

Abstract
Depending upon the drug and drug delivery platform, species-specific physiological differences can lead to errors in the interspecies extrapolation of drug performance. This manuscript provides an overview of the species-specific physiological variables that can influence the performance of parenteral dosage forms such as in situ forming delivery systems, nanoparticles, microspheres, liposomes, targeted delivery systems, lipophilic solutions, and aqueous suspensions. Also discussed are those factors that can influence the partitioning of therapeutic compounds into tumors, the central nervous system and the lymphatics. Understanding interspecies differences in the movement and absorption of molecules is important to the interpretation of data generated through the use of animal models when studying parenteral drug delivery.
KEY WORDS: animal model, interspecies differences, parenteral drug delivery, pharmacokinetics
INTRODUCTION
Animal testing supports the early assessment of a compound's therapeutic and toxicological potential. When preclinical species are used in the evaluation of human drug candidates, the accuracy of these predictions depends largely upon the interspecies similarities in drug metabolism, physiology, absorption, and distribution. Despite the availability of reviews on interspecies differences in drug pharmacokinetics (PK) (1–4), similar attention has not been adequately given to the interspecies differences that can influence the performance of parenteral drug delivery platforms and to those factors that may influence target site drug delivery
This manuscript provides an overview of the species-specific factors that can influence the performance of parenteral dosage forms such as in situ forming delivery systems, nanoparticles, microspheres, liposomes, targeted delivery systems, lipophilic solutions, and aqueous suspensions. Also discussed are those factors that can influence the partitioning of therapeutic compounds into tumors, the central nervous system (CNS) and the lymphatics. In some instances, literature discrepancies in interspecies differences are reported and discussed. These controversies support the goal of this review: to promote recognition of those variables that can bias the clinical interpretation of animal model data.
To describe the diverse array of physiological variables that can influence in vivo product performance, this manuscript is organized as follows:
Factors that can influence parenteral drug absorption, including interstitial tissue composition and the movement of drug from the interstitium to the blood.
Factors that can influence drug partitioning into and out of the blood, including interspecies differences in plasma/serum composition, plasma protein binding, and plasma lipoprotein composition.
Species-specific differences in inflammation and immune system responses.
Factors that can influence target tissue delivery, with the examples provided being tumor-directed drug delivery and drug delivery to the brain.
While it is impossible for a single review to provide an exhaustive summary of all factors that can influence drug absorption and partitioning, this manuscript highlights the many critical variable that can affect the inferential value of data derived from preclinical species.
DIFFERENCES IN PARENTERAL DRUG ABSORPTION
Interspecies differences in drug absorption have been observed with even relatively simple injectable formulations such as aqueous suspensions. For example, the comparative bioavailability (expressed as area under the concentration versus time curve, AUC) of two ampicillin formulations (ampicillin trihydrate aqueous versus ampicillin oily suspension) following intramuscular (IM) injections was equivalent in calves but was not equivalent in swine (5). Similarly, comparing two ivermectin formulations in swine versus cattle, Lifschitz et al. (6) observed nearly identical time to peak concentrations (Tmax) following subcutaneous (SC) injections of two formulations in cattle, but markedly different Tmax values when these two formulations were administered via SC injection to swine. In another example, cattle and swine exhibited very different in vivo drug release characteristics from an SC implant, even though these implants were administered in the identical anatomical location and at the same milligrams per kilogram dose (personal communication).
Tissue Fluid Volume
Depending upon drug aqueous solubility, differences in fluid volume at the injection site may influence in vivo dissolution characteristics and therefore, the rate at which the drug is absorbed (7,8). Accordingly, when using animal models to test the in vivo drug release characteristics of parenteral formulations, it is important to appreciate site-specific differences in local interstitial fluid volume (9). For example, differences in the fluid volume per wet weight in tissues of rats ranges from about 0.05 mL/g wet weight for muscles (e.g., gastrocnemius, semimembranous, and tibialis anterior muscle) to over 0.4 mL/g wet weight of skin (hindlimb skin and back skin).
Interstitial Matrix Composition
Drug absorption following IM and SC administration of solutions and suspension can be influenced by such factors as capillary permeability, local blood flow, interstitial diffusivity, and the characteristics of the tissue barrier to solute and solvent spread. This point was demonstrated by the changes in parenteral drug absorption occurring when the integrity of the interstitial network was altered through the addition of hyaluronidase, which degrades hyaluronic acid (HA), also known as hyaluronan. Hyaluronidase-induced degradation of HA at the IM injection site resulted in a marked increase in the rate of atropine rate of absorption in guinea pigs (10). Similarly, hyaluronidase significantly enhanced the absorption of IM administered [14C]inulin but had little influence on the absorption of [3H] water in the gastrocnemius muscle of rabbits (11). In fact today, hyaluronidase is used as an adjuvant to increase the absorption and dispersion of some injected drugs and radiopaque agents (12).
Along with collagen, the glycosaminoglycans (GAGs) restrict molecular movement via exerting a size exclusion function within the interstitial tissues. Among the GAGs, HA (non-sulfated GAG) significantly influences the hydration and physical properties of tissues, is integral to maintaining tissue structure and assembly, and is one of the chief components of the extracellular matrix. Although widely distributed throughout connective, epithelial, and neural tissues, HA content can differ across various anatomical regions or across animal species (within an anatomical region). Differences were observed in the HA content of rat skin versus muscle (9). In terms of interspecies differences, post-mortem HA content in the lamina propria of the vocal folds of dogs, humans, pigs, and ferrets were compared (13). The milligram amount of HA in pigs, dogs, and ferrets was approximately two-, three-, and fourfold greater than that observed in people. The corresponding distribution of HA across the top, middle, and lower-most layer of the lamina propria also differed across species. A markedly lower content was observed in the top layer of the lamina propria of dogs. A greater fraction was located in the middle lamina propria layer of the ferret. In contrast, the HA content is relatively evenly distributed across all three layers in people.
Although the composition of HA appears to be consistent across animal species, there may be differences in the molecular weight of this polymer across anatomical regions within an individual and between species (14,15). High-molecular-weight HA occurs in the loose connective tissue and has the role of maintaining cell integrity and water content. Whether or not IM or SC drug absorption characteristics vary as a function of HA, composition across animal species or administration site has not been clearly demonstrated. Nevertheless, depending upon the size, charge, and hydrophilicity of the drug molecule, an influence of these compositional differences on drug movement and absorption should not be discounted.
The smaller HA fragments form a highly complex intercellular signaling system (16). HA interacts with cells during healing, inflammation, and cancer by binding to cell surface receptors such as cluster determinant 44 (CD44) and receptor for HA-mediated motility. This HA–receptor interaction influences cell proliferation and movement and has implications in inflammation, allergic responses, neutrophil and T lymphocyte recruitment, atherosclerosis, injury repair, and autoimmune diseases, (17–19). CD44 has recently gained a great deal of attention owing to its apparent role in cell migration, tumorigenesis, metastasis, and regulation of immune responses (18,20). In fact, degradation of the HA matrix by exogenously added hyaluronidase has been observed to decrease tumor growth and facilitate anti-tumor activity of cancer therapeutics, and antibodies that block HA binding to CD44 can inhibit tumor growth and invasion (21).
HA is currently being explored as a backbone to which drugs (e.g., siRNA) can be conjugated with the purpose of targeted drug uptake into tissues that over-express HA receptors such as CD44. Once internalized, the drug can be released from HA through hydrolytic processes. Drug–HA conjugation can also be used to reduce drug toxicity, and to increase drug aqueous solubility (21). Targeted drug delivery systems, such as liposomes, dendrimers, micelles, and polymeric and ceramic nanoparticles, are being developed that bind to over-expressed CD44. Products are often formulated to provide an extended duration of residence within the systemic circulation and tissues or as extended release formulations, such as novel depot delivery systems (22). The ability of cells to interact with and internalize these systems will be a function of the magnitude of CD44 expression at the cell surface (23).
Clearly, interspecies differences in CD44 expression and binding affinity will influence the accuracy of preclinical species data generated during product development. Although published literature provided little insights into the species differences in the affinities and activities of these receptors, distinct area of low DNA sequence homology (within the region coding for the extracytoplamsic domain) has been identified when CD44 genes were compared in bovine, human, and mouse (24). It is thought that the low similarity within this region may contribute to species-specific types of cellular adhesion.
In addition to genetic coding, of importance to CD44 structure and function are species-specific splice variants and protein posttranslational modifications. Theoretically, hundreds of CD44 isoforms can be generated by alternative splicing where exons are inserted in different combinations between the two constant regions of the molecule. In addition to these splice variants, posttranslational modifications by glycosylation and GAG attachments can modify the function of the CD44 isoform (25,26). Thus, it is interesting to consider potential implications of the patterns of similarities and differences in some of the splice variants observed both across tissues of the same species (dog) or between species, such as the rat, dog, and human (27). In recognition of the potential involvement of CD44 variants in the progression of various types of tumors, since CD44 function is ultimately controlled by its posttranslational modifications (28), it is important to question whether or not (and the conditions under which) interspecies differences in CD44 primary structure can influence the accuracy of therapeutic predictions generated with preclinical species.
Movement of Drug from the Interstitium to Blood
Once a compound is solubilized, its uptake into the general circulation (transcellular transport) is primarily controlled by blood perfusion and not by the permeability of the capillary membrane (perfusion-limited transport). Conversely, small hydrophilic molecules are dependent upon paracellular transport processes. Therefore, the rate of tissue uptake is controlled by capillary membrane characteristics (e.g., pore diameter). These situations reflect diffusion-limited transport. For the latter, the magnitude of tissue uptake of the drug will vary across animal species and the concentration of the small molecule.
To demonstrate this point, Watson (29) modeled paired-tracer data (lactate and mannitol) in an effort to predict the likely differences in hindlimb skeletal muscle capillary permeability-surface area product (PS) in cats (their unpublished data) and rats (from previously published data). Because mannitol (molecular weight = 182) is twice the size of lactate (molecular weight = 89), mannitol is expected to diffuse more slowly than lactate from the blood. That is, mannitol is expected to have a lower permeability-surface area (PS) than lactate. The higher the PS, the more readily a drug will diffuse between blood and the interstitial fluids, and vice versa. With the assumption that the intracapillary concentrations equilibrate rapidly as compared with the extravascular concentrations, the amount of drug moved from the blood to the target tissue per unit time can be described as follows:
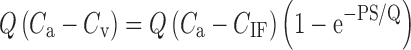 |
where Q = blood flow, Ca and Cv are the arterial and venous tracer concentrations, and CIF is the tracer concentration in the interstitial fluids (29). Applying this relationship, Watson showed that the PS for lactate and mannitol was approximately 2.6 times greater in the rat as compared to cats.
The relevance of capillary surface area in terms of its effects on drug tissue penetration was exemplified in the assessment of ciprofloxacin tissue to plasma ratios in lean versus obese human subjects. Ciprofloxacin was administered as a single intravenous (IV) infusion of 2.85 mg/kg over 20 min. Ciprofloxacin concentrations were measured both in plasma and interstitial fluids (microdialysis in the middle third of the anterior aspect of the right thigh). Markedly higher tissue penetration (based upon the ratio of ciprofloxacin concentrations in the interstitial fluids versus the plasma) was observed in the lean versus the obese patients. Consequently, the AUCtissue/AUCplasma ratio, was lower in obese than in lean subjects (0.45 ± 0.27 vs 0.82 ± 0.36, P < 0.01, for obese versus lean subjects, respectively). The comparative steady state volumes of distribution (expressed as liters), when corrected for kilogram body weight, were similar, indicating that the overall distribution volume was markedly lower in obese than in lean subjects. This difference in drug distribution volume was concluded to reflect differences in the capillary surface to tissue volume ratio and accordingly, lower tissue perfusion in the obese versus lean subjects (30).
