
- Ji C, Zhang J, Dioszegi M, Chiu S, Rao E, Derosier A, Cammack N, Brandt M, Sankuratri S. CCR5 small-molecule antagonists and monoclonal antibodies exert potent synergistic antiviral effects by cobinding to the receptor. Mol Pharmacol. 2007;72:18–28. doi: 10.1124/mol.107.035055. [DOI] [PubMed] [Google Scholar]
- Johnson VA, Barlow MA, Chou TC, Fisher RA, Walker BD, Hirsch MS, Schooley RT. Synergistic inhibition of human immunodeficiency virus type 1 (HIV-1) replication in vitro by recombinant soluble CD4 and 3′-azido-3′-deoxythymidine. J Infect Dis. 1989;159:837–844. doi: 10.1093/infdis/159.5.837. [DOI] [PubMed] [Google Scholar]
- Johnson VA, Barlow MA, Merrill DP, Chou TC, Hirsch MS. Three-drug synergistic inhibition of HIV-1 replication in vitro by zidovudine, recombinant soluble CD4, and recombinant interferon-alpha A. J Infect Dis. 1990;161:1059–1067. doi: 10.1093/infdis/161.6.1059. [DOI] [PubMed] [Google Scholar]
- Johnson VA, Merrill DP, Chou TC, Hirsch MS. Human immunodeficiency virus type 1 (HIV-1) inhibitory interactions between protease inhibitor Ro 31-8959 and zidovudine, 2′,3′-dideoxycytidine, or recombinant interferon-alpha A against zidovudine-sensitive or -resistant HIV-1 in vitro. J Infect Dis. 1992;166:1143–1146. doi: 10.1093/infdis/166.5.1143. [DOI] [PubMed] [Google Scholar]
- Johnson VA, Merrill DP, Videler JA, Chou TC, Byington RE, Eron JJ, D’Aquila RT, Hirsch MS. Two-drug combinations of zidovudine, didanosine, and recombinant interferon-alpha A inhibit replication of zidovudine-resistant human immunodeficiency virus type 1 synergistically in vitro. J Infect Dis. 1991;164:646–655. doi: 10.1093/infdis/164.4.646. [DOI] [PubMed] [Google Scholar]
- Kahle KM, Steger HK, Root MJ. Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog. 2009;5:e1000674. doi: 10.1371/journal.ppat.1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson Hedestam GB, Fouchier RA, Phogat S, Burton DR, Sodroski J, Wyatt RT. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat Rev Microbiol. 2008;6:143–155. doi: 10.1038/nrmicro1819. [DOI] [PubMed] [Google Scholar]
- Kennedy MS, Orloff S, Ibegbu CC, Odell CD, Maddon PJ, McDougal JS. Analysis of synergism/antagonism between HIV-1 antibody-positive human sera and soluble CD4 in blocking HIV-1 binding and infectivity. AIDS Res Hum Retroviruses. 1991;7:975–981. doi: 10.1089/aid.1991.7.975. [DOI] [PubMed] [Google Scholar]
- Ketas TJ, Klasse PJ, Spenlehauer C, Nesin M, Frank I, Pope M, Strizki JM, Reyes GR, Baroudy BM, Moore JP. Entry inhibitors SCH-C, RANTES, and T-20 block HIV type 1 replication in multiple cell types. AIDS Res Hum Retroviruses. 2003;19:177–186. doi: 10.1089/088922203763315678. [DOI] [PubMed] [Google Scholar]
- Ketas TJ, Kuhmann SE, Palmer A, Zurita J, He W, Ahuja SK, Klasse PJ, Moore JP. Cell surface expression of CCR5 and other host factors influence the inhibition of HIV-1 infection of human lymphocytes by CCR5 ligands. Virology. 2007;364:281–290. doi: 10.1016/j.virol.2007.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse PJ. Modeling how many envelope glycoprotein trimers per virion participate in human immunodeficiency virus infectivity and its neutralization by antibody. Virology. 2007;369:245–262. doi: 10.1016/j.virol.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse PJ, Moore JP. Quantitative model of antibody- and soluble CD4-mediated neutralization of primary isolates and T-cell line-adapted strains of human immunodeficiency virus type 1. J Virol. 1996;70:3668–3677. doi: 10.1128/jvi.70.6.3668-3677.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse PJ, Rosenkilde MM, Signoret N, Pelchen-Matthews A, Schwartz TW, Marsh M. CD4-Chemokine receptor hybrids in human immunodeficiency virus type 1 infection. J Virol. 1999;73:7453–7466. doi: 10.1128/jvi.73.9.7453-7466.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse PJ, Sanders RW, Cerutti A, Moore JP. How Can HIV-Type-1-Env Immunogenicity Be Improved to Facilitate Antibody-Based Vaccine Development? AIDS Res Hum Retroviruses. 2011 doi: 10.1089/aid.2011.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse PJ, Sattentau QJ. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J Gen Virol. 2002;83:2091–2108. doi: 10.1099/0022-1317-83-9-2091. [DOI] [PubMed] [Google Scholar]
