

Abstract
Dystrobrevin is a major component of a dystrophin-associated protein complex. It is widely expressed in mammalian tissues, including the nervous system, in which it is localized to the presynaptic nerve terminal with unknown function. In a genetic screen for suppressors of a lethargic phenotype caused by a gain-of-function isoform of SLO-1 in Caenorhabditis elegans, we isolated multiple loss-of-function (lf) mutants of the dystrobrevin gene dyb-1. dyb-1(lf) phenocopied slo-1(lf), causing increased neurotransmitter release at the neuromuscular junction, increased frequency of Ca2+ transients in body-wall muscle, and abnormal locomotion behavior. Neuron- and muscle-specific rescue experiments suggest that DYB-1 is required for SLO-1 function in both neurons and muscle cells. DYB-1 colocalized with SLO-1 at presynaptic sites in neurons and dense body regions in muscle cells, and dyb-1(lf) caused SLO-1 mislocalization in both types of cells without altering SLO-1 protein level. The neuronal phenotypes of dyb-1(lf) were partially rescued by mouse α-dystrobrevin-1. These observations revealed novel functions of the BK channel in regulating muscle Ca2+ transients and of dystrobrevin in controlling neurotransmitter release and muscle Ca2+ transients by localizing the BK channel.
Introduction
Dystrobrevins are cytosolic proteins that bind to dystrophin and are considered to be a major component of a dystrophin-associated protein complex (DAPC) that links the cytoskeleton to extracellular matrix (Davies and Nowak, 2006). There are two dystrobrevin genes (αDB and βDB) in human and mice (Rees et al., 2007) but only one such gene (dyb-1) in Caenorhabditis elegans (www.wormbase.org, WS225). In mouse, knock-out of αDB causes skeletal and cardiac myopathies and impaired nitric oxide-mediated signaling (Grady et al., 1999), and knock-out of both αDB and βDB causes synaptic defects and abnormal motor behavior (Grady et al., 2006). In C. elegans, mutations of either dyb-1 or the dystrophin gene dys-1 cause muscle degeneration in a sensitized genetic background as well as behavioral and pharmacological phenotypes suggestive of enhanced cholinergic transmission (Gieseler et al., 1999; Giugia et al., 1999). Intriguingly, similar phenotypes are observed in mutants of the BK channel gene slo-1 (Carre-Pierrat et al., 2006). The relationship between SLO-1 and the DAPC is not totally clear. A recent study shows that a membrane protein known as ISLO-1 is important to SLO-1 subcellular localization in C. elegans body-wall muscle by interacting with the DAPC (Kim et al., 2009).
The BK channel is a Ca2+ and voltage-gated K+ channel expressed in many tissues, including the nervous system (Wang, 2008) and skeletal muscle (Latorre et al., 1982; Blatz and Magleby, 1984; Knaus et al., 1995). In the nervous system, the BK channel colocalizes with voltage-gated Ca2+ channels at axon presynaptic sites (Robitaille et al., 1993; Yazejian et al., 2000) and serves as a key negative regulator of neurotransmitter release (Robitaille et al., 1993; Hu et al., 2001; