

Abstract
Botulinum neurotoxins (BoNTs) are the most lethal of biological substances, and are categorized as class A biothreat agents by the Centers for Disease Control and Prevention. There are currently no drugs to treat the deadly flaccid paralysis resulting from BoNT intoxication. Among the seven BoNT serotypes, the development of therapeutics to counter BoNT/A is a priority (due to its long half-life in the neuronal cytosol and its ease of production). In this regard, the BoNT/A enzyme light chain (LC) component, a zinc metalloprotease responsible for the intracellular cleavage of synaptosomal-associated protein of 25 kDa, is a desirable target for developing post-BoNT/A intoxication rescue therapeutics. In an earlier study, we reported the high throughput screening of a library containing 70,000 compounds, and uncovered a novel class of benzimidazole acrylonitrile-based BoNT/A LC inhibitors. Herein, we present both structure-activity relationships and a proposed mechanism of action for this novel inhibitor chemotype.
Keywords: Botulinum neurotoxin serotype A, Benzimidazole acrylonitrile, Structure-activity relationships, Molecular modeling, Time-dependent inhibition
1. Introduction
Botulinum neurotoxins (BoNTs) are among the most potent of known biological toxins1, 2, and are listed as category A biothreat agents by the Centers for Disease Control and Prevention. BoNTs are easily produced and may be delivered via an aerosol route2–4. Consequently, these toxins represent a serious threat to both military personnel and civilians5–7. Moreover, BoNTs are now widely used during both superficial cosmetic treatments and to ameliorate a range of physical ailments3, 8–15, making their attainment, misuse, and/or adverse side effects16 more likely. Importantly, the currently available BoNT toxoid vaccine, as well as experimental preventative antibodies, cannot counter postneuronal internalization of these toxins. This is a seminal point, as it is likely that BoNT-poisoned individuals will seek medical attention only after clinical symptoms of intoxication manifest (e.g., life-threatening paralysis). Currently, critical care mechanical ventilation is the only treatment option once neurons have been intoxicated and diaphragm muscles cease to function. However, long-term mechanical ventilation would be impractical for treating large numbers of intoxicated individuals. Therefore, there is an urgent need to identify and develop low molecular weight, non-peptidic BoNT inhibitors that will serve as both prophylactics and post-exposure ‘rescue’ therapeutics.
Of the seven BoNT serotypes (A–G), A, B, and E are known to cause botulism in humans1, 17, with the BoNT/A/LC metalloprotease exhibiting the longest duration of activity in the neuronal cytosol18–20. Hence, the vast majority of research to develop inhibitors to counter BoNT intoxication postneuronal internalization has focused on this LC serotype. Structurally, BoNT/A is composed of a 100 kDa heavy chain (HC) and a 50 kDa light chain (LC), which are joined by a disulfide bridge (until LC release upon neuronal internalization)21, 22. The LC component is a zinc-dependent endopeptidase.
Botulinum neurotoxin neuronal intoxication involves a stepwise process of cell surface binding, receptor-mediated endocytosis, pH-induced translocation, and cytosolic metalloendoprotease activity22. The HC serves as the toxin delivery system, as it binds specific neuronal surface receptors and translocates the LC into the cell cytosol via an endosomal mechanism. Once inside the cytosol, the BoNT/A/LC cleaves a 25 kDa synaptosomal-associated protein (SNAP-25)23, a component of the SNARE protein complex24, 25, which is responsible for transporting acetylcholine into neuromuscular junctions. The BoNT/A LC-mediated cleavage of SNAP-25 inhibits this neuronal function, to produce flaccid paralysis that can potentially lead to respiratory failure and death.
As indicated above, the identification and development of BoNT/A LC inhibitors is a high priority26. To this end, a substrate-to-inhibitor strategy was used to generate a potent peptide derivative, namely, 2-mercapto-3-phenylpropionyl-RATKML, which inhibits the BoNT/A LC with a Ki value of 330 nM27–30. Subsequently, a similar strategy was employed by Sukonpan et al.31 to develop 17-mer BoNT/A LC peptide inhibitors, the most active of which was an α-mercapto amide derivative possessing a Ki value of 300 nM31. Moreover, several high throughput screening (HTS) campaigns to identify small molecule inhibitors of the BoNT/A LC have been conducted, and various chemotypes have been identified. Burnett et al.4, 32, 33 identified a non-Zn(II) coordinating BoNT/A LC indole-based bis-amidine inhibitor (NSC 240898) via a combination of HTS and subsequent pharmacophore-based database mining. The chemical optimization of NSC 240898 (1, Figure 1) has produced a generation of inhibitors represented by compounds 2 and 3 (Figure 1), both of which are more potent than 1, which are enzyme specific and provide SNAP-25 protection in a chick neuronal assay34, 35,36. Additionally, Burnett and co-workers have employed their pharmacophore-based approach to discover and/or design several other, chemically diverse, non-Zn(II)-coordinating small molecule BoNT/A LC inhibitors possessing low µM to sub-µM Ki values37–40. Park et al.41 have reported the identification of a novel class of Zn(II)-coordinating thiophene ketone-based BoNT/A LC inhibitors (Ki ≥12 µM) employing computer-aided drug design, and have used synthetic modifications to improve the potencies (<1 µM) of these molecules42, 43. Boldt et al.44 have reported the synthesis of a Zn(II)-coordinating 2,4-dichlorocinnamic hydroxamic acid BoNT/A LC inhibitor that is reported to possess sub-µM in vitro activity, but possesses very minimal activity in vivo45. Furthermore, a series of benzylidene cyclopentenedione-based inhibitors have been reported to irreversibly inactivate BoNT/A LC in a biochemical assay by purportedly forming a covalent bond with the enzyme46. Finally, the HTS of a natural products library has led to the discovery that lomofungin is a noncompetitive inhibitor of BoNT/A LC (Ki = 6.7 ± 0.7 µM)47.
Figure 1.

Structures of BoNT/A LC inhibitors.
To identify novel small molecule inhibitors of BoNT/A LC, we screened a library of over 70,000 compounds using a fluorescence resonance energy transfer (FRET) assay to detect the inhibition of substrate proteolysis. Structurally novel benzimidazole acrylonitrile hit compound 4 (Figure 1) inhibited BoNT/A LC with low µM-range potency (IC50 = 7.2 µM). Modification of the benzimidazole acrylonitrile scaffold represented by compound 4 led to the generation of compound 5 (Figure 1), which displayed improved specificity for BoNT/A LC with respect to other metalloproteases, and provided good SNAP-25 protection in a neuronal assay for BoNT/A intoxication.48 Herein, structure-activity relationships, a proposed mechanism of action, and molecular modeling studies for the benzimidazole acrylonitrile series of BoNT/A LC inhibitors are reported.
2. Results and Discussion
2.1. Chemistry
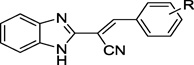
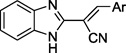
To develop structure-activity relationships (SARs) and potentially improve the potency and specificity of HTS hit compound 4, the structural modifications illustrated in Figure 2 were performed. Specifically, 1) substituents on the phenyl ring of 4 were modified (see Table 1 for the structures of the modifications) and 2) the phenyl ring of 4 was replaced with a variety of aromatic heterocycles (see Table 2 for the structures of the modifications). Of the derivatives, bisthiophene 5 was the most potent compound in vitro. Consequently, this derivative was subjected to further rounds of chemical modification to examine the effects of substituent and core scaffold changes on both in vitro potency and specificity (Figure 3) (see Table 4 for the structures of the modifications).
Figure 2.

Modifications of the hit structure 4.
Table 1.
Inhibitory activities of benzimidazole acrylonitriles 4, 8a–8k against BoNT/A LC and LF enzymes.
 |
|||
|---|---|---|---|
| Compound | R |
aBoNT/A LC IC50 (µM) |
bAnthrax LF IC50 (µM) |
| 4 | 3-I, 4-OMe | 7.2/10c | 74 |
| 8a | 3-I | >100 | >100 |
| 8b | 3-H, 4-OMe | >100 | >100 |
| 8c | 3-Br, 4-OMe | >100 | >100 |
| 8d | 3-Cl, 4-OMe | >100 | >100 |
| 8e | 3-F, 4-OMe | >100 | >100 |
| 8f | 3,4-di-OMe | >100 | >100 |
| 8g | 3-Cl, 4-OH, 5-OMe | 59 | >100 |
| 8h | 3-Br, 4-OH, 5-OMe | 94 | >100 |
| 8i | 4-Ph | 86 | >100 |
| 8j | 4-Imidazole | 73 | >100 |
| 8k | 3,4,5-tri-OMe | >100 | >100 |
Results obtained by average of two experiments in a FRET assay;
results obtained by FRET assay;
results obtained by HPLC assay.
