

Abstract
Background
Chronic electroconvulsive seizure (ECS), one of the most efficacious treatments for depressed patients, increases the levels of transcription factor CREB in rodent models and mediates the effects of chronic antidepressant treatment. The objective of this study was to determine the changes in CREB occupancy at gene promoters and subsequent gene expression changes induced by chronic ECS.
Methods
We use chromatin immunoprecipitation followed by microarray analysis (ChIP-chip) to identify CREB binding promoters that are influenced by chronic ECS (n=6 per group). Selected genes are confirmed by secondary validation techniques and the functional significance of one target was tested in behavioral models (n=8 per group) by viral mediated inhibition of gene expression.
Results
The results demonstrate that chronic ECS enhances CREB binding and activity at a select population of genes in the hippocampus, effects that could contribute to the efficacy of chronic ECS. Viral vector-mediated inhibition of one of the CREB-target genes regulated by chronic ECS, Fz6 produced anxiety and depressive-like effects in behavioral models of depression.
Conclusions
The results identify multiple gene targets differentially regulated by CREB binding in the hippocampus following chr-ECS and demonstrate the role of Fz6, a Wnt receptor in behavioral models of depression.
Keywords: Chr-ECS, CREB, Wnt signaling, Frizzled receptor, Depression, ChIP-chip
INTRODUCTION
Depression is a devastating illness affecting up to 17 percent of the population and one of the leading causes of disability worldwide. It likely results from a combination of genetic, biochemical, and environmental factors (1). Current chemical antidepressants have limited efficacy in approximately two thirds of depressed patients, and electroconvulsive therapy (ECT) is often successfully used to treat patients who are resistant to antidepressants. However, the molecular mechanisms underlying the therapeutic efficacy of ECT are not known. In rodents, both acute and repeated administration of electroconvulsive seizure (chr-ECS) have been shown to regulate the expression of a variety of genes, including angiogenic and neurotrophic factors (2-4), and to increase the proliferation of neural stem-like cells in the hippocampus (5).
In the central nervous system, the cAMP-response element binding (CREB) is a key modulator of various functions, including neuroplasticity, learning, and memory, and has been implicated in multiple psychiatric disorders (6, 7). In depression, the role for CREB is both regional and temporal specific and CREB has been shown to produce both pro-depressive and anti-depressive behaviors, depending on the brain regions examined (8, 9). In the hippocampus, CREB has been shown to mediate the effects of both chemical and non-chemical antidepressant treatments. Reduced CREB levels in postmortem brains of depressed patients, increased levels with antidepressant treatments (10-13) and increased antidepressant behavioral responses by induction of CREB in hippocampus of rodents (14), suggest an important role of CREB in the pathophysiology and treatment of depression.
CREB binds to the cAMP response element (CRE), and upon ser-133 phosphorylation, can lead to activation of multiple transcriptional pathways (15-17). ECS increases ser-133 phosphorylation of CREB in rodent brain (18). Using chromatin immunoprecipitation and microarray technology (ChIP-chip), we previously identified >860 CREB binding sites in rodent brain and altered CREB occupancy and/or ser-133 phosphorylation of CREB (pCREB) at a subset of these targets within 15 min of an initial (acute) ECS treatment (19). However, the underlying adaptive changes that contribute to the enhanced therapeutic efficacy produced by repeated ECS, similar to the time course for therapeutic intervention in depressed patients, are not fully understood. The current study addresses this question by determining the changes in CREB binding/activity resulting from chronic (chr)-ECS. The results demonstrate enhanced binding of CREB and pCREB at select gene promoters with chr-ECS relative to acute ECS. The functional consequences of altered CREB binding/activity were validated for a select number of genes, including Frizzled 6 (Fz6), one of the receptors involved in Wnt signaling.
Wnts are secreted glycoproteins that signal through the Fz receptors (20). Wnt2 was previously shown to be up regulated by different classes of antidepressants and viral-vector mediated over expression of Wnt2 has been shown to produce an antidepressant-like effect in rodents (21). Activation of the canonical Wnt signaling pathway results in inhibitory phosphorylation of GSK-3β resulting in increased accumulation of β-catenin in the nucleus leading to activation of Wnt target genes. Prior research has shown that the Wnt-glycogen-synthase kinase-3 (GSK-3) system is involved in the adaptive responses underlying antidepressant treatment (22-24) and selective inhibitors of GSK-3 have antidepressant efficacy in behavioral models of depression (25, 26). However, little is known about the Fz receptor subtypes that mediate the actions of Wnt signaling. After confirming that chr-ECS increased Fz6 mRNA and showing that chronic unpredictable stress (CUS) had the opposite effect, we determined the behavioral significance of viral-mediated knockdown of Fz6 expression in the hippocampus.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (180–220g for Chr-ECS treatment; 250-300g for adeno-associated virus (AAV) studies) (Charles River Labs, Wilmington, MA) were housed, two or three per cage, under standard illumination parameters (12h light/dark cycle) and were given free access to food and water. All procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Yale University Animal Care and Use Committee (YACUC).
