

Abstract
Enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively) are attaching and effacing (A/E) bacterial pathogens that cause severe diarrheal disease worldwide. To cause disease, A/E pathogens require a type III secretion system, which facilitates transport of bacterial effector proteins directly into infected host cells. One of these effector proteins translocated by the type III secretion system, EspZ, is essential for A/E pathogen infection and functions to prevent rapid death of EPEC-infected cells. We further investigated the mechanism of EspZ-mediated protection of infected host cells and found that a severe decrease in host mitochondrial membrane potential (Δψm) occurs concurrently with host cell lysis during infection with EPEC lacking EspZ (ΔespZ). It was also demonstrated that EspZ localizes to host cell mitochondria and interacts with the translocase of inner mitochondrial membrane 17b (TIM17b). In addition, host cell cytotoxicity was exacerbated in the absence of TIM17b during wild-type (WT) EPEC infection. The findings of this study together provide the first evidence that EspZ localizes to host mitochondria and that TIM17b contributes to protection against rapid cell death during EPEC infection.
INTRODUCTION
Enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively) are attaching and effacing (A/E) pathogens that are significant causes of morbidity and mortality worldwide. EPEC infection results in severe infantile diarrhea in developing countries, whereas EHEC is responsible for severe bloody diarrhea worldwide (8). A/E pathogens interact with host intestinal epithelial cells, where they are able to locally efface microvilli and attach to the surface of the host cell (8). The ability of these pathogens to cause disease is mediated by a type III secretion system (T3SS) encoded on a pathogenicity island termed the locus of enterocyte effacement (LEE). Intimate attachment to host cells is mediated by the translocated intimin receptor (Tir), which is inserted into the host cell plasma membrane and interacts with intimin, an A/E pathogen outer membrane protein (17). Subsequently, a plethora of effector proteins is translocated into the infected host cell by the T3SS, where they subvert cellular functions. EPEC encodes at least 21 effector proteins, whereas EHEC encodes at least 40 (15, 30). Although the functions of some of these effector proteins during A/E pathogen infection are becoming clearer, the roles of many remain unknown. It is also becoming increasingly apparent that many effector proteins have several functions during infection.
Host cell death during A/E pathogen infection is intricately regulated by several effector proteins translocated by the T3SS. The effector protein EspF was the first effector to be associated with increased apoptosis of infected epithelial cells. EspF localizes to the mitochondria and causes release of cytochrome c and depletion of antiapoptotic proteins, which initiate the intrinsic apoptosis cascade (14). The effector protein Map also localizes to mitochondria and facilitates organellar dysfunction and decreased mitochondrial membrane potential (Δψm), which lead to cell death (20, 25). Similarly, the effector Cif also facilitates cytochrome c release and apoptosis of infected host cells (26). In addition to proapoptotic effector proteins, A/E pathogens also inject several prosurvival factors. NleH1 and NleH2 inhibit apoptosis by interaction with Bax inhibitor-1 (BI-1), and NleD downregulates proapoptotic signaling by cleaving c-Jun N-terminal kinase (JNK) (2, 13). We have also shown that host cell death is strongly antagonized by the effector EspZ during EPEC infection (28).
EspZ is a 9-kDa LEE-encoded effector protein that contains two putative transmembrane domains and is essential for virulence of the murine A/E pathogen Citrobacter rodentium (10, 16, 28). We found that infection of cells in culture with an espZ-knockout strain of EPEC (ΔespZ) causes cytotoxicity of infected cells. We subsequently determined that EspZ upregulates phosphorylation of AKT and focal adhesion kinase (FAK) and acts partially through host CD98 to increase FAK activation (28). Since FAK activation enhances prosurvival signaling, this was one mechanism by which EspZ could dampen host cell death during EPEC infection. However, the EspZ-CD98 interaction was only partially responsible for the protective phenotype exhibited by EspZ since CD98 knockdown did not heavily influence cytotoxicity during infection with wild-type (WT) EPEC (28). We were therefore interested in further investigating the mechanisms of cell death antagonized by EspZ and identifying other host protein targets.
In our previous study, we identified the translocase of inner mitochondrial membrane 17b (TIM17b) to be a putative EspZ-interacting protein by yeast two-hybrid assay (28). TIM17 is a component of the TIM23 complex, which is responsible for transport of proteins into the inner mitochondrial membrane and the mitochondrial matrix (5, 11). This complex is composed of three inner membrane proteins (TIM50, TIM23, and TIM17) and is dependent on energy derived from the mitochondrial matrix motor, an intact Δψm, and ATP (5). Specifically, TIM17 is responsible for recruiting the presequence translocase-associated motor (PAM) and sorting preproteins in the inner mitochondrial membrane (IMM) (6).
In this study, we identified a second mechanism by which EspZ is able to antagonize host cell death. To further characterize the mechanism of host cell survival promoted by EspZ, we investigated whether EspZ functions at the mitochondrion, since this organelle plays a central role in host cell death and survival pathways. We determined that a significant decrease in Δψm accompanies cytotoxicity mediated by EPEC ΔespZ infection. We further found that EspZ localizes to host mitochondria and that it also interacts with TIM17b. The ability of EspZ to protect cells against death during EPEC infection was dampened following small interfering RNA (siRNA) knockdown of TIM17b. Collectively, this represents a second mechanism by which EspZ protects host cells against rapid cell death during EPEC infection.
MATERIALS AND METHODS
Tissue culture, bacterial strains, primers, and transfection and infection conditions.
