

Abstract
Periodontitis appears to promote chronic inflammatory diseases, including atherosclerosis, but relevant mechanisms need clarification. Oral bacteria induce antibodies that bind not only bacteria, but also oxLDL. Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans induce remarkable IgG responses that are dominated by IgG2, and IgG2 is IFN-γ-dependent and is promoted by dendritic cells (DCs). LDL-reactive antibodies induced by P. gingivalis and A. actinomycetemcomitans include anti-phosphorylcholine (α-PC) and β2-glycoprotein-1-dependent anticardiolipin (α-CL), and these antibodies may link chronic inflammatory diseases at a mechanistic level. Antibody-mediated uptake of oxLDL or bacteria dramatically enhances DC-IL-12, and DC-IL-12 induces NK-cell-IFN-γ responses that promote Th-1 responses and sustained inflammation. DCs may be derived from monocytes, and this is striking in cultures of aggressive periodontitis (AgP) monocytes, where DC numbers are about double control levels. Moreover, serum α-CL levels in individuals with AgP are frequently elevated, and these antibodies promote atherosclerosis in persons with antiphospholipid syndrome. Elevated serum levels of soluble-intercellular adhesion molecule, soluble-vascular cell adhesion molecule, and soluble-E-selectin are atherosclerosis-associated indicators of vascular inflammation, and these markers are elevated in the subset of AgP patients with high α-CL. We reason that periodontitis patients with elevated antibodies reactive with oxLDL could be a subgroup at high risk for cardiovascular sequelae.
Keywords: aggressive periodontitis, dendritic cells, oxidized low-density lipoprotein, α-oxLDL, chronic inflammation, antiphospholipid syndrome
Introduction
Millions of people are afflicted with chronic inflammatory diseases that involve prolonged suffering, economic loss, and, in some cases, premature death. The results reviewed here derive from studies of periodontal disease, but the mechanisms implicated may apply in atherosclerosis, arthritis, inflammatory bowel disease, and a host of autoimmune disorders, including antiphospholipid syndrome (APS). APS is caused by auto-antibodies produced against β2 glycoprotein I (α-CL), and this antibody is of special interest, given that it reacts with oxidized low-density lipoproteins (ox LDL) as well as P. gingivalis and A. actinomycetemcomitans. APS manifests as a disorder of coagulation resulting in blood clots and is associated with atherosclerosis as well as pregnancy-related complications, including preterm delivery and miscarriage. Epidemiological studies and studies of animals infected with periodontal pathogens suggest that periodontitis may promote atherosclerosis and subsequent coronary heart disease. An extensive review of this relationship has recently been published in the Journal of Dental Research (Kebschull et al., 2010). Nevertheless, mechanisms involved remain to be fully determined, although a variety of mechanisms has been suggested, ranging from increased systemic levels of inflammatory mediators to induction of atheromas as a consequence of infection of vascular cells with oral bacteria (Kebschull et al., 2010). Results reviewed here suggest an additional mechanism based on α-oxLDLs induced by oral bacteria. In brief, individuals with periodontitis frequently display elevated serum levels of α-PC and α-CL (Schenkein et al., 1999, 2001, 2003). Moreover, these antibodies react with oral bacteria as well as with oxLDL and serve to promote phagocytosis (Schenkein et al., 1999; Purkall et al., 2002; Kikuchi et al., 2010). The oral organisms include P. gingivalis and A. actinomycetemcomitans, which are potent immunogens and induce large quantities of specific IgG (Wilson and Hamilton, 1992; Lu et al., 1993; Califano et al., 1999). Moreover, α-CL is induced by immunization of animals with A. actinomycetemcomitans (Wang et al., 2008). In short, these antibodies form immune complexes (ICs) that stimulate dendritic cells (DCs) and macrophages more dramatically than oral bacteria or oxLDL alone, leading to production of pro-inflammatory cytokines and chemokines, both by these accessory cells and also by subsequently stimulated NK cells and T-cells.
Dendritic Cells in Aggressive Periodontitis
Dendritic cells (DCs) are located adjacent to epithelia of blood vessels and the mucosa that protects soft tissues from microbial invasion. Bacteria or antigen-antibody complexes of bacteria or bacterial products that breach these barriers will encounter DCs and induce DC responses. In brief, DCs are considered to be “sentinels” of the immune system because they are typically the first immune system cells to encounter and respond to invading micro-organisms (reviewed in Nestle et al., 2009). Moreover, the nature of this initial DC response has a great deal to do with the type of acquired immune response that is induced. Langerhans cells and dermal DCs may be found in large numbers in gingival tissue, and mature CD83+ DCs have been found in inflamed gingival tissue from individuals with periodontitis (Jotwani et al., 2001; Jotwani and Cutler, 2003). DCs have remarkable ability to stimulate both NK cells of the innate immune system and naïve T-cells of the acquired immune system and are uniquely able to initiate immune responses. The unique immunostimulatory activity of DCs derives from their ability to efficiently capture and present antigens with optimal expression of cytokines and co-stimulatory molecules (reviewed in Steinman and Hemmi, 2006). The presence of numerous DCs in periodontal lesions is just beginning to be appreciated, and the presence of mature DCs that are particularly immunostimulatory is noteworthy (Jotwani et al., 2001; Jotwani and Cutler, 2003). DCs in normal tissues are typically immature, but they mature as they migrate to secondary lymphoid tissues, where they may present antigens to naïve T-cells. However, immature and mature DCs may be found in chronic inflammatory lesions, including periodontal lesions and atherosclerotic plaques, and the presence of mature cells in sites of chronic inflammation is distinct from that in acute lesions, where DCs leave local sites and mature in the draining secondary lymphoid tissues, where they process and present antigens.
The Appendix Fig. illustrates the influence of dendritic cell cytokines on T-helper cell activation and differentiation. The legend includes references to critical reviews with helpful illustrations that provide background covering DC characteristics, DC subtypes, DC maturation/co-stimulatory markers, Th1, Th2, Th17 cells, classic cytokine responses, and activating Fc receptors with their URLs.