Table 2.
Inhibitory activities of benzimidazole acrylonitriles 10a–f against BoNT/A LC and LF enzymes.
 |
|||
|---|---|---|---|
| Compound | Ar |
aBoNT/A LC IC50 (µM) |
bAnthrax LF IC50 (µM) |
| 10a |  |
>100 | >100 |
| 10b |  |
>100 | >100 |
| 10c | 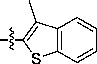 |
>100 | >100 |
| 10d | 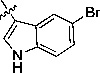 |
>100 | 60% inh.@100µM |
| 10e |  |
>100 | >100 |
| 10f | 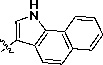 |
>100 | >100 |
| 5 | 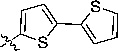 |
26 (29c) | >100 |
Results obtained by average of two experiments in a FRET assay;
results obtained by FRET assay;
results obtained by HPLC assay.
Figure 3.

Modification of compound 5.
Table 4.
Inhibitory activities of compounds 12a–e, 14a–d, and 15 against BoNT/A LC and LF enzymes.
| Compound | Ar |
aBoNT/A LC IC50 (µM) |
bLF IC50 (µM) |
|---|---|---|---|
| 12a | 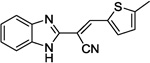 |
>100 | >100 |
| 12b | 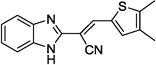 |
>100 | >100 |
| 12c |  |
>100 | >100 |
| 12d | 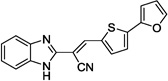 |
59 | >100 |
| 12e | 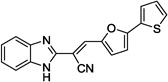 |
>100 | >100 |
| 14a | 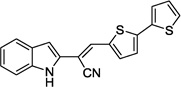 |
>100 | >100 |
| 14b |  |
>100 | >100 |
| 14c | 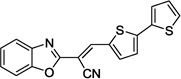 |
>100 | N.D. |
| 14d | 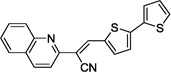 |
>100 | >100 |
| 15 |  |
>100 | N.D. |
Benzimidazole acrylonitrile compounds 8a, 8c–k, 5 and 10a–f were prepared by condensation of commercially available benzimidazole acetonitrile 6 with a variety of substituted aldehydes (Scheme 1). Specifically, to evaluate the impact of substituents with different chemical and steric properties, the phenyl ring of 4 was substituted with a variety of different substituents (see Table 1 for the structures of the modifications), as well as heteroaromatic rings (see Table 2 for the structures of the modifications).
Scheme 1.

Syntheses of benzimidazole acrylonitriles 8a–k, 10a–f, 5, and 12a–e. a Reagents and conditions: (a) NaOAc, HOAc, reflux, 2h.
To develop the SAR for bis-thiophene compound 5, acrylonitrile derivatives 12a–e and 14a–d were synthesized (Schemes 1 and 2, respectively) via acid-catalyzed condensation of aryl acetonitriles 6 and 13 with a variety of substituted aldehydes (e.g., 11 and 9g in Schemes 1 and 2, respectively) (see Table 4 for the structures of the modifications). Compound 15 was prepared by dimethylation of the benzimidazole ring of compound 5 using methyl iodide (Scheme 3) (see Table 4 for the structure of the modification).
Scheme 2.

Syntheses of 14a–d. a Reagents and conditions: (a) NaOAc, HOAc, reflux, 2h.
Scheme 3.

Synthesis of 15. a Reagents and conditions: (a) CH3I, DMF, 2h.
2.2. Biological evaluation
Synthesized derivatives of benzimidazole acrylonitrile 4 were evaluated in a fluorescence resonance energy transfer (FRET)-based recombinant BoNT/A LC assay for inhibitory potency,49 and counter screened in an anthrax Lethal Factor (LF) assay to provide preliminary indications of selectivity49, 50. Of the synthesized analogs, five provided BoNT/A LC inhibition (Tables 1 and 2). Importantly, no appreciable activity was observed when the derivatives were examined against LF (Tables 1 and 2).
All substituent modifications of structure 4 were detrimental to BoNT/A LC inhibitory potency. For example, removing the 4-OMe group (8a), removing the 3-iodo group (8b), or replacing it with smaller, and more electronegative halogen atoms (8c–e) eliminated inhibitory potency. Moreover, exchanging the 3-iodo substituent for a 3-OMe substituent (8f) also eliminated inhibitory potency, while tri-substitutions on the phenyl ring (8g, 8h, 8k) significantly decreased or diminished activity (e.g., with respect to 4). Two compounds with 5- or 6-membered aromatic rings appended to the 4-position of the phenyl group (8i, 8j) exhibited anti-BoNT/A LC activity, but also with significantly lower potency with respect to 4. Since modification of the substituents on the phenyl ring failed to improve inhibitory potency, we next examined replacement of the substituted phenyl ring with various aromatic heterocycles including pyridine (10a), pyrimidine (10b), benzothiophene (10c), indoles (10d–e), a fused tricyclic ring (10f) and bis-thiophene 5. Inhibition results for these derivatives are shown in Table 2. Only bis-thiophene 5 exhibited significant inhibitory activity against the BoNT/A LC (IC50 = 26 µM) in the FRET-based assay, which was confirmed in a secondary HPLC-based assay (IC50 = 29 µM) (Table 2).
Compounds 4 and 5 were subjected to advanced in vitro characterization to determine: 1) enzyme specificity (e.g., in addition to LF inhibition); 2) the possibility of Zn chelation; 3) cellular efficacy; and 4) potential thiol-inactivation (Table 3). With regard to specificity, neither 4 nor 5 inhibited the BoNT serotype B LC.48 And while compound 4 was found to modestly inhibit anthrax LF (IC50 = 74 µM), compound 5 did not inhibit this enzyme up to concentrations of 100 µM. Additionally, neither compound inhibited human MMP-1, MMP-2 or MMP-9. Overall, the results from the specificity assays clearly demonstrate that compounds 4 and 5 are highly specific for BoNT/A LC.
Table 3.
In vitro characterization of compounds 4 and 5.
| Compound 4 | Compound 5 | |
|---|---|---|
| Assay | IC50 (µM) or %inhibition |
IC50 (µM) or %inhibition |
| BoNT/A LC FRET | 7.2 | 26 |
| BoNT/A LC HPLC | 10 | 29 |
| BoNT/B LC FRET | >100 | >100 |
| Anthrax LF | 74 | >100 |
| MMP-1 | >100 | >100 |
| MMP-2 | >100 | >100 |
| MMP-9 | >100 | >100 |
| % Inhibition@30µM chick neuronal assay | <10% Inhibition | 59% Inhibition |
| Inactivated by zinc chelation | No | No |
| Inactivated by glutathione | Yes | No |
| Inactivated by cysteine | Yes | No |
Inactivation studies were performed by pre-incubating the potential inactivator (at 2.5 or 5 mM) with compound in the assay mixture for 15 minutes at 37 °C and then adding the 17-mer SNAP-25 substrate and BoNT/A LC.
Examination of the BoNT/A LC inhibitory potencies of 4 and 5 in the presence of 50 uM Zn indicated that neither are metal chelating agents. Neither of these inhibitors displayed a change in inhibitory potency when excess Zn was included in the FRET-based assay (Table 3). Interestingly, compound 4 was inactivated by thiol-containing glutathione and cysteine, while compound 5 showed no significant, non-specific reactivity towards these thiol-containing compounds (Table 3), suggesting that the electrophilicity of the β-carbon of benzimidazole acryloitrile 5 has been significantly reduced with respect to compound 4, so that the free sulfhydryl group of glutathione or cysteine is not able to undergo Michael addition to this β-carbon atom.