HeLa and HEK293T cells were purchased from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle medium (DMEM) high glucose (Thermo Scientific) supplemented with 10% fetal bovine serum (FBS; Thermo Scientific), 1% nonessential amino acids (NEAA; Gibco), and 1% GlutaMAX medium (Gibco). Cells were used from passages 5 to 20. Bacterial strains used in this study are listed in Table 1. Oligonucleotide primers used in this study are listed in Table 2, and plasmids used in this study are listed in Table 3.
Table 1.
Bacterial strains used in this study
| Strain | Source or reference |
|---|---|
| EPEC O127:H6 strain E2348/69 | |
| WT | 19 |
| ΔespZ | 28 |
| ΔespZ/espZ (MCE007) | This study |
| ΔespZ/espZ-HA (MCE008) | This study |
| ΔsepD | 9 |
| ΔsepD ΔespZ | 28 |
| ΔsepD ΔespZ/espZ (MCE010) | This study |
| ΔsepD ΔespZ/espZ-HA (MCE011) | This study |
| Escherichia coli | |
| SM10λpir | 22 |
| DH5αλpir | 22 |
| EC100Dpir+ | Epicentre Biotechnologies |
| ω7249 | 1 |
Table 2.
Oligonucleotide primers used in this study
| Name | Sequence (5′ → 3′)a |
|---|---|
| TIM17-F | AATTGCGGCCGCGGCACCATGGAGGAGTACGCTCGGGAGCCC |
| TIM17-R | AATAAGCTTGTGGTACTGATAGCTGGGG |
| espZ-f | CGACGGATCCTTAGTTATACTCTAAAGCAAACGTAAC |
| espZ-r | TAGAATTCTTAGGCATATTTCATCGCTAATC |
| espZHA-r | TAGAATTCTTAAGCGTAATCTGGAACATCGTATGGGTAGGCATATTTCATCGCTAATCCG |
Restriction endonuclease cleavage sites are in bold. Kozak sequences are underlined.
Table 3.
Plasmids used in this study
HeLa cells were transfected using FuGENE HD transfection reagent (Roche) as previously described (28) and were assayed at 24 to 36 h posttransfection. Lysates were subject to Western blot analysis to determine efficiency of transfection. HEK293T cells were transfected using calcium phosphate as described previously (28).
Infections were performed as previously described (28). For the 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) assay, a 1:600 dilution of overnight standing bacterial cultures was prepared in DMEM and used to infect HeLa cells in a 96-well plate.
Generation of ΔespZ/espZ and ΔespZ/espZ-HA complemented strains.
Complementation of espZ was achieved by chromosomal insertion with Tn7 into EPEC ΔespZ or ΔespZ ΔsepD as previously described (27). Briefly, espZ was PCR amplified from EPEC genomic DNA using oligonucleotides espZ-f and espZ-r. A hemagglutinin (HA)-tagged version of espZ (espZ-HA) was also created by PCR using oligonucleotides espZ-f and espZHA-r. These PCR products were sequenced at the Nucleic Acid Protein Service (NAPS) unit (University of British Columbia [UBC], Vancouver, British Columbia) and cloned into the BamHI/EcoRI site of pMAC5 prior to conjugation and site-specific integration into the chromosome of EPEC ΔespZ or ΔespZ ΔsepD. The complementing strains were designated MCE007 (ΔespZ/espZ), MCE008 (ΔespZ/espZ-HA), MCE010 (ΔespZ ΔsepD/espZ), and MCE011 (ΔespZ ΔsepD/espZ-HA).
Generation of TIM17b-FLAG fusion.
TIM17b cDNA was obtained from clones from the BD Matchmaker pretransformed HeLa cell cDNA library as previously described (28). The TIM17b cDNA lacking its stop codon was cloned into pCMV-TAG4A (pCMV4A) (Stratagene). Primers TIM17-F and TIM17-R were used to PCR amplify TIM17b cDNA with a Kozak sequence on the 5′ end. The purified PCR product was cloned as a NotI/HindIII fragment into the multiple-cloning site of pCMV4A in frame with the downstream FLAG tag sequence. The TIM17b gene sequence was confirmed by DNA sequencing.
JC-1 Δψm assay.
HeLa cells were plated at 5 × 104 cells/well in a 96-well black clear-bottom tissue culture-treated plate (Costar) at 24 h prior to infection. Cells were washed in phosphate-buffered saline with Ca2+ and Mg2+ (PBS+/+), and a 1:600 dilution of EPEC strains or uninoculated LB broth in DMEM was added to each well. Cells were infected for 2, 3, or 4 h, as indicated. The JC-1 mitochondrial membrane potential (Δψm) assay kit (Cayman Chemical Company) was used according to the manufacturer's instructions. Briefly, at 15 min prior to termination of the infection, 10 μl of prepared JC-1 reagent was added to each well and the contents were mixed by gently swirling the plate. The plate was centrifuged for 5 min at 400 relative centrifugal force (RCF) in a Beckman tabletop centrifuge, and the supernatant was aspirated. Two hundred microliters of assay buffer was added to each well, and the plate was centrifuged as described above. Wash steps were repeated, followed by addition of 100 μl assay buffer and analyses using a Tecan M200 microplate reader. JC-1 aggregates (J aggregates) and J monomers were quantified using excitation/emission wavelengths of 560/595 nm and 485/535 nm, respectively. Data were plotted as J aggregates/J monomers of samples in triplicate.