- Klasse PJ, Shattock R, Moore JP. Antiretroviral Drug-Based Microbicides to Prevent HIV-1 Sexual Transmission. Annu Rev Med. 2008;59:455–471. doi: 10.1146/annurev.med.59.061206.112737. [DOI] [PubMed] [Google Scholar]
- Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- Kuhmann SE, Platt EJ, Kozak SL, Kabat D. Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J Virol. 2000;74:7005–7015. doi: 10.1128/jvi.74.15.7005-7015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhmann SE, Pugach P, Kunstman KJ, Taylor J, Stanfield RL, Snyder A, Strizki JM, Riley J, Baroudy BM, Wilson IA, et al. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J Virol. 2004;78:2790–2807. doi: 10.1128/JVI.78.6.2790-2807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature. 2002;420:678–682. doi: 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laal S, Burda S, Gorny MK, Karwowska S, Buchbinder A, Zolla-Pazner S. Synergistic neutralization of human immunodeficiency virus type 1 by combinations of human monoclonal antibodies. J Virol. 1994;68:4001–4008. doi: 10.1128/jvi.68.6.4001-4008.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latinovic O, Le N, Reitz M, Pal R, Devico A, Foulke JS, Redfield RR, Heredia A. Synergistic inhibition of R5 HIV-1 by maraviroc and CCR5 antibody HGS004 in primary cells: implications for treatment and prevention. AIDS. 2011a;25:1232–1235. doi: 10.1097/QAD.0b013e3283471edb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latinovic O, Reitz M, Le NM, Foulke JS, Fatkenheuer G, Lehmann C, Redfield RR, Heredia A. CCR5 antibodies HGS004 and HGS101 preferentially inhibit drug-bound CCR5 infection and restore drug sensitivity of Maraviroc-resistant HIV-1 in primary cells. Virology. 2011b;411:32–40. doi: 10.1016/j.virol.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederman MM, Offord RE, Hartley O. Microbicides and other topical strategies to prevent vaginal transmission of HIV. Nat Rev Immunol. 2006;6:371–382. doi: 10.1038/nri1848. [DOI] [PubMed] [Google Scholar]
- Lee B, Sharron M, Blanpain C, Doranz BJ, Vakili J, Setoh P, Berg E, Liu G, Guy HR, Durell SR, et al. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J Biol Chem. 1999;274:9617–9626. doi: 10.1074/jbc.274.14.9617. [DOI] [PubMed] [Google Scholar]
- Li A, Baba TW, Sodroski J, Zolla-Pazner S, Gorny MK, Robinson J, Posner MR, Katinger H, Barbas CF, 3rd, Burton DR, et al. Synergistic neutralization of a chimeric SIV/HIV type 1 virus with combinations of human anti-HIV type 1 envelope monoclonal antibodies or hyperimmune globulins. AIDS Res Hum Retroviruses. 1997;13:647–656. doi: 10.1089/aid.1997.13.647. [DOI] [PubMed] [Google Scholar]
- Li A, Katinger H, Posner MR, Cavacini L, Zolla-Pazner S, Gorny MK, Sodroski J, Chou TC, Baba TW, Ruprecht RM. Synergistic neutralization of simian-human immunodeficiency virus SHIV- vpu+ by triple and quadruple combinations of human monoclonal antibodies and high-titer anti-human immunodeficiency virus type 1 immunoglobulins. J Virol. 1998;72:3235–3240. doi: 10.1128/jvi.72.4.3235-3240.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953;3:285–290. [PubMed] [Google Scholar]
- Magnus C, Regoes RR. Estimating the stoichiometry of HIV neutralization. PLoS Comput Biol. 2010;6:e1000713. doi: 10.1371/journal.pcbi.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus C, Rusert P, Bonhoeffer S, Trkola A, Regoes RR. Estimating the stoichiometry of human immunodeficiency virus entry. J Virol. 2009;83:1523–1531. doi: 10.1128/JVI.01764-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascola JR, Louder MK, VanCott TC, Sapan CV, Lambert JS, Muenz LR, Bunow B, Birx DL, Robb ML. Potent and synergistic neutralization of human immunodeficiency virus (HIV) type 1 primary isolates by hyperimmune anti-HIV immunoglobulin combined with monoclonal antibodies 2F5 and 2G12. J Virol. 1997;71:7198–7206. doi: 10.1128/jvi.71.10.7198-7206.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov. 2004;3:215–225. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- McKeating JA, Cordell J, Dean CJ, Balfe P. Synergistic interaction between ligands binding to the CD4 binding site and V3 domain of human immunodeficiency virus type I gp120. Virology. 1992;191:732–742. doi: 10.1016/0042-6822(92)90249-o. [DOI] [PubMed] [Google Scholar]
- Mkrtchyan SR, Markosyan RM, Eadon MT, Moore JP, Melikyan GB, Cohen FS. Ternary complex formation of human immunodeficiency virus type 1 Env, CD4, and chemokine receptor captured as an intermediate of membrane fusion. J Virol. 2005;79:11161–11169. doi: 10.1128/JVI.79.17.11161-11169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On The Nature Of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Montefiori DC, Graham BS, Zhou J, Bucco RA, Schwartz DH, Cavacini LA, Posner MR. V3-specific neutralizing antibodies in sera from HIV-1 gp160-immunized volunteers block virus fusion and act synergistically with human monoclonal antibody to the conformation-dependent CD4 binding site of gp120. NIH-NIAID AIDS Vaccine Clinical Trials Network. J Clin Invest. 1993;92:840–847. doi: 10.1172/JCI116658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Klasse PJ. Thermodynamic and kinetic analysis of sCD4 binding to HIV-1 virions and of gp120 dissociation. AIDS Res Hum Retroviruses. 1992;8:443–450. doi: 10.1089/aid.1992.8.443. [DOI] [PubMed] [Google Scholar]
- Moore JP, McKeating JA, Huang YX, Ashkenazi A, Ho DD. Virions of primary human immunodeficiency virus type 1 isolates resistant to soluble CD4 (sCD4) neutralization differ in sCD4 binding and glycoprotein gp120 retention from sCD4-sensitive isolates. J Virol. 1992;66:235–243. doi: 10.1128/jvi.66.1.235-243.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Sodroski J. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J Virol. 1996;70:1863–1872. doi: 10.1128/jvi.70.3.1863-1872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PL, Crooks ET, Porter L, Zhu P, Cayanan CS, Grise H, Corcoran P, Zwick MB, Franti M, Morris L, et al. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J Virol. 2006;80:2515–2528. doi: 10.1128/JVI.80.5.2515-2528.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga JD, Franti M, Pevear DC, Maddon PJ, Olson WC. Potent antiviral synergy between monoclonal antibody and small-molecule CCR5 inhibitors of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2006;50:3289–3296. doi: 10.1128/AAC.00699-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima KA, Thompson DA, Rosenfield SI, Maddon PJ, Dragic T, Olson WC. Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J Infect Dis. 2001;183:1121–1125. doi: 10.1086/319284. [DOI] [PubMed] [Google Scholar]
- Nakata H, Steinberg SM, Koh Y, Maeda K, Takaoka Y, Tamamura H, Fujii N, Mitsuya H. Potent Synergistic Anti-Human Immunodeficiency Virus (HIV) Effects Using Combinations of the CCR5 Inhibitor Aplaviroc with Other Anti-HIV Drugs. Antimicrob Agents Chemother. 2008;52:2111–2119. doi: 10.1128/AAC.01299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogert RA, Hou Y, Ba L, Wojcik L, Qiu P, Murgolo N, Duca J, Dunkle LM, Ralston R, Howe JA. Clinical resistance to vicriviroc through adaptive V3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5. Virology. 2010;400:145–155. doi: 10.1016/j.virol.2010.01.037. [DOI] [PubMed] [Google Scholar]
- Pancera M, Wyatt R. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology. 2005;332:145–156. doi: 10.1016/j.virol.2004.10.042. [DOI] [PubMed] [Google Scholar]