In chick neuron culture, compound 5 inhibited BoNT/A-mediated SNAP-25 cleavage by 58% at 30 uM concentration, while compound 4 provided approximately 10% inhibition of SNAP-25 cleavage at 30 µM.48 During the assay, compound 1 (NSC 240898), a previously reported indole-based BoNT/A LC inhibitor, was used as a positive control, and exhibited a 42% inhibitory activity against BoNT/A-mediated SNAP-25 cleavage at 30 µM.
Based on the high degree of specificity and the SNAP-25 protection provided by 5, closely related analogs were prepared (see Table 4 for the structures, and Figure 3 for the design strategy). Accordingly, compounds 12a–e, 14a–d and 15 were synthesized and their inhibitory efficacies against BoNT/A LC, as well as anthrax LF (for preliminary selectivity evaluation) were calculated (Table 4). Compounds containing only one thiophene ring, with various substituents, such as 5-Me (12a), 4,5-di-Me (12b), or a simple unsubstituted thiophene (12c) provided no BoNT/A LC inhibition, while compound 12d, which possesses a furan ring substituted on the 5-position of the central thiophene, was approximately two-fold less potent than 5 (Table 5). Interestingly, no activity was observed for compound 12e, which possesses a furan in place of the central thiophene ring in 5. The bis-thiophene moiety connected directly to the acrylonitrile component is, therefore, highly preferred for anti-BoNT/A LC potency, and the data clearly indicate that even subtle changes to the central thiophene ring have a profoundly negative impact on BoNT/A LC inhibitory potency.
Table 5.
Enzyme/Inhibitor “Rescue” studies.
| Experimental Condition | Activity Remaining (Fraction of Dialyzed Control) |
|---|---|
| 50µM 5 | 0.55 |
| 50µM 4 | 0.17 |
| 50µM 2 | 1.25 |
| 50µM 5/50µM 2 | 1.13 |
| 50µM 4/50µM 2 | 0.99 |
We next kept the bis-thiophene motif intact, and investigated the impact of modification/replacement of the benzimidazole component (Table 4). In particular, replacement of the benzimidazole ring with alternative heterocycles, such as indole (14a), benzothiazole (14b), benzoxazole (14c) and quinoline (14d) eliminated inhibitory potency. Moreover, dimethylation of the benzimidazole component (to provide 15) was also detrimental to potency. Thus, it appears that the benzimidazole core is necessary for BoNT/A LC inhibition with this chemotype.
2.3. Mechanism of action
Benzimidazole acrylonitriles 4 and 5 demonstrated noncompetitive kinetics with respect to the substrate (17-mer SNAPtide), which suggested that the inhibitors are not binding in the BoNT/A LC active site.48 Therefore, enzyme/inhibitor “rescue studies” were performed to further characterize the mechanism of action of 4 and 5 (Table 5). Inactivation was detected when BoNT/A LC was incubated with the benzimidazole acrylonitrile inhibitors and subsequently subjected to dialysis. Specifically, we found that the inhibitors were bound to the protein fraction, and could only be partially recovered. However, this inactivation was prevented when the BoNT/A LC was simultaneously incubated in the presence of a benzimidazole acrylonitrile inhibitor and known competitive inhibitor 2 (and then subjected to dialysis); benzimidazole acrylonitrile could be recovered during this dialysis experiment. Hence, this experiment indicated that even though benzimidazole acrylonitriles are not active site inhibitors, they may bind to a site close to the active site, and that the active site inhibitor may interfere with the binding of the benzimidazole acrylonitrile inhibitors by partially blocking their enzyme interaction site. Further analysis of the mechanism of action has revealed that compounds 4 and 5 inhibit BoNT/A LC in a time-dependent manner: when compound 5 was pre-incubated with enzyme for 90 minutes (assay time was 40 minutes), the IC50 value decreased from 26 µM (no preincubation; FRET assay) to 17 µM (FRET assay), suggesting that this compound is a time-dependent inhibitor. The specificity constant kinact/Ki values for compounds 4 and 5 were measured as 979 and 326 M−1s−1, respectively (Figures 4 and 5)46, 51. The time-dependent inactivation of BoNT/A LC by 4 and 5 suggests that covalent modification of the target enzyme may occur in their mechanism of action.
Figure 4.

Inactivation of BoNT/A LC with compound 4.
Figure 5.

Inactivation of BoNT/A LC with compound 5.
2.4. Molecular modeling
To identify a nucleophilic amino acid that could be responsible for the generation of a covalent adduct, we first examined the amino acid sequence of BoNT/A LC (425 AAs, PDB reference code 3BOK_A) and identified two cysteine residues located at positions 134 and 165 (Figure 6). Cysteine is the most intrinsically nucleophilic amino acid residue in proteins, and in addition to its role in catalysis, it is subject to posttranslational chemical modifications that are related to its nucleophilicity52. Examination of the crystal structure (PDB reference code: 2G7N) of BoNT/A LC indicates that positively charged Arg231 is located within 5 Å of Cys165, while Cys165 is negatively charged and coordinated with two silver cations in the crystal structure. No positively charged amino acid residues were observed within 5 Å of Cys134. Cys165 is located approximately 8 Å from the enzyme catalytic Zn2+ ion, while Cys134 is located on a β-sheet structure that is in the bottom bay area opposite to the entrance to the β-exosite53. Consequently, we hypothesized that benzimidazole acrylonitrile inhibitors could inactivate BoNT/A LC by forming a covalent bond with one of these two cysteine residues.
Figure 6.

Amino acid sequence of BoNT/A LC (amino acids 1–425).
Mechanistically, two steps are generally proposed for covalent bond formation. Initially, the inhibitor binds to the target enzyme to form a non-covalent Michaelis complex (EI*), which then proceeds through a second step to yield the covalent adduct (EI)54.With this knowledge, a computational modeling study was performed to identify which cysteine residue might be involved in the covalent modification process. Compound 5 was first non-covalently docked into sites in the proximity of both Cys134 and Cys165, and it displayed greater binding affinity for the Cys165 site than for the Cys134 site. Moreover, when compound 5 was docked non-covalently near the Cys165 residue (Figure 7), thereby mimicking the Michaelis complex (EI*), its electrophilic double bond was in a favorable orientation that would accommodate nucleophilic attack by the Cys165 sulfhydryl group. The S–Cβ distance is about 4.3 Å, which renders the nucleophilic Michael addition reaction plausible, without the need to overcome a high energy barrier. Interestingly, the guanidine group of Arg177 forms hydrogen bonds with one of the benzimidazole nitrogen atoms and the nitrile group of 5, which further stabilizes the noncovalent Michaelis complex. Conversely, in the best docking mode for compound 5 in the Cys134 site, the S–Cβ distance is about 9.1 Å, and the geometry of compound 5 is not favorable for covalent bond formation (Figure 8). We have demonstrated earlier that compound 5 does not react with free cysteine due to the reduced electrophilicity of the β-carbon. However, the pKa value of most protein cysteine thiols is between 8–9,55 while the Cys165 residue exists as a thiolate anion at neutral pH and is in close proximity to the positively charged Arg231 residue. The lower pKa value of the Cys165 allows it to be sufficiently nucleophilic to interact with compound 5 to form a covalent bond. The proximity of the nucleophile to the electrophile in the bound structure offers an entropic advantage to subsequent covalent bond formation. Taking this information into account, we propose that Cys165 is the preferred nucleophilic amino acid for covalent modification of compound 5, and that a Michael addition occurs between the sulfhydryl group of Cys165 and the acrylonitrile functionality of compound 5 to produce persistent inactivation of BoNT/A LC (Figure 9). We have termed the Cys165 site as the ϒ-exosite, to supplement the known substrate SNAP-25 α- and β-exosites53.
Figure 7.

Compound 5 non-covalently bound near the C165 site (ϒ-exosite).
Figure 8.

Molecular model of compound 5 in the Cys134 site of BoNT/A LC.
Figure 9.

Compound 5 is proposed to inactivate BoNT/A LC by forming a covalent bond with Cys165.