The relationship among DCs, aggressive periodontitis (AgP), and serum immunoglobulin G2 (IgG2) levels is summarized in Table 1. Serum levels of IgG2 in individuals with AgP are elevated over those in race- and age-matched non-periodontitis control individuals (NP controls) (Lu et al., 1994). Moreover, responses to A. actinomycetemcomitans and P. gingivalis in AgP are largely of the IgG2 subclass (Lu et al., 1993; Califano et al., 1999), with average serum IgG2 specific for A. actinomycetemcomitans serotype b at about 100 µg/mL vs. about 10 µg/mL for the typically more abundant IgG1 (Wilson and Hamilton, 1992; Lu et al., 1993). Similarly, individuals with AgP have high levels of serum IgG2, and 10-50 µg/mL is reactive with P. gingivalis serotype-specific antigens K1 through K6 (Califano et al., 1999). Specific recall responses for P. gingivalis in culture exhibit elevated IgG2 responses but only if DCs are present (Kikuchi et al., 2005). This elevated IgG2 response was apparent when AgP-PBLs were stimulated by the B-cell activator pokeweed mitogen (PWM). However, NP-lymphocytes could be converted into high IgG2 responders by the addition of AgP monocytes, indicating that IgG2 is regulated by cells in the monocyte population (Zhang et al., 1996).
Table 1.
Summary of Dendritic Cells and IgG2 in Individuals with Aggressive Periodontitis
| Feature of Immune Response | Status in AgP | References |
|---|---|---|
| 1- Immunoglobulin subclasses | IgG2 levels are elevated in AgP sera | Lu et al. (1994) |
| 2- Specific Ig responses | Responses to A. actinomycetemcomitans and P. gingivalis are dominated by IgG2 | Lu et al. (1993); Wilson and Hamilton (1992); Califano et al. (1999) |
| 3- Accessory cell activity | Dendritic cells promote IgG2 responses in AgP, including specific anti-P. gingivalis responses | Zhang et al. (1996); Barbour et al. (2002); Kikuchi et al. (2005) |
| 4- Dendritic cells vs. macrophages | Monocyte maturation in AgP is skewed toward dendritic cells | Barbour et al. (2002) |
In culture, some monocytes spontaneously mature into DCs, as indicated by the loss of CD14 together with expression of CD11c and the ability to stimulate potent mixed-leukocyte reactions. This spontaneous transformation is skewed in AgP-monocyte cultures, where, after 4 days, DC populations are approximately two-fold higher than in NP cultures (Barbour et al., 2002). However, DCs generated from NP monocytes, by use of GM-CSF and IL-4, also induce a marked increase in IgG2 in a DC-dose-dependent fashion when NP cultures are stimulated with PWM. In contrast, the IgG1 responses in the same cultures are not significantly altered (Barbour et al., 2002). In short, individuals with AgP have elevated DC activity, and DCs promote elevated IgG2 responses.
Pro-Inflammatory Cytokines and IgG2 Responses
Pro-inflammatory cytokines promote tissue destruction characteristic of chronic inflammatory diseases. They also help regulate local immune responses, and regulation involves DCs and NK cells (Table 2). Immunoglobulin G1 (IgG1) and IgE are promoted by Th2 cytokines, whereas IgG2 is Th1-dependent in humans and is promoted by pro-inflammatory IFN-γ (Tanaka et al., 2003). Interleukin-12 and IL-18 promote IFN-γ production, and IL-12 is a prominent DC product (Tanaka et al., 2006). A. actinomycetemcomitans is a potent inducer of DC-IL-12, whereas IL-10 is the dominant macrophage product under the same conditions (Kikuchi et al., 2004). Similarly, P. gingivalis will induce potent DC-IL-12 responses in the presence of IFN-γ, which also enhances A. actinomycetemcomitans-induced DC-IL-12 responses (Kikuchi et al., 2010). In blood, or in sites infiltrated with blood, NK cells are the source of this rapid IFN-γ (24-hour) that promotes DC-IL-12. NK cells need IL-12 for optimal IFN-γ, and it appears that P. gingivalis-DC-NK interactions result in reciprocal activation and increased DC-IL-12 and NK cell-IFN-γ (Kikuchi et al., 2004, 2005, 2010). Both DCs and NK cells are necessary to get the rapid 24-hour IFN-γ needed to induce the P. gingivalis-specific IgG2 in recall responses in PBL cultures (Kikuchi et al., 2005). Moreover, specific IgG2 responses appear to require a cocktail of pro-inflammatory cytokines, since neutralizing antibodies for IL-12, IFN-γ, and IL-1 as well as indomethacin inhibit A. actinomycetemcomitans- specific IgG2 recall responses (Tanaka et al., 2006). Interestingly, IL-17 levels are also elevated in the sera of individuals with AgP (Schenkein et al., 2010). In inflammatory diseases, Th17 cells are inducible by DCs, and IL-17 promotes germinal center formation and Ig class-switching, including enhanced IgG (Blaho et al., 2009). Thus, Th-17 cells like Th1 and Th2 cells may modulate B-cell function, and this pro-inflammatory cytokine could also promote the unusual IgG2 levels in AgP.
Table 2.
Summary of Dendritic Cells, NK Cells, Pro-inflammatory Cytokines, and IgG2
| Pro-inflammatory Cytokine | Relationship to AgP | References |
|---|---|---|
| 1- IL-12 | Oral bacteria and oxLDL induce dendritic cell-IL-12 responses | Kikuchi et al. (2004, 2010) |
| 2- IFN-γ | IL-12 induces rapid NK-cell-IFN-γ responses, and NK-cell-IFN-γ promotes IgG2 responses in culture | Kikuchi et al. (2004, 2005, 2010) |
| 3- Cytokine inhibition | α-IL12, α-IFN-γ, and α-IL-1 inhibit IgG2 responses in culture, suggesting a role in elevating serum IgG2 | Tanaka et al. (2006) |
| 4- IL-12, IFN-γ, and specific IgG2 responses | OxLDL, P. gingivalis, or A. actinomycetemcomitans-DC-NK cell interactions promote DC-IL-12 and rapid production of NK cell IFN-γ; optimal P. gingivalis-specific IgG2 depends on NK-cell-IFN-γ | Kikuchi et al. (2004, 2005, 2010) |
| 5- IL-17 | Serum IL-17 levels are elevated in individuals with AgP, and IL17 cells are inducible by DCs; IL-17 promotes Ig-class-switching and IgG production | Blaho et al. (2009); Schenkein et al. (2010) |
Oral Bacteria, Anti-oxLDLs, and Systemic Disease
Anti-phosphorylcholine (α-PC)
Normal human sera contain α-PC antibodies (Schenkein et al., 1999). These antibodies are primarily in the IgG2 and IgG1 subclasses and recognize both bacterial PC as well as many self-antigens which contain PC, including platelet-activating factor (PAF), apoptotic cells, and oxLDL (Shaw et al., 2003). It has been known for many years that the cell wall C-polysaccharide of S. pneumoniae contains PC (Tomasz, 1967), and that protective immune responses against murine S. pneumoniae infection may be mediated by naturally occurring IgM α-PC (McNamara et al., 1984; Briles et al., 1992). Similarly, LPS from strains of H. influenzae also contain PC (Weiser et al., 1997). While examining the unusual IgG2 responsiveness of individuals with AgP, we noted that sera from those with periodontitis contain elevated levels of α-PC in comparison with sera from individuals with no attachment loss (Schenkein et al., 1999). Subsequent studies revealed that a significant proportion of dental plaque bacteria, including strains of A. actinomycetemcomitans, Fusobacterium nucleatum, Streptococcus oralis, and Streptococcus sanguinis, react with TEPC-15, a mouse myeloma protein with specificity for PC (Gmür et al., 1999; Schenkein et al., 1999). Additional studies demonstrated that gingival crevicular fluid contains higher concentrations of IgG α-PC than serum from the same individuals, indicating that there is local production of α-PC that is likely induced by oral bacteria (Schenkein et al., 2004).