- Pantophlet R, Burton DR. GP120: target for neutralizing HIV-1 antibodies. Annu Rev Immunol. 2006;24:739–769. doi: 10.1146/annurev.immunol.24.021605.090557. [DOI] [PubMed] [Google Scholar]
- Platt EJ, Durnin JP, Kabat D. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J Virol. 2005;79:4347–4356. doi: 10.1128/JVI.79.7.4347-4356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poignard P, Fouts T, Naniche D, Moore JP, Sattentau QJ. Neutralizing antibodies to human immunodeficiency virus type-1 gp120 induce envelope glycoprotein subunit dissociation. J Exp Med. 1996a;183:473–484. doi: 10.1084/jem.183.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poignard P, Klasse PJ, Sattentau QJ. Antibody neutralization of HIV-1. Immunol Today. 1996b;17:239–246. doi: 10.1016/0167-5699(96)10007-4. [DOI] [PubMed] [Google Scholar]
- Poignard P, Moulard M, Golez E, Vivona V, Franti M, Venturini S, Wang M, Parren PW, Burton DR. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J Virol. 2003;77:353–365. doi: 10.1128/JVI.77.1.353-365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poignard P, Saphire EO, Parren PW, Burton DR. gp120: Biologic aspects of structural features. Annu Rev Immunol. 2001;19:253–274. doi: 10.1146/annurev.immunol.19.1.253. [DOI] [PubMed] [Google Scholar]
- Potts BJ, Field KG, Wu Y, Posner M, Cavacini L, White-Scharf M. Synergistic inhibition of HIV-1 by CD4 binding domain reagents and V3-directed monoclonal antibodies. Virology. 1993;197:415–419. doi: 10.1006/viro.1993.1604. [DOI] [PubMed] [Google Scholar]
- Reeves JD, Gallo SA, Ahmad N, Miamidian JL, Harvey PE, Sharron M, Pohlmann S, Sfakianos JN, Derdeyn CA, Blumenthal R, et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc Natl Acad Sci U S A. 2002;99:16249–16254. doi: 10.1073/pnas.252469399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht CR, Krarup A, Reynell L, Mann AM, Brandenberg OF, Berlinger L, Abela IA, Regoes RR, Gunthard HF, Rusert P, et al. MPER-specific antibodies induce gp120 shedding and irreversibly neutralize HIV-1. J Exp Med. 2011;208:439–454. doi: 10.1084/jem.20101907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safarian D, Carnec X, Tsamis F, Kajumo F, Dragic T. An anti-CCR5 monoclonal antibody and small molecule CCR5 antagonists synergize by inhibiting different stages of human immunodeficiency virus type 1 entry. Virology. 2006;352:477–484. doi: 10.1016/j.virol.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Sampah ME, Shen L, Jilek BL, Siliciano RF. Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc Natl Acad Sci U S A. 2011;108:7613–7618. doi: 10.1073/pnas.1018360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RW, Venturi M, Schiffner L, Kalyanaraman R, Katinger H, Lloyd KO, Kwong PD, Moore JP. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J Virol. 2002;76:7293–7305. doi: 10.1128/JVI.76.14.7293-7305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattentau QJ, Zolla-Pazner S, Poignard P. Epitope exposure on functional, oligomeric HIV-1 gp41 molecules. Virology. 1995;206:713–717. doi: 10.1016/s0042-6822(95)80094-8. [DOI] [PubMed] [Google Scholar]
- Scanlan CN, Pantophlet R, Wormald MR, Ollmann Saphire E, Stanfield R, Wilson IA, Katinger H, Dwek RA, Rudd PM, Burton DR. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of alpha1-->2 mannose residues on the outer face of gp120. J Virol. 2002;76:7306–7321. doi: 10.1128/JVI.76.14.7306-7321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, Ott RG, Anthony RM, Zebroski H, Hurley A, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009;458:636–640. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- Schonning K, Lund O, Lund OS, Hansen JE. Stoichiometry of monoclonal antibody neutralization of T-cell line- adapted human immunodeficiency virus type 1. J Virol. 