

Abstract
A complete molecular understanding of β-cell mass expansion will be useful for the improvement of therapies to treat diabetic patients. During normal periods of metabolic challenges, such as pregnancy, β-cells proliferate, or self-renew, to meet the new physiological demands. The transcription factor Forkhead box D3 (Foxd3) is required for maintenance and self-renewal of several diverse progenitor cell lineages, and Foxd3 is expressed in the pancreatic primordium beginning at 10.5 d postcoitum, becoming localized predominantly to β-cells after birth. Here, we show that mice carrying a pancreas-specific deletion of Foxd3 have impaired glucose tolerance, decreased β-cell mass, decreased β-cell proliferation, and decreased β-cell size during pregnancy. In addition, several genes known to regulate proliferation, Foxm1, Skp2, Ezh2, Akt2, and Cdkn1a, are misregulated in islets isolated from these Foxd3 mutant mice. Together, these data place Foxd3 upstream of several pathways critical for β-cell mass expansion in vivo.
Approximately 7% of pregnant women are affected by gestational diabetes mellitus (GDM), a disease resulting from the inability of the resident β-cell population to produce sufficient insulin during pregnancy (1). GDM increases the risk of complications to both mother and newborn child; the mother is more likely to develop type 2 diabetes later in life, and the child is more likely to be born with birth defects, macrosomia, and an increased risk of developing type 2 diabetes (2–6). During normal murine pregnancy, β-cells proliferate, or self-renew, thereby expanding the total β-cell mass to meet the mother's increasing demand for insulin (1, 7–12). This mechanism of β-cell mass expansion during human pregnancy remains controversial. Analogous measurements are not ethically feasible in humans. However, morphological analyses of human pancreata indicate that β-cell mass is increased during pregnancy (13, 14). Recent work analyzing pancreata from 38 cadaveric donors (18 pregnant, 20 controls) suggested that β-cells do not proliferate during human pregnancy. Instead, an increased number of smaller islets and insulin-positive cells was observed within the ductal epithelium (15). However, this work is not without caveats. It is well known that murine β-cells proliferate within a defined time window, and it is likely that the 18 pregnant human donors examined were not within an analogous gestational age (16, 17). This study also included donors with inflammatory disease that may have adversely affected β-cell mass expansion (18). It is important to note that the pregnancy-associated hormones prolactin, placental lactogen (PL), and human growth hormone all stimulate β-cell proliferation in islets isolated from mice, rats, and humans, suggesting that cell proliferation is a conserved mechanism of β-cell mass expansion during pregnancy (6). Although much of β-cell proliferation is controlled systemically (19), cell-autonomous control of β-cell proliferation is also a factor. Therefore, it is important to identify factors required for murine β-cell self-renewal, because the associated gene regulatory networks controlling β-cell mass expansion are likely to be conserved in human β-cells.
To date, there are only a few existing mouse models of GDM. A heterozygous deletion of the leptin receptor (Leprdb/+) causes extreme insulin resistance, and during pregnancy, this results in maternal hyperglycemia (20). A β-cell-specific deletion of Hepatic Nuclear Factor 4 alpha (Hnf4α) results in decreased β-cell proliferation, β-cell mass, pancreatic insulin content, and islet size resulting in impaired glucose tolerance (21). The transcription factor Forkhead box M1 (Foxm1) functions downstream of PL signaling. A pancreatic deletion of Foxm1 results in decreased β-cell proliferation and β-cell mass in adult mice, and pregnant mutant mice have GDM by 15.5 d gestation, two-thirds of the way through gestation (22). In addition, inhibition of serotonin synthesis, normally increased in response to PL signaling, results in decreased β-cell mass expansion and glucose intolerance during pregnancy (23). An additional mechanism for glucose homeostasis regulation and β-cell proliferation is at the epigenetic level. Menin1 functions as part of a histone methyltransferase complex to promote tri-methylation of histone 3 lysine 4, thereby maintaining expression of cell cycle inhibitors p27 and p18. Menin1 is normally down-regulated in β-cells during pregnancy, and artificially maintained expression during pregnancy causes decreased β-cell proliferation and maternal hyperglycemia (17). Recent work highlights two proteins of the Polycomb complex required for β-cell proliferation: enhancer of zeste homolog 2 (Ezh2) and Bmi1 polycomb ring finger oncogene (Bmi1) facilitate epigenetic modifications permitting β-cell proliferation during basal physiological conditions (24, 25).
Forkhead box D3 (Foxd3), a Forkhead transcriptional regulator, is critical for self-renewal of multiple progenitor cells (26–30). Foxd3 is expressed within the pancreatic primordium in two distinct cell populations: neural crest cells fated to innervate the pancreas (31) and pancreatic β-cells (32). Pancreatic coexpression of Foxd3 and insulin begins by 15.5 d post coitum, and in adults, Foxd3 is expressed in β-cells (32). This expression is also observed in human and rat islets, suggesting a conserved function among mammalian species (32, 33). Because Foxd3 is required for embryonic stem (ES) cell and neural crest progenitor cell self-renewal (26, 29, 34, 35), and β-cell mass expansion is primarily accomplished through self-renewal of existing β-cells (7–9), we hypothesized that Foxd3 is required for β-cell self-renewal and, by extension, β-cell mass expansion during pregnancy. Using a pancreatic and duodenal homeobox 1 (Pdx1)-Cre transgenic mouse, we deleted Foxd3 within the Pdx1-Cre expression domain, including the pancreas (36). Although these mice are unaffected under normal conditions, they suffer from glucose intolerance during pregnancy. In the absence of Foxd3 in the Pdx1 expression domain, Foxm1, Ezh2, S-phase kinase-associated protein 2 (Skp2), thymoma viral proto-oncogene 2 (Akt2), and cyclin-dependent kinase inhibitor (Cdkn)1a are misregulated, and the mice suffer from pregnancy-associated defects in β-cell proliferation, β-cell mass, and hyperglycemia.
Materials and Methods
Mouse strains
Mice with a Foxd3 null allele, Foxd3tm2.Lby (called Foxd3− throughout), and a conditional Foxd3 allele, Foxd3tm3.Lby (called Foxd3fl throughout), were bred to mice carrying a Pdx1-Cre transgene [Tg(Ipf1-cre)1Tuv] to generate mice lacking Foxd3 expression in the Pdx1-Cre expression domain (Foxd3fl/−;Pdx1-Cre) and littermate controls (Foxd3fl/+). The Foxd3− allele was used to minimize the number of recombination events mediated by Cre recombinase and allow direct comparisons between animals. These mouse lines and genotyping were previously described (29, 37, 38). For timed matings, females were checked daily for presence of a vaginal plug; noon on the day of an observed plug was considered 0.5 d gestation. Mice were housed on ALPHA-Dri bedding (Shepherd Specialty Papers, Chicago, IL) and fed Laboratory Rodent Diet 5001 (LabDiet, Richmond, IN). All mouse lines were maintained on an outbred genetic background consisting primarily of CD-1 and C57BL/6NHsd and handled in accordance with Association for Assessment and Accreditation of Laboratory Animal Care standards and protocols with approval from the Vanderbilt University Institutional Animal Care and Use Committee.