In support of our hypothesis, the inactivation of other protein targets via the covalent binding of an inhibitor to a specific, noncatalytic cysteine residue has been demonstrated. Examples include several kinase inhibitors56 and HCV protease inhibitors57. Interestingly, we noticed that nucleophilic cysteine residues involved in the covalent enzyme inactivation of other enzymes are generally located near their active sites or substrate binding sites. For example, a noncatalytic Cys797 is near the ATP binding site in Epidermal Growth Factor Receptor (EGFR) kinase58, and a Cys159 is close to the substrate binding site of HCV NS3/4a protease57. Both of these sites have been used to develop inhibitors that form a covalent bond with their respective target enzymes56, 57. Currently, we are unable to generate X-ray co-crystal structures of compound 5 with BoNT/A LC due to the low aqueous solubility of the compound, and a preliminary liquid chromatography-mass spectrometry (LC-MS) experiment was performed in an attempt to detect a covalent adduct, which was also unsuccessful.
With regard to compound 5, it is also important to note that the nucleophilic Michael addition occurring between the Cys165 sulfhydryl group and the acrylonitrile double bond generates two new chiral centers in the inhibitor structure. Consequently, four covalently-bound diastereomers are possible. Therefore, we modeled all four possible diastereomeric covalent adducts, and of the four, the covalent (2S, 3R)-5:BoNT/A LC adduct demonstrated the lowest binding energy compared to the other three adducts. The modeled structure of the (2S, 3R)-5:BoNT/A LC adduct is shown in Figure 10. The orientation of the inhibitor in this structure is similar to its orientation in the non-covalent Michaelis-like complex (Figure 7). The inhibitor benzimidazole ring is positioned near the catalytic site, and the bis-thiophene moiety is bound along the β-sheet structure containing Cys165. Interestingly, the nitrile group maintains its hydrogen bonding interaction with Arg177 (compare Figure 7 and Figure 10), which further enhances binding affinity. Finally, it is also important to note that, even though the actual function of Cys 165 is unknown, covalent modification of Cys 165 would prevent the BoNT/LC mediated SNAP-25 cleavage.
Figure 10.

Compound 5 covalently bound at the Cys165 site (ϒ-exosite).
3. Conclusion
A novel benzimidazole acrylonitrile BoNT/A LC inhibitor chemotype was identified during the HTS of a library containing 70,000 small molecules. Modification of hit compound 4 led to the generation of compound 5, which displays a high degree of specificity for BoNT/A LC with respect to other metalloproteases, and demonstrates SNAP-25 protection in neurons during BoNT/A challenge. Structure-activity relationship studies were performed to define the chemical features of compound 5 that are important for BoNT/A LC inhibitory potency. In summary, we have discovered a highly specific, time-dependent inhibitor that inactivates BoNT/A LC through the proposed covalent modification of a non-catalytic cysteine residue in a previously unidentified substrate binding site. Our ultimate goal for this class of inhibitor is to examine representative compounds in in vivo models of BoNT/A intoxication.
4. Experimental section
4.1 General procedures
All commercially obtained solvents and reagents were used as received. Melting points were determined in open capillary tubes with an EZ-Melt (Stanford Research Systems) apparatus and are uncorrected. 1H NMR spectra were determined on a Bruker 300 MHz instrument. Chemical shifts are given in δ values referenced to the internal standard tetramethylsilane. LC-MS analyses were performed by CreaGen Biosciences, Inc. (Woburn, MA) using a Shimadzu LC-10 AD VP HPLC and a Waters micromass quattro ultima triple-quad MS. All of the compounds tested in vitro possessed >95% purity based on LC-MS analyses. Only one regioisomer was observed for the benzimidazole acrylonitrile compounds based on the 1H NMR and LC-MS.
4.2. The general synthesis of acrylonitrile compounds
Aryl acetonitrile (3.2 mmol, 1.0 eq.), aldehyde (3.2 mmol, 1.0 eq.), and ammonium acetate (9.5 mmol, eq.) were mixed in glacial acetic acid (10 mL) and heated at reflux for 2 h. Following, the reaction mixture was cooled to room temperature. The precipitate that formed was collected by filtration, washed with water, followed by a small volume of methanol, and dried in a vacuum oven at 50 °C for 18 h.
2-(1-Cyano-2-phenylvinyl)benzimidazole (8a)
Light brown powder, Rf = 0.35 (1:4 EtOAc/hexane), mp = 259–260 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.09 (s, 1H), 8.34 (s, 1H), 8.28 (s, 1H), 7.96 (dd, J = 8.1, 12 Hz, 2H), 7.71 (d, J = 7.2 Hz, 1H), 7.58 (d, J = 5.7 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.3–7.24 (m, 2H). LC-MS (+ESI): m/z 372.07 (M+1)+.
2-(1-Cyano-2-phenylvinyl)benzimidazole (8b)
Yellow powder, Rf = 0.16 (1:4 EtOAc/hexane), mp = 223–224 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.98 (br, s, 1H), 8.28 (s, 1H), 8.01 (d, J = 8.1, 2H), 7.64 (br, s, 1H), 7.59 (br, s, 1H), 7.25 (d, J = 7.8 Hz, 1H), 7.17 (d, J = 8.1 Hz, 2H), 3.87 (s, 3H). LC-MS (+ESI): m/z 276.2 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(4-methoxy-3-bromophenyl)acrylonitrile (8c)
Yellow solid, Rf = 0.65 (1:1 EtOAc/hexane), mp = 253–254 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.99 (br, s, 1H), 8.28 (d, J = 2.4 Hz, 1H), 8.26 (s, 1H), 8.00 (dd, J = 2.1, 8.9 Hz, 1H), 7.66 (br, s, 1H), 7.58 (br, s, 1H), 7.35 (d, J = 8.7 Hz ,1H), 7.26 (br, d, J = 4.8 Hz, 2H), 3.97 (s, 3H). LC-MS (+ESI): m/z 354.1 (M+1)+.
(1H-Benzo[d]imidazol-2-yl)-3-(3-chloro-4-methoxyphenyl)acrylonitrile (8d)
Pale yellow solid, Rf = 0.71 (1:1 EtOAc/hexane), mp = 250–251 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.0 (s, 1H), 8.26 (s, 1H), 8.12(d, J = 2.4 Hz, 1H), 7.96 (dd, J = 2.1, 8.7 Hz, 1H), 7.69 (d, J = 7.2 Hz, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.38 (d, J = 8.7 Hz ,1H), 7.31-7.21 (m, 2H), 3.98 (s, 3H). LC-MS (+ESI): m/z 310.17 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(3-fluoro-4-methoxyphenyl)acrylonitrile (8e)
Light yellow solid, Rf = 0.50 (1:1 EtOAc/hexane), mp = 271–272 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.01 (s, 1H), 8.27 (s, 1H), 7.92 (dd, J = 2.1, 15 Hz, 1H), 7.81 (d, J = 8.4 Hz,1H), 7.62 (s, br, 2H), 7.39 (t, J = 8.7 Hz, 1H), 7.26 (dd, J = 3, 6 Hz, 2H), 3.96 (s, 3H). LC-MS (+ESI): m/z 294.17 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(3,4-dimethoxyphenyl)acrylonitrile (8f)
Yellow solid, Rf = 0.54 (1:1 EtOAc/hexane), mp = 184–185 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.98 (br, s, 1H), 8.27 (s, 1H), 7.74 (d, J = 2.1 Hz, 1H), 7.62 (br, s, 1H), 7.58 (dd, J = 2.1, 6.0 Hz, 2H), 7.62–7.23 (m, 2H), 7.19 (d, J = 8.7 Hz ,1H), 3.88 (s, 3H), 3.86 (s, 3H). LC-MS (+ESI): m/z 306.23 (M+1)+.
2-[1-Cyano-2-(3-chloro-5-methoxy-4-hydroxyphenyl)vinyl]benzimidazole (8g)
Light orange solid, Rf = 0.55 (80:18:2 CHCl3/CH3OH/CH3NH2), mp = 286–287 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.99 (br, s, 1H), 10.65 (br, s, 1H), 8.22 (s, 1H), 7.67 (s, 3H), 7.57 (br, s, 1H), 7.26 (d, J = 5.1 Hz, 2H), 3.93 (s, 3H). LC-MS (+ESI): m/z 326.18 (M+1)+.