1999;73:8364–8370. doi: 10.1128/jvi.73.10.8364-8370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, Zhou Y, Pitt E, Anderson KS, Acosta EP, et al. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat Med. 2008;14:762–766. doi: 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Siliciano RF. Achieving a quantitative understanding of antiretroviral drug efficacy. Clin Infect Dis. 2010;51:1105–1106. doi: 10.1086/656688. author reply 1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Dennison SM, Liu P, Gao F, Jaeger F, Montefiori DC, Verkoczy L, Haynes BF, Alam SM, Tomaras GD. Prolonged exposure of the HIV-1 gp41 membrane proximal region with L669S substitution. Proc Natl Acad Sci U S A. 2010;107:5972–5977. doi: 10.1073/pnas.0912381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sougrat R, Bartesaghi A, Lifson JD, Bennett AE, Bess JW, Zabransky DJ, Subramaniam S. Electron tomography of the contact between T cells and SIV/HIV-1: implications for viral entry. PLoS Pathog. 2007;3:e63. doi: 10.1371/journal.ppat.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, Hipkin RW, Chou CC, Pugliese-Sivo C, Xiao Y, et al. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2005;49:4911–4919. doi: 10.1128/AAC.49.12.4911-4919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhnel J. Parallel dose-response curves in combination experiments. Bull Math Biol. 1998;60:197–213. doi: 10.1006/bulm.1997.0009. [DOI] [PubMed] [Google Scholar]
- Thali M, Furman C, Wahren B, Posner M, Ho DD, Robinson J, Sodroski J. Cooperativity of neutralizing antibodies directed against the V3 and CD4 binding regions of the human immunodeficiency virus gp120 envelope glycoprotein. J Acquir Immune Defic Syndr. 1992;5:591–599. [PubMed] [Google Scholar]
- Tilley SA, Honnen WJ, Racho ME, Chou TC, Pinter A. Synergistic neutralization of HIV-1 by human monoclonal antibodies against the V3 loop and the CD4-binding site of gp120. AIDS Res Hum Retroviruses. 1992;8:461–467. doi: 10.1089/aid.1992.8.461. [DOI] [PubMed] [Google Scholar]
- Tremblay C, Merrill DP, Chou TC, Hirsch MS. Interactions among combinations of two and three protease inhibitors against drug-susceptible and drug-resistant HIV-1 isolates. J Acquir Immune Defic Syndr. 1999;22:430–436. doi: 10.1097/00126334-199912150-00002. [DOI] [PubMed] [Google Scholar]
- Tremblay CL, Giguel F, Chou TC, Dong H, Takashima K, Hirsch MS. TAK-652, a novel CCR5 inhibitor, has favourable drug interactions with other antiretrovirals in vitro. Antivir Ther. 2005a;10:967–968. [PubMed] [Google Scholar]
- Tremblay CL, Giguel F, Guan Y, Chou TC, Takashima K, Hirsch MS. TAK-220, a novel small-molecule CCR5 antagonist, has favorable anti-human immunodeficiency virus interactions with other antiretrovirals in vitro. Antimicrob Agents Chemother. 2005b;49:3483–3485. doi: 10.1128/AAC.49.8.3483-3485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay CL, Giguel F, Kollmann C, Guan Y, Chou TC, Baroudy BM, Hirsch MS. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob Agents Chemother. 2002;46:1336–1339. doi: 10.1128/AAC.46.5.1336-1339.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay CL, Kollmann C, Giguel F, Chou TC, Hirsch MS. Strong in vitro synergy between the fusion inhibitor T-20 and the CXCR4 blocker AMD-3100. J Acquir Immune Defic Syndr. 2000;25:99–102. doi: 10.1097/00042560-200010010-00001. [DOI] [PubMed] [Google Scholar]
- Trkola A, Kuhmann SE, Strizki JM, Maxwell E, Ketas T, Morgan T, Pugach P, Xu S, Wojcik L, Tagat J, et al. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc Natl Acad Sci U S A. 2002;99:395–400. doi: 10.1073/pnas.012519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolini S, Mondor I, Parren PW, Burton DR, Tilley SA, Klasse PJ, Sattentau QJ. Inhibition of virus attachment to CD4+ target cells is a major mechanism of T cell line-adapted HIV-1 neutralization. J Exp Med. 1997;186:1287–1298. doi: 10.1084/jem.186.8.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veazey RS, Klasse PJ, Schader SM, Hu Q, Ketas TJ, Lu M, Marx PA, Dufour J, Colonno RJ, Shattock RJ, et al. Protection of macaques from vaginal SHIV challenge by vaginally delivered inhibitors of virus-cell fusion. Nature. 2005;438:99–102. doi: 10.1038/nature04055. [DOI] [PubMed] [Google Scholar]