Chronic Electroconvulsive Shock (Chr-ECS)
Bilateral ECS was administered as described previously (2). For ChIP-chip experiments, animals were killed by decapitation 15 min after the tenth sham/ECS treatment (n=6) or 24h and 15 min after the ninth ECS treatment with sham treatment 15 min prior to sacrifice (n=6) (Fig. 1A). For RT-PCR, insitu hybridization and immunmohistochemistry experiments, a different cohort of animals were killed by decapitation 6 h after the last ECS treatment.
Figure 1. Identification of Chr-ECS-induced CREB occupied promoters following Chr-ECS.

(A) Schematic diagram showing details of the experimental timeline for analysis of hippocampus. (B) Venn diagram of the overlap between promoters enriched 1.5-fold, p<0.05 relative to pre-immune IgG by total CREB antibody 15 min after a single ECS (acute), pCREB antibody 15 min after acute ECS, and total CREB antibody 15 min after the 10th ECS (chronic). (C) Bar graph indicates number of promoters in the hippocampus with ECS/sham differences in total CREB (C) or pCREB (D) occupancy of >1.3-fold, p<0.05 (black) or <−1.3 fold, p<0.05 (white) following acute or Chr-ECS. (E) Box plots indicate average log10 ratio (ECS/sham) for total CREB antibody following acute ECS (bottom), total CREB antibody following Chr-ECS (lower middle), pCREB antibody following acute ECS (upper middle) and pCREB antibody following Chr-ECS (top) for the 95 promoters exhibiting >1.3-fold, p<0.05 enhancement of total CREB occupancy or the 78 promoters exhibiting >1.3-fold, p<0.05 enhancement of pCREB occupancy relative to sham treatment following Chr-ECS.
ChIP-chip processing, hybridization and analysis
ChIP-chip experiments, quality control and analysis were performed exactly as described (19). Briefly, chromatin isolated from 10 mg tissue was sonicated into 200-500 bp fragments. Resulting samples were immunoprecipitated with 4 μg total CREB antibody (Upstate #06863), 2 μg pCREB antibody (Upstate #06519) or 5 μg normal rabbit IgG (Sigma I8140). ChIP DNA was amplified to linear phase by ligation-mediated PCR and amplified DNA was labeled using Genesphere’s DNA900 labeling system and hybridized to DNA microarrays from the Beta Cell Biology Consortium containing 1 Kb sequences immediately upstream of the transcription start site (TSS) of 12,000 genes and 2 Kb sequences from 3 Kb to 1 Kb upstream of the TSS for half of the represented genes (www.betacell.org). Mean Cy3/Cy5 ratios from 6 independent animal/ChIP/array replicates were calculated on a per spot, per array basis using GenePix Pro 6.0 (Axon instruments) and Genespring 7.2 (Silcon Genetics) following intensity dependent Lowess normalization. Analysis of ratio data was performed in Matlab R2007a (Mathworks).
Quantitative Real-Time RT-PCR
Total RNA from hippocampus stored at −80C was purified using RNAqueous according to the manufacturer’s directions (Ambion, Austin, TX, U SA). One microgram of total RNA was reverse transcribed as before (2). Gene specific primers were designed using PRIMER3 software.
In situ hybridization analysis
In situ hybridization was conducted on coronal brain sections (14μ) by hybridization with the 35S-labeled riboprobes for Fz6, Slc6a9 and Per1 as previously described (2, 27). Gene specific primers were designed using PRIMER3 software (SI). Relative gene expression changes were determined using the software IMAGEJ (http://rsb.info.nih.gov/ij/).
Primers to make riboprobes for Insitu hybridization
Fz6 F; gaactagcacgggcgtgacc
Fz6 R; ccaagccttctaatacgactcactatagggagatgcactccatcaggccagtc
Slc6a9 F; gccaaagggatgctgaatgg
Slc6a9 R; ccaagccttctaatacgactcactatagggagagcaaactggccgaaggagag
Per1 F; caaacccggaactggaggtg
Per1 R; ccaagccttctaatacgactcactatagggagatggctccttccgaggagttg
Chronic unpredictable stress
Samples tested for CUS were obtained from the rats that were subjected to the CUS procedure which included briefly exposure to a variable sequence of 12 different stressors, two per day, for 35 days as described previously (28).
Design and construction of AAV-Fz6shRNA plasmids
Hairpin RNA was designed to specifically target Fz6 mRNA. 24bp oligonucleotide sequence within the coding region of Fz6 gene was identified using published criteria (29, 30). Hairpins were designed such that the antisense strand came before the sense strand during transcription. Two sets of oligonucleotides were synthesized (Integrated DNA Technologies) for cloning: Fz6shRNA top: 5′-TTTGAATTGTGCTTCAGGAAGAACTCACTTCCTGTCATGAGTTCTTCCTGAAGCACAATTATTTTT - 3′; bottom 5′- CTAGAAAAATAATTGTGCTTCAGGAAGAACTCATGACAGGAAGTGAGTTCTTCCTGAAGCACAATTC-3′ and scrshRNA top: 5′- TTTGTGGAGCCGAGTTTCTAAATTCCGCTTCCTGTCACGGAATTTAGAAACCCGGC TCCAATTTTT-3′; bottom, 5′- CTAGAAAAATTGGAGCCGGGTTTCTAAATTCCGTGACAGGAAGCGGAAT TTAGAAACTCGGCTCCAC-3′. The oligonucleotides had Sap1 and Xba1 overhangs to allow for ligation into mU6pro region of the modified pAAV-MCS vector, pAAV-shRNA. The pAAV-shRNA plasmid was designed to coexpress shRNA and EGFP under the control of an independent RNA polymerase II promoter for EGFP and U6 promoter for shRNA. All final clones were verified for sequencing. Four sets of oligonucleotides were designed for Fz6shRNA and one set was finally used in behavioral experiments after ensuring efficient knockdown of Fz6 mRNA in primary cortical neuronal cultures and hippocampus (Fig. 5C).