2-[1-Cyano-2-(3-bromo-5-methoxy-4-hydroxyphenyl)vinyl]benzimidazole (8h)
Light orange solid, Rf = 0.61 (80:18:2 CHCl3/CH3OH/CH3NH2), mp = 268–269 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.98 (br, s, 1H), 10.69 (br, s, 1H), 8.23 (s, 1H), 7.796 (d, J = 2.1 Hz ,1H), 7.706 (d, J = 1.8 Hz ,1H), 7.67 (br, s, 1H), 7.57 (br, s, 1H), 7.3–7.2 (m, 2H), 3.93 (s, 3H). LC-MS (+ESI): m/z 372.16 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(biphenyl-4-yl)acrylonitrile (8i)
Yellow solid, Rf = 0.33 (1:4 EtOAc/hexane), mp = 267–268 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.11 (s, 1H), 8.39 (s, 1H), 8.11 (d, J = 8.4 Hz, 2H), 7.94 (d, J = 8.4 Hz, 2H), 7.80 (dd, J = 1.5, 7.7 Hz, 2H), 7.73–7.69 (m, 1H), 7.59–7.42 (m, 4H), 7.33–7.23 (m, 2H). LC-MS (+ESI): m/z 322.29 (M+1)+.
3-(4-(1H-Imidazol-1-yl)phenyl)-2-(1H-benzo[d]imidazol-2-yl)acrylonitrile (8j)
Light yellow solid. Rf = 0.67 (1:1 EtOAc/hexane), mp = 267–268 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.06 (br, s, 1H), 8.46 (s, 1H), 8.39 (s, 1H), 8.14 (d, J = 8.7 Hz, 2H), 7.95 (s, 1H), 7.92–7.91 (m, 2H), 7.65 (br, s, 2H), 7.29–7.26 (m, 2H), 7.18 (s, 1H). LC-MS (+ESI): m/z 312.26 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(3-hydroxy-4,5-dimethoxyphenyl)acrylonitrile (8k)
Yellow powder, Rf = 0.81 (80:18:2 CHCl3/CH3OH/CH3NH2), mp = 260–261 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.98 (br s, 1H), 9.79 (br s, 1H), 8.19 (s, 1H), 7.63 (br s, 2H), 7.28-7.25 (m, 2H), 7.22 (d, 2H), 3.86 (s, 3H), 3.79 (s, 3H). LCMS (+ESI): m/z 322.18 (M+1)+.
2-[1-Cyano-2-(2-chloro-5-phenylpyridin-3-yl)vinyl]-benzimidazole (10a)
Bright yellow solid, Rf = 0.82 (1:1 EtOAc/hexane), mp = 270–271 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.43 (br, 1H), 8.88 (dd, J = 2.4, 12.6 Hz, 2H), 8.49 (s, 1H), 7.83 (d, J = 7.2 Hz, 2H), 7.68 (br, 2H), 7.61–7.49 (m, 3H). LC-MS (+ESI): m/z 355.2 (M–1)−.
2-[1-Cyano-2-(2,4-dichloropyrimidin-3-yl)vinyl]-benzimidazole (10b)
Light orange solid, Rf = 0.01 (1:1 EtOAc/hexane), mp >300 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.42 (br, 1H), 8.85 (d, J = 8.1 Hz ,1H), 8.69 (s, 1H), 8.56 (s, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.62 (t, J = 7.2 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H).
2-(1H-Benzo[d]imidazol-2-yl)-3-(3-methylbenzo[b]thiophen-2-yl)acrylonitrile (10c)
Light orange solid, Rf = 0.78 (1:1 EtOAc/hexane), mp = 267–268 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.19 (br, 1H), 8.64 (s, 1H), 8.11 (dd, J = 1.2, 6.7 Hz, 1H), 7.99 (dd, J = 2.1, 6.9 Hz, 1H), 7.71 (br, 1H), 7.59–7.49 (m, 3H), 7.30–7.25 (m, 2H), 2.74 (s, 3H). LC-MS (+ESI): m/z 316.48 (M+1)+.
1H-Benzo[d]imidazol-2-yl)-3-(5-bromo-1H-indol-3-yl)acrylonitrile (10d)
Bright yellow solid, Rf = 0.61 (1:1 EtOAc/hexane), mp >300 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.86 (br, s 1H), 12.42 (br, s 1H), 8.59 (s, 1H), 8.52 (s, 1H), 8.21 (s, 1H), 7.66 (br, s, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.42 (dd, J = 1.8, 8.7 Hz, 1H), 7.24 (br, s, 2H). LC-MS (+ESI): m/z 363.14 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(4-nitro-1H-indol-3-yl)acrylonitrile (10e)
Orange yellow solid, Rf = 0.20 (1:1 EtOAc/hexane), mp >300 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.93 (br, s, 2H), 8.69 (d, J = 8.1 Hz, 2H), 8.05 (dd, J = 8.1, 12.3 Hz, 2H), 7.64 (br, s, 2H), 7.45 (t, J = 15.9 Hz, 1H), 7.28–7.25 (m, 2H). LC-MS (+ESI): m/z 330.2 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(1H-benzo[g]indol-3-yl)acrylonitrile (10f)
Yellow solid, Rf = 0.77 (1:1 EtOAc/hexane), mp >300 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.04 (br, s, 2H), 8.75 (s, 1H), 8.54 (s, 1H), 8.48 (d, J = 8.1 Hz, 1H), 8.13 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 9 Hz, 1H), 7.69-7.64 (m, 3H), 7.54 (t, J = 7.2 Hz, 1H), 7.27–7.24 (m, 2H). LC-MS (+ESI): m/z 335.3 (M+1)+.
3-(2,2'-Bithiophen-5-yl)-2-(1H-benzo[d]imidazol-2-yl)acrylonitrile (5)
Orange brown solid, Rf = 0.69 (1:1 EtOAc/hexane), mp = 271–272 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.00 (s, 1H), 8.51 (s, 1H), 7.79 (d, J = 3.9 Hz, 1H), 7.71-7.66 (m, 2H), 7.58 (dd, J = 1.2, 3.0 Hz, 1H), 7.54 (d, J = 4.2 Hz, 2H), 7.28–7.23 (m, 2H), 7.21–7.18 (m, 1H). LC-MS (+ESI): m/z 334.15 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(5-methylthiophen-2-yl)acrylonitrile (12a)
Bright orange solid, Rf = 0.38 (1:4 EtOAc/hexane), mp = 279–280 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.45 (s, 1H), 7.66 (d, J = 3.6 Hz, 2H), 7.54 (br, 1H), 7.23 (d, J = 4.8 Hz, 2H), 7.06 (dd, J = 0.9, 3.6 Hz, 1H), 2.59 (s, 3H). LC-MS (+ESI): m/z 266.44 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(4,5-dimethylthiophen-2-yl)acrylonitrile (12b)
Brown yellow solid, Rf = 0.44 (1:4 EtOAc/hexane), mp = 291–292 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.92 (s, 1H), 8.36 (s, 1H), 7.59 (br, 1H), 7.54 (s, 1H), 7.23 (dd, J = 2.7, 6.0 Hz, 2H), 2.43 (s, 3H), 2.16 (s, 3H). LC-MS (+ESI): m/z 280.5 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(thiophen-2-yl)acrylonitrile (12c)
Bright yellow solid, Rf = 0.38 (1:4 EtOAc/hexane), mp = 222–223 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.02 (br, 1H), 8.56 (s, 1H), 8.07 (d, J = 4.8 Hz, 1H), 7.86 (d, J = 3.6 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.35 (dd, J = 3.6, 4.9 Hz, 1H), 7.30–7.21 (m, 2H). LC-MS (+ESI): m/z 252.3 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(5-(furan-2-yl)thiophen-2-yl)acrylonitrile (12d)
Dark brown solid, Rf = 0.79 (1:1 EtOAc/hexane), mp = 273–274 °C; 1H NMR (300 MHz, DMSO-d6) δ 12.99 (br, 1H), 8.52 (s, 1H), 7.87 (d, J = 1.8 Hz, 1H), 7.81 (d, J = 4.5 Hz 1H), 7.66 (br, 1H), 7.58-7.53 (m, 2H), 7.26–7.24 (m, 2H), 7.11 (d, J = 3.6 Hz, 1H), 6.70 (dd, J = 1.8, 4.5 Hz, 1H). LC-MS (+ESI): m/z 318.44 (M+1)+.