Antibodies reactive with LDL have also been identified as important factors in development of, and resistance against, cardiovascular diseases. In the ApoE-/- mouse model of atheroma formation, it has been demonstrated that production of α-oxLDL accompanies development of atherosclerosis (Shaw et al., 2000). Immunization of mice with S. pneumoniae capsular polysaccharide to induce high levels of IgM α-PC results in diminished atheroma formation, due to the ability of this antibody to block access of LDL to macrophage scavenger and LDL receptors (Binder et al., 2003). It has been further demonstrated that a major component of the α-oxLDL response in this model is comprised of IgM α-PC (Binder and Silverman, 2005).
The role of α-PC in human atherosclerosis is not clear, and contrasting results may relate to the isotype or immunoglobulin subclass involved. Individuals with histories of CVD have elevated serum levels of α-oxLDL antibodies and may also have circulating ICs containing oxLDL (Lehtimaki et al., 1999). This antibody, which binds to neoantigens exposed on LDL following oxidation or other chemical modifications, appears to be protective in that it blocks the uptake of LDL into macrophages (Palinski and Witztum, 2000). Clinical studies have indicated that low serum levels of IgM α-PC are associated with increased cardiovascular risk, including atherosclerosis and ischemic stroke (Su et al., 2006; de Faire and Frostegard, 2009; Sjoberg et al., 2009).
Studies examining cross-reactivity of human α-PC revealed that α-PC binds to both a variety of oral bacteria and to oxLDL (Schenkein et al., 2001). Human IgG α-PC binds to Streptococcus oralis, Streptococcus sanguinis, Haemophilus aphrophilus, Actinomyces naeslundii, Fusobacterium nucleatum, and A. actinomycetemcomitans. Furthermore, purified α-PC bound to a PC-bearing strain of A. actinomycetemcomitans. OxLDL absorbs α-PC from human sera, and purified α-PC binds directly to oxLDL but not to LDL. Finally, PC-bearing strains of A. actinomycetemcomitans are capable of invading human endothelial cells via the PAF receptor, demonstrating a potential route of access to the systemic circulation for oral-bacteria-bearing PC (Schenkein et al., 2000).
We have hypothesized that human dental plaque, containing a large proportion of bacteria containing PC antigens, and capable of entering the systemic circulation by various means, can induce elevated systemic α-PC responses that might influence the course of cardiovascular diseases.
Anti-cardiolipin (α-CL)
Anti-cardiolipin antibodies are characteristically found in sera of patients with APS and systemic lupus erythematosis (Levine et al., 2002). The major target antigen of autoimmune α-CL is a neo-epitope on the phospholipid-binding protein β2-glycoprotein-1 (β2GPI) that appears consequent to its binding to phospholipids (Galli et al., 1990; Viard et al., 1992). The pathogenicity of α-CL is thought to be related to its ability to interfere with the regulatory function of β2GPI in prevention of the initiation of coagulation (Shoenfeld, 2003; Mehdi et al., 2010).
APS is clinically characterized by vascular thrombosis and pregnancy complications such as fetal involution and spontaneous abortion (Levine et al., 2002). In addition, clinical studies have suggested that APS patients experience accelerated atherosclerosis, and analyses of α-CL demonstrate that these antibodies bind to oxLDL/β2GPI complexes and enhance uptake of oxLDL into macrophages (Ames et al., 2008; Matsuura et al., 2009). Thus, some investigators characterize α-CL as prothrombotic antibodies.
It has been proposed that infection with a variety of viral and bacterial pathogens can induce pathogenic α-CL via molecular mimicry (Blank et al., 2002; Avcin et al., 2008; Garcia-Carrasco et al., 2009). Some microbial pathogens contain antigens with peptide sequences with sufficient similarity to the critical TLRVYK sequence in β2GPI so as to induce cross-reactive α-CL that can then induce fetal involution in pregnant mice (Blank et al., 2002). In addition, there are several studies showing associations between infection and triggering of APS and other rheumatic diseases, suggesting that molecular mimicry may be a mechanism for the induction of α-CL in human disease.
Associations between periodontal disease and α-CL have been demonstrated. Assessment of α-CL in individuals with and without periodontitis demonstrated a higher prevalence of those with chronic and AgP having elevated IgG and IgM α-CL titers than healthy control individuals (Schenkein et al., 1999). In this study, 15-20% of such persons tested positive for α-CL. According to the Swiss-Prot database, at least 3 periodontal disease pathogens contain homologous sequences to TLRVYK, including P. gingivalis (TLRIYT), Treponema denticola (TLALYK), and A. actinomycetemcomitans (SIRVYK). In addition, other oral organisms may have homologous sequences and contribute to α-CL responses. Wang and co-workers (Wang et al., 2008) have shown elevated anti-SIRVYK antibodies in persons with chronic and AgP compared with control individuals, a significant correlation between anti-TLRVYK and anti-SIRVYK, and elevated anti-SIRVYK in A. actinomycetemcomitans-positive compared with A. actinomycetemcomitans-negative persons. They therefore argue that A. actinomycetemcomitans induces antibodies that mimic β2GPI. Additionally, this group has demonstrated a strong association between α-CL and periodontitis in individuals with the thrombotic Buerger’s disease (Chen et al., 2009).