Although interspecies differences in body composition may likewise lead to differences in blood–tissue drug exchange, less variability may be anticipated when comparisons are considered relative to lean body mass. Schmittmann and Rohr (31) suggested that the PS of skeletal muscle capillaries tends to be very similar across several species. These investigators observed that in humans and animal species, there is virtually the same negative linear correlation between PS and molecular weight. However, interspecies differences are seen in lymphatic drug absorption (32–34). For example:
Rats: Macromolecules tend to be absorbed primarily via capillaries.
Sheep, dogs, and pigs: Macromolecule uptake is often highly dependent upon lymphatic absorption.
These observed interspecies differences may be attributable to a variety of factors including (32–34):
Site of administration (e.g., thigh of rat versus interdigital space of the hind limb of sheep).
Actual dose-corrected amount of drug (where higher concentrations of molecules such as insulin used in sheep can lead to aggregate formation, which then encourages lymphatic absorption).
Lymphatic vessel used for sampling.
Use of anesthesia.
Freedom of movement.
In regard to the impact of administration site, using radio-labeled liposomes, Oussoren et al. (35) demonstrated that in rats, the percentage of residual liposomes at the injection site was highly dependent upon the site of injection (flank, dorsal side of the foot, footpad). The greatest amount of residual drug remaining at the injection site occurred when the liposomes were administered into the flank region. The highest percent absorbed occurred when the drug was administered into the footpad. These differences reflected the magnitude of lymphatic flow at the various sites. When the pressure in the interstitial fluid increases above that present within the lymphatic capillaries, the intercellular junctions in the lymphatic capillaries open up, leading to an increase in lymph flow (36). The injected fluid itself can lead to an increase in interstitial pressure. Therefore, the footpad and dorsal aspect of the rat foot, which has little adipose tissue, exhibits a substantial increase in interstitial pressure as a result of the injection. On the other hand, the tissue in the flank region is loose, allowing for negligible changes in tissue pressure upon injection and therefore, a lesser degree of liposomal uptake into the lymphatics.
Similarly, in sheep, injection site was associated with substantial differences in the absorption rate of darbepoetin alfa (37), a protein whose interstitial absorption occurs primarily via the lymphatics. In fact, at least in part, marked discrepancies between the time to peak systemic darbepoetin alfa exposure in sheep versus humans appears to be related to the difference in site of injection used in the animal model (time to peak concentrations (Tmax) = 8 h) versus human subjects (Tmax = 32–72 h). When darbepoetin alfa was injected into the interdigitial space (which is where it is usually administered when using the sheep model) or in the shoulder, Tmax occurred at approximately 8 h following SC injection. However, peaks occurred within 18–72 h after SC administration into the sheep abdomen. Human SC administration of this compound is typically in the thigh, shoulder, or abdomen. Moreover, comparing the relative fraction of the administered dose absorbed over 90% was absorbed when injected into the interdigital space while only about 67% was absorbed after abdominal injection. Lymphatic uptake remained the primary route of absorption at both injection sites. It was hypothesized that this site-dependent difference in uptake may reflect the level of muscle activity associated with the various regions, with higher blood and lymph flow occurring at sites with high muscle activity. Thus again, we see the possible bias in drug absorption kinetics occurring as a function of injection site.
Site dependence also varies as a function of animal species (36). For example, lymphatic absorption and lymph node uptake after SC injection into the flank of guinea pigs is much higher as compared to that observed when the liposomes are injected into the flank region of rats. This difference in lymphatic uptake is believed to reflect structural differences in the interstitial tissue, where the rat provides less dispersion area as compared to the guinea pig.
Impact of Interstitial Differences on the Absorption of Large Molecules
With this background in mind, it is not surprising that the absorption characteristics of some human parenteral formulations may be poorly predicted from data generated in preclinical species. The PK of human recombinant-erythropoetin-β (rHEPO) following IV and SC injection was studied in dogs, rats, and mice. These results were compared to data previously collected in humans (38). Although the slow absorption observed in dogs was the most similar to human absorption kinetics, the estimated absolute bioavailability (F) of the SC dose was markedly higher in dogs as compared to humans. These relationships are presented in Table I.
Table I.
PK of rHEPO Following IV or SC Injection (Values Fitted to a Simple One-Compartment Model)
| Species | Dog (IU/kg) | Tmax (h) | T1/2 elimination (h) | %F | T1/2 absorption (h) | |
|---|---|---|---|---|---|---|
| IV | SC | |||||
| Dog | 500 (0.5 mL/kg) | 18.1 | 6.7 | 24.6 | 80.3 | 27.7 |
| Rat | 1,000 (1 mL/kg) | 12 | 13.8 | 12.6 | 76 | 5.4 |
| Mouse | 50 (10 mL/kg) | 7 | 11 | 17.7 | 73 | 0.8 |
| Mouse | 150 (10 mL/kg) | 3 | 7.7 | 9.7 | 68 | |
| Mana | (see note in legend) | 7.1 | 34.6 | 22.8 | ||
In all cases, serum concentration was corrected for background levels of endogenous erythropoietin. Animal results are compared against published information on the PK of this compound in humans. Based upon the information provided by Bleuel et al. (7). Note that in reviewing available information in the citations used to derive human dose, it would appear that most of the modeling was based upon doses of approximately 250 IU/kg or less
aWeighted average of means from 13 publications (original data and calculations not provided)
As we examine the interspecies differences in Table I, there are some important caveats to consider when assessing the relevance of these reported interspecies differences. The observed interspecies differences in F may be related to the anatomical site of administration (note that the injection sites used in the investigation by Bleuel et al. were not provided in the study report). Furthermore, while these authors suggest that the human bioavailability is only about 34.6%, Woo and Jusko (39) suggest that F can go as high as 100% in humans and monkeys when the SC dose of rHEPO-α ≥ 2,400 IU/kg. Woo and Jusko observed that F in humans and monkeys increase with dose. However, a similar observation was not observed in rats (where F did not exceed 58%, regardless of dose). Interestingly, the Woo and Jusko estimate of F for rats was somewhat lower than the 76% estimated and reported by Bleuel et al. (38). In this regard, it should be noted that the two evaluations employed different forms of rHEPO. Although the chemical characteristics of rHEPO-β, which was used in the study by Bleuel et al., and rHEPO-α, studied by Woo and Jusko (39), are highly similar (40); we cannot exclude the possibility of differences in absorption kinetics of the α and β forms of this recombinant proteins.
Nevertheless, the importance of potential mechanistic differences in drug absorption, as a potential source of bias when extrapolating bioavailability across animal species, was underscored by the study of Woo and Jusko (39). In fitting previously published PK data, they observed a biphasic SC drug absorption process: a zero-order component which is believed to be related to the lymphatic uptake of this therapeutic protein and a first-order process that appeared to be related to tissue size and blood flow. Unlike the zero-order component, the first-order process appeared to be inversely related to body weight. However, the lymphatic uptake was difficult to predict across animal species. Their evaluation suggested that >68% of the SC dose was absorbed by the zero-order process in rats and people but only about 35% of the total absorption was associated with a zero-order process in monkeys. In sheep, about 75% of SC-administered rHEPO-α is believed to be absorbed via the lymphatics (41). Because lymphatic flow is highly dependent upon vessel density and muscle movement, the zero-order absorption component is likewise highly dependent upon animal species, muscular activity, and site of drug administration.
Interspecies differences in SC absorption were also observed when insulin aspart (IA) and human-soluble insulin (HI) were administered via IV and SC injection to rats, dogs, and pigs (42). Because of the degree of similarity of pig and human SC lipid structure, it was predicted that pigs would provide a satisfactory model of human drug absorption. Similar to what was observed in people, the investigators note that swine SC IA was absorbed more rapidly than SC HI. Although these two insulins depleted at different rates both in swine and humans, the terminal elimination half-lives (T1/2) of IA and HI following SC injection were nearly identical both in dogs and rats. It was suggested that the interspecies difference in drug-related SC absorption kinetics reflected the difference in amount of SC fat. It was suggested that the lower amount of SC fat in dogs and rats allowed for the faster absorption of IA as compared to what was observed when IA was administered to pigs.
FACTORS INFLUENCING DRUG MOVEMENT INTO AND OUT OF THE BLOOD
Plasma/Serum Composition
Interactions between drugs and drug delivery systems with host serum components can influence systemic drug exposure which, in turn, can influence product safety and/or effectiveness. Therefore, unique species plasma lipid and protein characteristics can likewise lead to species-specific differences in drug binding, serum transport, and ultimately its PK. In this section, evidence supporting the importance of recognizing the potential impact of these species-specific plasma/serum attributes is discussed.
Interspecies differences in plasma composition include the relative concentration of albumin and α1-glycoprotein (43) (Fig. 1). Although total plasma protein content is greatest in the dog, the rat and mouse have much higher levels of α1-glycoprotein.
Fig. 1.

Interspecies comparison of plasma protein composition. Based upon the information from Davies and Morris (43)
Brocks et al. (44) examined the lipoprotein cholesterol (esterified and unesterified), triglyceride, and protein concentrations (milligrams per deciliter) in the various fractions of fasted, normolipidemic human, dog, and rat plasma samples. The dog possessed noticeably higher total protein levels in the lipoprotein-deficient fraction (LPDP) and in the high-density lipoprotein (HDL) fractions as compared to that observed in rats and humans (Fig. 2). Differences in plasma lipoprotein levels and the potential ramifications of these differences are expanded upon later in this section.
Fig. 2.

Mean plasma lipoprotein composition in dogs, rats, and humans (Y-axis decreased for visualization purposes. Dog HDL = 171.8 mg/dL). Based upon information by Brocks et al. (44) and Ramaswamy et al. (56). TRL triglyceride-rich lipoprotein fraction, LDL low-density lipoprotein fraction
Differences in the amount of total body water and extracellular body water (expressed as milliliters) can be explained on the basis of allometry. Because the observed allometric exponent approaches a value of 1.0, it would appear that extracellular and total body water varies in direct proportion to species body weight.
Plasma Protein Binding
With the 80% homology of serum albumin amino acid sequences of humans, rats, bovine, rabbit, and dogs, interspecies differences in binding site activity has been observed. Whether such differences are an experimental artifact or a valid representation of in vivo binding differences remains a point of debate. Using equilibrium dialysis for approximately 12 h, the binding of marker compounds (phenylbutazone, ibuprofen, diazepam, and warfarin) were explored (45). The binding to site 1 (by hydrophobic or by static interactions) for warfarin and phenylbutazone appeared to be effectively absent in dogs, a finding that was attributed to the canine tertiary structure near that binding domain. However, site I binding was high in humans. In contrast, with respect to site II binding kinetics for drugs such as diazepam and ibuprofen, the albumin derived from cattle, rats, and rabbits were markedly different from that of dogs and humans. Canine and human site II binding appeared to be very similar.
Interestingly, in some cases, observed interspecies differences in plasma protein binding may be an experimental artifact. Sample processing can lead to the loss of CO2 which in turn can markedly increase the pH of ex vivo blood samples. This progressive increase in the alkalinity can occur within only a few hours after blood has been collected (46). Kochansky et al. (47) showed that the pH of fresh rat plasma (incubated at 37°C) increased from 7.2 at time zero to 8.0 (within 60 min of incubation), and it went up to 8.7 at 250 min of incubation. Similar changes were observed in samples of frozen human plasma.
Using plasma from humans, rats, and dogs, Kochansky et al. (47) compared the relationship between the free fraction (of 55 drugs in human plasma and 14 drugs in dog and rat plasma) when the plasma samples were incubated with 10% CO2versus the free incubated in air. Thus, in this study, the fu,p ratio effectively examined the ratio of the unbound fraction estimated at pH 7.2 versus 8.8. The effect of pH on plasma protein binding of the drug (fu,p) showed greater similarity between humans and dogs than it did between humans and rats. The greater human–dog versus human–rat correlation appeared to be primarily a function of the unbound fraction at pH 7.2. The magnitude of correlation observed in both sets of interspecies comparisons decreased substantially when the pH was allowed to increase to 8.6. Thus, a failure to adequately control the pH of the incubated samples may contribute to differences in the free faction of drug observed in human versus animal plasma. Since anionic compounds tend to present with lower fu,p ratio than do the cationic compounds, this disparity may be a particularly important source of bias when evaluating the percent protein binding of anionic compounds.