2-(1H-Benzo[d]imidazol-2-yl)-3-(5-(thiophen-2-yl)furan-2-yl)acrylonitrile hemiacetate (12e)
Orange solid, Rf = 0.59 (1:2 EtOAc/hexane), mp = 243–244 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.0 (s, 1H), 11.98 (s, 0.5 H), 8.14 (s, 1H), 7.76 (dd, J = 0.9, 4.9 Hz, 1H), 7.67(dd, J = 1.2, 3.6 Hz, 2H), 7.53 (d, J = 7.5 Hz, 1H), 7.42 (d, J = 3.6 Hz, 1H), 7.26–7.22 (m, 3H), 7.17 (d, J = 3.6 Hz, 1H), 1.92 (s, 1.5 H). LC-MS (+ESI): m/z 318.44 (M+1)+.
3-(2,2'-Bithiophen-5-yl)-2-(1H-indol-2-yl)acrylonitrile (14a)
Orange solid, Rf = 0.38 (1:4 EtOAc/hexane), mp = 245–246 °C; 1H NMR (300 MHz, DMSO-d6) δ 11.72 (br, s, 1H), 8.08 (s, 1H), 7.65 (dd, J = 0.9, 5.1 Hz, 1H), 7.59–7.57 (m, 2H), 7.51 (dd, J = 0.9, 3.6 Hz, 1H), 7.47 (d, J = 3.9 Hz, 1H), 7.41 (dd, J = 0.3, 9 Hz, 1H), 7.22–7.15 (m, 2H), 7.05 (ddd, J = 0.9, 8.1, 15 Hz, 1H), 6.77 (s, 1H). LC/MS (+ESI): m/z 333.3 (M+1)+.
3-(2,2'-Bithiophen-5-yl)-2-(benzo[d]thiazol-2-yl)acrylonitrile (14b)
Red solid, Rf = 0.60 (1:4 EtOAc/hexane), mp = 188–189 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.61 (s, 1H), 8.16 (dd, J = 0.6, 7.9 Hz, 1H), 8.05 (dd, J = 0.6, 7.8 Hz, 1H), 7.99 (d, J = 4.5 Hz, 1H), 7.73 (dd, J = 0.9, 5.1 Hz, 1H), 7.60 (dd, J = 1.2, 3.8 Hz, 1H), 7.57 (d, J = 3.9 Hz, 1H), 7.53 (dd, J = 1.2, 9.6 Hz, 1H), 7.48 (dd, J = 0.9, 7.4 Hz, 1H), 7.19 (dd, J = 3.9, 5.1 Hz, 1H). LC-MS (+ESI): m/z 351.25 (M+1)+.
3-(2,2'-Bithiophen-5-yl)-2-(benzo[d]oxazol-2-yl)acrylonitrile (14c)
Dark red solid, Rf = 0.84 (1:2 EtOAc/hexane), mp = 192–193 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.67 (s, 1H), 8.0 (d, J = 3.9 Hz, 1H), 7.80–7.69 (m, 3H), 7.66 (dd, J = 3.2, 0.9 Hz, 1H), 7.56 (d, J = 3.9 Hz, 1H), 7.47–7.39 (m, 2H), 7.18 (dd, J = 4.5, 1.2 Hz, 1H). LC-MS (+ESI): m/z 335.43 (M+1)+.
3-(2,2'-Bithiophen-5-yl)-2-(quinolin-2-yl)acrylonitrile (14d)
Brown solid, Rf = 0.87 (1:4 EtOAc/hexane), mp = 174–175 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.49 (d, J = 8.7 Hz, 1H), 8.07–8.0 (m, 3H), 7.90 (d, J = 3.9 Hz, 1H), 7.83 (ddd, J = 1.5, 7.2, 15.6 Hz, 1H), 7.69 (dd, J = 1.2, 5.1 Hz, 1H), 7.64 (ddd, J = 1.2, 8.1, 15.0 Hz, 1H), 7.58 (dd, J = 1.2, 3.6 Hz, 1H), 7.55 (d, J = 3.9 Hz, 1H), 7.19 (dd, J = 3.6, 4.9 Hz, 1H). LC-MS (+ESI): m/z 345.5 (M+1)+.
2-(2-(2,2'-Bithiophen-5-yl)-1-cyanovinyl)-1,3-dimethyl-1H-benzo[d]imidazol-3-ium iodide (15)
A mixture of 2-[1-cyano-2-(2,2'-bithiophen-5-yl)vinyl]benzimidazole (150 mg, 0.45 mmol) and NaH (60% in oil) (270 mg, 0.67 mmol) was stirred in DMF (5 mL) at 43 °C for 15 min. Methyl iodide (960 mg, 42 µL, 0.67 mol) was added and the reaction mixture was stirred for 24 h. The solution was poured into excess water, in which produced a sticky brown precipitate. EtOAc was added and the mixture was stirred for 1 h. The precipitate turned into a brick red solid, which was collected by filtration, washed with water and dried at 50 °C under vacuum overnight to give the desired product (70 mg, 32%), Rf = 0.73 (1:5 MeOH/CH2Cl2), mp = 232–233 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.49 (s, 1H), 8.14 (d, J = 3.3 Hz, 1H), 8.12 (d, J = 3.3 Hz, 1H), 8.03 (d, J = 4.2 Hz, 1H), 7.81 (d, J = 5.1 Hz, 1H), 7.78 (d, J = 3.3 Hz, 1H), 7.76 (d, J = 3.3 Hz, 1H), 7.72 (d, J = 3.6 Hz, 1H), 7.69 (d, J = 3.9 Hz, 1H), 7.25 (dd, J = 1.2, 4.5 Hz, 1H), 4.13 (s, 6H). LC-MS (+ESI): m/z 362.3 (M)+.
4.3. BoNT/A LC FRET-based assay
The fluorescence resonance energy transfer (FRET)-based assay used in these studies has been described previously7, 8. Briefly, 20 µM SNAP-25 substrate (AA 187–203) of sequence SNRTRIDEAN[DnpK]RA[daciaC]RML (Peptides International, Kentucky) was incubated at 37 °C for 40 min in the presence of 2 nM BoNT/A LC (List Biologicals, California). The fluorescence of the cleaved substrate was measured at 485 nm following excitation at 398 nm in a Victor 2 (Perkin Elmer, Waltham, MA) plate reader. IC50 values were calculated as the concentration of compound that produced 50% inhibition in the assay.
4.4. BoNT/A LC HPLC-based assay
The BoNT/A LC HPLC-based assay to quantitate inhibition is a modified version of a published protocol49. Incubation conditions were the same as for the FRET-bsed assay, except that the enzyme concentration was 6 nM and the substrate (17-mer SNAPtide) concentration was 60 µM. The samples were analyzed by reverse-phase HPLC (Gilson, Wisconsin) using a C18 column (Grace Alltima, UK). The effluent was monitored at 365 nm and the resultant peaks quantified by integration utilizing Gilson Trilution® software.
4.5. Anthracis Lethal Factor (LF) assay
A LF FRET-based assay was performed using peptide substrate (MCA-KKVYPYPME[dnp]K amide), and 5.55 nM BaLF (List Biological, California), and incubating at 37 °C for 30 min as described previously49, 50.
4.6. Enzyme inhibitor inactivation studies
BoNT/A LC was incubated with select concentrations of inhibitor at various time points. At the end of the incubation period, remaining enzyme activity was assessed using the BoNT/A LC FRET-based assay (described above). Results were plotted and Kobs/[I] were determined utilizing a method by Adam et al51.
4.7. Enzyme/Inhibitor “rescue” studies
BoNT/A LC was co-incubated with DMSO equivalent (control) or inhibitor at 37 °C for 90 minutes. The experimental samples (500 µL) were then loaded into dialysis cassettes (Pierce 66330 cassettes 3,500 molecular weight cutoff) and dialyzed against 1L of 20 mM HEPES pH 7.4, 10 % Glycerol, and 0.05 % Tween-20 for 20–24 h at 2–8 °C. The experimental samples were then assessed for remaining activity using the standard FRET-based assay (indicated above). The enzyme-inhibitor activity that remained was compared to either the DMSO equivalent or the DMSO equivalent/50 µM compound 2 (acetic acid salt form) for the competitive inhibitor protection studies.