5-Bromo-2-deoxyuridine (BrdU) incorporation and immunohistochemistry
For BrdU incorporation analyses on pregnant mice, mice were injected with 50 mg of BrdU per kg body weight at noon at 13.5- and 14.5-d gestation. Similarly, to analyze BrdU incorporation in postnatal d 14 mice, mice were injected with BrdU at 13 d of age. To assess BrdU incorporation in 8-wk-old mice, BrdU was placed in the drinking water (0.8 mg/ml) for 1 wk and treated water replaced after 4 d to ensure BrdU activity. Pancreata were dissected and fixed overnight in 4% paraformaldehyde in PBS and histological experiments performed using standard procedures (28). For BrdU detection and β-cell size analyses, immunohistochemistry was performed as described (9) with rat anti BrdU (1:250; Accurate Chemical, Westbury, NY), guinea pig antiinsulin (1:500; Millipore, Billerica, MA), Cy2-donkey antiguinea pig (1:500; Jackson ImmunoResearch, West Grove, PA), Cy3-donkey antirat (1:500; Jackson ImmunoResearch), and Cy5-donkey antiguinea pig (1:500; Jackson ImmunoResearch) antibodies. To detect Foxd3 protein, slides were incubated in citrate unmasking solution (Vector Laboratories, Burlingame, CA) for 2 h at high pressure. Antibody for Foxd3 was as described (28) and detected with donkey antibiotin secondary antibody (Jackson ImmunoResearch) followed by Tyramide Signal Amplification (TSA kit no. 22; Invitrogen, Carlsbad, CA). Sections were counterstained with 4′, 6-diamidino-2-phenylindole (1:2000; Sigma, St. Louis, MO). For β-cell mass analyses, immunohistochemistry was performed using guinea pig antiinsulin (1:500; Millipore) and biotin-donkey antiguinea pig (1:500; Jackson ImmunoResearch) antibodies. The signal was amplified using Ready-to-Use (RTU) (Vector Laboratories) and 3′3 diaminobenzidine (Vector Laboratories). Sections were counterstained with hematoxylin (Richard Allen Scientific, Kalamazoo, MI). Terminal deoxynucleotidyl transferase 2′-deoxyuridine, 5′-triphosphate nick end labeling analysis was completed with the In Situ Cell Death kit (Roche, Basel, Switzerland).
Quantitative analyses and imaging
For BrdU incorporation, islets from five sections evenly spaced throughout the pancreas (accounting for approximately 5–8% of total pancreatic mass) were imaged using a Zeiss AxioSkop2 Plus equipped with a Q Imaging Retiga 2000R camera (Carl Zeiss, Oberkochen, Germany). The total number of BrdU-positive β-cells was divided by the total number of β-cells. For β-cell mass, the pancreas was embedded in a manner to ensure equal representation of the dorsal and ventral pancreas. Five evenly spaced pancreatic sections (accounting for 5–8% of the total pancreatic mass) were imaged using an Ariol SL-50 imaging system. Insulin-positive area and total pancreatic area were determined using ImageJ software (National Institutes of Health, Bethesda, MD) and β-cell mass determined by dividing total β-cell-positive area by total pancreatic area multiplied by pancreatic wet weight. To determine β-cell size, every islet from four evenly spaced sections (accounting for ∼4–6% of total pancreatic mass) was imaged as above (39). The total insulin-positive area from each section was calculated using Adobe Photoshop. The area was then divided by the total number of β-cells per section. At least 5000 β-cells per animal were counted.
Gene expression analyses by TaqMan low density arrays and quantitative real-time PCR (qRT-PCR)
Islets were isolated following standard procedures by the Vanderbilt Islet Procurement and Isolation Core (40). To assay Foxd3 and Skp2 expression in the presence of PL, islets were cultured in 50 ng/ml PL for 4 d (22). Total RNA was collected with the RNeasy Mini kit (QIAGEN, Valencia, CA), contaminating DNA removed with Turbo DNase (Ambion, Austin, TX) and cDNA generated from 400 ng of total RNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Genes included in this array are shown in Table 1; 100 ng of cDNA were prepared for analysis using TaqMan Universal PCR Master Mix (Applied Biosystems) and analyzed on a custom TaqMan Low Density Array (Applied Biosystems). The sequences for the primers and probes were previously published (41). The relative amount of RNA was determined with 18S rRNA as a reference. For qRT-PCR, 4 ng of cDNA per sample were prepared with Power SYBR Green PCR Master Mix (Applied Biosystems). All samples were run in duplicate, and the relative amount of RNA was determined by comparison with Hypoxanthine guanine phosphoribosyl transferase (Hprt) mRNA. The following primer sequences were used:
Table 1.
Genes assayed by TaqMan low density array
| Insulin signaling | Cell cycle | β-Cell function | Pancreas development |
|---|---|---|---|
| Akt1 | Akt1 | Foxo1 | Arx |
| Akt2 | Akt2 | Gjd2 | Foxa2 |
| Foxo1 | Bmi1 | Glut2 | Gcg |
| Irs2 | Ccnd2 | Ins1 | Hnf4a |
| Prkca | Cdc25a | Ins2 | Isl1 |
| Pten | Cdk4 | MafA | MafA |
| Rictor | Cdkn1a | Nkx2.2 | MafB |
| Sgk1 | Cdkn1b | Pdx1 | Neurog3 |
| Cdkn2a | Snap25 | Nkx2.2 | |
| Cdkn2b | Stx1 | Nkx6.1 | |
| Cdkn2c | Stxbp1 | Pax6 | |
| Ezh2 | Vamp2 | Pdx1 | |
| Foxm1 | Ptf1a | ||
| Men1 | |||
| Prdm16 | |||
| Pten | |||
| Rictor | |||
| Skp2 |
Relative mRNA levels of the listed genes were assayed using a TaqMan low density array. Genes were clustered into groups based on the function of the produced protein. Note, some genes were included in multiple categories.
Foxd3, 5′-GTCCGCTGGGAATAACTTTCCGTA-3′ and 5′-ATGTACAAAGAATGTCCCTCCCACCC-3′;
Skp2, 5′-AGCCGAGGTGAATGAGAGTT-3′ and 5′-AGCATGAATGCTCCACAAAG-3′;
HPRT, 5′-TACGAGGAGTCCTGTTGAATGTTGC-3′ and 5′-GGGACGCAGCAACTGACATTTCTA-3′.
Intraperitoneal glucose tolerance testing (IPGTT)
After a 16-h fast, mice were subjected to an ip injection of 2 mg of dextrose per gram of body weight. Glucose measurements were obtained with blood samples from the tail vein using a glucometer (FreeStyle) before injection and at 15, 30, 60, 90, and 120 min after injection.
Serum insulin assays
Blood samples obtained from the saphenous vein were collected in a Microvette CB 300 (Fisher Scientific, Pittsburgh, PA) capillary tube before injection of dextrose, as above, and 15 and 30 min after injection. Serum insulin content was measured using Insulin Ultra-Sensitive ELISA (Alpco Diagnostics, Salem, NH). All samples were run in duplicate.
Islet perifusions
Islet perifusions were performed as described using 16.7 mm glucose and 20 mm KCl in 5.6 mm glucose as stimuli (42). Insulin was extracted from perifusates in acid alcohol for 48 h and insulin levels measured using RIA performed by the Vanderbilt Hormone Assay and Analytical Services Core. The total amount of insulin was normalized to 100 islet equivalents to standardize the data for comparison.
Statistical significance
Two-tailed Student's t tests were used to determine statistical significance for each assay except IPGTT. In the case of IPGTT, statistical significance was determined using repeated measures ANOVA with Bonferoni post hoc tests.