LDH release assay.
For analysis of lactate dehydrogenase (LDH) presence in supernatants, a CytoTox96 nonradioactive cytotoxicity assay (Promega Corporation, Madison, WI) was used according to the manufacturer's instructions. The absorbance at 492 nm was read in a Tecan plate reader. Percent cytotoxicity was measured by normalizing values at 492 nm to the values for a 100% cytotoxicity control and uninfected controls according to the manufacturer's instructions. LDH release from bacterial strains alone was quantified and found to be negligible.
Confocal microscopy.
HeLa cells were plated on sterile glass coverslips at 24 h prior to transfection. Cells were washed in PBS+/+ and fixed in 3.7% paraformaldehyde (Sigma) for 20 min at room temperature. Coverslips were washed 3× 10 min in PBS without Ca2+ and Mg2+ (PBS−/−), and cells were permeabilized in 0.2% Triton X-100 for 5 min at room temperature. Coverslips were inverted on 50 μl blocking buffer (5% normal goat serum [NGS] in PBS with 0.5% Tween 20 [PBST] and 0.1% bovine serum albumin [BSA], 50 mM NH4Cl) for 30 min under humidified conditions at room temperature. Coverslips were incubated in primary antibody (1:200 dilution of mouse anti-HA.11 [Sigma], rabbit anti-cytochrome c oxidase IV [COX IV; Cell Signaling Technologies], or anti-TIM17b [ProteinTech]) in 1% NGS-PBST-BSA overnight at 4°C. When MitoTracker Deep Red stain (Invitrogen) was used, 500 nM reagent was added to cells at 20 min prior to harvesting of coverslips. Coverslips were washed 3 times for 10 min each time in PBST-BSA, followed by inversion on secondary antibody solution (Alexa 488-conjugated goat antimouse or Alexa 633-conjugated goat antirabbit antibodies with 1% NGS in PBST-BSA) in the dark under humidified conditions at room temperature for 90 min. Coverslips were washed in the dark 3 times for 10 min each time in PBST-BSA and mounted on clean glass slides in ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) mounting medium. Coverslips were sealed with nail polish and imaged on an Olympus Fluoview 10i laser scanning confocal microscope at the UBC BioImaging Facility (Vancouver, British Columbia, Canada). Images were assembled using ImageJ and Adobe Photoshop software.
Immunoprecipitation.
HEK293T cell lysates were generated using NP-40 lysis buffer (1% [vol/vol] Nonidet P-40, 20 mM Tris [pH 7.5], 150 mM NaCl, 10 mM sodium pyrophosphate, and 50 mM NaF all supplemented with 1 mM Na3VO4, and protease inhibitor cocktail [Roche]) and cleared by centrifugation as described previously (28). Protein G-conjugated DynaL beads (Invitrogen) were bound to mouse anti-FLAG M2 (Sigma) or normal mouse IgG isotype control (IC; Jackson ImmunoResearch Labs). Immunoprecipitations were performed according to the manufacturer's instructions. Twenty microliters of 3× Laemmli sample buffer was used to resuspend beads prior to Western blot analysis.
Western blotting.
Proteins separated by denaturing SDS-PAGE were transferred to pure nitrocellulose or methanol-activated polyvinylidene difluoride membranes (Bio-Rad) using a wet transfer cell (Bio-Rad). Membranes were blocked for 30 min in blocking buffer (5% nonfat milk in Tris-buffered saline [TBS] plus 0.1% Tween 20 [TBST]) with rocking at room temperature. Primary antibodies were diluted in blocking buffer as described below, and membranes were incubated in this solution overnight at 4°C with rocking. Wash steps were performed in TBST 3 times for 10 min each time, followed by incubation in a 1:5,000 dilution of goat anti-mouse or goat anti-rabbit horseradish peroxidase-conjugated antibodies for 60 min at room temperature with rocking. Membranes were washed as described above, followed by addition of enhanced chemiluminescence reagent (Amersham) and chemiluminescent imaging on Kodak BioMax film. Primary antibodies were diluted as follows: 1:1,000 for anti-HA.11 (Sigma), 1:1,000 for anti-TIM17b (ProteinTech), 1:1,000 for anti-green fluorescent protein (anti-GFP; Abcam), 1:2,500 for anticalnexin (Enzo Pharmaceuticals), 1:1,000 for anti-β-tubulin (Sigma), 1:1,000 for anti-COX IV (Cell Signaling Technologies), and 1:1,000 for anti-FLAG M2 (Sigma).
Isolation of cellular mitochondria.
Mitochondrion-enriched fractions were isolated from HeLa cells either infected with EPEC strains or transfected with pcDNA3::HA2espZ or pcDNA3::HA2 using a mitochondrion isolation kit for cultured cells (Pierce; Thermo Scientific). Briefly, 2 × 107 cells were pelleted, resuspended in mitochondrial isolation reagent (MIR) A (Pierce, Thermo Scientific), and vortexed at medium speed for 5 s, followed by incubation on ice for 2 min. MIR B (Pierce; Thermo Scientific) was added, and the tubes were incubated on ice for 5 min with 5 s of vortex mixing every minute. MIR C (Pierce; Thermo Scientific) was added, and the tubes were inverted to mix, followed by centrifugation at 700 RCF for 10 min at 4°C. Supernatants were transferred to new tubes and centrifuged at 12,000 RCF for 15 min at 4°C. Supernatants (cytosolic fraction) were transferred to a new tube and stored at −20°C until use. The pellet (mitochondrion-enriched fraction) was washed in MIR C and centrifuged at 12,000 RCF for 5 min. Mitochondria were solubilized in 100 μl 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate in TBS. Cytosolic and mitochondrial protein concentrations were quantified using the Coomassie Plus protein assay (Pierce; Thermo Scientific), and cytosolic and mitochondrial proteins were resuspended in 3× Laemmli sample buffer prior to protein separation by SDS-PAGE and Western blot analysis.