4.8. Molecular modeling
Docking calculations were performed using Schrödinger Glide 5.0 software (Schrödinger, New York). The covalent adducts were constructed using the ‘best’ docking mode and the energy of the inhibitor-enzyme adduct was fully minimized until RMSD < 0.3 Å.
Acknowledgments
This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (5U01AI070430). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services. The authors thank Dr. James C. Burnett for helpful discussions and Dr. Donald T. Moir for assistance with the kinetic analyses. The authors thank CreaGen Biosciences, Inc., for the preparation of starting material, e.g., 2-(1H-indol-2-yl)acetonitrile (6) and for the scale-up of compound 14a.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. Botulinum toxin as a biological weapon: medical and public health management. Jama. 2001;285:1059–1070. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 2.Paddle BM. Therapy and prophylaxis of inhaled biological toxins. J. Appl. Toxicol. 2003;23:139–170. doi: 10.1002/jat.903. [DOI] [PubMed] [Google Scholar]
- 3.Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. The evolving field of biodefense: therapeutic developments and diagnostics. Nat. Rev. Drug Discov. 2005;4:281–297. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burnett JC, Schmidt JJ, McGrath CF, Nguyen TL, Hermone AR, Panchal RG, Vennerstrom JL, Kodukula K, Zaharevitz DW, Gussio R, Bavari S. Conformational sampling of the botulinum neurotoxin serotype A light chain: implications for inhibitor binding. Bioorg. Med. Chem. 2005;13:333–341. doi: 10.1016/j.bmc.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Josko D. Botulin toxin: a weapon in terrorism. Clin. Lab. Sci. 2004;17:30–34. [PubMed] [Google Scholar]
- 6.Clarke SC. Bacteria as potential tools in bioterrorism, with an emphasis on bacterial toxins. Br. J. Biomed. Sci. 2005;62:40–46. doi: 10.1080/09674845.2005.11732685. [DOI] [PubMed] [Google Scholar]
- 7.Hicks RP, Hartell MG, Nichols DA, Bhattacharjee AK, van Hamont JE, Skillman DR. The medicinal chemistry of botulinum, ricin and anthrax toxins. Curr. Med. Chem. 2005;12:667–690. doi: 10.2174/0929867053202223. [DOI] [PubMed] [Google Scholar]
- 8.Shukla HD, Sharma SK. Clostridium botulinum: a bug with beauty and weapon. Crit. Rev. Microbiol. 2005;31:11–18. doi: 10.1080/10408410590912952. [DOI] [PubMed] [Google Scholar]
- 9.Comella CL, Pullman SL. Botulinum toxins in neurological disease. Muscle Nerve. 2004;29:628–644. doi: 10.1002/mus.20033. [DOI] [PubMed] [Google Scholar]
- 10.Glogau RG. Review of the use of botulinum toxin for hyperhidrosis and cosmetic purposes. Clin. J. Pain. 2002;18:S191–S197. doi: 10.1097/00002508-200211001-00012. [DOI] [PubMed] [Google Scholar]
- 11.Marks JD. Medical aspects of biologic toxins. Anesthesiol. Clin. North America. 2004;22:509–532. vii.. doi: 10.1016/j.atc.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Montecucco C, Molgo J. Botulinal neurotoxins: revival of an old killer. Curr. Opin. Pharmacol. 2005;5:274–279. doi: 10.1016/j.coph.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Bhidayasiri R, Truong DD. Expanding use of botulinum toxin. J. Neurol. Sci. 2005;235:1–9. doi: 10.1016/j.jns.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Bigalke H, Rummel A. Medical aspects of toxin weapons. Toxicology. 2005;214:210–220. doi: 10.1016/j.tox.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 15.Foster KA. A new wrinkle on pain relief: re-engineering clostridial neurotoxins for analgesics. Drug Discov. Today. 2005;10:563–569. doi: 10.1016/S1359-6446(05)03389-1. [DOI] [PubMed] [Google Scholar]
- 16.Cote TR, Mohan AK, Polder JA, Walton MK, Braun MM. Botulinum toxin type A injections: adverse events reported to the US Food and Drug Administration in therapeutic and cosmetic cases. J. Am. Acad. Dermatol. 2005;53:407–415. doi: 10.1016/j.jaad.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 17.Foran PG, Mohammed N, Lisk GO, Nagwaney S, Lawrence GW, Johnson E, Smith L, Aoki KR, Dolly JO. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J. Biol. Chem. 2003;278:1363–1371. doi: 10.1074/jbc.M209821200. [DOI] [PubMed] [Google Scholar]
- 18.Greenfield RA, Brown BR, Hutchins JB, Iandolo JJ, Jackson R, Slater LN, Bronze MS. Microbiological, biological, and chemical weapons of warfare and terrorism. Am. J. Med. Sci. 2002;323:326–340. doi: 10.1097/00000441-200206000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Rosenbloom M, Leikin JB, Vogel SN, Chaudry ZA. Biological and chemical agents: a brief synopsis. Am. J. Ther. 2002;9:5–14. doi: 10.1097/00045391-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Meunier FA, Lisk G, Sesardic D, Dolly JO. Dynamics of motor nerve terminal remodeling unveiled using SNARE-cleaving botulinum toxins: the extent and duration are dictated by the sites of SNAP-25 truncation. Mol. Cell Neurosci. 2003;22:454–466. doi: 10.1016/s1044-7431(02)00016-7. [DOI] [PubMed] [Google Scholar]
- 21.Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- 22.Simpson LL. Identification of the major steps in botulinum toxin action. Annu. Rev. Pharmacol. Toxicol. 2004;44:167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 23.Binz T, Blasi J, Yamasaki S, Baumeister A, Link E, Sudhof TC, Jahn R, Niemann H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J. Biol. Chem. 1994;269:1617–1620. [PubMed] [Google Scholar]
- 24.Singh BR. Intimate details of the most poisonous poison. Nat. Struct. Biol. 2000;7:617–619. doi: 10.1038/77900. [DOI] [PubMed] [Google Scholar]
- 25.Turton K, Chaddock JA, Acharya KR. Botulinum and tetanus neurotoxins: structure, function and therapeutic utility. Trends Biochem. Sci. 2002;27:552–558. doi: 10.1016/s0968-0004(02)02177-1. [DOI] [PubMed] [Google Scholar]
- 26.Li B, Peet NP, Butler MM, Burnett JC, Moir DT, Bowlin TL. Small molecule inhibitors as countermeasures for botulinum neurotoxin intoxication. Molecules. 2011;16:202–220. doi: 10.3390/molecules16010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt JJ, Stafford RG. A high-affinity competitive inhibitor of type A botulinum neurotoxin protease activity. FEBS Lett. 2002;532:423–426. doi: 10.1016/s0014-5793(02)03738-9. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt JJ, Bostian KA. Proteolysis of synthetic peptides by type A botulinum neurotoxin. J. Protein Chem. 1995;14:703–708. doi: 10.1007/BF01886909. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt JJ, Bostian KA. Endoproteinase activity of type A botulinum neurotoxin: substrate requirements and activation by serum albumin. J. Protein Chem. 1997;16:19–26. doi: 10.1023/a:1026386710428. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt JJ, Stafford RG, Bostian KA. Type A botulinum neurotoxin proteolytic activity: development of competitive inhibitors and implications for substrate specificity at the S1' binding subsite. FEBS Lett. 1998;435:61–64. doi: 10.1016/s0014-5793(98)01041-2. [DOI] [PubMed] [Google Scholar]
- 31.Sukonpan C, Oost T, Goodnough M, Tepp W, Johnson EA, Rich DH. Synthesis of substrates and inhibitors of botulinum neurotoxin type A metalloprotease. J. Pept. Res. 2004;63:181–193. doi: 10.1111/j.1399-3011.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- 32.Burnett JC, Ruthel G, Stegmann CM, Panchal RG, Nguyen TL, Hermone AR, Stafford RG, Lane DJ, Kenny TA, McGrath CF, Wipf P, Stahl AM, Schmidt JJ, Gussio R, Brunger AT, Bavari S. Inhibition of metalloprotease botulinum serotype A from a pseudo-peptide binding mode to a small molecule that is active in primary neurons. J. Biol. Chem. 2007;282:5004–5014. doi: 10.1074/jbc.M608166200. [DOI] [PubMed] [Google Scholar]