In addition to the percent protein binding, for highly bound compounds, binding affinities need to be considered. If sufficiently low, the high protein binding could have a beneficial effect, serving as a mechanism for systemic transport of the drug. Conversely, a tightly bound compound could render the drug molecule ineffective. Pistolozzi and Bertucci (48) examined the relative binding affinities of a variety of compounds to albumin derived from humans, dogs, rats, and bovine. They observed that the relative binding affinities varied both as a function of drug, binding site, and animal species. They also observed a species-specific stereoselectivity of drug binding to the albumin (which in turn is a function of the protein conformational change induced by the ligand binding). For example, the binding affinity of phenylbutazone to the serum albumin of cattle and rats were approximately tenfold greater than that of humans and dogs. Nearly opposite relationships were observed for the binding affinity of diazepam (where the binding to dog and human albumin was approximately tenfold greater than that observed in cattle and rats). The authors noted that the use of circular dichroism spectroscopy enabled them to selectively monitor binding to stereospecific sites on the albumin.
Other examples of differences in plasma protein binding affinities are provided below.
Insulin
The prolonged residence of fatty acid-acylated insulin has been attributed to its slow dissociation from host albumin. The magnitude of this binding affinity varies markedly across animal species. Comparing insulin binding to the albumin from humans, pigs, and rabbits (expressed relative to the binding affinity to human albumin), the relative binding varied from 1:1.5:35, respectively. The much higher binding affinity of this compound to the serum albumin of rabbits, as compared to that of other species, is believed to be responsible for the diminished but prolonged effects in rabbits as compared to those observed in pigs following SC injection. These findings represent the importance of species differences in ligand binding when evaluating highly albumin-bound peptide derivatives (49).
Contrast Agents
Understanding interspecies differences in the binding affinities to the various protein constituents is particularly important when developing radiopharmaceuticals. For example, the interaction between neutral lipophilic copper chelates with serum albumin was found to be highly compound and animal species specific. Using ultrafiltration binding assays, Basken et al. (50) observed that binding of pyruvaldehyde bis(N4–methylthiosemicarbazonato)copper (II) (Cu–PTSM) to pig serum substantially exceeded the binding to pig albumin (binding evaluations conducted under room air). There were also tremendous interspecies differences in the relative drug binding to the various serums versus to albumins (Fig. 3). These interspecies differences in protein binding affected not only the tissue distribution and half-life of these compounds, but also the signal to noise ratio associated with the use of these contrast agents (51).
Fig. 3.

Interspecies differences in the relationship between in vitro serum and serum albumin binding of Cu-PTSM. Based upon the data from Basken et al. (50)
Propanfenone
Propanfenone is an anti-arrhythmic drug that binds largely to α1-glycoproteins. Markedly different in vitro protein binding was seen in the plasma of eight mammalian species (including humans). At concentrations of 250 ng/mL, in vitro free drug concentrations ranged from as low as 1.5% (mouse, rat, dog, and horse) to as high as 6% (rabbit). At that concentration, human free drug fraction was approximately 2.5%. At higher drug concentration (2,000 ng/mL plasma), in vitro free drug concentrations remained below 3% in the rat, dog, and cow, but rose to as high as 14% in goats. Thus, nonlinear protein binding was observed in some (e.g., mouse, goat, horse) but not in other (e.g., human, dog, rabbit, bovine) species (52). All studies were conducted under room air conditions.
Alendronate
Differences in serum drug–protein interactions may also be attributable to the presence of substances that compete for the protein binding site. Alendronate (an inhibitor of osteoclast-mediated bone reabsorption) binds both to plasma proteins and to bone. Irreversible binding to bone constitutes the primary mechanism of drug clearance from the central compartment (53). At pH 7.4, relatively low plasma protein binding was seen in dogs, but high protein binding was observed in rats. This interspecies difference in protein binding was, at least in part, attributable to the apparent presence of dialyzable displacers(s) in dogs that were absent in the blood of rats (based upon in vitro experiments). The addition of calcium to the dog plasma sample diminished the effect of the displacer(s).
It is interesting to note that this difference in factors influencing drug displacement may affect not only interspecies predictions but also predictions within species. For example, in an abstract presented at the 2009 the International Conference on Antimicrobial Agents and Chemotherapy, Bonapace et al. (54) reported that there were apparent differences between total calcium concentrations versus ceftriaxone precipitation in the plasma of neonates versus adults. In neonatal plasma, a substantially greater proportion of drug precipitated in vitro as compared to that observed in vitro in the plasma of human adults. Reasons for this observed disparity were not determined.
Plasma Lipoprotein Composition
The various plasma lipoproteins include:
High-density lipoproteins (HDL)
Low-density lipoproteins (LDL)
Lipoprotein-deficient plasma (LPDP)
Triglyceride-rich lipoproteins (TRL)
The interaction between lipoprotein fraction and lipophilic compounds can influence drug PK. This may be due to lipoprotein-drug binding, which can facilitate receptor-mediated uptake of drug into tissues or can lead to restricted availability to the various tissues (55). Thus, it can lead to disease-related changes in PK in people or to interspecies differences in drug PK. For example:
Although still controversial, there is evidence suggesting that hypolipidemia (particularly hypocholesterolaemia) can lead to increased cyclosporine toxicity while hyperlipidemia can lead to a decrease in drug effectiveness.
Amiodarone and halofantrin exhibit significant decreases in drug elimination rate and tissue distribution in the presence of elevated plasma lipoprotein levels.
The renal toxicity of amphotericin B is increased in the presence of hyperlipidemia, presumably due to decreased drug tissue distribution and consequently, increased amounts of drug being presented to the kidney.
Although free concentrations of nifedipine decreases in the presence of hyperlipidemia, it also leads to a prolongation of systemic residence time which, in turn, allows for improved drug effectiveness.
These three drugs are discussed in more detail below.
Halofantrine
In studying the distribution of halofantrine (HF), a highly lipophilic compound possessing a chiral center, marked interspecies difference in the distribution pattern of R(−) and S(+) enantiomers across the various lipoprotein fractions were observed in fasted, normolipodemic people, dogs, and rats (44). Although all three species had a majority of the (−)HF enantiomer in LPDP, which includes α-glycoprotein and albumin, far more (−)HF was present in the lipoprotein-containing fractions of human as compared to that in dog. In the HDL fractions of two out of the five rats examined, concentrations of both enantiomers were below the analytical limit of quantification (25 ng/mL). For the (+) enantiomer, there were no consistent tends in the disposition within the plasma of rat, dog, and human. While (+)HF was recovered primarily in the lipoprotein rich fractions of dog and people, it was recovered primarily in the LPDP fraction in rats. Across all three species, the dog exhibited the greatest stereospecificity in drug distribution (Fig. 4a–c).
Fig. 4.

Distribution of halofantine (HF) enantiomers following in vitro incubation in normolipodemic human, dog, and rat plasma (values expressed as percent initial HF concentration at beginning of incubation). a distribution of (+/−) HF; b distribution of (−)HF; c distribution of (+)HF. Based upon the work by Brocks et al. (44). HDL high-density lipoprotein; TRL triglyceride-rich lipoprotein. LDL low-density lipoprotein. LPDP lipoprotein-deficient plasma
Nystatin
Interspecies differences in the lipoprotein composition of the various plasma fractions can affect the partitioning of free and liposomal nystatin concentrations across the various lipoprotein fractions of rats, dogs, and humans (56). As the amount of total triglyceride in the LDL fraction increased (rat being lowest, human being highest), the amount of nystatin recovered within the LDL fraction proportionally decreased. In general, both free and liposomal nystatin distributed primarily in the LPDP fraction. However, a greater percentage of nystatin was in the HDL when formulated as a liposomal preparation as compared to when it was presented as the free drug. Moreover, owing to differences in the composition of the various lipoprotein fractions, statistically significant differences between dogs and humans were observed when examining the percent of free and liposomal drugs in the LPDP (human greater than dog) and in the HDL (dog greater than human) fraction (56).
Amphotericin B
It has been proposed that formulation-related differences in drug partitioning across the various lipoprotein fractions may, at least in part, be responsible for amphotericin B formulation-related differences in drug safety and effectiveness (55). The use of a liposomal formulation (consisting of amphotericin B and dimyrystioylphosphatidyline glycerol, ABLC) resulted in more than 90% of the ABLC being associated with the HDL fraction when incubated in human plasma. In contrast, 75% of the amphotericin B, formulated as a deoxycholate dispersion, was associated with the LPDP when incubated in human plasma. When tested in rabbits, the ABLC showed no evidence of renal changes but those animals receiving the deoxycholate dispersion did present with a decrease in renal function, suggesting a protective role of the lipid formulation. Interestingly, the toxicity of the deoxycholate dispersion was attenuated by the administration of a high-cholesterol diet, suggesting that the decrease in renal toxicity was associated with a re-distribution of the drug from the LPDP to the TRL fraction.
Encapsulation of amphotericin B into unilamellar vesicles resulted in higher plasma drug concentrations, a lower volume of distribution, and a substantial reduction in both renal and biliary amphotericin clearances (57). To more fully explore the implications of this relationship, Hong et al. (58) evaluated the in vitro and ex vivo distribution of ABLC across the various lipoprotein fractions, the plasma protein fraction distribution of the deoxycholate formulation, and the in vivo PK characteristics of the ABLC. These investigators found a statistically significant negative correlation existed between the ABLC clearance and the fraction of the total amphotericin in the HDL fraction. Conversely, a significant positive correlation was observed between the ABLC clearance and its distribution in the VLDL.
Self-Assembled Drug Delivery Systems
Interspecies differences in plasma composition can influence the stability of novel drug delivery platforms. Jin et al. (59) examined the relative stability of self-assembled drug delivery systems (SADDS) as a potential mechanism for enhancing the delivery of didanosine as a first-line defense for the treatment of human immunodeficiency virus. The objective of the SADDS technology is to provide selective drug delivery with high drug load, no drug leakage, and an extended duration of drug release at the target site.
Jin et al. (59) studied the stability of cholesteryl-succinyl didanosine nanoparticles, a type of SADDS, in the plasma from five different species, including rats, mice, rabbits, dogs, and human. The resulting T1/2 values in plasma varied from 217 h (rats) to 990 h (human), with dog plasma being the species most similar to human (dog T1/2 = 866 h). This species-specific degradation rate has also been observed by Jamal and Hawthorne (60) who noted a tenfold higher plasma esterase activity in rats versus humans. Similarly, Minagawa et al. (61) found that for certain esters, the esterase activity in rat plasma can be several hundredfold higher than what is observed in humans. Clearly, such interspecies differences could bias predictions of product performance when evaluating targeted drug delivery systems.
SPECIES-SPECIFIC DIFFERENCES IN INFLAMMATION, IMMUNE RESPONSES, AND TOXICITY
Regardless of the targeting mechanism used for site-specific drug delivery, the interaction between a particle and the host tissues will determine the successful delivery of drug to the site of action. Influencing the biological properties of these particles (which, in turn, influences the safety and effectiveness of these formulations) is the initiation of host immune-mediate responses (such as complement activation and the uptake by host phagocytic cells), the induction of hemolysis (which could be immune-mediated or related to a direct drug-erythrocyte interaction), or particle-induced thrombogenesis (which may include a secondary immune-mediated component). Particle uptake into host tissues can occur in the blood by the monocytes, platelets, and leukocytes. Within tissues, particles can interact with dendritic cells or with tissue-specific resident phagocytes (62).