Additional studies have demonstrated that α-CL found in individuals with periodontitis may have functional biological consequences. It has been shown that pathogenic α-CL from individuals with APS binds to endothelial cells, causing cytokine production and up-regulation of serum adhesion molecules (Pierangeli et al., 1999; Cho et al., 2002; Espinola et al., 2003). We found a significant association between elevated α-CL and elevated serum markers of vascular inflammation in individuals with periodontitis, particularly in those with AgP (Schenkein et al., 2007). This relationship is illustrated in Table 3. We studied serum samples from periodontally healthy persons (NP) as well as those with chronic and generalized AgP, measuring concentrations of IgG and IgM α-CL as well as soluble-intercellular adhesion molecule-1 (sICAM-1), soluble-vascular cell adhesion molecule-1, (sVCAM-1), and sE-selectin. We noted significant associations between elevated α-CL levels (> 15 U/mL) and levels of all 3 inflammatory markers in sera from those with generalized AgP that were not apparent in other studied groups. This association was not influenced by smoking, since the same associations were found in subsets of smokers and non-smokers. We further noted that statistical adjustment for periodontal measures and demographic variables did not alter these relationships (Table 4).
Table 3.
Serum sICAM-1, sVCAM-1, and sE-selectin Levels in Individuals with Elevated or Normal Anti-CL within Diagnostic Categories
| Concentrations (ng/mL) | |||||
|---|---|---|---|---|---|
| Adhesion Molecule | Anti-CL | N | Mean | SE | |
| Periodontally healthy individuals | |||||
| sICAM-1 | normal | 76 | 259.0 | 11.4 | p = 0.93 |
| elevated | 14 | 261.6 | 26.6 | ||
| sVCAM-1 | normal | 76 | 661.1 | 25.0 | p = 0.91 |
| elevated | 14 | 654.2 | 58.3 | ||
| sE-selectin | normal | 76 | 64.4 | 3.3 | p = 0.49 |
| elevated | 14 | 70.1 | 7.6 | ||
| Individuals with CP | |||||
| sICAM-1 | normal | 74 | 280.6 | 16.8 | p = 0.41 |
| elevated | 26 | 253.1 | 28.3 | ||
| sVCAM-1 | normal | 74 | 676.8 | 22.5 | p = 0.62 |
| elevated | 26 | 698.9 | 38.0 | ||
| sE-selectin | normal | 74 | 77.8 | 4.6 | p = 0.98 |
| elevated | 26 | 77.6 | 7.80 | ||
| Individuals with GAgP | |||||
| sICAM-1 | normal | 66 | 280.9 | 15.4 | p = 0.03 |
| elevated | 24 | 343.8 | 25.5 | ||
| sVCAM-1 | normal | 66 | 610.6 | 33.62 | p = 0.005 |
| elevated | 24 | 800.1 | 55.7 | ||
| sE-selectin | normal | 66 | 63.3 | 4.0 | p = 0.0002 |
| elevated | 24 | 94.2 | 6.7 | ||
This Table is from Schenkein et al. (2007) and is presented here with permission from the American Academy of Periodontology The bolded numbers highlight statistically significant differences.
Table 4.
Multiple Regression Analyses of Associations of Study Variables with sICAM-1, sVCAM-1, and E-selectin Levels in Individuals with Aggressive Periodontitis
| sICAM-1 | sVCAM-1 | sE-selectin | ||||
|---|---|---|---|---|---|---|
| Variable | F Ratio | Prob > F | F Ratio | Prob > F | F Ratio | Prob > F |
| Age | 0.7693 | 0.3830 | 0.8780 | 0.3515 | 0.0442 | 0.8339 |
| Race | 10.6658 | 0.0016 | 13.4365 | 0.0004 | 0.9414 | 0.3348 |
| Sex | 0.1957 | 0.6594 | 0.0212 | 0.8846 | 2.2841 | 0.1346 |
| Attachment loss | 0.1163 | 0.7339 | 0.1500 | 0.6995 | 0.2287 | 0.6337 |
| Pocket depth | 0.3481 | 0.5568 | 0.2340 | 0.6299 | 0.9434 | 0.3343 |
| Number of teeth | 2.5842 | 0.1118 | 0.2941 | 0.5891 | 2.4993 | 0.1177 |
| Anti-CL | 3.0716 | 0.0834 | 8.9383 | 0.0037 | 11.9450 | 0.0009 |
This Table is from Schenkein et al. (2007) and is presented here with permission from the American Academy of Periodontology The bolded numbers highlight statistically significant differences.
Blank and colleagues (Blank et al., 2002) used a murine model to implicate molecular mimicry in the induction of APS. Naïve mice were immunized with microbial pathogens that share structural homology with the TLRVYK peptide. Various levels of mouse α-CL antibodies were observed following immunization with different pathogens, including Haemophilus influenza and Neisseria gonorrheae, that gave high α-CL titers. The mouse antibodies were purified from the sera of these immunized mice and used for passive immunization of naïve mice at day 0 of pregnancy. These pregnant mice developed increased fetal loss, similar to that of a control group of mice passively immunized with known pathogenic α-CL antibodies. We reasoned that molecular mimics on P. gingivalis would induce α-CL and that α-CL antibodies from immune animals would induce adverse pregnancy outcomes similar to those described by Blank and colleagues (Blank et al., 2002). Analysis of recent data indicated that immunization of rabbits or mice with P. gingivalis induced antibodies that recognized the cardiolipin/β2GPI complex in immunoassays. Furthermore, these antibodies induced fetal involution and fetal growth restriction in a mouse model, confirming the findings from Blank et al. (2002) and extending the results to P. gingivalis (Schenkein et al., unpublished observations).
In summary, individuals with periodontitis have elevated levels of 2 antibodies, α-PC and α-CL, which have the common attributes that they can be induced by and bind to oral bacteria, and promote uptake of oral bacteria and oxLDL, and they may be clinically associated with atherosclerosis risk or development.