Figure 5. Experimental outline and Influence of Fz6 shRNA knockdown in models of depression.

(A) Schematic diagram showing details of experimental timeline. (B) AAV-GFP-Scr (Control) or AAV-GFP-Fz6shRNA was infused into the hippocampus. Representative image showing expression of GFP in the hippocampus (inlet, dentate gyrus) of animal infused with AAV-GFP-Fz6shRNA. (C) Data represent mean ± SEM of percent of Fz6mRNA expression relative to AAV scrshRNA at 2 weeks post injection (n=2). Rats were injected with AAVFz6shRNA on one side and AAV-scrshRNA on the other side of the brain. Primary cortical neuronal cultures were incubated with AAV-scrshRNA and 2 weeks post infection levels of Fz6 mRNA were determined. Results are the mean ± SEM of percent of Fz6mRNA expression relative AAV-scrshRNA controls (n=2). The influence of AAV-Fz6shRNA expression on different behavioral models was examined, including sucrose (D), total consumption (E) and sucrose preference (F) in the sucrose preference (SPT) test, # of open arm (G), closed arm (H) entries and time spent in open arms (I) in elevated plus maze (EPM) task, latency to feed in the novelty suppressed feeding (NSF) (J), and escape failures (K) and latency to escape (L) in the learned helplessness-active avoidance test. Results are the mean ± SEM of AAV-scr (n=8) vs. AAV-Fz6shRNA (n=8). ns=not significant; *p < 0.05 compared to AAV-scr (Student’s t-test); #=not significant but significant trend observed at one measure (one-way ANOVA).
Viral production and purification
The virus was generated using a triple-transfection, helper-free method, and purified with a modified version of a published protocol (30). Briefly, HEK 293 cells were cultured in five 150 ×25 mm cell culture dishes and transfected with pAAV-scr/Fz6shRNA, pHelper and pAAV-RC plasmids (Stratagene) using a standard calcium phosphate method and subsequent purification was done as described previously (30, 31).
Stereotaxic surgery and infusions
Rats were anesthetized with xylazine (6 mg/kg, i.m., Lloyd laboratories, Shenandoar, IA) and ketamine (80 mg/kg i.m., Fort Dodge Animal Health, Overland Park, KS). Bilateral viral injections were performed as described previously {Paxinos, 1985 #206}. After behavioral testing, 40μ sections were cut using a microtome for GFP visualization.
Novelty Suppressed Feeding (NSF)
NSF was conducted as previously described with minor modification of allowing the test to continue for 20 min (32).
Sucrose Preference test (SPT)
All behavioral testing was performed during the light cycle as previously described (32, 33). SPT was performed before NSF on day 21 by habituating the animals to 1% sucrose solution for 48h. Due to the variability within the groups and absence of preference in control animals, the test was suspended for 3 days and repeated after performing NSF. On day 25 after the NSF, animals were habituated to a sucrose solution (2%, Sigma, St. Louis, MO) for 48h, followed by 4h fluid deprivation on the test day. Rats were presented with two identical bottles of sucrose and water for 1h and sucrose and water consumption were calculated. Sucrose preference was calculated as the percentage of sucrose consumed over total fluid consumption.
Elevated plus maze (EPM)
Rats were placed in the center of the maze facing an open arm and were allowed to explore the maze for 5 minutes. A blinded observer recorded the total time spent in each arm as well as the number of entries into each arm. Entries were operational defined as occurring when all four paws crossed into a particular arm (pillow, 1985) EPM was conducted in a different cohort of animals (AAV-Control:n=6, AAV-Fz6shRNA: n=8). Prior to EPM, the animals were tested for total locomotor activity and there was no significant difference between the groups. NSF was also performed in the same group as a positive control and similar results were obtained as the first cohort of animals i.e., there was significant increase in latency to feed in animals infused with AAV-Fz6shRNA as compared to controls.
Forced swim test (FST)
FST was conducted as previously described(32, 33). On the test day, rats were placed for 10 min in a clear cylinder with water (24±1°C, 45 cm depth). The sessions were recorded from the side and the time spent immobile was calculated by a blind observer.
Active avoidance (AA) paradigm
The AA procedure was performed in custom built, 2-chambered shuttle boxes (Med Associates, Vermont) and AA testing was performed as previously described (33). Results are expressed as the number of escape failures observed in the 30 trial period. Latency to escape was also calculated for every 5 escape trials.