Results
Foxd3 is not required to maintain euglycemia under basal physiological conditions
To analyze the function of Foxd3 in β-cells, we used a Pdx1-Cre transgene to generate mice lacking pancreatic Foxd3 (Foxd3fl/−;Pdx1-Cre) along with control (Foxd3fl/+) and heterozygous littermates (Foxd3fl/− and Foxd3fl/+;Pdx1-Cre) (29, 37, 38), referred to as mutant, control, and heterozygote, respectively. We chose Pdx1-Cre to delete Foxd3 because Pdx1-Cre mice are euglycemic (22, 43–45). All Foxd3 expressing cells within the pancreatic endoderm are contained within the Pdx1 expression domain (32). Using qRT-PCR, we verified that Foxd3 mRNA was decreased approximately 3-fold in mutant islets, confirming that Pdx1-Cre deleted the Foxd3 coding region (Fig. 1A). It is important to note that Foxd3 is also expressed in neural crest derivatives that innervate the islets (29, 31, 34); therefore, residual Foxd3 expression is likely due to expression within this lineage. Additionally, using immunofluorescence analysis of Foxd3 protein expression, we could not detect the protein in the nucleus of the islets from mutant mice (Fig. 1C) compared with controls (Fig. 1B). Because Pdx1-Cre is active in the hypothalamus (36, 46, 47), a region of the central nervous system critical for metabolic control, we analyzed Foxd3 mRNA expression in the hypothalamus of control animals using RT-PCR and determined that Foxd3 mRNA was not detectable in this region (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). Additionally, Foxd3 expression in both forms of heterozygous mice (Foxd3fl/− and Foxd3fl/+;Pdx1-Cre) is not statistically different than Foxd3fl/+ controls (Supplemental Fig. 2). The observation that Foxd3 expression in heterozygotes is similar to control mice is consistent with previous reports suggesting that Foxd3 functions as a transcriptional repressor (48), and our data suggest the hypothesis that Foxd3 can autoregulate its own transcription. To determine the requirement for Foxd3 in maintaining euglycemia under basal physiological conditions, we performed IPGTT on 38-wk-old (data not shown) and 52-wk-old mice (Supplemental Fig. 3) fed a normal chow (5.1% fat/kcal). At both ages and for both genders, glucose clearance curves showed no significant differences between mutants, heterozygotes, and controls, suggesting that Foxd3 is not required to maintain glucose tolerance under normal physiological conditions.
Fig. 1.

Validation of loss of Foxd3 and Foxd3 expression in pancreatic islets during pregnancy. A, qRT-PCR data showed that Foxd3 expression was decreased in mutant islets (white bars) using Pdx1-Cre (left graph). Foxd3 mRNA levels were decreased in islets isolated from 15.5-d gestation (D.G.) control (black bars) and mutant (white bars) mice compared with islets from control virgin mice (right graph). Foxd3 expression in virgin controls was arbitrarily set to 1. There was no statistical difference between the Foxd3 mRNA levels in virgin mutants compared with controls or mutants at 15 D.G. B–E, Immunohistochemistry for Foxd3 demonstrated that Foxd3 (red) was detected in the nuclei of insulin-expressing β-cells (green) from control animals (B). B' and C', Red channel alone. Foxd3 protein was not detected in β-cell nuclei in mutant animals (C). Nuclear expression of Foxd3 is indicated by arrows (B' inset), and nonspecific cytoplasmic staining is indicated by arrowheads (C' inset). Additionally, immunofluorescent analyses confirmed that Foxd3 is not expressed in β-cells during pregnancy. Foxd3 protein (red) was not detected in the nuclei of β-cells (insulin, green) in mutant (E and E') or control animals (D and D') at 15.5 D.G. F. Foxd3 expression decreased, whereas Skp2 expression increased in islets isolated from wild-type females cultured with human PL (white bars). Foxd3 and Skp2 mRNA levels in islets cultured without PL was arbitrarily set to 1 (black bars). n = 3–4 mice in each group. Error bars indicate sem; *, P < 0.05; **, P < 0.01.
Differential expression of Foxd3 during pregnancy
Because Foxd3 is required for proliferation and/or self-renewal of other cell lineages (28, 29, 34, 35, 37), and it is expressed in adult β-cells (32), we hypothesized that Foxd3 plays a similar role in β-cells. To test this, we used pregnancy as a model, because during pregnancy, murine β-cells self-renew to expand β-cell mass (1, 12, 16, 17). To determine whether Foxd3 is differentially regulated during pregnancy, we analyzed Foxd3 expression in islets from control and mutant females at 15.5 d gestation and found that, surprisingly, Foxd3 mRNA levels were approximately 5-fold down-regulated in wild-type females at 15.5-d gestation (Fig. 1A). These levels were similar to the levels detected in the mutant tissue, suggesting an almost complete loss of Foxd3 transcription. Additionally, Foxd3 protein was not detected in the nucleus of β-cells from control or mutant mice during pregnancy (Fig. 1, D and E). These data indicate that Foxd3 is negatively regulated in response to pregnancy cues. During pregnancy, PL, growth hormone, and prolactin are up-regulated and stimulate β-cell mass expansion. Although prolactin and growth hormone regulate β-cell proliferation, the most potent stimulus for β-cell proliferation during pregnancy is PL (1, 12, 49). Therefore, to determine whether Foxd3 expression decreases in response to PL, we cultured islets isolated from wild-type mice in the presence and absence of PL. To establish that our culture system worked as expected, we analyzed expression of Skp2 after 4 d in culture. Skp2 is a target of Foxm1 that increases expression in response to PL (22). We found that Skp2 expression was increased, thereby validating this system (Fig. 1F). At the same time, Foxd3 mRNA levels were decreased approximately 6.7-fold, supporting the notion that PL is a potent regulator of Foxd3 expression during pregnancy (Fig. 1F).
Requirement for Foxd3 to maintain euglycemia during pregnancy
To assess the ability of β-cells from mutant mice to meet the metabolic challenges of pregnancy, we analyzed pregnant females for their ability to clear glucose. To establish a baseline, IPGTT on 8-wk-old virgin control and mutant mice demonstrated that, as expected, Foxd3 was not required to maintain glucose tolerance (Fig. 2A). However, when the same mice were tested during pregnancy, mutants showed severe defects in glucose tolerance at 15.5-d gestation compared with controls, illustrated by both glucose tolerance curves and the calculated area under the curve (Fig. 2, B and C). There were no defects observed in heterozygous mice at either stage (Supplemental Fig. 4, A and B).
Fig. 2.