Immune System Physiology and Morphology
With regard to immune responses and hypersensitivity reactions, large interspecies differences in lymphatic tissue anatomy, histology, and cellular contents can lead to challenges in extrapolating these responses across animal species. Contradictory results in immune responses can occur not only between human versus preclinical species but also across such laboratory species as mice, rats, and guinea pigs. Many of these differences are described in the review by Haley (63). For example, the pulmonary alveolar macrophages (PAMs) of humans are larger than are those of the laboratory species examined, with human macrophages having greater phagocytic capability (with regard to size of particle) as compared to that of rodents, dogs, or non-human primate species. The PAMs from mice, rats, and dogs are morphologically similar, each presenting with a narrow size frequency distribution. In contrast, while the PAMs from humans are larger than are those from the non-human primates, both species exhibit a heterogonous size distribution. The number of macrophages per unit lung area may also account for the greater ability of human PAMs to sequester foreign particles. It further appears that for some compounds, the greater the phagocytic activity, the greater the sensitivity to potential cytotoxicity.
Immunogenic responses can present as hypersensitivity reactions. These reactions often involve mass cell degranulation, which can lead to potentially life-threatening responses. Again, interspecies differences exist, with species-related variations in mast cell contents and distribution (63). While these cells have also been identified in the broncho-alveolar lavage (BAL) of normal non-human primates, normal humans, and dogs, an increase in mast cell numbers in BAL samples have been seen only in people presenting with pulmonary disease. In addition to cell number, cell contents can all vary across species. Human mast cells have the same amount of histamine regardless of anatomical location, whereas in the rat, connective tissue mast cells can have up to ten times the histamine content of rat mucosal mast cells.
With regard to anaphylactic responses and delayed hypersensitivity responses, species differences are seen in the primary shock organ, responsible antibody, and vasoactive amine (63). Thus, anaphylaxis represents a qualitatively different phenomenon across species. For example, the principal shock organ of the rat is the intestine and liver, for the mouse is vasculature and intestine, dogs are the splancnic blood vessels and for humans it is the lung, larynx, and vasculature. While the primary vasoactive amine includes histamine across all preclinical species and humans, the involvement of other vasoactive amines exhibit species specificity. In rabbits, mice, and mice, serotonin is an important mediator (not a key player in human shock response). Mice, human, guinea pigs, and rats (but not rabbits or dogs) shock responses are associated with the slow reactive substance of anaphylaxis. Kinins can also be involved in human, dog, mouse, and rats. In terms of the pivotal antibodies associated with shock, IgE is involved in all species. However, in rodents, pivotal antibodies also included various forms of IgG, the specific isoform varying in a species-specific manner.
The Immunogenicity of Therapeutic Proteins
Brinks et al. (64) recently reviewed the use of animal models to assess the immunogenicity of therapeutic proteins, noting that while most therapeutic proteins can elicit an antibody response, the clinical implications of these responses can vary dramatically. Examples of types of clinically relevant reactions include loss of compound efficacy, neutralization of the endogenous counterpart, and immune-mediated adverse events. One of the inherent problems associated with the use of animal models is the lack of cross-species protein conservation. To overcome this problem, the use of the transgenic mouse model and the use of non-human primates may be of value. However, even in these situations, the interpretation of experimental results needs to proceed with caution, with some types of predictions being more robust than others. Transgenic mice and non-human primates may have a positive predictive value for identifying the immunogenicity of neo-epitopes, the relative immunogenicity of compounds, and the breaking of tolerance (i.e., a cessation of immunological non-reactivity to a protein, often due to previous exposure to the endogenous counterpart of that protein). However, despite efforts to maximize similarity to the human, these animal models may render false conclusions with regard to the immunosensitivity potential in the patient population, the human population frequency of immunogenicity reactions, or the clinical consequence of an immune response.
Brinks et al. (64) also warned that the predictive value of animal models may be linked to potential differences in the mechanisms underlying the immune response and the lack of genetic diversity of the animal model being employed. In some case, the mouse genetic line itself may render offspring that are immunologically insensitive to a specific therapeutic protein. The authors also noted the importance of considering the experimental design, where dose, route of administration, dose frequency, and protein impurities can alter experimental outcome. Similarly, the formation of aggregates can influence immune reactions, where aggregate formation tends to render proteins more immunogenic as compared to that of the original monomer. Lastly, the authors raise the issue of antibody assays, which due to a lack of assay standardization, can (in and of itself) lead to interstudy inconsistencies in experimental outcomes.
The Complement System
Potential interspecies differences in immune responses are important to consider when animal models are used to examine the efficiency of methods for masking or camouflaging nanoparticles and liposomes from reticuloendothelial cells. Particle size and composition can lead to the binding of organ-specific opsonins, resulting in organ-specific drug sequestration (65,66). Opsonization involves the adsorption of blood components to exogenous substance (e.g., particles, micro-organisms). Complement proteins such as C3, C4 and C5, and immunoglobulins are typically involved in particle opsonization (66). Liposomal opsonins can include complement proteins and immunoglobulins (leading to uptake by the macrophages) or non-immune opsonins, which serve as ligands that interact with cellular receptors (e.g., on hepatocytes). Other variables that can influence particle immune system uptake include vesicle morphology, surface curvature and charge, lipid composition, bilayer packing, temperature-dependent packing defects, and vesicle dose (67,68).
The relationship between liposome composition and its clearance from the circulation was shown to be markedly different in rats and mice (69). In mice, the circulation time for different combinations of phosphatidylcholine (PC), phosphatidylserine (PS) dioleolphosphatidylethanolamine-N-(poly(ethylene glycol) 5000) (PEG5000-PE), cholesterol (Chol), and monosialoganglioside (GM1) was in the order of PC/Chol/GMI > PC/Chol/PEG5000-PE > PC/Chol > PC/Chol/PS. In these mice, liposomes containing 10% of GM1 or PEG5000-PE exhibited a single decline phase and terminal half-lives of about 16 h. In contrast, the same formulations in rats showed a biphasic decline (with an initial half-life of about 10 min, followed by a secondary slower rate of decline). Furthermore, at 4-h post-dose, there was a remarkable species difference in the uptake of the various liposomal formulations in the liver and spleen (Fig. 5), a finding that has been attributed to interspecies differences in the mechanisms stimulating phagocytic uptake. In mice, liver uptake did not involve the presence of specific opsonins while in rats, uptake was highly dependent upon liposomal opsonization. Furthermore, there appears to be an interspecies difference in the opsonization activity of serum, as was demonstrated by differences in the percent liver uptake in rats and mice when the various liposomes were pre-incubated in serum from mouse, rat, human, or bovine. The degree to which serum influenced uptake depended upon liposome formulation and diameter.
Fig. 5.

Relationship between liposome composition and its hepatic versus splenic uptake in rats versus mice at 4-h post-dose. Based upon the work of Liu et al. (67)
Huong et al. (70) studied the relationship between liposome diameter and cholesterol content versus complement fixation to the liposome and its subsequent systemic (renal and hepatic) clearance when administered to rats and guinea pigs (Fig. 6). Regarding in vitro interaction between the liposomes and serum, degradation occurred rapidly in rat serum but negligible in vitro degradation occurred in guinea pig serum. In contrast, despite negligible in vitro degradation in guinea pig serum, pronounced liposome degradation in guinea pigs occurred after IV injection, and this degradation was inversely related to cholesterol content (i.e., greater degradation for low cholesterol liposomes). In contrast, the IV injection of low cholesterol liposomes in rats remained in the circulation for a longer duration than did the high-cholesterol liposomes (sixfold difference in AUC values). To understand reasons for these discrepancies, rats and guinea pig liposomal clearances were evaluated with and without prior IV injection of K-76, a compound that inhibits complement fixation (C5 and C3). Although K-76 successfully exerted an anti-complement effect in both animal species, it only impacted the renal and hepatic clearance of the liposomes in rats, indicating that influence of C3 and/or C5 on liposomal opsonization and systemic clearance differed in rats and guinea pigs.
Fig. 6.

Parameter values associated with the administration of liposomes in guinea pigs and rats. Based upon data by Huong et al. (68). AUC (% administered dose × min/mL), CL (milligrams per minute); CLh hepatic clearance; CLr renal clearance; Low HC low-cholesterol liposome, size = 621–800 nm; composition ratio 6/2/1. High HC high-cholesterol liposome, size = 441–800 nm; composition ratio = 4/4/1. Ratio = hydrogenated egg phosphatidylcholine/cholesterol/diacetylphosphate
Species difference in the complement system can influence nanoparticles that are intended for targeted delivery of anticancer therapies. Palmitoyl rhizoxin, a potent anti-tumor compound, was prepared as an emulsion containing particles of varying diameter. Although the clearance of the drug (when administered as an IV colloidal solution) showed a strong allometric relationship in rats, dogs, mice, and rabbits, it was cleared far more rapidly in dogs as compared to the other animal species when it was injected as an oil in water emulsion. Nearly 100% of the injected emulsion accumulated in the canine liver within 6 h after injection, while less than 20% of the administered dose was recovered in the livers or mice and rats. The mechanism for this difference was evaluated by comparing the process of emulsion opsonization when incubated in fresh plasma from mice, rats, dogs, rabbits, and humans (5 min at 37°C). The resulting opsonized particles were subsequently injected into rats or rabbits, and the systemic clearance and tissue accumulation (liver and spleen) was studied. Only those particles incubated in dog plasma significantly increased particle clearance above that observed when the emulsion was injected IV into rats and rabbits. The accumulation of the canine-opsonized emulsions in the liver of rats and rabbits paralleled that observed when the emulsion was administered via IV injection to dogs, suggesting that dog plasma, as compared to that of the other species examined (including human), possessed strong opsonizing activities for the emulsion. Consequently, there are marked interspecies PK differences when palmitoyl rhizoxin is administered as an emulsion (71).
Formulation-by-species interactions can also result from differences in complement-activation-related pseudoallergy (CAPRA), a non-IgE-mediated hypersensitivity non-allergic reaction that can lead to anaphylaxis. Interspecies differences in CAPRA-mediated anaphylactic responses include (72,73):
A much a greater hypersensitivity to Cremophor EL (CrEl) and polysorbate 80 (Tween 80) in dogs as compared to that observed in other species.
A substantially lower risk of CrEl-induced cardiovascular distress in pigs, even when administered large amounts of CrEl. However, pigs tend to exhibit the greatest hypersensitivity reaction to multi-lamellar phospholipid vesicles (liposomes) and liposome encapsulated hemoglobin (e.g., cardiovascular reaction, anaphylaxis, death).
A dose-associated hypersensitivity reaction to liposomal phospholipids (e.g., Doxil).
IV injection of some liposomal drugs, diagnostic agents, micelles, and lipid-based nanoparticles are known to cause CAPRA in some human patients due to activation of the complement system, leading to C5a and C3a liberation. Observed anaphylactic reactions happened rapidly after injection. As shown in Fig. 7, in an interstudy comparison of anaphylactic reactions to various doses of Doxil in rats, dogs, people, and swine, it was found that rats were markedly less sensitive to liposomal phospholipids as compared to dogs, swine, and people (73). This assessment was based upon the phospholipid to doxorubicin weight ratio in Doxil (7:1), the range of doses tested (across multiple investigations within each species) and the corresponding estimate of the total amount of phospholipid in the circulation.
Fig. 7.