Locomotor Activity (LA)
Ambulatory locomotor activity was assessed in clear plastic boxes fitted to automated activity meters (MED Associates, St. Albans, VT) consisting of two parallel rows of photosensors (16 sensors per row, 2.5 cm apart). Locomotor activity was recorded in 5-min bins for a total of 30 min by using Med PC software.
RESULTS
Identification of CREB target genes regulated by chr-ECS
To identify CREB gene targets in hippocampus following Chr-ECS, we first determined the promoters enriched by total CREB ChIP relative to pre-immune IgG. The results demonstrate that 373 of the 19,536 promoter regions assayed were enriched by total CREB ChIP >1.5-fold (t-test, p<0.05 relative to pre-immune IgG, t-test). The identified CREB targets showed a significant overlap (hypergeometric p-value <1e−130, representation factor 19.4) with the CREB targets identified previously following acute ECS (Fig. 1B). Likewise, there was strong overlap with the phosphorylated Ser133 CREB (pCREB) targets identified previously in hippocampus following acute ECS (hypergeometric p-value <1e−98, representation value 18.6) (Fig 1B).
Next, we compared the relative effects of acute and Chr-ECS versus sham treatment on CREB binding and activity at the 586 CREB occupied promoters identified in hippocampus after either acute or Chr-ECS. Promoters that were increased >1.3 fold (p<0.05) and decreased >1.3-fold (p<0.05) were identified. Fifteen minutes after the last Chr-ECS treatment, CREB binding was increased at 95 (16.2%) CREB targets and decreased at 18 (3.1%) CREB targets relative to sham treatment (Fig 1c) compared to just 52 (8.9%) and 4 (0.7%) CREB targets 15 min after a single ECS. Similar results were observed with the pCREB antibody. After Chr-ECS, occupancy of pCREB was increased at 78 (13.3%) CREB targets and decreased at 17 (2.9%) CREB targets (Fig. 1d) compared to 60 (10.2%) and 7 (1.2%) CREB targets 15 min after an initial ECS treatment.
We then used a metagene approach to compare the average enrichment of the promoter sets identified in each experiment. Specifically, the average promoter enrichment within a particular CREB target set was determined and was compared across experiments. When comparing the 125 total CREB targets identified in both the acute and chr-ECS experiments, there was no difference in the average fold enrichment, indicating equivalent ChIP efficiencies across experiments (Fig S1A in the Supplement). When considering the 95 promoters affected (fold change >1.3, p<.05) by Chr-ECS, there was a robust (3.3-fold, p=0.0002) enhancement in the average total CREB-occupancy change relative to acute ECS (Fig. 1E). Similar to total CREB, when considering the 78 promoters affected (pCREB fold change >1.3, p<.05) by Chr-ECS, there was a significant enhancement (2.3-fold, p=0.003) in the average pCREB occupancy relative to acute ECS (Fig. 1E). There was little to no difference in the overall magnitude of change between acute and chr-ECS when considering the significant ECS induced changes identified in the acute ECS experiment (Fig S1B in the Supplement). These results show an induction of CREB binding and phosphorylation at a select set of promoters following Chr-ECS. The vast majority of these changes were stably maintained between daily treatments as demonstrated by comparing these results to those in animals who received 9 ECS treatments and a sham treatment on the 10th day (24h) (Fig S2 in the Supplement).
Expression levels of ECS-CREB targets
To validate the ChIP-chip data and to determine whether chr-ECS-induced CREB binding and phosphorylation has an effect on the transcript levels of these target genes, quantitative RT-PCR was performed on 10 genes of interest, DEGS1, FZ6, SLC6A9, CYP8B1, GDNF, DKK2, HSD3B, PER1, VEGFb and NRG3. Each of these genes exhibited significantly (p<0.05) increased CREB binding and phosphorylation 15 min or 24 hr after chr-ECS with the exception of PER1 which showed significantly decreased CREB binding and activity and NRG3 which exhibited significant increases in CREB phosphorylation but no consistent change in CREB binding (Fig. 2A, B). Levels of FZ6 and SLC6A9 mRNA were significantly increased, and levels of PER1 were decreased by chr-ECS, in agreement with the observed changes in CREB occupancy and phosphorylation (Fig. 3). PER2, which was not identified as a CREB target in our studies, exhibited no transcriptional response to chr-ECS (Fig. S3 in the Supplement). We observed significant down regulation of DKK2 mRNA after Ch-ECS despite a significant increase in CREB occupancy and phosphorylation at its promoter following acute and Ch-ECS suggesting that additional regulatory events or feedback loops impact DKK2 expression. BDNF, although not present on the promoter array, was included as a positive control as it is reported to be increased by ECS(34) (Fig. 3).
Figure 2. Validation of ECS-regulated CREB target genes.