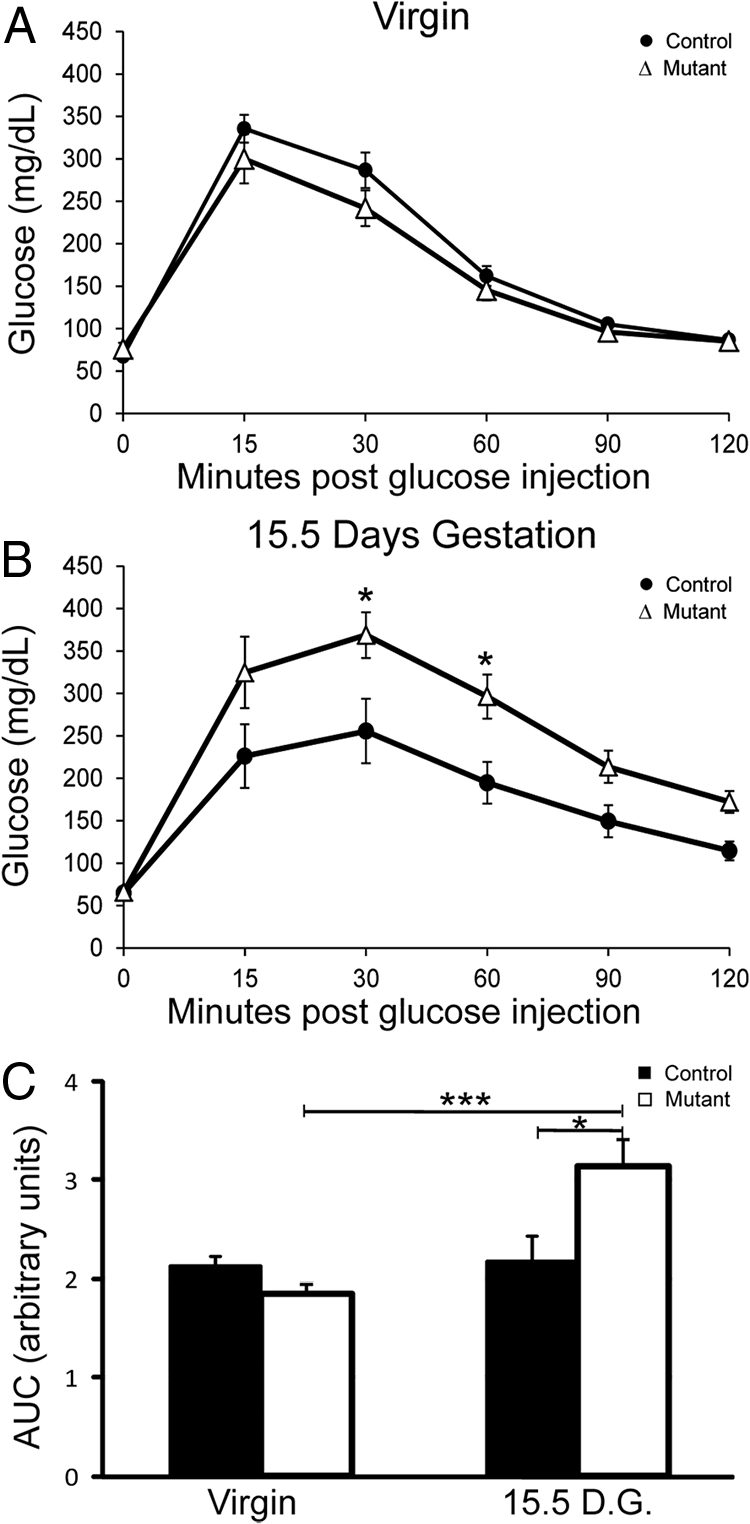
Glucose tolerance was impaired in Foxd3flox/−;Pdx1-Cre mice during pregnancy. A, IPGTT demonstrated that 8-wk-old virgin Foxd3flox/−;Pdx1-Cre (Δ, mutant) and Foxd3flox/+ (●, control) female mice exhibited normal glucose tolerance. B, IPGTT on the same mice at 15.5-d gestation (D.G.) showed mutant mice had impaired glucose tolerance, whereas controls did not. C, The area under the curve (A.U.C.) of mutant mice (white bars) at 15.5-d gestation was greater than pregnant control (black bars) mice and virgin control and mutant mice. n = 10 mice in each group. Error bars indicate sem; *, P < 0.05; ***, P < 0.001.
To determine whether impaired glucose tolerance in mutant mice resulted from reduced levels of secreted insulin, we measured serum insulin levels during an IPGTT from both virgin and pregnant mice. As expected, mutant and control serum insulin levels were similar before pregnancy (Fig. 3A), confirming that insulin production and secretion is not impaired in virgin Foxd3 mutant mice. However, during pregnancy, mutant mice had decreased serum insulin levels in response to a glucose stimulus (Fig. 3B), suggesting that Foxd3 is required to produce sufficient insulin levels to maintain glucose homeostasis in response to metabolic demand.
Fig. 3.
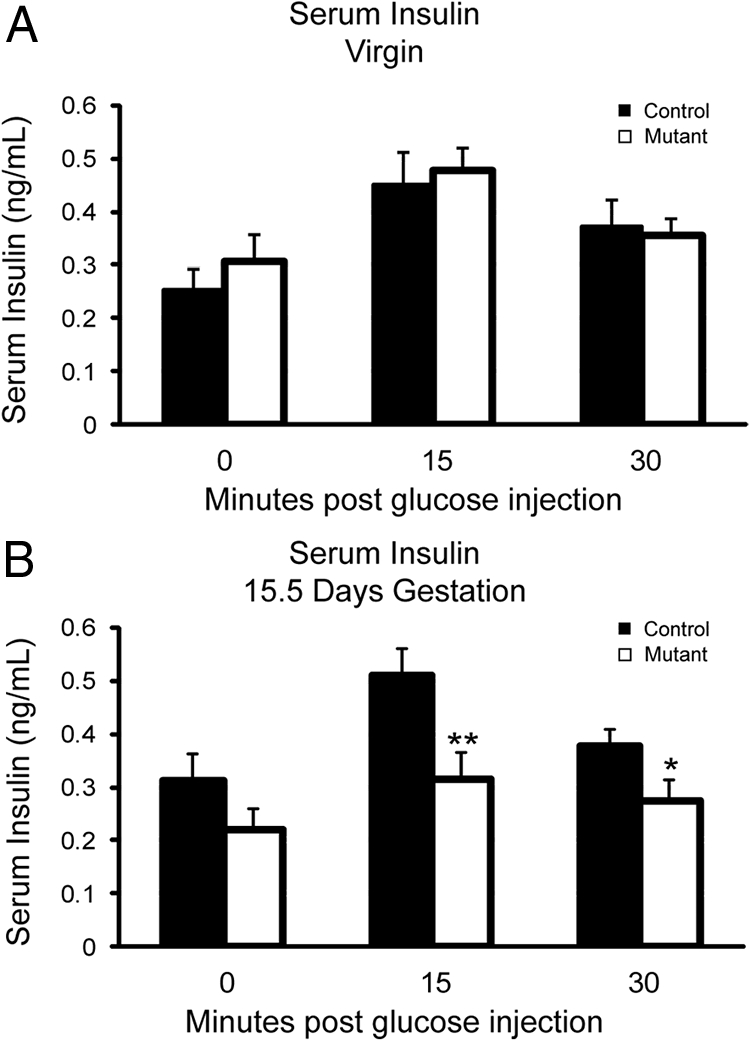
Serum insulin levels were decreased in mutant mice at 15.5-d gestation. A, ELISA using serum collected from mutant and control virgin mice demonstrated that virgin mutant mice (white bars) had serum insulin levels similar to control mice (black bars). n = 9–10 animals in each group. B, In contrast, mutant mice at 15.5-d gestation had reduced insulin serum levels 15 and 30 min after a glucose injection compared with controls. n = 9–10 animals in each group. Error bars indicate sem; *, P < 0.05; **, P < 0.01.
Genes required for cell proliferation and β-cell function are misregulated in Foxd3 mutant mice
To understand where Foxd3 functions in the known molecular pathways regulating glucose homeostasis, and to characterize genes misexpressed in mutant mice, we used TaqMan Low Density Arrays to evaluate expression of 47 genes (Table 1) implicated in either β-cell mass expansion/proliferation, β-cell function, insulin signaling, and/or pancreas development (Fig. 4). Several genes that regulate β-cell proliferation, including Cdkn1a, Cdkn2a, Ezh2, Foxm1, and Skp2, were misregulated in virgin mutant animals compared with virgin controls. There was a slight, but statistically significant, reduction in Cdkn1b expression. Additionally, Ezh2 and Akt2 were also severely down-regulated in pregnant mutants compared with pregnant controls, whereas Skp2 was up-regulated in pregnant mutants. These data suggest impaired β-cell proliferation that will impact β-cell mass expansion. In addition to expression changes in genes regulating β-cell proliferation, several genes required for glucose-stimulated insulin secretion were also misregulated. In virgin mutants, v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A (MafA), Forkhead box A2 (Foxa2), and Neurokinin 2 transcription factor related, locus 2 (Nkx2.2) mRNA levels were significantly repressed compared with virgin controls, suggesting there may be defects in glucose-stimulated insulin secretion in Foxd3 mutant animals. Interestingly, in the pregnant mutants, both MafA and Foxa2 were up-regulated, whereas the mRNA encoding synaptosomal-associated protein 25 (Snap25) was significantly decreased. Both Ins2 and vesicle-associated membrane protein 2 (Vamp2) were slightly decreased in pregnant mutants compared with controls (<20% decrease in gene expression), but this change in expression is not likely to be biologically relevant. Lastly, a few genes required for pancreatic development were misregulated in the virgin Foxd3 mutant animals. Both sex determining region of Chr Y box containing gene 9 (Sox9) and Gcg were up-regulated in the absence of Foxd3, although we did not detect changes in α-cell mass (data not shown). An increase in these Sox9 and Gcg genes may indicate the onset of an embryonic developmental program to compensate for defects in the adult mice. Although there was a slight reduction of insulin receptor substrate 2 (Irs2) and serum/glucocorticoid regulated kinase 1 (Sgk1) (<20% change in gene expression) in pregnant mutant animals, the mRNA levels of these genes were very similar to those of controls, suggesting that there were no defects in insulin signaling in islets from mutant animals. Together, these data suggest misregulation of genes known to control β-cell proliferation, and β-cell function may account for glucose intolerance during pregnancy in Foxd3 mutant animals.