Interstudy comparison of reported phospholipid doses that have been associated with hypersensitivity reactions. In contrast to dogs, swine, and humans, rats do not exhibit anaphylactic responses until the doses are markedly elevated. Figures are based upon data reported by Szebeni et al. (73). The primary graph provides the lipid dose dependence of hypersensitivity reaction to phospholipids injection in humans, dogs, and pigs. Rats are not included in that comparison to allow for visualization of the differences between these three species. However, the insert is included to demonstrate the relative insensitivity of rats to the injected phospholipids
Liu et al. (74) studied interspecies differences in complement-mediated immune damage to hydrogenated phosphatidylcholine-based liposome by examining the damage to liposomes (expressed as percent drug released) occurring when liposomes (identical in terms of lipid composition, surface property, size, and charge) were incubated in fresh serum from rabbits, guinea pigs, mice, dogs, rats, bovine, and humans. Obvious leakage occurred when the liposomes were incubated in fresh rat or bovine serum. However, when incubated with fresh serum from humans, rabbits, guinea pigs, mice, and dogs, almost no liposomal leakage was observed. Leakage in rat and bovine serum was completely inhibited by heating at 56°C for 30 min or by treatment with EDTA. However, neither was inhibited by treatment with EGTA/Mg2+, suggesting that the liposomal lysis was attributable to complement activation via the alternative pathway rather than via the classical pathway. Furthermore, these same authors demonstrated that the in vivo uptake of liposomes by liver Kupffer cells differed among mice, rats, and rabbits due to species-related differences in the uptake characteristics of the Kupffer cells rather than to species-related differences in the density of the Kupffer cells (75).
Biocompatibility and Inflammatory Responses
Upon product exposure to the interstitial fluids, the excipient, N-methyl-2-pyrrolidone (NMP) precipitates into a semisolid mass that traps the drug, leading to slow and sustained drug release from this in situ-formed implant. Although NMP is well tolerated in Rhesus monkey (only a mild local tissue response without visual inflammatory effects) it is very poorly tolerated when administered to dogs and cats (76). The in situ polymer precipitating solution, NMP plus dimethyl sulfoxide, exhibited myotoxic potential in rats but not in rhesus monkeys (77).
Difference in inflammatory responses can influence the viability and release characteristics of implanted drug delivery systems. Using tissue cage models, Leppert et al. (78) explored the relative plasma concentrations of cefoxitin (hydrophilic) or ivermectin (lipophilic) following either subcutaneous injection or drug release from an implanted tissue cage that was primed with drug on days 11, 32, and 60 following its implantation in sheep, cattle, dogs, and rat. Cefoxitin and ivermectin bioavailability from the tissue cage progressively decreased in dogs and rats as a function of the in vivo tissue cage residence time due to the formation of dense, avascular fibrous tissue surrounding the interior and exterior surfaces of the tissue cage. Conversely, the bioavailability of these two compounds was not compromised in sheep. In fact, an increase in drug bioavailability was observed in sheep, particularly in the case of ivermectin. The authors suggest that the increase in ovine drug absorption may reflect a high degree of neovascularization during wound healing and during tissue growth inside of the cage. Ultimately, these observed interspecies differences in foreign body responses could have an important influence on the interspecies extrapolation of material biocompatibility.
FACTORS THAT CAN INFLUENCE ORGAN AND TARGET TISSUE DRUG DELIVERY
Tumor-Directed Drug Delivery
One of the factors influencing drug delivery to tumors is the difference between vascular hydrostatic pressure versus the tissue osmotic pressure (i.e., the colloidal osmotic pressure) within the tumor. In fact, it is the interstitial fluid pressure (IFP) that is believed to be one of the primary factors limiting the delivery of large therapeutic molecules to solid tumors, including those in the brain (79). In humans, the normal tissue IFP is approximately 0 mm Hg while that of the various carcinoma types ranges from 14 to 30 mm Hg. This pressure is uniformly high throughout the tumor core, but drops precipitously towards the tumor periphery or in the normal tissue immediately surrounding the tumor (80–82). Therefore, understanding the pressure differentials between interstitial versus microvascular spaces and how these differences vary across animal species and tumor types is one factor to consider when using animal models to assess the targeted delivery of cancer therapeutics.
Osmotic pressure is influenced by protein composition and type. Normally, large proteins are trapped in blood and cannot cross into the interstitium, thereby allowing for the development of a blood-tissue osmotic pressure gradient. However, the factors influencing pressure changes in cancerous tumors are not simple and therefore will influence the success of targeted drug delivery. The relationship between protein molecular weight, protein concentration, and tumor type was examined by Stohrer et al. (82) using implanted human tumor cell lines in nude mice. Protein concentrations (using a wick method) were evaluated when the resulting tumors reached a diameter of 8.5 to 10 mm. Generally, a gradient of the large protein molecules (>25,000 molecular weight) remained between blood and tissue, with blood concentrations tending to exceed that in tissues. However, this gradient tended to be reversed (i.e., higher in tumor tissues as compared to blood) for the very small protein molecules (<25,000 molecular weight). The greater concentration of smaller molecular weight proteins in tumors likely reflected protein degradation within necrotic tumor regions and other tumor-associated proteins. Osmotic pressure and the molecular weight of the protein fraction tended to vary as a function of tumor type.
One of the concerns expressed by Stohrers et al. (82) was that literature examples of osmotic pressure in tumors were previously examined using an empirical equation that failed to acknowledge interspecies differences in the proportions of the various proteins. Based upon work published in the 1970s it is clear that the relative amounts of the various proteins (and therefore plasma osmotic pressure) tend to be animal species specific. Across a variety of plasma samples including that from cattle, horses, dogs, and cats, albumin exerted over fourfold greater colloidal osmotic pressure as compared the γ-globuins. In fact, the concentration/pressure relationship of both proteins tended to increase in a greater than linear manner, with the relationship between albumin osmotic pressure being markedly steeper than the corresponding relationship is for the γ-globulins. Nevertheless, for any given total protein concentration, there was an apparent plateau in the relationship between the albumin/globulin ratio and the corresponding osmotic pressure (83).
Use of algorithms for estimating this osmotic pressure, such as the Landis-Pappenheimer equation, relies upon an assumption that this pressure varies only as a function of the total protein content. Accordingly, it fails to consider interspecies differences in plasma protein composition. This resulted in flawed predictions when applied across animal species. As shown in Fig. 8, for a variety of animal species, plasma osmotic pressure varies from as low as 12 cm H2O in the Himalayan tapir goat to as high 36 cm H2O in the orangutan and dolphin. The colloidal osmotic pressure in the plasma of cats, dogs, rabbits, and rats are lower than that of in humans (84) In another study, the ratio of albumin to globulin content of plasma samples and the colloidal osmotic pressure from plasma samples derived from dogs, rats, and humans were found to differ. The albumin: globulin ratio of rats, dogs, and people were 1.4, 0.59, and 2.1, respectively, with corresponding differences in colloidal osmotic pressure. The latter was predicted to affect glomerular filtration dynamics (85).
Fig. 8.

Estimates of colloidal osmotic pressure in plasma taken from venipuncture of anasthetized subjects (zoo animals and humans) or from an arterial indwelling catheter of pentobarbital-anesthetized laboratory animals (asterisk). Based upon data reported by Zweifach and Intaglietta (84)
Organ Blood Flow
The rate and extent of target site drug distribution may be influenced by the blood flow characteristics to that organ. A comparison of the percent cardiac output to various organ systems of the rat, dog, and human is provided in Fig. 9 [based upon the data from Brown et al. (86)]. As seen in this figure, with the exception of the brain where humans have the highest distribution of cardiac output, dog, and humans have similar proportions of their total cardiac output delivered to the other organ systems. In this respect, less similarity is observed between humans and rodents.
Fig. 9.

Comparison of regional blood flow distribution expressed as percent cardiac output in unanesthetized animals. Based upon the data from Brown et al. (86)
Direct Brain Administration of Therapeutics
Non-autologous somatic gene therapy is currently being explored using genetically modified universal recombinant cell lines that can be implanted into patients to deliver a therapeutic product (87). To minimize rejection, microencapsulation methods are used. Microencapsulated cells are being explored as a mechanism for delivering genetic products that can counteract deficiencies of these products due to defective genes.
Microcapsule implantation is being tested as a method for targeted delivery of these therapeutics to the CNS. For example, animal models were used to explore the use of non-autologous somatic gene therapy for counteracting congenital deficiencies of a lysosomal enzyme that can occur in people, mice, and dogs. When alginate-encapsulated engineered mouse myoblasts were implanted into the ventricles of the brain of affected mice, the pathologic manifestations and neurologic performance of these mice were dramatically improved (88). However, scaling from small animal models to humans presented an array of challenges due to variations in the development, and architecture of the mouse CNS as compared to that of humans. Similar challenges are associated with data extrapolation from mouse to animal species such as dogs and cats.
As reviewed elsewhere (89), there is a substantial body of research using large animal models to explore CNS-directed gene therapies for a number of human diseases. When the same laboratory that conducted the aforementioned mouse study evaluated the identical therapy in dogs, similar successes were not observed (90). Encapsulated Madin-Darby Canine Kidney cells transfected with the canine gene for α-iduronidase was encapsulated with poly-L-lysine and alginate. These cells were introduced via stainless steel cannula, into the left and right lateral ventricles of the brains of dogs expressing this deficiency [mucopolysaccharidosis I (MPS I-dogs)]. Although the brains appeared normal on microscopic examination, areas that were in contact with the microcapsules exhibited mild to severe inflammatory reactions. This immune response was not seen with empty microcapsules, indicating that the reactions were attributable to foreign antigens being introduced into the brain of animals deficient in this enzyme. Barsoun et al. (90) suggest that the results obtained in the canine study may be indicative of a fundamental problem associated with the introduction of gene product in large animals expressing a null phenotype. They ascribe part of the scale-up problem to the following:
As compared to mice, the larger brain size of dogs causes the surgery to be more invasive and less precise. In this regard, it is noted that this problem may be overcome by the more precise stereotactic methods used during human CNS surgery.
The gene product is less well dispersed in the brain of larger animals due to diffusional difficulties associated with the brain size of larger species (diffusional distances) and the volume of cerebral spinal fluid (CSF).
Thus, while intraventricular drug delivery was capable of delivering drug to surrounding brain tissues of mice, delivery distance appeared to emerge as a limiting factor in dogs. It should be noted that similar failures in the targeted therapies for MPS I-dogs was observed by others (91,92).
Interpretation of mechanisms underlying the observed failure in dogs is challenged by variable outcomes in primate (including human) studies. Implantation therapies have been met with signs of therapeutic success in a cynomologous model of Parkinson's disease (93). In addition, six human amyotrophic lateral sclerosis patients were successfully implanted into the lumbar intrathecal space with polymer capsules containing genetically engineered baby hamster kidney (BHK) cells releasing human ciliary neurotropic factor (CNTF). The implants successfully released drugs in all patients, although the clinical relevance of this therapy could not be established. No evidence of bio-incompatibility was observed (94). However, there were reports of adverse responses (depression), even in the absence of obvious drug toxicity (95). Bloch et al. (95) used intracerebral administration to deliver polymer-encapsulated BHK cell line. The biological activity of the delivery system was variable and 13 of the 24 implanted capsules failed to release significant amounts of CNTF after retrieval.
Blood-CNS Partitioning: Passive Versus Active Transport
Kawakami et al. (96) demonstrated a statistically significant correlation in the log–log relationship of brain weight versus CSF volume, brain weight versus bulk CSF flow rate, and brain weight versus the weight of the choroid plexuses based upon a literature review of data generated in rats, rabbits, dogs, cats, and humans. With the exclusion of brain weight, CFS volume, and bulk flow rate varied in direct proportion to body weight (i.e., the allometric exponent was approximately 1). Using published fluoroquinolone (FQ) data on blood and CSF concentrations, Kawakami et al. (96) employed physiologically based PK models to estimate the diffusion of the FQ from the blood into the CSF and the unidirectional drug efflux clearance from the CSF into the blood. Based upon the estimated allometric relationships, these investigators concluded that the transmembrane diffusional surface area increased in proportion to brain weight. This outcome suggests that the passive entry of lipophilic molecules into the brain is governed by the diffusional surface area which, in turn, is directly proportional to brain weight. In contrast, the pseudoequilibrium FQ concentrations in the CSF is less than the unbound FQ concentrations in serum or plasma, suggesting that clearance from the CSF was governed by active efflux mechanisms (which, as discussed below, may differ in a drug-dependent manner across animal species). The magnitude of this efflux was observed to decrease as brain weight increased. Thus, predicting human brain access of therapeutic molecules needs to factor the possible involvement and interspecies differences in the activity and/or tissue expression of influx and efflux transporters.