- 33.Burnett JC, Schmidt JJ, Stafford RG, Panchal RG, Nguyen TL, Hermone AR, Vennerstrom JL, McGrath CF, Lane DJ, Sausville EA, Zaharevitz DW, Gussio R, Bavari S. Novel small molecule inhibitors of botulinum neurotoxin A metalloprotease activity. Biochem. Biophys. Res. Commun. 2003;310:84–93. doi: 10.1016/j.bbrc.2003.08.112. [DOI] [PubMed] [Google Scholar]
- 34.Butler MM, Cardinale SC, Li B, Pai R, Ruthel G, Nuss JE, Wanner LM, Park J-B, Rich C, Basu A, Mills D, Peet NP, Moir D, Bavari S, Bowlin TL. Unpublished results. [Google Scholar]
- 35.Li B, Pai R, Cardinale SC, Butler MM, Peet NP, Moir DT, Bavari S, Bowlin TL. Synthesis and Biological Evaluation of Botulinum Neurotoxin A Protease Inhibitors. J. Med. Chem. 2010;53:2264–2276. doi: 10.1021/jm901852f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burnett JC, Li B, Pai R, Cardinale SC, Butler MM, Peet NP, Moir D, Bavari S, Bowlin T. Analysis of botulinum neurotoxin serotype A metalloprotease inhibitors: Analogs of a chemotype for therapeutic development in the context of a three-zone pharmacophore. Open Access Bioinformatics. 2010;2010:11–18. doi: 10.2147/OAB.S7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burnett JC, Wang C, Nuss JE, Nguyen TL, Hermone AR, Schmidt JJ, Gussio R, Wipf P, Bavari S. Pharmacophore-guided lead optimization: the rational design of a non-zinc coordinating, sub-micromolar inhibitor of the botulinum neurotoxin serotype a metalloprotease. Bioorg. Med. Chem. Lett. 2009;19:5811–5813. doi: 10.1016/j.bmcl.2009.01.111. [DOI] [PubMed] [Google Scholar]
- 38.Hermone AR, Burnett JC, Nuss JE, Tressler LE, Nguyen TL, Solaja BA, Vennerstrom JL, Schmidt JJ, Wipf P, Bavari S, Gussio R. Three-dimensional database mining identifies a unique chemotype that unites structurally diverse botulinum neurotoxin serotype A inhibitors in a three-zone pharmacophore. ChemMedChem. 2008;3:1905–1912. doi: 10.1002/cmdc.200800241. [DOI] [PubMed] [Google Scholar]
- 39.Nuss JE, Dong Y, Wanner LM, Ruthel G, Wipf P, Gussio R, Vennerstrom JL, Bavari S, Burnett JC. Pharmacophore refinement guides the rational design of nanomolar-range inhibitors of the botulinum neurotoxin serotype A metalloprotease. ACS Med. Chem. Lett. 2010;1:301–305. doi: 10.1021/ml100056v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solaja BA, Opsenica D, Smith KS, Milhous WK, Terzic N, Opsenica I, Burnett JC, Nuss J, Gussio R, Bavari S. Novel 4-aminoquinolines active against chloroquine-resistant and sensitive P. falciparum strains that also inhibit botulinum serotype A. J. Med. Chem. 2008;51:4388–4391. doi: 10.1021/jm800737y. [DOI] [PubMed] [Google Scholar]
- 41.Park JG, Sill PC, Makiyi EF, Garcia-Sosa AT, Millard CB, Schmidt JJ, Pang Y-P. Serotype-selective, small-molecule inhibitors of the zinc endopeptidase of botulinum neurotixin serotype A. Bioorg. Med. Chem. 2006;14:395–408. doi: 10.1016/j.bmc.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 42.Tang J, Park JG, Millard CB, Schmidt JJ, Pang YP. Computer-aided lead optimization: improved small-molecule inhibitor of the zinc endopeptidase of botulinum neurotoxin serotype A. PLoS One. 2007;2:e761. doi: 10.1371/journal.pone.0000761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pang YP, Vummenthala A, Mishra RK, Park JG, Wang S, Davis J, Millard CB, Schmidt JJ. Potent new small-molecule inhibitor of botulinum neurotoxin serotype A endopeptidase developed by synthesis-based computer-aided molecular design. PLoS One. 2009;4:e7730. doi: 10.1371/journal.pone.0007730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boldt GE, Kennedy JP, Janda KD. Identification of a potent botulinum neurotoxin a protease inhibitor using in situ lead identification chemistry. Org. Lett. 2006;8:1729–1732. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc. Natl. Acad. Sci. U. S. A. 2007;104:2602–2607. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capkova K, Hixon MS, Pellett S, Barbieri JT, Johnson EA, Janda KD. Benzylidene cyclopentenediones: First irreversible inhibitors against botulinum neurotoxin A's zinc endopeptidase. Bioorg. Med. Chem. Lett. 2009;20:206–208. doi: 10.1016/j.bmcl.2009.10.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eubanks LM, Silhar P, Salzameda NT, Zakhari JS, Xiaochuan F, Barbieri JT, Shoemaker CB, Hixon MS, Janda KD. Identification of a natural product antagonist against the botulinum neurotoxin light chain protease. ACS Med. Chem. Lett. 2010;1:268–272. doi: 10.1021/ml100074s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardinale SC, Butler MM, Ruthel G, Nuss JE, Wanner LM, Li B, Pai R, Peet NP, Bavari S, Bowlin TL. Novel benzimidazole inhibitors of botulinum neurotoxin/A display enzyme and cell based potency. The Botulinum J. 2011;2:16–29. doi: 10.1504/tbj.2011.041813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt JJ, Stafford RG. Fluorogenic substrates for the protease activities of botulinum neurotoxins, serotypes A, B, and F. Appl. Environ. Microbiol. 2003;69:297–303. doi: 10.1128/AEM.69.1.297-303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Panchal RG, Hermone AR, Nguyen TL, Wong TY, Schwarzenbacher R, Schmidt J, Lane D, McGrath C, Turk BE, Burnett J, Aman MJ, Little S, Sausville EA, Zaharevitz DW, Cantley LC, Liddington RC, Gussio R, Bavari S. Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 2004;11:67–72. doi: 10.1038/nsmb711. [DOI] [PubMed] [Google Scholar]
- 51.Adam GC, Cravatt BF, Sorensen EJ. Profiling the specific reactivity of the proteome with non-directed activity-based probes. Chem. Biol. 2001;8:81–95. doi: 10.1016/s1074-5521(00)90060-7. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Y, Jensen ON. Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics. 2009;9:4632–4641. doi: 10.1002/pmic.200900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Breidenbach MA, Brunger AT. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature. 2004;432:925–929. doi: 10.1038/nature03123. [DOI] [PubMed] [Google Scholar]
- 54.Scott CJ, McDowell A, Martin SL, Lynas JF, Vandenbroeck K, Walker B. Irreversible inhibition of the bacterial cysteine protease-transpeptidase sortase (SrtA) by substrate-derived affinity labels. Biochem. J. 2002;366:953–958. doi: 10.1042/BJ20020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim JR, Yoon HW, Kwon KS, Lee SR, Rhee SG. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal. Biochem. 2000;283:214–221. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- 56.Singh J, Petter RC, Kluge AF. Targeted covalent drugs of the kinase family. Curr. Opin. Chem. Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- 57.Hagel M, Niu D, St Martin T, Sheets MP, Qiao L, Bernard H, Karp RM, Zhu Z, Labenski MT, Chaturvedi P, Nacht M, Westlin WF, Petter RC, Singh J. Selective irreversible inhibition of a protease by targeting a noncatalytic cysteine. Nat. Chem. Biol. 2010;7:22–24. doi: 10.1038/nchembio.492. [DOI] [PubMed] [Google Scholar]
- 58.Waterson AG, Petrov KG, Hornberger KR, Hubbard RD, Sammond DM, Smith SC, Dickson HD, Caferro TR, Hinkle KW, Stevens KL, Dickerson SH, Rusnak DW, Spehar GM, Wood ER, Griffin RJ, Uehling DE. Synthesis and evaluation of aniline headgroups for alkynyl thienopyrimidine dual EGFR/ErbB-2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2009;19:1332–1336. doi: 10.1016/j.bmcl.2009.01.080. [DOI] [PubMed] [Google Scholar]