Fig. 4.

Gene expression profiling in Foxd3 mutant and control pancreata. A TaqMan low density array was used to analyze gene expression from mutant and control pancreata before and during pregnancy. The heat map illustrates relative changes in gene expression in mutant compared with controls at each stage (P < 0.10). Gray rectangles indicate no significant change in gene expression (P > 0.10). n = 4 mice in each group. D.G., Days gestation; NC, no change.
β-Cell mass, proliferation, and size are decreased in mutant mice
Several of the misregulated genes (Ezh2, Foxm1, Skp2, and Akt2) are required for β-cell proliferation and β-cell mass expansion. Therefore, we quantified β-cell mass in 2-wk-old and 8-wk-old virgin and 10-wk-old pregnant mice at 15.5-d gestation with and without the deletion of Foxd3. At 2 wk of age, β-cell mass was similar in mutant and control mice. However, by 8 wk of age, β-cell mass in mutant mice was decreased 23% compared with controls (Fig. 5A), although these mice were euglycemic (Fig. 2A). Unsurprisingly, β-cell mass was decreased 30% in pregnant mutant mice compared with control mice (Fig. 5A). Predictably, β-cell mass was unchanged in heterozygous animals at 15.5-d gestation compared with controls (Supplemental Fig. 4C). These data demonstrate that, in agreement with previous studies, a 25–30% decrease in β-cell mass can be tolerated under basal physiological conditions in virgin mice (43, 50). However, our data indicate that during pregnancy, a 30% decrease in β-cell mass may contribute to impaired glucose tolerance. Consistent with decreased β-cell mass, we detected an increase in the α- to β-cell area at 15.5-d gestation (Fig. 5B). This change in proportion between α- and β-cell area during pregnancy may also contribute to the defects in glucose tolerance.
Fig. 5.

β-Cell mass, β-cell proliferation, and β-cell size were decreased in mutant mice. A, The total β-cell mass was calculated and was similar between female mutant (white bars) and control (black bars) mice at 2 wk of age. However, mutant mice had decreased β-cell mass compared with controls at both 8 wk of age and 15.5-d gestation (D.G.). B, The α- to β-cell area was calculated by measuring the total glucagon-positive area divided by the total insulin-positive area. In virgin animals, the α- to β-cell area ratio was similar between controls and mutants. However, the α- to β-cell area was increased in pregnant mutants compared with pregnant control littermates, suggesting a deficiency in the β-cell mass. C–E, BrdU incorporation between postnatal d 13 and 14 (C), 7 and 8 wk of age (D), and 13.5 and 15.5 d gestation (E) was calculated by quantifying the number of BrdU/insulin double-positive cells. BrdU incorporation between postnatal d 13 and 14 and 7 and 8 wk of age was unchanged. In contrast, during pregnancy, BrdU incorporation between 13.5 and 15.5 d gestation was decreased approximately 2-fold in mutant mice compared with controls. n = 3–4 animals per group. F, β-Cell size was calculated by measuring the insulin-positive area divided by the total number of β-cells from evenly spaced sections throughout the pancreas. In both virgin and pregnant females, β-cell size was decreased in mutant animals compared with control littermates. n = 4–6 mice in each group. Error bars indicate sem; *, P < 0.05.
Because we observed a defect in β-cell mass in 8-wk-old virgin and 10-wk-old pregnant mice, we assessed β-cell proliferation at these stages. β-Cells proliferate slowly in virgin mice at 8-wk of age (reviewed in Ref. 51); therefore, we measured BrdU incorporation for 1 wk. The percent of β-cells that incorporated BrdU was similar between mutant and control mice (Fig. 5D). This result was surprising, because β-cell mass was decreased by 23% in mutant mice at 8 wk of age (Fig. 5A). To determine whether there was a defect in β-cell proliferation at earlier developmental stages, we assessed BrdU incorporation in younger mice (between 13 and 14 d), because β-cells proliferate more rapidly in neonatal rodents than adult rodents (52). At this time, BrdU incorporation in β-cells was similar between mutant and control animals (Fig. 5C), supporting the notion that proliferation defects are not the fundamental cause of the slightly decreased β-cell mass in virgin mutants. During pregnancy, murine β-cells proliferate to compensate for increased metabolic demand (1, 12, 13), and it is possible that different transcriptional networks and signals regulate this compensatory proliferation. Because β-cell proliferation during murine pregnancy peaks just before 15.5-d gestation (16, 17, 22), we monitored BrdU incorporation between 13.5- and 15.5-d gestation. The percent of β-cells colabeled with BrdU was approximately 2-fold lower in mutants compared with controls (Fig. 5E and Supplemental Fig. 5, A and B), indicating that although Foxd3 was not absolutely required for β-cell proliferation, during pregnancy, fewer Foxd3 mutant β-cells proliferate compared with β-cells from control littermates. Another potential cause of decreased β-cell mass is increased β-cell apoptosis. To determine whether Foxd3 is required for β-cell survival, we analyzed apoptosis at 15.5-d gestation, and in all samples analyzed, we were unable to detect any terminal deoxynucleotidyl transferase 2′-deoxyuridine, 5′-triphosphate nick end labeling-positive β-cells (Supplemental Fig. 5, C and D). Another factor affecting β-cell mass is the size of individual cells. To determine whether decreased hypertrophy contributes to decreased β-cell mass in both virgin and pregnant mutants, we analyzed β-cell size by measuring the total insulin-positive area divided by the total number of β-cells. We found that β-cell size was decreased in both virgin and pregnant mutants compared with control littermates (Fig. 5F). This suggests that in virgin mutant mice, decreased hypertrophy contributed to decreased β-cell mass. However, in mutant mice, both decreased hypertrophy and proliferation combine to impact β-cell mass during pregnancy. Together with the results above, these data suggest that Foxd3 is required to maintain β-cell proliferation and size during pregnancy.
Foxd3 is not required to regulate glucose-stimulated insulin secretion
Another physiological change caused by pregnancy is the response of β-cells to glucose. During pregnancy, β-cells become more sensitive to blood glucose levels and secrete more insulin when presented with lower glucose concentrations (1, 6, 12). Because several genes required for β-cell function (MafA, Snap25, Foxa2, and Nkx2.2) were misregulated in the absence of Foxd3, we analyzed glucose-stimulated insulin secretion using islet perfusion assays at 15.5-d gestation. The amount of secreted insulin was similar between mutant and control animals using both high glucose (16.7 mm) and KCl (20 mm in 5.6 mm glucose) to elicit a response (Fig. 6). This suggests that although genes required for glucose-stimulated insulin secretion were misregulated in the mutant animals, Foxd3 is not required for enhanced glucose-stimulated insulin secretion during pregnancy. These data further indicate that mutant β-cells function normally on a per cell basis, and impaired glucose-stimulated insulin secretion is not the underlying cause of glucose intolerance during pregnancy. Together, all of our data point to the conclusion that decreased β-cell mass in the absence of Foxd3 results in decreased serum insulin levels and glucose intolerance during pregnancy.
Fig. 6.