Examples of interspecies comparisons in CNS transporter activity include the following:
Interspecies differences (mouse, rat, and human) in the various the gamma aminobutyric acid (GABA) transporters were observed in terms of the transporter primary sequence, distribution, and pharmacological properties (97).
Although the binding kinetics (affinity and capacity) of the striatal dopamine transport complex differed in the dog, rat, guinea pig, and mouse, the rank order of binding inhibitors was the same across all four animal species (98).
In some cases, transporters may be confined to the brain of one animal species but not in that of others. The Glucose-3 Transporter (GLUT-3) is expressed solely in the CNS of rodents, but can also be found in some human peripheral tissues (99). In fact, there is tremendous diversity in its distribution of this transporter in people, with two- to threefold higher concentrations existing in the cerebral cortex of adults as compared to neonates but similar adult–neonate levels being observed in the cerebellum (100). GLUT-3 is a high affinity glucose transporter that is most abundant in those brain regions associated with high synaptic density. In dogs, rodents, and people, it is primarily located in the grey matter of the cerebral cortex, portions of the hypothalamus, medulla, thalamus, and the parallel fibers of the cerebellum. Species differences in GLUT-3 distribution were also seen in blood brain barrier (BBB) endothelial cells, being present in the cerebro-vascular walls of dogs and people but absent in the vascular walls of rats (99,100).
An overview of differences in the distribution of various active transport systems in human versus rat BBB and brain-cerebral spinal fluid barriers are summarized elsewhere (101).
Exploring interspecies differences in those transporter that may be involved in drug efflux at the BBB, Warren et al. (102) measured mRNA expression of the various ABC transporters (based upon quantitative PCR) in the brain and brain microvessel endothelia cells from human, rat, mouse, bovine, and pigs. Through this work, they identified 41 transporter genes in humans, 39 in rats, 38 in mouse, 21 in cattle, and 21 in swine. These investigators concluded that interspecies differences in transporter expression could be an important consideration when selecting an animal model for CNS drug development (102).
Beyond the issue of transporter distribution, the drug affinities of homologous transporter proteins need to be considered. Differences in substrate recognition and transport efficiency was observed between human and mouse P-glycoprotein (P-gp), leading to challenges in the use of the mouse as a model for extrapolating the accessibility of antiepileptic drugs to the CNS (103). Numerous substances that are P-gp substrates in rodent brain tissues, and therefore exhibit poor CNS penetration, achieve high concentrations in the primate brain. Some radioligands that are P-gp substrates in rodents gain access to the primate CNS (104). In fact, certain radioligands were found to have a brain to plasma ratio that was 8.6-fold greater in human versus rats, and threefold higher in humans versus cynomolgus monkeys.
Even when P-gp activity is inhibited by the co-administration of cyclosporine, the large species differences in brain concentrations for the various ligands were not abolished, indicating that P-gp transporter capacity is not the sole mechanism for the observed interspecies differences in compound access into the CNS. Nevertheless, interspecies differences in P-gp transporter efficiencies appear to be largely responsible for the higher radioligand BBB penetration observed in humans as compared to rats, guinea pigs, and cynomolous monkeys (104). Furthermore, despite the numerous drug transporters that have been identified at the BBB, P-gp continues to be considered the transporter which is primarily responsible for limiting the access of drugs into the CNS (105). Along those lines, it is of interest to note that relative to the human, the percent homology in P-gp amino acid composition is 82% (guinea pig), 85% (rat), 87% (dog and mouse), 93% (rhesus monkey), and 97% (chimpanzee) [104]. While percent amino acid homology alone does not dictate the nature of the corresponding functional consequences in protein activity, it is interesting to note that it is consistent with the greater similarity in radioligand efflux observed between humans and non-human primates versus humans and rodents.
Finally, active transport systems aside, species differences in molecular size-dependent discrimination at the BBB appears to have a role in some of the unique species BBB characteristics. For example, as compared to the normal guinea pigs, the normal human BBB exhibits greater size discrimination (humans tend to exclude large molecules from entering into the CNS). This point was evidenced by differences in brain/plasma ratios of IgG versus to albumin across animal species (106).
Despite the many dissimilarity described above, animal models remain invaluable for demonstrating potential noninvasive methods for CNS drug delivery in humans. When intranasally administered, some CNS-targeted peptides and proteins are proving to be efficacious in human clinical trials, an outcome that is consistent with some rodent studies (107).
Rodent models may also provide valuable information regarding the delivery of drugs to human patients suffering from conditions that alter the BBB. Human neurological disorders due to diseases such as glioma, multiple sclerosis, viral encephalitis, traumatic brain injury, and stroke are known to be associated with a leaky BBB. In these situations, animal models can be used for evaluating noninvasive drug delivery or the delivery of contrast agents to human patients. The feasibility of distribution of magnetic residence probes into the brain via the lymphatic system was evaluated by administering contrast agents by either intraperitoneal injection or eye drop solution to the conjunctival sac of mice that had experimentally induced BBB leakage. By evaluating the use of BBB leakage in mice, the feasibility of using noninvasive methods for the delivery of contrast probes to the human brain via the lymphatic system was confirmed (108).
Binding to CNS Tissues
Unlike the observed interspecies differences in plasma protein binding there appears to be tremendous interspecies similarity in the molecular binding of drugs to brain tissue (109). This was demonstrated by examining the binding of 47 diverse compounds representing a range of physico-chemical characteristics with a range of logD values ranging from −1.43 to 6.01. These compounds were incubated with brain homogenates of derived from Sprague–Dawley rats, Wistar Han rats, CD-1 mice, Hartley guinea pigs, beagle dogs, humans, and cynomolous monkeys. Using an orthogonal regression of the log transformed estimates of unbound fraction (pairwise comparison across species), the correlation coefficient was consistently greater than 0.90. It was suggested that in contrast to the diversity of protein binding in plasma, this species similarity the drug binding to brain homogenates may be due to either the higher lipid content of the brain (as compared to plasma), or to relatively low concentrations of protein in the brain (thereby minimizing selective protein binding for the compounds of interest).
CONCLUDING THOUGHTS
Depending upon the drug and drug delivery platform, species-specific physiological differences can lead to errors in the interspecies extrapolation of drug performance. Underlying these discussions is a simple question: can interspecies differences in product performance alert us to possible variation in product performance when administered to the physiologically compromised human patient? Can the lessons learned be instructional for product developers or prescribing practitioners? Ultimately, as our drug delivery platforms and therapeutic molecules become increasingly complex, the ability to address potential sources of PK variation becomes increasingly important. By viewing animal model data from the perspective of enhancing our understanding of physiological influences on drug delivery, we can acquire a greater appreciation of those factors impacting drug formulation development and the possible impact of pathophysiological states on human drug product performance.
References
- 1.Martinez M. Interspecies differences in physiology and pharmacology: extrapolating preclinical data to human populations. In: Rogge MC, Taft DR, editors. Preclinical Drug Development. 2. Boca Raton: Taylor and Francis Group; 2009. pp. 35–70. [Google Scholar]
- 2.Kararli TT. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos. 1995;16(5):351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 3.de Zwart LL, Rompelberg CJM, Sips AJAM, Welink J, and van Engelen JGM. Anatomical and physiological differences between various species used in studies on the pharmacokinetics and toxicology of xenobiotics. A review of literature. RIVM report 623860 010: October 1999. http://www.rivm.nl/bibliotheek/rapporten/623860010.pdf. Accessed 1 Oct 2011.
- 4.Martignoni M, Groothuis GMM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition, and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875–894. doi: 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- 5.Martinez MN, Pedersoli WM, Ravis WR, Jackson JD, Cullison R. Feasibility of interspecies extrapolation in determining the bioequivalence of animal products intended for intramuscular administration. J Vet Pharmacol Ther. 2001;24(2):125–135. doi: 10.1046/j.1365-2885.2001.00316.x. [DOI] [PubMed] [Google Scholar]
- 6.Lifschitz A, Pis A, Alvarez L, Virkel G, Sanchez S, Sallovitz J, et al. Bioequivalence of ivermectin formulations in pigs and cattle. J Vet Pharmacol Ther. 1999;22(1):27–34. doi: 10.1046/j.1365-2885.1999.00172.x. [DOI] [PubMed] [Google Scholar]
- 7.Hirano K, Yamada H. Studies on the absorption of practically water-insoluble drugs following injection VI: subcutaneous absorption from aqueous suspensions in rats. J Pharm Sci. 1982;71(5):500–505. doi: 10.1002/jps.2600710506. [DOI] [PubMed] [Google Scholar]
- 8.Larsen C, Larsen SW, Jensen H, Yaghmur A, Ostergaard J. Role of in vitro release models in formulation development and quality control of parenteral depots. Expert Opin Drug Deliv. 2009;6(12):1283–1295. doi: 10.1517/17425240903307431. [DOI] [PubMed] [Google Scholar]
- 9.Wiig H, Reed RK, Tenstad O. Interstitial fluid pressure, composition of interstitium, and interstitial exclusion of albumin in hypothyroid rats. Am J Physiol Heart Circ Physiol. 2000;278(5):H1627–H1639. doi: 10.1152/ajpheart.2000.278.5.H1627. [DOI] [PubMed] [Google Scholar]
- 10.Schriftman H, Kondritzer AA. Absorption of atropine from muscle. Am J Physiol. 1957;191(3):591–594. doi: 10.1152/ajplegacy.1957.191.3.591. [DOI] [PubMed] [Google Scholar]
- 11.Saikawa EA, Masegi M, Hashida M, Sezaki H. Contribution of interstitial diffusion in drug absorption from perfused rabbit muscle: effect of hyaluronidase on absorption. Chem Pharm Bull (Tokyo) 1992;40(3):737–740. doi: 10.1248/cpb.40.737. [DOI] [PubMed] [Google Scholar]