Results were confirmed by RT-PCR analysis and demonstrate enrichment of selected target genes by CREB (A) or pCREB (B) ChIP. FZ6 (Frizzled-6), DEGS1 (Degenerative spermatocyte homolog1), SLC6A9 (Solute carrier family 6, member 9 or Glycine transporter gene 1), CYP8B1 (Cytochrome p450 family member), GDNF (Glial-derived neurotrophic factor), DKK2 (Dickkopf 2), HSD3B (Hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase4), PER1 (Period1), VEGFb (Vascular endothelial growth factorb), BDNF (Brain-derived neurotrophic factor). Data represent mean ± SEM of Log10 (ECS/Sham) total enrichment.
Figure 3. Expression of ECS-regulated CREB target genes.
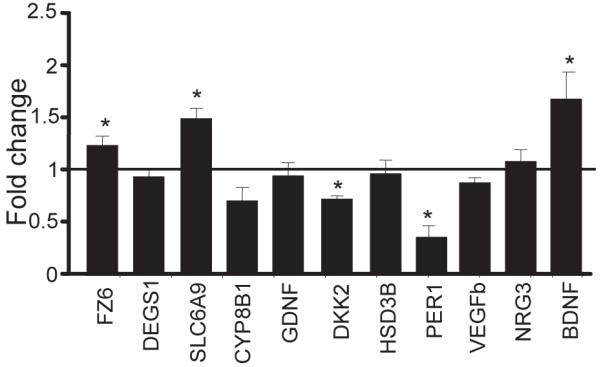
a.Levels of hippocampal mRNA for selected target genes was determined by RT-PCR. Data represent mean ± SEM of fold change in mRNA, n=5 per group. Fold change of 1 indicates no change in expression in Chr-ECS-treated rats over sham. Student’s t-test; * p<0.05.
Regulation of FZ6, SLC6A9 and PER1 mRNA expression was also examined by in situ hybridization analysis. Chr-ECS increased levels of FZ6 mRNA expression in the dentate gyrus (DG) granule cell layer, and the CA3, but not in the CA1 pyramidal cell layer of hippocampus (Fig. 4A). There was a trend for increased expression of SLC6A9 mRNA in DG, CA1 and CA3 cell layers (Fig. 4B). Levels of PER1 mRNA were significantly decreased in DG, CA1 and CA3 hippocampal subfields in response to Chr-ECS (Fig. 4C).
Figure 4. Regulation of Fz6, Slc6a9 and Per1 mRNA in the sub regions of hippocampus by chr-ECS and CUS.

Representative in situ hybridization autoradiograms showing Fz6 (A), Slc6a9 (B) and Per1 (C) mRNA in various regions of the rat brain. Coronal section of brain from sham (left) or Chr-ECS-treated rat (right). Data represent mean ± SEM of fold change in mRNA, n=5 per group, and are presented as fold change relative to control (value of 1 indicates no change in expression over control). * p<0.05 (t-test). (D) The influence of chronic unpredictable stress (CUS) on levels of Fz6, Slc6a9 and Per1 mRNA in hippocampus was determined by RT-PCR. Data represent mean ± SEM of fold change in mRNA, n=6 per group. Student’s t-test; * p<0.05 compared to non stressed controls.
We also examined the expression of FZ6, PER1 and SLC6A9 after exposure to chronic unpredictable stress (CUS), a rodent model of depression that results in behavioral abnormalities observed in depressed patients, most notably anhedonia (32, 33). CUS exposure for 5 weeks decreases the expression of Fz6 mRNA in the hippocampus, but did not significantly influence levels of Per1 or Slc6a9 (Figure 4D). We have also conducted studies to examine levels of Fz6 by western blot and immunohistochemistry, to confirm that the changes observed at the mRNA level result in altered protein levels. However, we have not been able to reliably detect Fz6 protein by either technique and with four different commercial antibodies. Future studies with better antibodies will be needed to further examine this issue.
Effect of Fz6shRNA on animal models of depression
Since chr-ECS increased and CUS decreased the expression of Fz6 in rat hippocampus, studies were conducted to determine the influence of altered Fz6 in behavioral models of depression. An shRNA strategy was used in combination with an AAV-2 vector to achieve targeted knockdown of Fz6 in the hippocampus. AAV-Fz6shRNA or scrambled shRNA control (AAV-GFP-Scr) was bilaterally injected into adult rat hippocampus. The influence of AAV-Fz6shRNA on levels of Fz6 mRNA was determined both in vivo and in cultured cells (Fig. 5C). Behavioral testing and expression analysis was initiated 3 weeks after the infusions to allow optimal expression of Fz6shRNA and efficient knockdown (Fig. 5A). A representative image (Fig. 5B) shows expression of GFP in the DG cell layer after infusion of AAV-Fz6shRNA, which resulted in 58% knockdown of Fz6 mRNA (Fig. 5C, which shows 42% remaining relative to control). In the sucrose preference test (SPT), AAV-Fz6shRNA significantly decreased the consumption and preference for sucrose (Fig. 5D, F), but did not alter water or total fluid consumption (Fig. 5E), suggesting that knockdown of Fz6 causes anhedonic behavior. The lack of sucrose preference observed in AAV-Fz6shRNA (~50%) is similar to the levels observed in chronically stressed animals. In the novelty suppressed feeding (NSF) test, in which latency to feed is a measure of anxiety-like behavior that is reversed by chronic antidepressant administration, AAV-Fz6shRNA significantly increased the latency (55%) (Fig. 5J). There was no significant difference in home cage food consumption showing that there was no overall effect on appetite (Fig. S4A in the Supplement). We examined a second anxiety-related task, the elevated plus maze (EPM), in another cohort of animals; AAV-Fz6shRNA significantly decreased the number of open arm (Fig. 5G), but not closed arm entries (Fig. 5H), and decreased the time spent in the open arms (Fig. 5I). This data strongly indicates an anxiety-like effect elicited by Fz6shRNA. In the learned helplessness paradigm, AAV-Fz6shRNA resulted in a trend for an increase in the number of escape failures (Fig. 6K) and latency to escape (Fig. 6L), measures of helplessness, although these effects did not reach significance. There was no significant effect of AAV-Fz6shRNA in the forced swim test (FST) (Fig. S4B in the Supplement), an acute antidepressant-screening test. Finally, there was no effect of AAV-FzshRNA on total locomotor activity (Fig. S4C in the Supplement).