Foxd3 is not required for glucose-stimulated insulin secretion during pregnancy. Islet perifusion assays were used to analyze insulin secretion in response to various stimuli (low glucose, high glucose, and KCl), and insulin levels from perifusates were normalized to 100 islet equivalents (IEQ). The normalized amount of secreted insulin was similar between mutants (Δ) and controls (●). Note, all mice were at 15.5-d gestation. G 16.7, 16.7 mm glucose; G 5.6, 5.6 mm glucose; KCl 20, 20 mm KCl in 5.6 mm glucose. n = 2 animals per genotype.
Discussion
Foxd3 is dramatically down-regulated during pregnancy in response to PL, and loss of Foxd3 results in impaired glucose tolerance during pregnancy, suggesting an important role for this protein during this metabolically challenging process. Here, we characterized the role of Foxd3 in maintaining glucose homeostasis and β-cell mass expansion during pregnancy. Although β-cell mass was normal at postnatal d 14, and mutant mice were euglycemic under basal physiological conditions, pregnant mutant females were glucose intolerant during pregnancy, had decreased β-cell mass due to decreased β-cell proliferation and β-cell size, and several genes required to regulate cell proliferation were misregulated. These data clearly show that a pancreas-specific deletion of Foxd3 is a novel mouse model of glucose intolerance during pregnancy.
The primary mechanism of β-cell mass expansion during normal homeostasis and pregnancy is β-cell proliferation, or self-renewal, of existing β-cells (7–9). β-Cells from Foxd3 mutant mice showed decreased BrdU incorporation during pregnancy, indicating that loss of Foxd3 functionally impacts β-cell self-renewal at a time when β-cells must rapidly replicate to compensate for metabolic demand. Because Foxd3 is expressed in embryonic insulin-producing cells and the mutant pancreas develops normally, Foxd3 must not control the cell cycle directly and must not regulate embryonic β-cell proliferation. This finding is not surprising, because postnatal and embryonic β-cells use different molecular pathways to regulate proliferation (53–57). In addition, our data raise the possibility that Foxd3 regulates a “replication refractory period” that normally prevents serial β-cell proliferation (9, 58), and Foxd3 may repress genes necessary to shorten the replication refractory period and therefore decrease the proportion of proliferating β-cells.
To identify the pathways through which Foxd3 function impacts β-cell proliferation, we analyzed gene expression differences between mutant and control pancreata and identified several misregulated genes required for cell proliferation. Foxm1 transcription was decreased 2.3-fold in virgin Foxd3 mutants; Foxm1 is required for β-cell proliferation after a 60% partial pancreatectomy and during pregnancy as a downstream mediator of PL signaling (43, 51, 53). Additionally, there is a large variation in β-cell size in Foxm1 mutant mice (53), suggesting that decreased Foxm1 expression may be partially responsible for decreased β-cell size in Foxd3 mutant animals. Another down-regulated transcript, Skp2, is a target of Foxm1 (59), supporting the notion that the Foxm1 pathway was affected in Foxd3 mutants. Cdkn1a was drastically up-regulated in islets isolated from mutant animals. Cdkn1a inhibits cyclin dependent kinase 4 (CDK4) activity causing cell cycle arrest at G1 (60), and increased levels of Cdkn1a may restrict β-cell proliferation in Foxd3 mutants. The Ezh2 was also down-regulated in islets from both virgin and, importantly, pregnant mutant mice. This protein is a histone methyltransferase required to repress the Ink4a/Arf locus that in turn regulates Foxm1 transcriptional activity and inhibits β-cell proliferation (25, 61). A β-cell-specific deletion of Ezh2 resulted in decreased β-cell proliferation and hyperglycemia (25). Although its role in regulating β-cell proliferation during pregnancy has not been analyzed, it is likely that pregnant Ezh2 mutant mice will continue to have severe defects in glucose homeostasis. In addition to genes misregulated in virgin mutants, Akt2 was down-regulated in pregnant Foxd3 mutants, and a compound mutation of Akt2 and thymoma viral proto-oncogene 1 (Akt1) results in β-cell proliferation defects (62). Because Ezh2 and Akt2 were down-regulated in mutant mice during pregnancy, it is likely that they contribute to the proliferation defects observed during pregnancy. These data suggest that alteration of multiple parallel and interacting pathways in Foxd3 mutants decreases proliferation, and Foxd3 impacts several factors that together control β-cell proliferation, β-cell size, and β-cell mass expansion during pregnancy (for a model, see Fig. 7).
Fig. 7.

Model of Foxd3 function in the β-cell. Foxd3 regulates several factors that control β-cell proliferation, including Foxm1, Skp2, Ezh2, Akt2, and Cdkn1a. Arrows do not imply direct interactions.
Interestingly, Foxd3 mutant mice exhibit a physiological defect during pregnancy, despite the observation that Foxd3 is no longer expressed in maternal islets by 15.5-d gestation. This suggests that Foxd3 is required before pregnancy to permit β-cell proliferation during gestation and does not agree with models in which Foxd3 acts as a direct activator of genes required for β-cell proliferation. In ES cells, Foxd3 is required to maintain multipotency by inhibiting differentiation (35). At a mechanistic level, Foxd3 “poises” the silent albumin (Alb1) locus for transcription (30). Even though ES cells do not express Alb1, the locus is epigenetically modified in the pluripotent stem cells compared with primary fibroblasts that lack the potential to generate endoderm and express Alb1 (30). Similarly, Foxd3 represses transcription of the B cell-specific lambda 5 preB1 locus in ES cells, maintaining the locus in an inactive state despite active epigenetic marks. As cells differentiate toward a B cell fate, the Foxd3-mediated repression is relieved, concomitant with a different Forkhead protein replacing Foxd3 at this binding site (63). A similar mechanism may occur in the pancreatic β-cell. Similar to the case of Alb1, Foxd3 may establish or maintain epigenetic mark(s) critical for up-regulation of genes controlling β-cell proliferation when needed or, as in the lambda 5 preB1 locus, remain as the last barrier to active transcription, so that when needed, downstream genes can be activated quickly. When β-cells are stimulated to proliferate during pregnancy, Foxd3 may then be replaced by another Forkhead family member (perhaps Foxa1, Foxa2, or Foxm1) to activate expression of target genes regulating β-cell proliferation. Foxa1 and Foxa2 are required for activation of Pdx1, and through Pdx1, they regulate pancreatic differentiation and growth (64), whereas Foxm1 regulates β-cell proliferation (22, 43, 53). Therefore, it is feasible that this mechanism is conserved in β-cells, and Foxd3 is required to poise target genes, whereas other Forkhead transcription factors are required for direct activation of gene expression. Because Foxd3 was deleted from the pancreatic epithelium early in development before β-cell differentiation, target loci may never be poised, and other Forkhead proteins may not have access to promote transcription of genes required for proliferation. Unfortunately, testing this model with the available reagents is not feasible. Ideally, to determine whether Foxd3 is required to poise a locus for transcription, we would acutely ablate Foxd3 expression before pregnancy using a Tamoxifen-inducible Cre recombinase. However, we found that although both control and conditionally mutant female mice injected with Tamoxifen to activate Cre could mate, they were unable to become pregnant in a predictable manner (n = 9 females) even 6 wk after the Tamoxifen injection. These findings are consistent with previously published data; rats injected with Tamoxifen had smaller uteri and ovaries, and their estrous cycles were arrested in diestrus (65–67). Therefore, this model awaits identification of direct targets of Foxd3, an active goal of our future work.
In summary, we have shown here that Foxd3 is required to regulate glucose homeostasis, β-cell proliferation, and β-cell mass during pregnancy. We have identified genes implicated in cell proliferation that are misregulated in the absence of Foxd3, and these data suggest that identification of molecules, such as Foxd3, that control β-cell proliferation could be extended and coopted for therapeutic applications requiring precise control of in vivo or in vitro expansion of existing β-cells.