siRNA.
siGENOME oligonucleotides targeting TIM17b (SMARTpool TIMM17B) or nontargeting oligonucleotides (control; SMARTpool Pool 1) were purchased from Dharmacon (Thermo Scientific). Oligonucleotides were suspended in 1× siRNA buffer (Dharmacon) and stored at −80°C until use. siRNA (100 nM) was transfected into HeLa cells at 30% confluence using Oligofectamine (Invitrogen) according to the manufacturer's directions, and media were changed at 24 h posttransfection. At 48 h posttransfection, cells were infected or transfected and assayed as described above. Lysates were collected for Western blot analysis to confirm knockdown.
Type III secretion assay.
Proteins secreted by the EPEC type III secretion system were assayed as previously described (9). Briefly, bacteria were subcultured from overnight shaking cultures into prewarmed DMEM and incubated in an incubator at 37°C in 5% CO2 for 6 h. Bacterial pellets were saved, and supernatants were filtered, followed by precipitation with trichloroacetic acid (TCA) overnight at 4°C. Secreted proteins were pelleted by centrifugation and resuspended in 3× Laemmli sample buffer. Saturated Tris solution was added to adjust the pH. Proteins (secreted and pelleted) were separated by SDS-PAGE, followed by either staining with Coomassie brilliant blue G250 or Western blot analysis.
Statistical analysis.
All statistical analyses were performed using an unpaired two-tailed t test with a 95% confidence interval. In all experiments, error bars denote standard deviation (±SD) of samples tested in duplicate or triplicate.
RESULTS
Chromosomal insertion of espZ can complement ΔespZ and preserve type III secretion of other effector proteins.
EspZ is an essential virulence factor in A/E pathogens. We have previously determined, using an espZ-knockout strain (ΔespZ), that it acts to protect host cells from rapid cell death (10, 28). In our previous study, we used cell lines stably expressing EspZ-GFP to complement ΔespZ since overexpression of espZ on a plasmid interfered with secretion of other effector proteins (28). For this study, we generated EPEC ΔespZ strains containing a single chromosomal copy of espZ either epitope tagged with HA or untagged. We then utilized an EPEC ΔespZ ΔsepD double knockout to visualize secretion of effector proteins, as performed previously (9, 28). Type III secretion assays were then used to visualize proteins secreted by various EPEC strains. We found that the EPEC ΔespZ/espZ and ΔespZ/espZ-HA strains exhibited a normal translocator profile since EspBD and EspA were all secreted (Fig. 1A). The ΔespZ ΔsepD strains all had normal effector secretion profiles, indicated by the presence of Tir and NleA (Fig. 1A) (9). EspZ could be visualized only in the supernatants of EPEC ΔsepD; however, the chromosomally inserted espZ-HA strain secreted EspZ, as detected by Western blot analysis, demonstrating that chromosomal copies of espZ in the ΔespZ background enable secretion of the EspZ protein (Fig. 1B). We then utilized this ΔespZ/espZ strain as a tool to further understand the mechanism(s) by which EspZ protects infected host cells from rapid cell death.
Fig. 1.
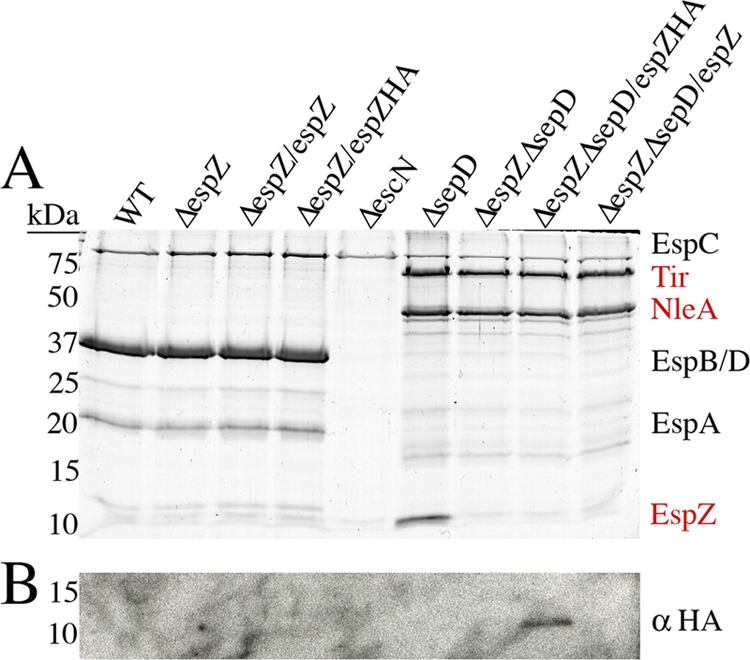
Chromosomal insertion of espZ preserves type III secretion system of effectors and translocators from EPEC. (A) Secreted proteins from EPEC strains were separated by SDS-PAGE and visualized by Coomassie brilliant blue G250 staining. Translocator (black text) and effector (red text) secretion was analyzed using ΔespZ, ΔespZ/espZ, and ΔespZ/espZ-HA in a WT or ΔsepD background. EPEC ΔescN was used as a control for no type III secretion system, since only EspC, a non-type III system-secreted protein, is observed. (B) Secreted proteins were subjected to Western blot analysis with anti-HA antibodies. An ∼11-kDa HA-reactive band was observed in the secreted protein fraction of the ΔsepD ΔespZ/espZ-HA strain.