DISCUSSION
The results demonstrate that there is a shift in CREB occupancy at hippocampal gene promoters between acute and chr-ECS, with a higher percentage of significant changes in total CREB and pCREB occupancy at gene promoters after chr-ECS. On average, the ECS-induced changes in CREB occupancy or phosphorylation identified after acute ECS were maintained (within 1.5 fold) following chronic treatment. In contrast, the average changes in CREB occupancy or phosphorylation in response to chr-ECS were 3.3- and 2.3-fold greater, respectively. Taken together, these experiments indicate that chr-ECS has a greater effect on CREB binding and activity at gene promoters than acute ECS. We also observed little difference between 15 min or 24 hr after the last ECS, indicating that the chr-ECS-induced enhancement of CREB occupancy/phosphorylation at gene promoters was stable for at least 24 hours.
There were only a few examples of reduced CREB binding and phosphorylation following ECS, such as at the Per1 promoter. CREB binding and Ser-133 phosphorylation is necessary but not sufficient for the induction of CREB mediated transcription (35, 36). Expression levels of some, but not all of the genes exhibiting changes in CREB occupancy were altered (i.e., FZ6, SLC6A9, PER1) by chr-ECS, consistent with the evidence that CREB binding and Ser-133 phosphorylation is necessary but not sufficient for the induction of CREB mediated transcription (15, 35, 36). Both FZ6 and PER1 also showed significant changes in mRNA expression in subregions of the hippocampus.
Per1 is a transcription factor and contributes to a regulatory loop in the suprachiasmatic nucleus (SCN) that maintains circadian rhythm. We found that total CREB binding at the PER1 promoter in the hippocampus was significantly decreased in response to chronic, but not acute ECS, whereas pCREB enrichment at the PER1 promoter was decreased with both acute and chr-ECS. The decreased enrichment of CREB and pCREB correlated with a significant decrease in PER1 mRNA in response to chr-ECS in the hippocampus. The link between depression and sleep homeostasis in humans has been well documented and use of antidepressants has been shown to significantly reduce depressive symptoms and improve quality and latency to sleep (37-41). Recent studies also demonstrate that disruption in circadian rhythm using genetic models (e.g., ClockΔ19 and Per2Brdm1 mutant mice) results in decreased immobility in the forced swim test and increased exploratory behaviors that can be interpreted as ‘less depressed’ and ‘manic’(42-44). Since Per1 is regulated by chronic ECS and not by chronic stress, it raises the possibility that Per1 may be involved in treatment response, but may not be involved in the pathophysiology of stress related illnesses. Further studies are needed to test these possibilities.
FZ6 is a seven transmembrane-spanning receptor involved in Wnt signaling, which includes activation of FZ/β-catenin, FZ/Ca+2 and FZ/planar cell polarity signaling pathways (45) and inhibited by Dkk family members(46, 47). Increased Fz6 expression combined with decreased Dkk2 indicates that Wnt-FZ6 signaling is increased by chr-ECS. We have recently reported that chronic administration of ECS, as well as chemical antidepressants, increases Wnt2 expression in the hippocampus, and that viral expression of Wnt2 is sufficient to produce antidepressant actions in behavioral models of depression (21).
We also found that Fz6 expression was decreased in response to exposure to CUS, a rodent model that results in depressive behaviors, including anhedonia, a core symptom of depression (32, 33). This suggests that decreased levels of Fz6 could contribute to the behavioral effects of CUS. In support of this hypothesis, we found that Fz6 knockdown is sufficient to decrease sucrose preference (an anhedonic response) and to increase anxiety in the novelty suppressed feeding test and elevated plus maze, behaviors that are elicited by CUS exposure (32, 33). There was a trend for decreased escapes in the learned helplessness test (a measure of despair), and no effect on immobility in the FST. The lack of significant effects in these latter models could result from behavior specific actions of Fz6 (e.g., anhedonia and anxiety, but not despair) and/or the validity of the models as a measure of depression (e.g., FST is a rapid antidepressant screen, but has limited use as a model of depression). There was also no effect on total locomotor activity indicating that the behavioral changes observed are not due to a nonspecific alteration of ambulation. It would be interesting to determine if Fz6 knockdown also blocks the responses to antidepressant treatments, although the interpretation of these experiments would be complicated because of the depressive/anxiogenic phenotype produced by knockdown alone.