Supplementary Material
Acknowledgments
We thank Billy Carver and Dr. Brian Nelms, Dr. Yuping Yang, and Dr. Maureen Gannon for their careful reading of this manuscript; Dr. David Tuveson for the Pdx1-Cre transgenic mice; Dr. Maureen Gannon for contributing human PL; and Dr. Christopher Wright, Dr. Anna Means, and Dr. Marcela Brissova for scientific discussions about this project.
Islets were isolated by the Vanderbilt Isolation Procurement and Analysis Core supported by the Vanderbilt Diabetes Research and Training Center (Grant P60 DK020593); pancreatic sections used for β-cell mass were imaged with the assistance of the Vanderbilt Epithelial Biology Center Imaging Resource; RIA were completed by the Hormone Assay and Analytical Services Core, Mouse Metabolic Phenotyping Core (MMPC), Vanderbilt University (Grants P60 DK59637 and P60 DK020593); P.A.L. was supported by the Juvenile Diabetes Research Foundation Grant 1-2006-219, the National Institutes of Health Grant RO1 HD036720, a pilot and feasibility grant through the Vanderbilt Diabetes Research and Training Center (P60 DK020593), and the Vanderbilt University Medical Center Academic Program Support. M.A.M. is supported by Grant U19 DK042502. J.L.P. was supported by the Molecular Endocrinology Training Program Grant (T32 DK07563-10) and by the American Heart Association Predoctoral Fellowship 10PRE4500024.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BrdU
- 5-Bromo-2-deoxyuridine
- Cdkn
- cyclin-dependent kinase inhibitor
- ES
- embryonic stem
- Ezh2
- enhancer of zeste homolog 2
- Foxa2
- Forkhead box A2
- Foxd3
- Forkhead box D3
- Foxm1
- forkhead box M1
- GDM
- gestational diabetes mellitus
- IPGTT
- ip glucose tolerance testing
- Mafa
- v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A
- Nkx2-2
- Neurokinin 2 transcription factor related, locus 2
- Pdx1
- pancreatic and duodenal homeobox1
- PL
- placental lactogen
- qRT-PCR
- quantitative real-time PCR
- Skp2
- S-phase kinase-associated protein 2.
References
- 1. Parsons JA, Brelje TC, Sorenson RL. 1992. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 130:1459–1466 [DOI] [PubMed] [Google Scholar]
- 2. Bellamy L, Casas JP, Hingorani AD, Williams D. 2009. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet 373:1773–1779 [DOI] [PubMed] [Google Scholar]
- 3. Feig DS, Zinman B, Wang X, Hux JE. 2008. Risk of development of diabetes mellitus after diagnosis of gestational diabetes. Cmaj 179:229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lindsay RS. 2008. Commentary: type 2 diabetes and birth weight–genetic and environmental effects. Int J Epidemiol 37:192–193 [DOI] [PubMed] [Google Scholar]
- 5. Mathiesen ER, Vaz JA. 2008. Insulin treatment in diabetic pregnancy. Diabetes Metab Res Rev 24(Suppl 2):S3–S20 [DOI] [PubMed] [Google Scholar]
- 6. Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, Friesen HG, Sorenson RL. 1993. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 132:879–887 [DOI] [PubMed] [Google Scholar]
- 7. Dor Y, Brown J, Martinez OI, Melton DA. 2004. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46 [DOI] [PubMed] [Google Scholar]
- 8. Dor Y, Melton DA. 2008. Facultative endocrine progenitor cells in the adult pancreas. Cell 132:183–184 [DOI] [PubMed] [Google Scholar]
- 9. Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. 2007. Growth and regeneration of adult β cells does not involve specialized progenitors. Dev Cell 12:817–826 [DOI] [PubMed] [Google Scholar]
- 10. Freemark M, Avril I, Fleenor D, Driscoll P, Petro A, Opara E, Kendall W, Oden J, Bridges S, Binart N, Breant B, Kelly PA. 2002. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology 143:1378–1385 [DOI] [PubMed] [Google Scholar]
- 11. Vasavada RC, Garcia-Ocaña A, Zawalich WS, Sorenson RL, Dann P, Syed M, Ogren L, Talamantes F, Stewart AF. 2000. Targeted expression of placental lactogen in the β cells of transgenic mice results in β cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem 275:15399–15406 [DOI] [PubMed] [Google Scholar]
- 12. Sorenson RL, Brelje TC. 1997. Adaptation of islets of Langerhans to pregnancy: β-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Hormone Metab Res 29:301–307 [DOI] [PubMed] [Google Scholar]
- 13. Van Assche FA, Aerts L, De Prins F. 1978. A morphological study of the endocrine pancreas in human pregnancy. BJOG 85:818–820 [DOI] [PubMed] [Google Scholar]
- 14. Butler PC, Meier JJ, Butler AE, Bhushan A. 2007. The replication of β cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab 3:758–768 [DOI] [PubMed] [Google Scholar]
- 15. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. 2010. Adaptive changes in pancreatic β cell fractional area and β cell turnover in human pregnancy. Diabetologia 53:2167–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rieck S, Kaestner KH. 2010. Expansion of β-cell mass in response to pregnancy. Trends Endocrinol Metab 21:151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, Fontaine M, Yen MH, Kim SK. 2007. Menin controls growth of pancreatic β-cells in pregnant mice and promotes gestational diabetes mellitus. Science 318:806–809 [DOI] [PubMed] [Google Scholar]
- 18. Genevay M, Pontes H, Meda P. 2010. β-Cell adaptation in pregnancy: a major difference between humans and rodents? Diabetologia 53:2089–2092 [DOI] [PubMed] [Google Scholar]
- 19. Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, Dadon D, Granot Z, Ben-Hur V, White P, Girard CA, Karni R, Kaestner KH, Ashcroft FM, Magnuson MA, Saada A, Grimsby J, Glaser B, Dor Y. 2011. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab 13:440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamashita H, Shao J, Ishizuka T, Klepcyk PJ, Muhlenkamp P, Qiao L, Hoggard N, Friedman JE. 2001. Leptin administration prevents spontaneous gestational diabetes in heterozygous Lepr(db/+) mice: effects on placental leptin and fetal growth. Endocrinology 142:2888–2897 [DOI] [PubMed] [Google Scholar]
- 21. Gupta RK, Gao N, Gorski RK, White P, Hardy OT, Rafiq K, Brestelli JE, Chen G, Stoeckert CJ, Jr, Kaestner KH. 2007. Expansion of adult β-cell mass in response to increased metabolic demand is dependent on HNF-4α. Genes Dev 21:756–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang H, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, Gannon M. 2010. Gestational diabetes mellitus resulting from impaired β-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes 59:143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, Fujitani Y, Kawamori R, Miyatsuka T, Kosaka Y, Yang K, Honig G, van der Hart M, Kishimoto N, Wang J, Yagihashi S, Tecott LH, Watada H, German MS. 2010. Serotonin regulates pancreatic β cell mass during pregnancy. Nat Med 16:804–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dhawan S, Tschen SI, Bhushan A. 2009. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic β-cell proliferation. Genes Dev 23:906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. 2009. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 23:975–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nelms BL, Pfaltzgraff ER, Labosky PA. 2011. Functional interaction between Foxd3 and Pax3 in cardiac neural crest development. Genesis 49:10–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Labosky PA, Kaestner KH. 1998. The winged helix transcription factor Hfh2 is expressed in neural crest and spinal cord during mouse development. Mech Dev 76:185–190 [DOI] [PubMed] [Google Scholar]
- 28. Tompers DM, Foreman RK, Wang Q, Kumanova M, Labosky PA. 2005. Foxd3 is required in the trophoblast progenitor cell lineage of the mouse embryo. Dev Biol 285:126–137 [DOI] [PubMed] [Google Scholar]