EspZ protects EPEC-infected cells from severe loss of Δψm.
We previously demonstrated that EspZ protects EPEC-infected cells from premature cell death by interaction with host CD98 (28). However, the EspZ-CD98 interaction does not account for all of the protection elicited by EspZ, suggesting that other mechanisms by which EspZ is protecting host cells exist (28). Since cell death caused by an espZ-null (ΔespZ) strain was characterized by rapid cell lysis, characteristic of necrotic cell death, we examined the mitochondrial inner membrane potential (Δψm) in HeLa cells infected with EPEC strains. To observe changes in Δψm over time during the course of EPEC infection, we utilized the J-aggregate-forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) (29). JC-1 preferentially localizes to mitochondria and is used to measure Δψm (7, 29). In healthy cells with a high Δψm, JC-1 forms aggregates (J aggregates), which emit a red fluorescence; however, when Δψm is low, JC-1 is present in a monomeric form (J monomers), which emits a green fluorescence. In this assay, Δψm can be quantified by plotting the ratio of J aggregates/J monomers. HeLa cells were infected with either EPEC WT, ΔespZ, or ΔespZ/espZ or were left uninfected for 2, 3, or 4 h, followed by staining with JC-1. Fluorescence intensity was measured at excitation/emission wavelengths of 560/595 nm and 485/535 nm for monomers and aggregates, respectively. Aggregate/monomer ratios were normalized to 1.0 in uninfected cells (Fig. 2A). Cells infected with WT EPEC had decreased Δψm compared to uninfected cells (Fig. 2A), as previously demonstrated (20). However, EPEC ΔespZ infection caused a significant decrease in the aggregate/monomer ratio compared to WT and ΔespZ/espZ infection at 3 and 4 h postinfection, indicating that Δψm is greatly decreased by EPEC ΔespZ infection (P < 0.05) (Fig. 2A). Supernatants were taken from infected cells, and LDH was quantified. Untreated cells were lysed using Triton X-100, and the LDH present in the supernatants of these cells was set at 100% cytotoxicity (see Materials and Methods). As observed previously, significantly greater LDH levels were present in the supernatants of HeLa cells infected with EPEC ΔespZ than cells infected with WT at 3 and 4 h postinfection (P < 0.01) (Fig. 2B). Infection with EPEC ΔespZ/espZ resulted in LDH release similar to that caused by WT EPEC infection at 3 h postinfection (Fig. 2B). These data demonstrate that EPEC ΔespZ infection causes a severe loss in Δψm that occurs concurrently with host cell lysis.
Fig. 2.

Infection with EPEC ΔespZ causes severe loss of Δψm. (A) HeLa cells were infected with either EPEC WT, ΔespZ, or ΔespZ/espZ or left uninfected (UI). JC-1 reagent was added to cells 15 min prior to termination of infection, and cells were washed with assay buffer three times. Fluorescence intensity was measured at excitation/emission wavelengths of 560/595 nm and 485/535 nm for J monomers (low Δψm) and J aggregates (high Δψm), respectively. Δψm was plotted as the ratio of J aggregates/J monomers. Asterisks indicate statistical significance (P < 0.05) by Student's t test for samples tested in triplicate. p.i., postinfection. (B) Supernatants of HeLa cells infected for 2, 3, or 4 h with either EPEC WT, ΔespZ, or ΔespZ/espZ, left uninfected, or treated with Triton X-100 (100% cytotoxicity; data not shown) were assayed for the presence of LDH. Values were normalized against those for Triton X-100-treated cells (100% cytotoxicity) and uninfected cells (1% cytotoxicity). Asterisks indicate statistical significance (P < 0.05) by Student's t test for samples tested in triplicate. These data are representative of three independent experiments.
EspZ interacts with TIM17b.
In a previous study, we identified TIM17b to be a putative host protein partner binding to EspZ by yeast two-hybrid assay (28). TIM17b is a mitochondrial inner membrane protein that is responsible for voltage gating of the TIM23 protein import complex and relies on an intact Δψm to function (4, 21). We thus aimed to confirm our previous yeast two-hybrid assay data indicating that EspZ interacts with host TIM17b. Coimmunoprecipitation experiments were performed to determine if a TIM17b-FLAG fusion protein could immunoprecipitate EspZ-enhanced GFP (eGFP). We used an anti-FLAG antibody to immunoprecipitate proteins interacting with TIM17b-FLAG and normal mouse IgG as an IC. We were able to specifically immunoprecipitate EspZ-eGFP but not eGFP alone with TIM17b-FLAG (Fig. 3A). Input lysates were analyzed by Western blotting to confirm expression of TIM17b-FLAG, EspZ-eGFP, and eGFP (Fig. 3B). As suggested by our previous yeast-two hybrid assay data (28), EspZ does indeed interact with TIM17b.
Fig. 3.

EspZ interacts with TIM17b. (A) HEK293T cells were cotransfected with pCMV4A::TIM-17b, pCMV-Tag4A, pEHespZ-NGFP, or pEGFP-N1, as indicated in the text. Cells were lysed and subject to immunoprecipitation with anti-FLAG antibodies or mouse IgG IC, as described in the text. Precipitated proteins were subjected to Western blot analysis. EspZ-eGFP and TIM17-FLAG are indicated with arrows, and nonspecific antibody bands are indicated with asterisks. (B) Lysates from HEK293T cells were subjected to Western blot analysis to confirm transfection efficiency. EspZ-eGFP, eGFP, and TIM17-FLAG are indicated with arrows.
EspZ localizes to mitochondria.
We have previously demonstrated that EspZ localizes to host cell membranes (28), and on the basis of the interaction between EspZ and TIM17b, we hypothesized that EspZ likely also localizes to host mitochondria. To test this hypothesis, we first infected HeLa cells with EPEC ΔespZ/espZ-HA and attempted to visualize EspZ using anti-HA staining by immunofluorescence microscopy. However, we were unable to observe EspZ staining by this method (data not shown). As an alternative method, we transfected HeLa cells with pcDNA3::HA2espZ and stained these cells for COX IV, which is a mitochondrial inner membrane protein (18). We used Alexa 633-conjugated goat anti-rabbit antibodies to image COX IV (red) and mouse anti-HA.11, followed by Alexa 488-conjugated goat anti-mouse antibodies to image EspZ (green). We observed similar staining patterns in the anti-COX IV and anti-HA images, suggesting that EspZ localized similarly to COX IV (Fig. 4A). We were also able to observe costaining between EspZ-eGFP and the mitochondrion-specific stain MitoTracker (data not shown).
Fig. 4.

EspZ localizes to host cell mitochondria. (A) HeLa cells were plated on glass coverslips and transfected with pcDNA3::HA2espZ. Cells were stained with rabbit anti-COX IV (red) and mouse anti-HA.11 (green), followed by Alexa 633-conjugated goat antirabbit and Alexa 488-conjugated goat antimouse antibodies, respectively. Coverslips were mounted in ProLong Gold with DAPI to image cellular nuclei (blue). (B) HeLa cells were transfected with either pcDNA3::HA2espZ or pcDNA3::HA2, and mitochondrial fractions were isolated as described in the text. Antibodies that recognize cytochrome c and β-tubulin were used as controls for mitochondrial (Mito) and cytoplasmic (Cyto) fractions, respectively. Anti-HA antibodies were used to visualize HA2EspZ.
To further confirm that EspZ localizes to mitochondria, in addition to other cellular membranes, we isolated mitochondria from HeLa cells expressing HA2EspZ and performed Western blot analysis on the cytosolic and mitochondrial fractions. COX IV and β-tubulin were used as markers for the mitochondrial and cytosolic fractions, respectively (Fig. 4B). We observed HA2EspZ in mitochondrially enriched but not cytosolic fractions (Fig. 4B), which provides further evidence that EspZ can localize to host cell mitochondria, in addition to other cellular membranes.
siRNA knockdown of TIM17b dampens EspZ-mediated survival during EPEC infection.
EspZ protects EPEC-infected host cells from premature cell death and is essential for successful colonization of C. rodentium in vivo (10, 28). To determine if TIM17b contributes to EspZ-mediated protection from cell death during EPEC infection, we performed LDH release assays using HeLa cells treated with TIM17b-specific (TIM17b) or nontargeting (control) siRNAs. HeLa cells transfected with TIM17b and control siRNAs were infected with EPEC WT or EPEC ΔespZ for 2, 3, or 4 h. TIM17b knockdown resulted in significantly greater HeLa cell cytotoxicity during WT EPEC infection compared to that by the nontargeting siRNA control at all time points examined (P < 0.05; Fig. 5A). However, knockdown of TIM17b did not influence cytotoxicity levels during infection with EPEC ΔespZ (Fig. 5B). Uninfected and detergent-treated cells were used as controls for spontaneous and total lysis, respectively. Lysates from infected cells treated with TIM17b and control siRNA were collected and subjected to Western blot analysis to confirm TIM17b knockdown. Indeed, TIM17b-specific siRNA resulted in a reduction of TIM17b protein (Fig. 5C). Calnexin was used as a loading control (Fig. 5C). Together, these data suggest that in the presence of EspZ, TIM17b contributes to host cell survival during EPEC infection.
Fig. 5.

siRNA knockdown of TIM17b dampens the ability of EspZ to protect HeLa cells from EPEC-mediated cell lysis. HeLa cells treated with either TIM17b-specific or nontargeting (control) siRNA were infected with EPEC WT (A) or ΔespZ (B) for 2, 3, or 4 h. LDH present in cellular supernatants was quantified and plotted as percent cytotoxicity. Asterisks indicate statistical significance of samples tested in triplicate (P < 0.05). Data are representative of two independent experiments. (C) Lysates were generated from EPEC-infected and uninfected cells and subjected to Western blot analysis using anti-TIM17b and anticalnexin (loading control) antibodies to demonstrate siRNA knockdown efficiency.
DISCUSSION
Diarrheagenic Escherichia coli relies on a plethora of virulence factors secreted by a T3SS to efficiently hijack host cell signaling pathways. Specifically, several effector proteins have been shown to intricately regulate cytotoxicity during EPEC infection by promoting antagonistic prosurvival and prodeath pathways. We had previously demonstrated that the EspZ effector protein translocated by the T3SS protects EPEC-infected cells from premature cytotoxicity partially due to interaction with host CD98 (28). Since CD98 could account for only partial protection against cell death elicited by EspZ, we investigated further mechanisms of EspZ-mediated cell survival.