The mechanisms by which Fz6 influences these behaviors are not known. There are several Wnt ligands and Fz receptor subtypes in the hippocampus, and the exact ligand for Fz6 and consequently the Wnt-Fz6 signaling pathways involved has not been determined. Disruption of neurogenesis and neuronal plasticity are two important cellular functions involved in the pathophysiology of depression. Role for Wnt3a in adult hippocampal neurogenesis (48) and for Wnt7a in promoting synaptogenesis and dendrite maturitation and strength was demonstrated previously (49, 50). Wnt7a has also been associated with increasing number of excitatory synapses (49). Role of β-catenin in dendritic arborization (51)and Dvl, a downstream component of Wnt-Fz interaction, in axon differentiation provide increasing evidence of the role of Wnt signaling in synaptic biology and neurogenesis. . In addition, recent genetic studies have demonstrated associations between gray matter volume changes in MDD patients and polymorphisms in several GSK-3β substrates and genes involved in Wnt signaling in the cerebral cortex implicating the changes in Wnt signaling in other brain regions in addition to hippocampus (52). It will also be interesting in future studies to determine if Fz6 plays a role in the known cellular actions of antidepressants and Wnt signaling, such as hippocampal neurogenesis, synaptic plasticity and regulation of growth factors.
Activation of CREB is mediated by multiple, converging signaling pathways, either phosphorylation-dependent or independent, and the antidepressant effects of ECS depend on a particular set of CREB-activated genes. The current study provides insight into the global changes in gene transcription mediated by Chr-ECS, and suggests that Fz6 signaling could represent a novel target for development of novel antidepressants.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Mounira Banasr for providing us with CUS samples and for assistance with the interpretation and discussion of behavioral results of the study. We also thank Dr. Ralph J DiLeone for providing us with the pAAV-shRNA and pAAV-GFP-scr plasmids.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
B.V, K.Q.T, S.S.N. and R.S.D designed the research. B.V performed the research and wrote the paper. K.Q.T performed, analyzed the data and wrote the results for ChIP-chip part of the study.
FINANCIAL DISCLOSURE
The authors report no biomedical financial interests or potential conflicts of interest pertaining to this work.
REFERENCES
- 1.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA: the journal of the American Medical Association. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, et al. Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J Neurosci. 2003;23:10841–10851. doi: 10.1523/JNEUROSCI.23-34-10841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ploski JE, Newton SS, Duman RS. Electroconvulsive seizure-induced gene expression profile of the hippocampus dentate gyrus granule cell layer. Journal of neurochemistry. 2006;99:1122–1132. doi: 10.1111/j.1471-4159.2006.04156.x. [DOI] [PubMed] [Google Scholar]
- 4.Altar CA, Laeng P, Jurata LW, Brockman JA, Lemire A, Bullard J, et al. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J Neurosci. 2004;24:2667–2677. doi: 10.1523/JNEUROSCI.5377-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segi-Nishida E, Warner-Schmidt JL, Duman RS. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc Natl Acad Sci U S A. 2008;105:11352–11357. doi: 10.1073/pnas.0710858105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 7.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 8.Carlezon WA, Jr., Duman RS, Nestler EJ. The many faces of CREB. Trends in neurosciences. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N, et al. Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:10883–10890. doi: 10.1523/JNEUROSCI.22-24-10883.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet. 1998;352:1754–1755. doi: 10.1016/S0140-6736(05)79827-5. [DOI] [PubMed] [Google Scholar]
- 11.Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biological psychiatry. 2001;50:260–265. doi: 10.1016/s0006-3223(01)01083-6. [DOI] [PubMed] [Google Scholar]
- 12.Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006;59:1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Yuan P, Zhou R, Wang Y, Li X, Li J, Chen G, et al. Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J Affect Disord. 124:164–169. doi: 10.1016/j.jad.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen AC, Shirayama Y, Shin KH, Neve RL, Duman RS. Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biological psychiatry. 2001;49:753–762. doi: 10.1016/s0006-3223(00)01114-8. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayr BM, Canettieri G, Montminy MR. Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci U S A. 2001;98:10936–10941. doi: 10.1073/pnas.191152098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 18.Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, et al. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanis KQ, Duman RS, Newton SS. CREB binding and activity in brain: regional specificity and induction by electroconvulsive seizure. Biol Psychiatry. 2008;63:710–720. doi: 10.1016/j.biopsych.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kikuchi A, Yamamoto H, Kishida S. Multiplicity of the interactions of Wnt proteins and their receptors. Cell Signal. 2007;19:659–671. doi: 10.1016/j.cellsig.