- Vermeire K, Princen K, Hatse S, De Clercq E, Dey K, Bell TW, Schols D. CADA, a novel CD4-targeted HIV inhibitor, is synergistic with various anti-HIV drugs in vitro. Aids. 2004;18:2115–2125. doi: 10.1097/00002030-200411050-00003. [DOI] [PubMed] [Google Scholar]
- Verrier F, Nadas A, Gorny MK, Zolla-Pazner S. Additive effects characterize the interaction of antibodies involved in neutralization of the primary dualtropic human immunodeficiency virus type 1 isolate 89.6. J Virol. 2001;75:9177–9186. doi: 10.1128/JVI.75.19.9177-9186.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijh-Warrier S, Pinter A, Honnen WJ, Tilley SA. Synergistic neutralization of human immunodeficiency virus type 1 by a chimpanzee monoclonal antibody against the V2 domain of gp120 in combination with monoclonal antibodies against the V3 loop and the CD4-binding site. J Virol. 1996;70:4466–4473. doi: 10.1128/jvi.70.7.4466-4473.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011 doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Simek MD, Priddy F, Gach JS, Wagner D, Zwick MB, Phogat SK, Poignard P, Burton DR. A limited number of antibody specificities mediate broad and potent serum neutralization in selected HIV-1 infected individuals. PLoS Pathog. 2010:6. doi: 10.1371/journal.ppat.1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- Weiss JN. The Hill equation revisited: uses and misuses. Faseb J. 1997;11:835–841. [PubMed] [Google Scholar]
- Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- Xu W, Smith-Franklin BA, Li PL, Wood C, He J, Du Q, Bhat GJ, Kankasa C, Katinger H, Cavacini LA, et al. Potent neutralization of primary human immunodeficiency virus clade C isolates with a synergistic combination of human monoclonal antibodies raised against clade B. J Hum Virol. 2001;4:55–61. [PubMed] [Google Scholar]
- Yang X, Kurteva S, Lee S, Sodroski J. Stoichiometry of antibody neutralization of human immunodeficiency virus type 1. J Virol. 2005;79:3500–3508. doi: 10.1128/JVI.79.6.3500-3508.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lipchina I, Cocklin S, Chaiken I, Sodroski J. Antibody binding is a dominant determinant of the efficiency of human immunodeficiency virus type 1 neutralization. J Virol. 2006;80:11404–11408. doi: 10.1128/JVI.01102-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh P, Kishony R. Networks from drug-drug surfaces. Mol Syst Biol. 2007;3:85. doi: 10.1038/msb4100133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura K, Shibata J, Kimura T, Honda A, Maeda Y, Koito A, Murakami T, Mitsuya H, Matsushita S. Resistance profile of a neutralizing anti-HIV monoclonal antibody, KD-247, that shows favourable synergism with anti-CCR5 inhibitors. AIDS. 2006;20:2065–2073. doi: 10.1097/01.aids.0000247587.31320.fe. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhou Y, Alcock C, Kiefer T, Monie D, Siliciano J, Li Q, Pham P, Cofrancesco J, Persaud D, et al. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J Virol. 2004;78:1718–1729. doi: 10.1128/JVI.78.4.1718-1729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick MB, Burton DR. HIV-1 neutralization: mechanisms and relevance to vaccine design. Curr HIV Res. 2007;5:608–624. doi: 10.2174/157016207782418443. [DOI] [PubMed] [Google Scholar]
- Zwick MB, Wang M, Poignard P, Stiegler G, Katinger H, Burton DR, Parren PW. Neutralization synergy of human immunodeficiency virus type 1 primary isolates by cocktails of broadly neutralizing antibodies. J Virol. 2001;75:12198–12208. doi: 10.1128/JVI.75.24.12198-12208.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.