Host cell death resulting from EPEC ΔespZ infection had characteristics of necrotic cell death. Cell lysis, characterized by the presence of LDH in cell supernatants, occurs rapidly following necrotic cell death and was displayed by EPEC ΔespZ-infected cells. Rapid cell lysis resulting from necrosis generally occurs after mitochondrial swelling, ATP depletion, and decreased mitochondrial membrane potential (12). We initially examined Δψm as a measure of necrotic cell death during EPEC infection and found that Δψm decreased significantly during EPEC ΔespZ infection concurrently with LDH release. Infection with a chromosomally complemented ΔespZ/espZ strain resulted in greater Δψm than infection with a ΔespZ strain, which suggests that in the absence of EspZ, EPEC causes necrotic cell death.
In our previous study, we identified several putative host partners binding to EspZ via stable isotope labeling with amino acids in cell culture (SILAC) and yeast-two hybrid assay screening. TIM17b was found to interact with EspZ by yeast-two hybrid assay but not SILAC (28). Since TIM17b contributes to voltage gating in mitochondria and requires intact Δψm to function, we tested whether EspZ does indeed interact with TIM17b. We initially found that EspZ bearing an N-terminal epitope tag does not interact with TIM17b (data not shown), which demonstrates why TIM17b did not immunoprecipitate with HA2EspZ in the SILAC experiments. However, EspZ with a C-terminal GFP epitope tag does interact with TIM17b by coimmunoprecipitation. We were thus able to validate the interaction between TIM17b and EspZ using a second method.
TIM17b is an IMM protein, and we therefore hypothesized that EspZ likely localizes to host mitochondria. Initially, we used EspZ-GFP to attempt to localize EspZ within host cells; however, we observed heavy background staining in the cell cytosol, likely due to the size of the epitope tag (data not shown). Consequently, we transiently expressed HA2EspZ in HeLa cells. We observed that HA2EspZ localized similarly to the mitochondrial inner membrane protein COX IV and attempted to further confirm EspZ localization using organellar fractionation. We observed EspZ localization in mitochondrion-enriched fractions of HeLa cells expressing HA2EspZ, which further suggests that ectopically expressed EspZ localizes to mitochondria. Interestingly, EspZ does not encode a canonical mitochondrion-targeting sequence (MTS). However, the effector EspJ also localizes to mitochondria in the absence of a known MTS (18).
Other EPEC effectors localize to and affect host cell mitochondria. Map and EspF are LEE-encoded effector proteins that localize to host mitochondria and impair their function by dampening Δψm, with the latter also causing cytochrome c release, resulting in host cell apoptosis (23–25). Although Map has not been linked directly with host cell apoptosis during EPEC infection, it does translocate into the mitochondrial matrix. Papatheodorou and colleagues found that Map transport into the mitochondria was dependent on translocase of outer mitochondrial membrane 22 (TOM22), TOM44, mitochondrial heat shock protein 70 (mtHsp70), and the Δψm (25). Conversely, EspF did not alter mitochondrial morphology (25). Both Map and EspF encode a canonical MTS in their N-terminal regions (23–25).
TIM17b is part of the TIM23 IMM translocase complex responsible for transporting proteins into the inner mitochondrial membrane or mitochondrial matrix (3). We therefore aimed to determine if EspZ requires TIM17b to gain access to host mitochondria. Interestingly, siRNA knockdown of TIM17b did not impact localization of EspZ to host mitochondria (data not shown), which implies that EspZ either utilizes another host protein for import or encodes a noncanonical mitochondrial targeting sequence, similar to EspJ (18). Interaction with TIM17b likely occurs following EspZ localization to mitochondria and may regulate mitochondrial localization of Map and EspF. Further investigation is required to determine if the EspZ-TIM17b interaction is required for import of other effectors into host mitochondria.
To determine if the EspZ-TIM17b interaction contributes to EspZ-mediated protection against host cell death, we utilized siRNA to knock down TIM17b. We observed increased cytotoxicity during infection with EPEC WT but not EPEC ΔespZ following siRNA knockdown, which suggests that TIM17b plays a role in protection against premature cytotoxicity in the presence of EspZ. TIM17b has not been implicated in cell death; however, its function is strongly correlated with an intact Δψm (21). We have examined the status of cytochrome c at host mitochondria during infection and found that infection with EPEC ΔespZ does not alter its localization (data not shown). This further supports the notion that EspZ protects EPEC-infected cells against necrotic cell death. Further studies are required to reveal the precise mechanisms by which EspZ is utilizing TIM17b to protect host cells against rapid cell death.
It has become increasingly apparent that many effector proteins translocated by the T3SS carry out several functions during A/E pathogen infection. By further investigating the roles of effector proteins, we are uncovering mechanisms by which these pathogens are able to efficiently cause disease and incapacitate their hosts. Regulation of host cell death is a critical function of several A/E pathogen effector proteins, and understanding the mechanisms by which they manipulate cell death pathways will enhance our understanding of their virulence strategies.
ACKNOWLEDGMENTS
We thank R. J. Law for constructive comments and critical reading of the manuscript.