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Okamoto H, Voleti B, Banasr M, Sarhan M, Duric V, Girgenti MJ, et al. Wnt2 Expression and Signaling Is Increased by Different Classes of Antidepressant Treatments. Biol Psychiatry. 2010;68:521–527. doi: 10.1016/j.biopsych.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chuang D, Manji HK. In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol Psych. 2007;62:4–6. doi: 10.1016/j.biopsych.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva R, Mesquita AR, Bessa J, Sousa JC, Sotiropoulos I, Leão P, Almeida OF, Sousa N. Lithium blocks stress-induced changes in depressive-like behavior and hippocampal cell fate: the role of glycogen-synthase-kinase-3beta. Neurosci. 2008;152:656–669. doi: 10.1016/j.neuroscience.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 25.Gould T, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol. 2004;7:387–390. doi: 10.1017/S1461145704004535. [DOI] [PubMed] [Google Scholar]
- 26.Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol Psych. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Madsen TM, Newton SS, Eaton ME, Russell DS, Duman RS. Chronic electroconvulsive seizure up-regulates beta-catenin expression in rat hippocampus: role in adult neurogenesis. Biol Psychiatry. 2003;54:1006–1014. doi: 10.1016/s0006-3223(03)00700-5. [DOI] [PubMed] [Google Scholar]
- 28.Duric V, Banasr M, Licznerski P, Schmidt H, Stockmeier C, Simen A, Newton SS, Duman RS. Negative regulator of MAP kinase is increased in depression and is necessary and sufficient for depressive behavior. Nat Med. 2010;16:1328–1332. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 30.Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ. Local gene knockdown in the brain using viral-mediated RNA interference. Nat Med. 2003;9:1539–1544. doi: 10.1038/nm964. [DOI] [PubMed] [Google Scholar]
- 31.Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- 32.Banasr M, Valentine GW, Li XY, Gourley SL, Taylor JR, Duman RS. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biol Psychiatry. 2007;62:496–504. doi: 10.1016/j.biopsych.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64:863–870. doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, et al. TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci U S A. 2003;100:12147–12152. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fava M, Thase ME, DeBattista C, Doghramji K, Arora S, Hughes RJ. Modafinil augmentation of selective serotonin reuptake inhibitor therapy in MDD partial responders with persistent fatigue and sleepiness. Ann Clin Psychiatry. 2007;19:153–159. doi: 10.1080/10401230701464858. [DOI] [PubMed] [Google Scholar]
- 38.Kupfer DJ, Gillin JC, Coble PA, Spiker DG, Shaw D, Holzer B. REM sleep, naps, and depression. Psychiatry Res. 1981;5:195–203. doi: 10.1016/0165-1781(81)90049-4. [DOI] [PubMed] [Google Scholar]
- 39.Modell S, Huber J, Holsboer F, Lauer CJ. The Munich Vulnerability Study on Affective Disorders: risk factors for unipolarity versus bipolarity. J Affect Disord. 2003;74:173–184. doi: 10.1016/s0165-0327(02)00010-1. [DOI] [PubMed] [Google Scholar]
- 40.Rao U, Dahl RE, Ryan ND, Birmaher B, Williamson DE, Rao R, et al. Heterogeneity in EEG sleep findings in adolescent depression: unipolar versus bipolar clinical course. J Affect Disord. 2002;70:273–280. doi: 10.1016/s0165-0327(01)00396-2. [DOI] [PubMed] [Google Scholar]
- 41.Wulff K, Gatti S, Wettstein JG, Foster RG. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci. 11:589–599. doi: 10.1038/nrn2868. [DOI] [PubMed] [Google Scholar]
- 42.Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci U S A. 2007;104:6406–6411. doi: 10.1073/pnas.0609625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Easton A, Arbuzova J, Turek FW. The circadian Clock mutation increases exploratory activity and escape-seeking behavior. Genes Brain Behav. 2003;2:11–19. doi: 10.1034/j.1601-183x.2003.00002.x. [DOI] [PubMed] [Google Scholar]
- 44.Zueger M, Urani A, Chourbaji S, Zacher C, Lipp HP, Albrecht U, et al. mPer1 and mPer2 mutant mice show regular spatial and contextual learning in standardized tests for hippocampus-dependent learning. J Neural Transm. 2006;113:347–356. doi: 10.1007/s00702-005-0322-4. [DOI] [PubMed] [Google Scholar]
- 45.Schulte G, Bryja V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:518–525. doi: 10.1016/j.tips.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 46.He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development. 2004;131:1663–1677. doi: 10.1242/dev.01117. [DOI] [PubMed] [Google Scholar]
- 47.Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- 48.Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 49.Sahores M, Gibb A, Salinas PC. Frizzled-5, a receptor for the synaptic organizer Wnt7a, regulates activity-mediated synaptogenesis. Development. 2010;137:2215–2225. doi: 10.1242/dev.046722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciani L, Boyle KA, Dickins E, Sahores M, Anane D, Lopes DM, et al. Wnt7a signaling promotes dendritic spine growth and synaptic strength through Ca2+/Calmodulin-dependent protein kinase II. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10732–10737. doi: 10.1073/pnas.1018132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nature neuroscience. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- 52.Inkster B, Nichols TE, Saemann PG, Auer DP, Holsboer F, Muglia P, et al. Pathway-based approaches to imaging genetics association studies: Wnt signaling, GSK3beta substrates and major depression. NeuroImage. 2010;53:908–917. doi: 10.1016/j.neuroimage.2010.02.065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.