Antibodies and Pro-Inflammatory Cytokine Production
We reasoned that enhanced uptake of ox-LDL and bacteria mediated by α-PC or α-CL would enhance DC-stimulation and pro-inflammatory cytokine production. ox-LDL alone did not stimulate DC-IL-12 (Fig. 1A). However, DC-IL-12 was apparent when purified IgG α-PC was added, and that response correlated with increased ox-LDL uptake (Kikuchi et al., 2010). Similarly, α-CL promoted a potent DC-IL-12 response when incubated with A. actinomycetemcomitans (Fig. 1B), although A. actinomycetemcomitans alone was adequate to stimulate DC-IL-12. The concentration of oral bacteria in blood was likely low, and the dose of A. actinomycetemcomitans used was 10,000 cells to model a number that might be obtained in the blood in vivo. However, even 1000 organisms were sufficient to induce a detectible DC-IL-12 response in this system when incubated with α-PC. Our major hypothesis was that α-PC/α-CL would promote pro-inflammatory cytokine production, including NK-cell-IFN-γ that is released by 24 to 48 hrs after engaging IL-12-producing DCs (Kikuchi et al., 2004). However, DC-IL-10 is anti-inflammatory, and the IL-12/IL-10 ratio is important in determining whether a given stimulus is pro-inflammatory or anti-inflammatory. Anti-PC did not significantly alter DC-IL-10 levels, but increased the level of IL-12, and a corresponding increase in the DC-IL-12/IL-10 ratio that correlated with the production of NK-cell-IFN-γ (Fig. 1C, upper panel). The importance of NK cells in the rapid 24-hour IFN-γ response was confirmed in PBL cultures where removal of CD56+ cells essentially eliminated this rapid IFN-γ response (Kikuchi et al., 2004).
Figure 1.
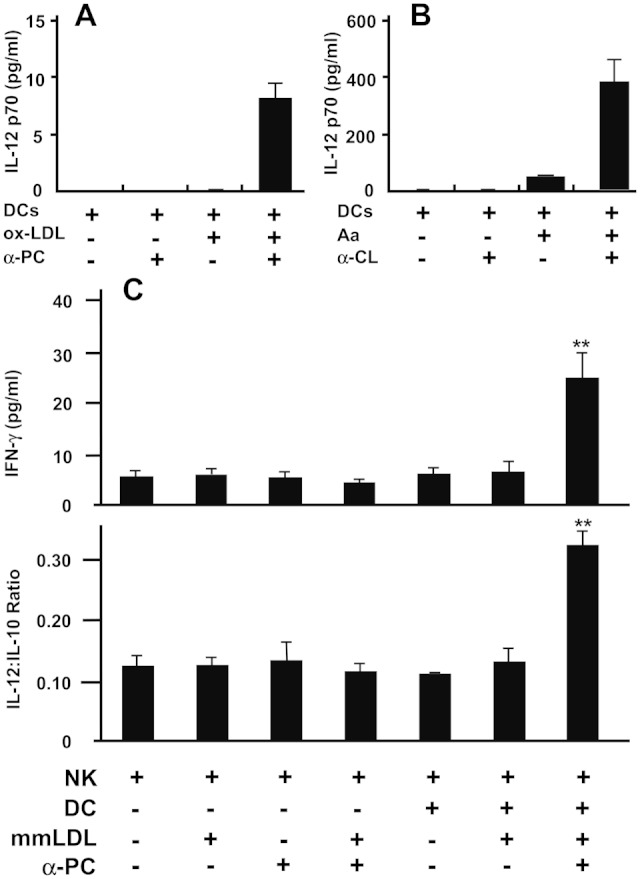
Antibodies reactive with both oral bacteria and oxLDL promote pro-inflammatory cytokine production. (A) Dendritic cells (DCs) from a periodontally healthy individual were stimulated with 2 µg/mL of oxLDL in the presence or absence of 2 µg/mL of purified α-PC of the IgG isotype. Supernatant fluids were collected at 24 hrs, and IL-12p70 production was measured. Cultures were set up in triplicate and used to calculate the standard deviations illustrated; the experiments were repeated 3 times. (B) Dendritic cells (DCs) at 1 x 105 per culture from a periodontally healthy individual were stimulated with A. actinomycetemcomitans. The concentration of oral bacteria in the blood was likely low, and the dose of A. actinomycetemcomitans was 10,000 cells to model a number that might be obtained in the blood in vivo. This would give a ratio of 1 bacterium per 10 DCs. Note that the optimal response to A. actinomycetemcomitans (Aa) occurred when treated with α-CL. This experiment was repeated 3 times, and the cultures were set up in triplicate and used to calculate the standard deviations illustrated. (C) Illustration of the enhanced ability of mmLDL to induce DC-IL-12 and promote NK-cell-IFN-γ when treated with α-PC. The LDL used was freshly isolated, and the designation “mmLDL” is used to indicate that the only modifications to the LDL were the minimal modifications that would occur as the LDL interacts with immune cells and oxygen in culture. NK cells and DCs were stimulated with mmLDL alone or with α-PC-treated mmLDL. The supernatant fluids were collected at 48 hrs, and the IL-10, IL-12, and IFN-γ levels were determined. Addition of α-PC to LDL increased IL-12 levels without significantly altering IL-10 levels, resulting in a higher IL-12/IL-10 ratio that corresponded with elevated IFN-γ responses. **p < 0.01. This experiment was repeated 3 times, and the cultures were set up in triplicate and were used to calculate the standard deviations illustrated. This Fig. (from Kikuchi et al., 2010) is presented here with the permission of the J Periodontal Res.
Relationships among DCs, α-oxLDLs, and Atherosclerosis
As described above, α-CL is elevated in individuals with APS and a significant number of those with periodontitis (Levine et al., 2002; Schenkein et al., 2003), and both diseases are associated with increased risk of atherosclerosis. Dendritic cells reside in the subendothelial layer of arterial walls in numbers comparable with Langerhans cells in the skin, and these DCs are in a favorable position to interact with antigens in the blood. The vascular DCs accumulate most densely in arterial regions with turbulent blood flow, and these sites are predisposed to the development of atherosclerosis (Lord and Bobryshev, 1999). It is of interest that these sites also develop arteritis as a consequence of IC-deposition in diseases such as serum sickness. Moreover, serum sickness induces vessel damage as a consequence of ICs being deposited between endothelial cells, and that stimulus promotes atherosclerosis in animals when combined with a lipid imbalance (Wissler and Group, 1996). Immune complexes of oxLDL and A. actinomycetemcomitans induced DC-IL-12 responses in culture (Fig. 1). This prompted reasoning that engagement of DCs in the subendothelial layer of arterial walls with ICs of micro-organisms, microbial products, or oxLDL from the blood likely triggers IL-12 production.