- 12.Dunn AL, Heavner JE, Racz G, Day M. Hyaluronidase: a review of approved formulations, indications and off-label use in chronic pain management. Expert Opin Biol Ther. 2010;10(1):127–131. doi: 10.1517/14712590903490382. [DOI] [PubMed] [Google Scholar]
- 13.Hanhn MS, Kobler JB, Starcher BC, Zeitels SM, Langer R. Quantitative and comparative studies of the vocal fold extracellular matrix. I: elastic fibers and hyaluronic acid. Ann Otol Rhinol Laryngol. 2006;115(2):156–164. doi: 10.1177/000348940611500213. [DOI] [PubMed] [Google Scholar]
- 14.Laurent UB, Granath KA. The molecular weight of hyaluronate in the aqueous humour and vitreous body of rabbit and cattle eyes. Exp Eye Res. 1983;36(4):481–492. doi: 10.1016/0014-4835(83)90042-8. [DOI] [PubMed] [Google Scholar]
- 15.Kuo JW. Practical aspects of hyaluronoan-based medical products. New York: CRC Press; 2006. [Google Scholar]
- 16.Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information-rich system. Eur J Cell Biol. 2006;85(8):699–715. doi: 10.1016/j.ejcb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4(7):528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 18.Toole BP, Slomiany MG. Hyaluronan: a constitutive regulator of chemoresistance and malignancy in cancer cells. Semin Cancer Biol. 2008;18(4):244–250. doi: 10.1016/j.semcancer.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson P, Ruffell B. CD44 and its role in inflammation and inflammatory diseases. Inflamm Allergy Drug Targets. 2009;8(3):208–220. doi: 10.2174/187152809788680994. [DOI] [PubMed] [Google Scholar]
- 20.Gee K, Kryworuchko M, Kumar A. Recent advances in the regulation of CD44 expression and its role in inflammation and autoimmune diseases. Arch Immunol Ther Exp (Warsz) 2004;52(1):13–26. [PubMed] [Google Scholar]
- 21.Platt VM, Szoka FC., Jr Anticancer therapeutics: targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Mol Pharm. 2008;5(4):474–486. doi: 10.1021/mp800024g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh EJ, Park K, Kim KS, Kim J, Yang JA, Kong JH, et al. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Control Release. 2010;141(1):2–12. doi: 10.1016/j.jconrel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 23.Eliaz RE, Szoka FC., Jr Liposome-encapsulated doxorubicin targeted to CD44: a strategy to kill CD44-overexpressing tumor cells. Cancer Res. 2001;61(6):2592–2601. [PubMed] [Google Scholar]
- 24.Bosworth BT, St John T, Gallatin WM, Harp JA. Sequence of the bovine CD44 cDNA: comparison with human and mouse sequences. Mol Immunol. 1991;28(10):1131–1135. doi: 10.1016/0161-5890(91)90028-I. [DOI] [PubMed] [Google Scholar]
- 25.Nedvetzki S, Golan I, Assayag N, Gonen E, Caspi D, Gladnikoff M, et al. A mutation in a CD44 variant of inflammatory cells enhances the mitogenic interaction of FGF with its receptor. J Clin Invest. 2003;111(8):1211–1220. doi: 10.1172/JCI17100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? Int J Biochem Cell Biol. 2007;39(7–8):1432–1449. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Milde KF, Alejandro R, Pastori RL. Expression of CD44 variant transcripts in dog lymphatic tissue. Immunogenetics. 1994;40(6):437–444. doi: 10.1007/BF00177826. [DOI] [PubMed] [Google Scholar]
- 28.Misra S, Heldin P, Hascall VC, Karamanos NK, Skandalis SS, Markwald RR, et al. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011. doi:10.1111/j.1742-4658.2011.08071.x. [DOI] [PMC free article] [PubMed]
- 29.Watson PD. Analysis of the paired-tracer method of determining cell uptake. Am J Physiol. 1998;275(2 Pt 1):E366–E371. doi: 10.1152/ajpendo.1998.275.2.E366. [DOI] [PubMed] [Google Scholar]
- 30.Hollenstein UM, Brunner M, Schmid R, Müller M. Soft tissue concentrations of ciprofloxacin in obese and lean subjects following weight-adjusted dosing. Int J Obes Relat Metab Disord. 2001;25(3):354–358. doi: 10.1038/sj.ijo.0801555. [DOI] [PubMed] [Google Scholar]
- 31.Schmittmann G, Rohr UD. Comparison of the permeability surface product (PS) of the blood capillary wall in skeletal muscle tissue of various species and in vitro porous membranes using hydrophilic drugs. J Pharm Sci. 2000;89(1):115–127. doi: 10.1002/(SICI)1520-6017(200001)89:1<115::AID-JPS12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 32.Porter CJ, Edwards GA, Charman SA. Lymphatic transport of proteins after s.c. injection: implications of animal model selection. Adv Drug Deliv Rev. 2001;50(1–2):157–171. doi: 10.1016/S0169-409X(01)00153-3. [DOI] [PubMed] [Google Scholar]
- 33.Edwards GA, Porter CJ, Caliph SM, Khoo SM, Charman WN. Animal models for the study of intestinal lymphatic drug transport. Adv Drug Deliv Rev. 2001;50(1–2):45–60. doi: 10.1016/S0169-409X(01)00148-X. [DOI] [PubMed] [Google Scholar]
- 34.Kagan L, Gershkovich P, Mendelman A, Amsili S, Ezov N, Hoffman A. The role of the lymphatic system in subcutaneous absorption of macromolecules in the rat model. Eur J Pharm Biopharm. 2007;67(3):759–765. doi: 10.1016/j.ejpb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Oussoren C, Zuidema J, Crommelin DJA, Storm G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. I. Influence of the anatomical site of injection. J Liposome Res. 1997;7(1):85–99. doi: 10.3109/08982109709035487. [DOI] [Google Scholar]
- 36.Oussoren C, Storm G. Liposomes to target the lymphatics by subcutaneous administration. Adv Drug Deliv Rev. 2001;50(1–2):143–156. doi: 10.1016/S0169-409X(01)00154-5. [DOI] [PubMed] [Google Scholar]
- 37.Kota J, Machavaram KK, McLennan DN, Edwards GA, Porter CJ, Charman SA. Lymphatic absorption of subcutaneously administered proteins: influence of different injection sites on the absorption of darbepoetin alfa using a sheep model. Drug Metab Dispos. 2007;35(12):2211–2217. doi: 10.1124/dmd.107.015669. [DOI] [PubMed] [Google Scholar]
- 38.Bleuel H, Hoffmann R, Kaufmann B, Neubert P, Ochlich PP, Schaumann W. Kinetics of subcutaneous versus intravenous epoetin-beta in dogs, rats and mice. Pharmacology. 1996;52(5):329–338. doi: 10.1159/000139398. [DOI] [PubMed] [Google Scholar]
- 39.Woo S, Jusko WJ. Interspecies comparisons of pharmacokinetics and pharmacodynamics of recombinant human erythropoietin. Drug Metab Dispos. 2007;35(9):1672–1678. doi: 10.1124/dmd.107.015248. [DOI] [PubMed] [Google Scholar]
- 40.Caldini A, Moneti G, Fanelli A, Bruschettini A, Mercurio S, Pieraccini G, et al. Epoetin alpha, epoetin beta and darbepoetin alfa: two-dimensional gel electrophoresis isoforms characterization and massspectrometry analysis. Proteomics. 2003;3(6):937–941. doi: 10.1002/pmic.200300405. [DOI] [PubMed] [Google Scholar]
- 41.McLennan DN, Porter CJH, Edwards GA, Martin SW, Heatherington AC, Charman SA. Lymphatic absorption is the primary contributor to the systemic availability of epoetin alfa following subcutaneous administration to sheep. J Pharmacol Exp Ther. 2005;313(1):345–351. doi: 10.1124/jpet.104.078790. [DOI] [PubMed] [Google Scholar]
- 42.Plum A, Agerso H, Andersen L. Pharmacokinetics of the rapid-acting insulin analog, insulin aspart, in rats, dogs, and pigs, and pharmacodynamics of insulin aspart in pigs. Drug Metab Dispos. 2000;28(2):155–160. [PubMed] [Google Scholar]
- 43.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 44.Brocks DR, Ramaswamy M, MacInnes AI, Wasan KM. The stereoselective distribution of halofantrine enantiomers within human, dog, and rat plasma lipoproteins. Pharm Res. 2000;17(4):427–431. doi: 10.1023/A:1007524919865. [DOI] [PubMed] [Google Scholar]
- 45.Kosa T, Maruyama T, Sakai N, Yonemura N, Yahara S, Otagiri M. Species differences of serum albumins: I: drug binding sites. Pharm Res. 1998;14(11):1607–1612. doi: 10.1023/A:1012138604016. [DOI] [PubMed] [Google Scholar]
- 46.Fura A, Harper TW, Zhang H, Fung L, Shyu WC. Shift in pH of biological fluids during storage and processing: effect on bioanalysis. J Pharm Biomed Anal. 2003;32(3):513–522. doi: 10.1016/S0731-7085(03)00159-6. [DOI] [PubMed] [Google Scholar]
- 47.Kochansky CJ, McMasters DR, Lu P, Koeplinger KA, Kerr HH, Shou M, et al. Impact of pH on plasma protein binding in equilibrium dialysis. Mol Pharm. 2008;5(3):438–448. doi: 10.1021/mp800004s. [DOI] [PubMed] [Google Scholar]
- 48.Pistolozzi M, Bertucci C. Species-dependent stereoselective drug binding to albumin: a circular dichronism study. Chirality. 2008;20(3–4):552–558. doi: 10.1002/chir.20521. [DOI] [PubMed] [Google Scholar]
- 49.Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Ribel U, Markussen J. Albumin binding and time action of acylated insulins in various species. J Pharm Sci. 1996;85(3):304–308. doi: 10.1021/js950412j. [DOI] [PubMed] [Google Scholar]
- 50.Basken NE, Mathias CJ, Lipka AE, Green MA. Species dependence of [64Cu]Cu–Bis(thiosemicarbazone) radiopharmaceutical binding to serum albumins. Nucl Med Biol. 2008;35(3):281–286. doi: 10.1016/j.nucmedbio.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giron MC. Radiopharmaceutical pharmacokinetics in animals: critical considerations. Q J Nucl Med Mol Imaging. 2009;53(4):359–364. [PubMed] [Google Scholar]
- 52.Puigdemont A, Arboix M, Gaspari F, Bortolotti A, Bonati M. In-vitro plasma protein binding of propafenone and protein profile in eight mammalian species. Res Commun Chem Pathol Pharmacol. 1989;64(3):435–440. [PubMed] [Google Scholar]
- 53.Lin JH, Chen IW, Deluna FA. Nonlinear kinetics of aldronate. Plasma protein binding and bone uptake. Drug Metab Disp. 1994;22(3):400–405. [PubMed] [Google Scholar]
- 54.Bonapace CR, Fowler S, Laessig KA, Lazor JA, Nambiar S. Ceftriaxone and calcium-containing fluids-rationale for product label changes, abstr. A1-008. Abstr. 49th Intersci. Conf. Antimicrob. Agents Chemother. Washington: American Society for Microbiology; 2009. [Google Scholar]
- 55.Wasan KM, Brocks DR, Lee SD, Sachs-Barrable K, Thornton SJ. Impact of lipoproteins on the biological activity and disposition of hydrophobic drugs: implications for drug discovery. Nat Rev Drug Discov. 2008;7(1):84–99. doi: 10.1038/nrd2353. [DOI] [PubMed] [Google Scholar]
- 56.Ramaswamy M, Wallace TL, Cossum PA, Wasan KM. Species differences in the proportion of plasma lipoprotein lipid carried by high-density lipoproteins influence the distribution of free and liposomal nystatin in human, dog, and rat plasma. Antimicrob Agents Chemother. 1999;43(6):1424–1428. doi: 10.1128/aac.43.6.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ. Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob Agents Chemother. 2002;46(3):828–833. doi: 10.1128/AAC.46.3.828-833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hong Y, Shaw PJ, Tattam BN, Nath CE, Earl JW, Stephen KR, et al. Plasma protein distribution and its impact on pharmacokinetics of liposomal amphotericin B in paediatric patients with malignant diseases. Eur J Clin Pharmacol. 2007;63(2):165–172. doi: 10.1007/s00228-006-0240-x. [DOI] [PubMed] [Google Scholar]
- 59.Jin Y, Ai P, Xin R, Tian Y, Dong J, Chen D, et al. Self-assembled drug delivery systems: Part 3. In vitro/in vivo studies of the self-assembled nanoparticulates of cholesteryl acyl didanosine. Int J Pharm. 2009;368(1–2):207–214. doi: 10.1016/j.ijpharm.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 60.Jemal M, Hawthorne DJ. Quantitative determination of BMS-186716, a thiol compound, in rat plasma by high-performance liquid chromatography-positive ion electrospray mass spectrometry after hydrolysis of the methyl acrylate adduct by the native esterases. J Chromatogr B: Biomed Sci Appl. 1997;698(1–2):123–132. doi: 10.1016/S0378-4347(97)00292-2. [DOI] [PubMed] [Google Scholar]