S.R.S. was supported by a National Sciences and Engineering Research Council of Canada (NSERC) graduate scholarship for a portion of this work. M.A.C. is supported by Canadian Institutes of Health Research (CIHR), Canadian Association of Gastroenterology (CAG), and Ferring Pharmaceuticals postdoctoral fellowships. B.B.F. is the UBC Peter Wall Distinguished Professor. This work was funded by operating grants held by B.B.F. from the Canadian Institutes of Health Research.
Footnotes
Published ahead of print on 26 September 2011.
REFERENCES
- 1. Babic A., Guerout A. M., Mazel D. 2008. Construction of an improved RP4 (RK2)-based conjugative system. Res. Microbiol. 159:545–549 [DOI] [PubMed] [Google Scholar]
- 2. Baruch K., et al. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30:221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bauer M. F., et al. 1999. Genetic and structural characterization of the human mitochondrial inner membrane translocase. J. Mol. Biol. 289:69–82 [DOI] [PubMed] [Google Scholar]
- 4. Bomer U., et al. 1996. The preprotein translocase of the inner mitochondrial membrane: evolutionary conservation of targeting and assembly of Tim17. J. Mol. Biol. 262:389–395 [DOI] [PubMed] [Google Scholar]
- 5. Chacinska A., Koehler C. M., Milenkovic D., Lithgow T., Pfanner N. 2009. Importing mitochondrial proteins: machineries and mechanisms. Cell 138:628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chacinska A., et al. 2005. Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 120:817–829 [DOI] [PubMed] [Google Scholar]
- 7. Cossarizza A., Baccarani-Contri M., Kalashnikova G., Franceschi C. 1993. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′, 6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem. Biophys. Res. Commun. 197:40–45 [DOI] [PubMed] [Google Scholar]
- 8. Croxen M. A., Finlay B. B. 2010. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8:26–38 [DOI] [PubMed] [Google Scholar]
- 9. Deng W., et al. 2005. Regulation of type III secretion hierarchy of translocators and effectors in attaching and effacing bacterial pathogens. Infect. Immun. 73:2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deng W., et al. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U. S. A. 101:3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frazier A. E., et al. 2003. Mitochondria use different mechanisms for transport of multispanning membrane proteins through the intermembrane space. Mol. Cell. Biol. 23:7818–7828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Golstein P., Kroemer G. 2007. Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32:37–43 [DOI] [PubMed] [Google Scholar]
- 13. Hemrajani C., et al. 2010. NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc. Natl. Acad. Sci. U. S. A. 107:3129–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Holmes A., Muhlen S., Roe A. J., Dean P. 2010. The EspF effector, a bacterial pathogen's Swiss army knife. Infect. Immun. 78:4445–4453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iguchi A., et al. 2009. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 191:347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanack K. J., Crawford J. A., Tatsuno I., Karmali M. A., Kaper J. B. 2005. SepZ/EspZ is secreted and translocated into HeLa cells by the enteropathogenic Escherichia coli type III secretion system. Infect. Immun. 73:4327–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kenny B., et al. 1997. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91:511–520 [DOI] [PubMed] [Google Scholar]
- 18. Kurushima J., Nagai T., Nagamatsu K., Abe A. 2010. EspJ effector in enterohemorrhagic E. coli translocates into host mitochondria via an atypical mitochondrial targeting signal. Microbiol. Immunol. 54:371–379 [DOI] [PubMed] [Google Scholar]
- 19. Levine M. M., et al. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i:1119–1122 [DOI] [PubMed] [Google Scholar]
- 20. Ma C., et al. 2006. Citrobacter rodentium infection causes both mitochondrial dysfunction and intestinal epithelial barrier disruption in vivo: role of mitochondrial associated protein (Map). Cell. Microbiol. 8:1669–1686 [DOI] [PubMed] [Google Scholar]
- 21. Martinez-Caballero S., Grigoriev S. M., Herrmann J. M., Campo M. L., Kinnally K. W. 2007. Tim17p regulates the twin pore structure and voltage gating of the mitochondrial protein import complex TIM23. J. Biol. Chem. 282:3584–3593 [DOI] [PubMed] [Google Scholar]
- 22. Miller V. L., Mekalanos J. J. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagai T., Abe A., Sasakawa C. 2005. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J. Biol. Chem. 280:2998–3011 [DOI] [PubMed] [Google Scholar]
- 24. Nougayrede J. P., Donnenberg M. S. 2004. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell. Microbiol. 6:1097–1111 [DOI] [PubMed] [Google Scholar]
- 25. Papatheodorou P., et al. 2006. The enteropathogenic Escherichia coli (EPEC) Map effector is imported into the mitochondrial matrix by the TOM/Hsp70 system and alters organelle morphology. Cell. Microbiol. 8:677–689 [DOI] [PubMed] [Google Scholar]
- 26. Samba-Louaka A., Nougayrede J. P., Watrin C., Oswald E., Taieb F. 2009. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infect. Immun. 77:5471–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sham H. P., et al. 2011. The attaching/effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-{kappa}B and p38-MAP kinase activation. Infect. Immun. 79:3552–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shames S. R., et al. 2010. The pathogenic E. coli type III effector EspZ interacts with host CD98 and facilitates host cell prosurvival signaling. Cell. Microbiol. 12:1322–1339 [DOI] [PubMed] [Google Scholar]
- 29. Smiley S. T., et al. 1991. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. U. S. A. 88:3671–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tobe T., et al. 2006. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U. S. A. 103:14941–14946 [DOI] [PMC free article] [PubMed] [Google Scholar]