An illustration summarizing DC, NK cell, T-cell, and α-PC/α-CL interactions with oral organisms that may induce and sustain periodontitis, as well as other forms of chronic inflammation including atherosclerosis, is presented in Fig. 2. These antibodies bind PC or TLRVYK on oxLDL and TLRVYK-like sequences on A. actinomycetemcomitans and P. gingivalis (Fig. 2A). These interactions result in ICs that bind activating-Fc receptors on DCs and promote engulfment of the ICs, antigen processing, and production of the IL-12 and IL-18 that promotes early-IFN-γ production by NK cells and later by T-cells in inflamed sites (Figs. 2B, 2C). Numerous pro-inflammatory chemokines and cytokines are produced by stimulated T-cells and macrophages that promote atherosclerosis (Fig. 2D). Production of pro-inflammatory cytokines by macrophages is well-known, although IFN-γ production is controversial. Nevertheless, numerous studies have reported macrophage-IFN-γ responses, and this may occur after treatment with IL-12 and IL-18 (Gerdes et al., 2002). Moreover, IFN-γ-producing macrophages are found in endarterectomy specimens (Gerdes et al., 2002), and we have confirmed that observation (unpublished). Pro-inflammatory cytokines and chemokines may up-regulate adhesion molecules on the epithelium and help sustain the influx of macrophages and other inflammatory cells (Fig. 2D). Macrophages activated by IFN-γ may promote plaque destabilization (Hansson and Libby, 2006). Disruption of atherosclerotic plaque and thrombus formation may result in embolization that may cause myocardial infarction, stroke, or peripheral arterial occlusive disease. Thus, engagement of DCs in the subendothelial layer of arterial walls with α-PC/α-CL bound to micro-organisms, microbial products, or oxLDL from the blood may trigger a pathway in atheromas involving DC-IL-12 production, antigen presentation, IFN-γ production, macrophage activation, plaque disruption, embolization, and, ultimately, life-threatening disease.
Figure 2.
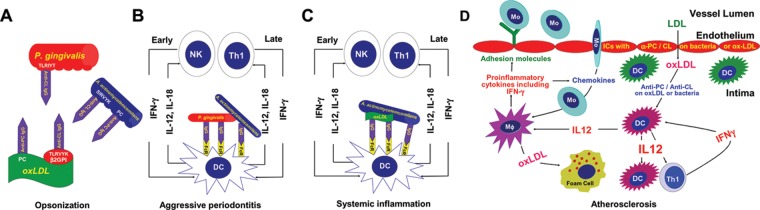
Role for dendritic cells and α-PC and/or α-CL in the induction of pro-inflammatory cytokine production that promotes chronic inflammation including atherosclerosis. (A) Illustrates interactions of α-PC with PC on oxLDL and A. actinomycetemcomitans and α-CL binding TLRVYK on β2-glycoprotein-1 associated with oxLDL, as well as sequences homologous to TLRVYK on P. gingivalis and A. actinomycetemcomitans. (B) Immune complexes of bacteria interacting with DC, leading to early production of IL-12 and IL-18 and early production of IFN-γ (< 24 hrs) by NK cells. The IFN-γ helps activate DCs as well as macrophages and promotes inflammation. Later, as chronic inflammation is developing, T-cells would be expected to replace the NK cells as major IFN-γ producers. (C) The same events described in panel B would be expected systemically and in other sites where oral bacteria and oxLDL and α-PC and/or α-CL would be available to interact and engage DCs. (D) Vascular DCs accumulate in arterial regions that are subject to turbulent blood flow, and these sites are predisposed to the development of atherosclerosis (Lord and Bobryshev, 1999). These are also sites where arteritis develops as a consequence of IC depositions that occur in serum sickness (Wissler and Group, 1996). In cell cultures, oxLDL-ICs and oral bacteria-ICs induced DC-IL-12 responses, prompting us to reason that engagement of DCs in the subendothelial layer of arterial walls with ICs of micro-organisms or oxLDL from the blood likely triggers IL-12 production. These DCs would process antigen, and T-cells would respond to the processed IC-derived peptides on the stimulated IL-12-producing DCs by producing IFN-γ. Monocytes also respond to the ICs and DC-IL-12 by maturing into macrophages and producing chemokines and cytokines that would promote increased levels of endothelium-adhesion molecules and the accumulation of more pro-inflammatory cells, including more monocytes and IFN-γ-activated macrophages. Antibody-bound oxLDL is taken up by macrophages including foam cells by a combination of Fc, complement, and scavenging receptors. The accumulated activated macrophages and rich combination of pro-inflammatory cytokines are thought to promote plaque instability and thrombosis (Hansson and Libby, 2006).
Conclusions
Anti-CL promotes atherosclerosis and adverse pregnancy outcomes, and these problems are associated with both APS and periodontitis. A. actinomycetemcomitans and P. gingivalis are potent immunogens, and 10 and 100 µg/mL of specific serum IgG are typically induced in infected persons, most of which is the DC-pro-inflammatory cytokine-associated IgG2. These antibodies correlate with resistance to periodontal destruction, but the level of protection is modest (Califano et al., 1996). However, these organisms also contain molecular mimics that appear to induce elevated α-PC/α-CL levels, and this is striking in AgP. The lack of better protection against periodontal destruction may relate to the ability of these antibodies to promote chronic inflammation. The A. actinomycetemcomitans- and P. gingivalis-induced antibodies can bind both oxLDL and oral bacteria and participate in DC-dependent mechanisms that promote inflammation. Elevated soluble-intercellular adhesion molecules are atherosclerosis-associated indicators of vascular inflammation, and these markers are elevated in AgP individuals with high α-CL. Thus, the subset of individuals with periodontitis with elevated α-CL levels may be at high risk of atherosclerosis and/or adverse pregnancy outcomes. Fortunately, serum α-PC and α-CL levels can be determined with simple blood tests, and individuals with high antibody levels may be assisted by reducing cholesterol levels to minimize atherosclerosis risk and by prenatal care to minimize adverse pregnancy outcomes. Interventions of this kind remain to be tested, but the α-oxLDL-associated pro-inflammatory pathway described here prompts us to believe that this approach is worthy of serious consideration.