- 61.Minagawa T, Kohno Y, Suwa T, Tsuji A. Species differences in hydrolysis of isocarbacyclin methyl ester (TEI-9090) by blood esterases. Biochem Pharmacol. 1995;49(10):1361–1365. doi: 10.1016/0006-2952(95)00071-7. [DOI] [PubMed] [Google Scholar]
- 62.Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5(4):487–495. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003;188(1):49–71. doi: 10.1016/S0300-483X(03)00043-X. [DOI] [PubMed] [Google Scholar]
- 64.Brinks V, Jiskoot W, Schellekens H. Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res. 2011;28(10):2379–2385. doi: 10.1007/s11095-011-0523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishida T, Harashima H, Kiwada H. Interactions of liposomes with cells in vitro and in vivo: opsonins and receptors. Curr Drug Metab. 2001;2(4):397–409. doi: 10.2174/1389200013338306. [DOI] [PubMed] [Google Scholar]
- 66.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 67.Moghimi SM, Hunter AC. Recognition by macrophages and liver cells of opsonized phospholipid vesicles and phospholipid headgroups. Pharm Res. 2001;18(1):1–8. doi: 10.1023/A:1011054123304. [DOI] [PubMed] [Google Scholar]
- 68.Yan X, Scherphof GL, Kamps JA. Liposome opsonization. J Liposome Res. 2005;15(1–2):109–139. doi: 10.1081/lpr-64971. [DOI] [PubMed] [Google Scholar]
- 69.Liu D, Hu Q, Song YK, Song Liposome clearance from blood: different animal species have different mechanisms. Biochemica et Biophysica Acta. 1995;1240(2):277–284. doi: 10.1016/0005-2736(95)00184-0. [DOI] [PubMed] [Google Scholar]
- 70.Huong TM, Harashima H, Kiwada H. In vivo studies on the role of complement in the clearance of liposomes in rats and guinea pigs. Biol Pharm Bull. 1999;22(5):515–520. doi: 10.1248/bpb.22.515. [DOI] [PubMed] [Google Scholar]
- 71.Kurihara A, Shibayama Y, Kasuya A, Ikeda M, Hisaoka M. Species variation in pharmacokinetics and opsonization of palmitoyl rhizoxin (RS-1541) incorporated in lipid emulsions. J Drug Target. 1998;5(6):491–505. doi: 10.3109/10611869808997875. [DOI] [PubMed] [Google Scholar]
- 72.Szebeni J. Complement activation-related pseudoallergy caused by amphiphilic drug carriers: the role of lipoproteins. Curr Drug Deliv. 2005;2(4):443–449. doi: 10.2174/156720105774370212. [DOI] [PubMed] [Google Scholar]
- 73.Szebeni J, Alving CR, Rosivall L, Bünger R, Baranyi L, Bedöcs P, et al. Animal models of complement-mediated hypersensitivity reactions to liposomes and other lipid-based nanoparticles. J Liposome Res. 2007;17(2):107–117. doi: 10.1080/08982100701375118. [DOI] [PubMed] [Google Scholar]
- 74.Liu S, Ishida T, Kiwada H. Effect of serum components from different species on destabilizing hydrogenated phosphatidylcholine-based liposomes. Biol Pharm Bull. 1997;20(8):874–880. doi: 10.1248/bpb.20.874. [DOI] [PubMed] [Google Scholar]
- 75.Harashima H, Komatsu S, Kojima S, Yanagi C, Morioka Y, Naito M, et al. Species difference in the disposition of liposomes among mice, rats, and rabbits: allometric relationship and species dependent hepatic uptake mechanism. Pharm Res. 1996;13(7):1049–1054. doi: 10.1023/A:1016058724452. [DOI] [PubMed] [Google Scholar]
- 76.Matschkea C, Iselea U, van Hoogevestb P, Fahrc A. Sustained-release injectables formed in situ and their potential use for veterinary products. J Control Release. 2002;85(103):1–15. doi: 10.1016/S0168-3659(02)00266-3. [DOI] [PubMed] [Google Scholar]
- 77.Packhaeuser CB, Schnieders J, Oster CG, Kissel T. In situ forming parenteral drug delivery systems: an overview. Eur J Pharm Biopharm. 2004;58(2):445–455. doi: 10.1016/j.ejpb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 78.Leppert PS, Cammack L, Cargill R, Coffman L, Cortese M, Engle K, et al. Interspecies differences in systemic drug availability following subcutaneous pulsatile administration in cattle, sheep, dogs, and rats. J Biomed Mater Res. 1994;28(6):713–722. doi: 10.1002/jbm.820280608. [DOI] [PubMed] [Google Scholar]
- 79.Ali MJ, Navalitloha Y, Vavra MW, Kang EW, Itskovich AC, Molnar P, et al. Isolation of drug delivery from drug effect: problems of optimizing drug delivery parameters. Neuro Oncol. 2006;8(2):109–118. doi: 10.1215/15228517-2005-007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47(12):3039–3051. [PubMed] [Google Scholar]
- 81.Netti PA, Hamberg LM, Babich JW, Kierstead D, Graham W, Hunter GJ, et al. Enhancement of fluid filtration across tumor vessels: implication for delivery of macromolecules. Proc Natl Acad Sci U S A. 1999;96(6):3137–3142. doi: 10.1073/pnas.96.6.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stohrer M, Boucher Y, Stangassinger M, Jain RK. Oncotic pressure in solid tumors is elevated. Cancer Res. 2000;60(15):4251–4255. [PubMed] [Google Scholar]
- 83.Thomas LA, Brown SA. Relationship between colloid osmotic pressure and plasma protein concentration in cattle, horses, dogs, and cats. Am J Vet Res. 1992;53(12):2241–2244. [PubMed] [Google Scholar]
- 84.Zweifach BW, Intaglietta M. Measurement of blood plasma colloid osmotic pressure: II. Comparative study of different species. Microvasc Res. 1971;3(1):83–88. doi: 10.1016/0026-2862(71)90009-4. [DOI] [PubMed] [Google Scholar]
- 85.Navar PD, Navar LG. Relationship between colloid osmotic pressure and plasma protein concentration in the dog. Am J Physiol. 1977;233(2):H295–H298. doi: 10.1152/ajpheart.1977.233.2.H295. [DOI] [PubMed] [Google Scholar]
- 86.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13(4):407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- 87.Chang PL. Non-autologous somatic gene therapy. In: Chang PL, editor. Somatic gene therapy. Boca Raton: CRC Press, Inc; 1995. pp. 203–224. [Google Scholar]
- 88.Ross CJD, Ralph M, Chang PL. Somatic gene therapy for a neurodegenerative disease using microencapsulated recombinant cells. Exp Neurol. 2000;166(2):276–286. doi: 10.1006/exnr.2000.7531. [DOI] [PubMed] [Google Scholar]
- 89.Gagliardi C, Bunnell BA. Large animal models of neurological disorders for gene therapy. ILAR J. 2009;50(2):128–143. doi: 10.1093/ilar.50.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barsoum SC, Milgram W, Mackay W, Coblentz C, Delaney KH, Kwiecien JM, et al. Delivery of recombinant gene product to canine brain with the use of microencapsulation. J Lab Clin Med. 2003;142(6):399–413. doi: 10.1016/j.lab.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 91.Shull R, Lu X, Dube I, Lutzko C, Kruth S, Abrams-Oqq A, et al. Humoral immune response limits gene therapy in canine MPS I. Blood. 1996;88(1):377–379. [PubMed] [Google Scholar]
- 92.Shull RM, Lu X, McEntee MF, Bright RM, Pepper KA, Kohn DB. Myoblast gene therapy in canine mucopolysaccharidosis. I. Abrogation by an immune response to alpha-L-iduronidase. Hum Gene Ther. 1996;7(13):1595–1603. doi: 10.1089/hum.1996.7.13-1595. [DOI] [PubMed] [Google Scholar]
- 93.Aebischer P, Goddard M, Signore AP, Timpson RL. Functional recovery in hemiparkinsonian primates transplanted with polymer-encapsulated PC12 cells. Exp Neurol. 1994;126(2):151–158. doi: 10.1006/exnr.1994.1053. [DOI] [PubMed] [Google Scholar]
- 94.Aebischer P, Schluep M, Déglon N, Joseph JM, Hirt L, Heyd B, et al. Intrathecal delivery of CNTF using encapsulated genetically modified xenogeneic cells in amyotrophic lateral sclerosis patients. Nat Med. 1996;2(6):696–699. doi: 10.1038/nm0696-696. [DOI] [PubMed] [Google Scholar]
- 95.Bloch J, Bachoud-Lévi AC, Déglon N, Lefaucheur JP, Winkel L, Palfi S, et al. Neuroprotective gene therapy for Huntington's disease, using polymer-encapsulated cells engineered to secrete human ciliary neurotrophic factor: results of a phase I study. Hum Gene Ther. 2004;15(10):968–975. doi: 10.1089/hum.2004.15.968. [DOI] [PubMed] [Google Scholar]
- 96.Kawakami J, Yamamoto K, Sawada Y, Iga T. Prediction of brain delivery of ofloxacin, a new quinolone, in the human from animal data. J Pharmacokinet Biopharm. 1994;22(3):207–227. doi: 10.1007/BF02353329. [DOI] [PubMed] [Google Scholar]
- 97.Smith EK, Gustafson EL, Borden LA, Dhar TGM, Durkin MM, Vaysse PJJ, et al. Heterogeneity of brain GABA transporters. In: Tanaka C, Bowery NG, et al., editors. GABA: receptors, transporters, and metabolism. Switzerland: Birkauser Verlag; 1996. pp. 63–72. [Google Scholar]
- 98.Sharif NA, Nunes JL, Michel AD, Whiting RL. Comparative properties of the dopamine transport complex in dog and rodent brain: striatal [(3)H]GBR12935 binding and [(3)H]dopamine uptake. Neurochem Int. 1989;15(3):325–332. doi: 10.1016/0197-0186(89)90140-X. [DOI] [PubMed] [Google Scholar]
- 99.Gerhart DZ, Leino RL, Borson ND, Taylor WE, Gronlund KM, McCall AL, et al. Localization of glucose transporter GLUT 3 in brain: comparison of rodent and dog using species-specific carboxyl-terminal antisera. Neuroscience. 1995;66(1):237–246. doi: 10.1016/0306-4522(94)00544-F. [DOI] [PubMed] [Google Scholar]
- 100.Mantych GJ, James DE, Chung HD, Devaskar SU. Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology. 1992;131(3):1270–1278. doi: 10.1210/en.131.3.1270. [DOI] [PubMed] [Google Scholar]
- 101.Westerhout J, Danhof M, De Lange EC. Preclinical prediction of human brain target site concentrations: considerations in extrapolating to the clinical setting. J Pharm Sci. 2011. doi:10.1002/jps. [DOI] [PubMed]
- 102.Warren MS, Zerangue N, Woodford K, Roberts LM, Tate EH, Feng B, et al. Comparative gene expression profiles of ABC transporters in brain microvessel, endothelial cells and brain in five species including human. Pharmacol Res. 2009;59(6):404–413. doi: 10.1016/j.phrs.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 103.Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Löscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. 2007;52(2):333–346. doi: 10.1016/j.neuropharm.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 104.Syvänen S, Lindhe O, Palner M, Kornum BR, Rahman O, Långström B, et al. Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Dispos. 2009;37(3):635–643. doi: 10.1124/dmd.108.024745. [DOI] [PubMed] [Google Scholar]
- 105.Liu X, Chen C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharm Exp Ther. 2008;325(2):349–356. doi: 10.1124/jpet.107.130294. [DOI] [PubMed] [Google Scholar]
- 106.Reiber H, Thiele P. Species-dependent variables in blood cerebrospinal fluid barrier function for proteins. J Clin Chem Clin Biochem. 1983;21(4):199–202. doi: 10.1515/cclm.1983.21.4.199. [DOI] [PubMed] [Google Scholar]
- 107.Dhuria SV, Hanson LR, Frey WH., 2nd Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 108.Liu CH, You Z, Ren JQ, Kim YR, Eikermann-Haerter K, Liu PK. Noninvasive delivery of gene targeting probes to live brains for transcription MRI. FASEB J. 2008;22(4):1193–1203. doi: 10.1096/fj.07-9557com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Di L, Umland JP, Chang G, Huang Y, Lin Z, Scott DO, et al. Species independence in brain tissue binding using brain homogenates. Drug Metab Dispos. 2011;39(7):1270–1277. doi: 10.1124/dmd.111.038778. [DOI] [PubMed] [Google Scholar]