- 29. Teng L, Mundell NA, Frist AY, Wang Q, Labosky PA. 2008. Requirement for Foxd3 in the maintenance of neural crest progenitors. Development 135:1615–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu J, Watts JA, Pope SD, Gadue P, Kamps M, Plath K, Zaret KS, Smale ST. 2009. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev 23:2824–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Plank JL, Mundell NA, Frist AY, LeGrone AW, Kim T, Musser MA, Walter TJ, Labosky PA. 2011. Influence and timing of arrival of murine neural crest on pancreatic β cell development and maturation. Dev Biol 349:321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perera HK, Caldwell ME, Hayes-Patterson D, Teng L, Peshavaria M, Jetton TL, Labosky PA. 2006. Expression and shifting subcellular localization of the transcription factor, Foxd3, in embryonic and adult pancreas. Gene Expr Patterns 6:971–977 [DOI] [PubMed] [Google Scholar]
- 33. White P, May CL, Lamounier RN, Brestelli JE, Kaestner KH. 2008. Defining pancreatic endocrine precursors and their descendants. Diabetes 57:654–668 [DOI] [PubMed] [Google Scholar]
- 34. Mundell NA, Labosky PA. 2011. Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development 138:641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Labosky PA. 2008. Regulation of embryonic stem cell self-renewal and pluripotency by Foxd3. Stem Cells 26:2475–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, Bernal-Mizrachi E, Elghazi L, Roe MW, Labosky PA, Myers MG, Jr, Gannon M, Powers AC, Dempsey PJ. 2010. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59:3090–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanna LA, Foreman RK, Tarasenko IA, Kessler DS, Labosky PA. 2002. Requirement for Foxd3 in maintaining pluripotent cells of the early mouse embryo. Genes Dev 16:2650–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4:437–450 [DOI] [PubMed] [Google Scholar]
- 39. Crawford LA, Guney MA, Oh YA, Deyoung RA, Valenzuela DM, Murphy AJ, Yancopoulos GD, Lyons KM, Brigstock DR, Economides A, Gannon M. 2009. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and β-cell proliferation during embryogenesis. Mol Endocrinol 23:324–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brissova M, Fowler M, Wiebe P, Shostak A, Shiota M, Radhika A, Lin PC, Gannon M, Powers AC. 2004. Intraislet endothelial cells contribute to revascularization of transplanted pancreatic islets. Diabetes 53:1318–1325 [DOI] [PubMed] [Google Scholar]
- 41. Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. 2011. Rictor/mTORC2 is essential for maintaining a balance between β-cell proliferation and cell size. Diabetes 60:827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tweedie E, Artner I, Crawford L, Poffenberger G, Thorens B, Stein R, Powers AC, Gannon M. 2006. Maintenance of hepatic nuclear factor 6 in postnatal islets impairs terminal differentiation and function of β-cells. Diabetes 55:3264–3270 [DOI] [PubMed] [Google Scholar]
- 43. Ackermann Misfeldt A, Costa RH, Gannon M. 2008. β-Cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes 57:3069–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xie T, Chen M, Weinstein LS. 2010. Pancreas-specific Gsα deficiency has divergent effects on pancreatic α- and β-cell proliferation. J Endocrinol 206:261–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morioka T, Asilmaz E, Hu J, Dishinger JF, Kurpad AJ, Elias CF, Li H, Elmquist JK, Kennedy RT, Kulkarni RN. 2007. Disruption of leptin receptor expression in the pancreas directly affects β cell growth and function in mice. J Clin Invest 117:2860–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Honig G, Liou A, Berger M, German MS, Tecott LH. 2010. Precise pattern of recombination in serotonergic and hypothalamic neurons in a Pdx1-cre transgenic mouse line. J Biomed Sci 17:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song J, Xu Y, Hu X, Choi B, Tong Q. 2010. Brain expression of Cre recombinase driven by pancreas-specific promoters. Genesis 48:628–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Steiner AB, Engleka MJ, Lu Q, Piwarzyk EC, Yaklichkin S, Lefebvre JL, Walters JW, Pineda-Salgado L, Labosky PA, Kessler DS. 2006. FoxD3 regulation of nodal in the Spemann organizer is essential for Xenopus dorsal mesoderm development. Development 133:4827–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nielsen JH, Galsgaard ED, Moldrup A, Friedrichsen BN, Billestrup N, Hansen JA, Lee YC, Carlsson C. 2001. Regulation of β-cell mass by hormones and growth factors. Diabetes 50(Suppl 1):S25–S29 [DOI] [PubMed] [Google Scholar]
- 50. Peshavaria M, Larmie BL, Lausier J, Satish B, Habibovic A, Roskens V, Larock K, Everill B, Leahy JL, Jetton TL. 2006. Regulation of pancreatic β-cell regeneration in the normoglycemic 60% partial-pancreatectomy mouse. Diabetes 55:3289–3298 [DOI] [PubMed] [Google Scholar]
- 51. Ackermann AM, Gannon M. 2007. Molecular regulation of pancreatic β-cell mass development, maintenance, and expansion. J Mol Endocrinol 38:193–206 [DOI] [PubMed] [Google Scholar]
- 52. Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. 1997. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 138:1736–1741 [DOI] [PubMed] [Google Scholar]
- 53. Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, Costa RH, Gannon M. 2006. The FoxM1 transcription factor is required to maintain pancreatic β-cell mass. Mol Endocrinol 20:1853–1866 [DOI] [PubMed] [Google Scholar]
- 54. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. 1999. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nat Genet 22:44–52 [DOI] [PubMed] [Google Scholar]
- 55. Georgia S, Bhushan A. 2004. β-Cell replication is the primary mechanism for maintaining postnatal β cell mass. J Clin Invest 114:963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Georgia S, Bhushan A. 2006. p27 Regulates the transition of β-cells from quiescence to proliferation. Diabetes 55:2950–2956 [DOI] [PubMed] [Google Scholar]
- 57. Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. 2005. Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Mol Cell Biol 25:3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salpeter SJ, Klein AM, Huangfu D, Grimsby J, Dor Y. 2010. Glucose and aging control the quiescence period that follows pancreatic β cell replication. Development 137:3205–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. 2005. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol 25:10875–10894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816 [DOI] [PubMed] [Google Scholar]
- 61. Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P, Costa RH. 2004. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev 18:830–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen WS, Peng XD, Wang Y, Xu PZ, Chen ML, Luo Y, Jeon SM, Coleman K, Haschek WM, Bass J, Philipson LH, Hay N. 2009. Leptin deficiency and β-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol Cell Biol 29:3151–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liber D, Domaschenz R, Holmqvist PH, Mazzarella L, Georgiou A, Leleu M, Fisher AG, Labosky PA, Dillon N. 2010. Epigenetic priming of a pre-B cell-specific enhancer through binding of Sox2 and Foxd3 at the ESC stage. Cell Stem Cell 7:114–126 [DOI] [PubMed] [Google Scholar]
- 64. Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. 2008. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev 22:3435–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tower AM, Trinward A, Lee K, Joseph L, Jacobson HI, Bennett JA, Andersen TT. 2009. AFPep, a novel drug for the prevention and treatment of breast cancer, does not disrupt the estrous cycle or fertility in rats. Oncol Rep 22:49–56 [DOI] [PubMed] [Google Scholar]
- 66. Orcheson LJ, Rickard SE, Seidl MM, Thompson LU. 1998. Flaxseed and its mammalian lignan precursor cause a lengthening or cessation of estrous cycling in rats. Cancer Lett 125:69–76 [DOI] [PubMed] [Google Scholar]
- 67. Kim HS, Shin JH, Moon HJ, Kim TS, Kang IH, Seok JH, Kim IY, Park KL, Han SY. 2002. Evaluation of the 20-day pubertal female assay in Sprague-Dawley rats treated with DES, tamoxifen, testosterone, and flutamide. Toxicol Sci 67:52–62 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.