Supplementary Material
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
This work was supported by USPHS research grants DE017223 (JGT) and DE018125 (HAS) from the National Institute of Dental and Craniofacial Research.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Ames PR, Scenna G, Antinolfi I, Lopez L, Iannaccone L, Matsuura E, et al. (2008). Atherosclerosis in primary antiphospholipid syndrome. Expert Rev Clin Immunol 4:53-60. [DOI] [PubMed] [Google Scholar]
- Avcin T, Canova M, Guilpain P, Guillevin L, Kallenberg CG, Tincani A, et al. (2008). Infections, connective tissue diseases and vasculitis. Clin Exp Rheumatol 26(1 Suppl 48):18S-26S. [PubMed] [Google Scholar]
- Barbour SE, Ishihara Y, Fakher M, Al Darmaki S, Caven TH, Shelburne CP, et al. (2002). Monocyte differentiation in localized juvenile periodontitis is skewed toward the dendritic cell phenotype. Infect Immun 70:2780-2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder CJ, Silverman GJ. (2005). Natural antibodies and the autoimmunity of atherosclerosis. Springer Semin Immunopathol 26:385-404. [DOI] [PubMed] [Google Scholar]
- Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, et al. (2003). Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 9:736-743. [DOI] [PubMed] [Google Scholar]
- Blaho VA, Buczynski MW, Dennis EA, Brown CR. (2009). Cyclooxygenase-1 orchestrates germinal center formation and antibody class-switch via regulation of IL-17. J Immunol 183:5644-5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank M, Krause I, Fridkin M, Keller N, Kopolovic J, Goldberg I, et al. (2002). Bacterial induction of autoantibodies to beta2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. J Clin Invest 109:797-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briles DE, Forman C, Crain M. (1992). Mouse antibody to phosphocholine can protect mice from infection with mouse-virulent human isolates of Streptococcus pneumoniae. Infect Immun 60:1957-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano JV, Gunsolley JC, Nakashima K, Schenkein HA, Wilson ME, Tew JG. (1996). Influence of anti-Actinobacillus actinomycetemcomitans Y4 (serotype b) lipopolysaccharide on severity of generalized early-onset periodontitis. Infect Immun 64:3908-3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano JV, Schifferle RE, Gunsolley JC, Best AM, Schenkein HA, Tew JG. (1999). Antibody reactive with P. gingivalis serotypes K1-6 in adult and generalized early-onset periodontitis. J Periodontol 70:730-735. [DOI] [PubMed] [Google Scholar]
- Chen YW, Nagasawa T, Wara-Aswapati N, Ushida Y, Wang D, Takeuchi Y, et al. (2009). Association between periodontitis and anti-cardiolipin antibodies in Buerger disease. J Clin Periodontol 36:830-835. [DOI] [PubMed] [Google Scholar]
- Cho CS, Cho ML, Chen PP, Min SY, Hwang SY, Park KS, et al. (2002). Antiphospholipid antibodies induce monocyte chemoattractant protein-1 in endothelial cells. J Immunol 168:4209-4215. [DOI] [PubMed] [Google Scholar]
- de Faire U, Frostegard J. (2009). Natural antibodies against phosphorylcholine in cardiovascular disease. Ann NY Acad Sci 1173:292-300. [DOI] [PubMed] [Google Scholar]
- Espinola RG, Liu X, Colden-Stanfield M, Hall J, Harris EN, Pierangeli SS. (2003). E-Selectin mediates pathogenic effects of antiphospholipid antibodies. J Thromb Haemost 1:843-848. [DOI] [PubMed] [Google Scholar]
- Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, Breda-Vriesman PJ, et al. (1990). Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 335:1544-1547. [DOI] [PubMed] [Google Scholar]
- Garcia-Carrasco M, Galarza-Maldonado C, Mendoza-Pinto C, Escarcega RO, Cervera R. (2009). Infections and the antiphospholipid syndrome. Clin Rev Allergy Immunol 36:104-108. [DOI] [PubMed] [Google Scholar]
- Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schonbeck U. (2002). Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med 195:245-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmür R, Thurnheer T, Guggenheim B. (1999). Dominant cross-reactive antibodies generated during the response to a variety of oral bacterial species detect phosphorylcholine. J Dent Res 78:77-85. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Libby P. (2006). The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol 6:508-519. [DOI] [PubMed] [Google Scholar]
- Jotwani R, Cutler CW. (2003). Multiple dendritic cell (DC) subpopulations in human gingiva and association of mature DCs with CD4+ T-cells in situ. J Dent Res 82:736-741. [DOI] [PubMed] [Google Scholar]
- Jotwani R, Palucka AK, Al Quotub M, Nouri-Shirazi M, Kim J, Bell D, et al. (2001). Mature dendritic cells infiltrate the T cell-rich region of oral mucosa in chronic periodontitis: in situ, in vivo, and in vitro studies. J Immunol 167:4693-4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull M, Demmer RT, Papapanou PN. (2010). “Gum bug, leave my heart alone!”—epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res 89:879-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Hahn CL, Tanaka S, Barbour SE, Schenkein HA, Tew JG. (2004). Dendritic cells stimulated with Actinobacillus actinomycetemcomitans elicit rapid gamma interferon responses by natural killer cells. Infect Immun 72:5089-5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Willis DL, Liu M, Purkall DB, Sukumar S, Barbour SE, et al. (2005). Dendritic-NK cell interactions in P. gingivalis-specific responses. J Dent Res 84:858-862. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, El Shikh MM, El Sayed RM, Purkall DB, Elaasser MM, Sarraf A, et al. (2010). Anti-phosphorylcholine-opsonized low-density lipoprotein promotes rapid production of proinflammatory cytokines by dendritic cells and natural killer cells. J Periodontal Res 45:720-730. [DOI] [PubMed] [Google Scholar]
- Lehtimaki T, Lehtinen S, Solakivi T, Nikkila M, Jaakkola O, Jokela H, et al. (1999). Autoantibodies against oxidized low density lipoprotein in patients with angiographically verified coronary artery disease. Arterioscler Thromb Vasc Biol 19:23-27. [DOI] [PubMed] [Google Scholar]
- Levine JS, Branch DW, Rauch J. (2002). The antiphospholipid syndrome. N Engl J Med 346:752-763. [DOI] [PubMed] [Google Scholar]
- Lord RS, Bobryshev YV. (1999). Clustering of dendritic cells in athero-prone areas of the aorta. Atherosclerosis 146:197-198. [DOI] [PubMed] [Google Scholar]
- Lu H, Califano JV, Schenkein HA, Tew JG. (1993). Immunoglobulin class and subclass distribution of antibodies reactive with the immunodominant antigen of Actinobacillus actinomycetemcomitans serotype b. Infect Immun 61:2400-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wang M, Gunsolley JC, Schenkein HA, Tew JG. (1994). Serum immunoglobulin G subclass concentrations in periodontally healthy and diseased individuals. Infect Immun 62:1677-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura E, Kobayashi K, Lopez LR. (2009). Atherosclerosis in autoimmune diseases. Curr Rheumatol Rep 11:61-69. [DOI] [PubMed] [Google Scholar]
- McNamara MK, Ward RE, Kohler H. (1984). Monoclonal idiotope vaccine against Streptococcus pneumoniae infection. Science 226:1325-1326. [DOI] [PubMed] [Google Scholar]
- Mehdi AA, Uthman I, Khamashta M. (2010). Antiphospholipid syndrome: pathogenesis and a window of treatment opportunities in the future. Eur J Clin Invest 40:451-464. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. (2009). Skin immune sentinels in health and disease. Nat Rev Immunol 9:679-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palinski W, Witztum JL. (2000). Immune responses to oxidative neoepitopes on LDL and phospholipids modulate the development of atherosclerosis. J Intern Med 247:371-380. [DOI] [PubMed] [Google Scholar]
- Pierangeli SS, Colden-Stanfield M, Liu X, Barker JH, Anderson GL, Harris EN. (1999). Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation 99:1997-2002. [DOI] [PubMed] [Google Scholar]
- Purkall D, Tew JG, Schenkein HA. (2002). Opsonization of Actinobacillus actinomycetemcomitans by immunoglobulin G antibody reactive with phosphorylcholine. Infect Immun 70:6485-6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkein HA, Gunsolley JC, Best AM, Harrison MT, Hahn CL, Wu J, et al. (1999). Antiphosphorylcholine antibody levels are elevated in humans with periodontal diseases. Infect Immun 67:4814-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkein HA, Barbour SE, Berry CR, Kipps B, Tew JG. (2000). Invasion of human vascular endothelial cells by Actinobacillus actinomycetemcomitans via the receptor for platelet-activating factor. Infect Immun 68:5416-5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkein HA, Berry CR, Purkall D, Burmeister JA, Brooks CN, Tew JG. (2001). Phosphorylcholine-dependent cross-reactivity between dental plaque bacteria and oxidized low-density lipoproteins. Infect Immun 69:6612-6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkein HA, Berry CR, Burmeister JA, Brooks CN, Barbour SE, Best AM, et al. (2003). Anti-cardiolipin antibodies in sera from patients with periodontitis. J Dent Res 82:919-922. [DOI] [PubMed] [Google Scholar]
- Schenkein HA, Berry CR, Burmeister JA, Brooks CN, Best AM, Tew JG. (2004). Locally produced anti-phosphorylcholine and anti-oxidized low-density lipoprotein antibodies in gingival crevicular fluid from aggressive periodontitis patients. J Periodontol 75:146-153. [DOI] [PubMed] [Google Scholar]
- Schenkein HA, Best AM, Brooks CN, Burmeister JA, Arrowood JA, Kontos MC, et al. (2007). Anti-cardiolipin and increased serum adhesion molecule levels in patients with aggressive periodontitis. J Periodontol 78:459-466. [DOI] [PubMed] [Google Scholar]
- Schenkein HA, Koertge TE, Brooks CN, Sabatini R, Purkall DE, Tew JG. (2010). IL-17 in sera from patients with aggressive periodontitis. J Dent Res 89:943-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, et al. (2000). Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest 105:1731-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PX, Goodyear CS, Chang MK, Witztum JL, Silverman GJ. (2003). The autoreactivity of anti-phosphorylcholine antibodies for atherosclerosis-associated neo-antigens and apoptotic cells. J Immunol 170:6151-6157. [DOI] [PubMed] [Google Scholar]
- Shoenfeld Y. (2003). Etiology and pathogenetic mechanisms of the anti-phospholipid syndrome unraveled. Trends Immunol 24:2-4. [DOI] [PubMed] [Google Scholar]
- Sjoberg BG, Su J, Dahlbom I, Gronlund H, Wikstrom M, Hedblad B, et al. (2009). Low levels of IgM antibodies against phosphorylcholine—a potential risk marker for ischemic stroke in men. Atherosclerosis 203:528-532. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hemmi H. (2006). Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol 311:17-58. [DOI] [PubMed] [Google Scholar]
- Su J, Georgiades A, Wu R, Thulin T, de Faire U, Frostegard J. (2006). Antibodies of IgM subclass to phosphorylcholine and oxidized LDL are protective factors for atherosclerosis in patients with hypertension. Atherosclerosis 188:160-166. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Barbour SE, Best AM, Schenkein HA, Tew JG. (2003). Prostaglandin E2-mediated regulation of immunoglobulin G2 via interferon gamma. J Periodontol 74:771-779. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Fakher M, Barbour SE, Schenkein HA, Tew JG. (2006). Influence of proinflammatory cytokines on Actinobacillus actinomycetemcomitans specific IgG responses. J Periodontal Res 41:1-9. [DOI] [PubMed] [Google Scholar]
- Tomasz A. (1967). Choline in the cell wall of a bacterium: novel type of polymer-linked choline in Pneumococcus. Science 157:694-697. [DOI] [PubMed] [Google Scholar]
- Viard JP, Amoura Z, Bach JF. (1992). Association of anti-beta 2 glycoprotein I antibodies with lupus-type circulating anticoagulant and thrombosis in systemic lupus erythematosus. Am J Med 93:181-186. [DOI] [PubMed] [Google Scholar]
- Wang D, Nagasawa T, Chen Y, Ushida Y, Kobayashi H, Takeuchi Y, et al. (2008). Molecular mimicry of A. actinomycetemcomitans with beta2 glycoprotein I. Oral Microbiol Immunol 23:401-405. [DOI] [PubMed] [Google Scholar]
- Weiser JN, Shchepetov M, Chong ST. (1997). Decoration of lipopolysaccharide with phosphorylcholine: a phase-variable characteristic of Haemophilus influenzae. Infect Immun 65:943-950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ME, Hamilton RG. (1992). Immunoglobulin G subclass response of localized juvenile periodontitis patients to Actinobacillus actinomycetemcomitans Y4 lipopolysaccharide. Infect Immun 60:1806-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissler RW, Group PD. (1996). Atheroarteritis: a combined immunological and lipid imbalance. Int J Cardiol 54(Suppl):37S-49S. [DOI] [PubMed] [Google Scholar]
- Zhang JB, Quinn SM, Rausch M, Gunsolley JC, Schenkein HA, Tew JG. (1996). Hyper-immunoglobulin G2 production by B cells from patients with localized juvenile periodontitis and its regulation by monocytes. Infect Immun 64:2004-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.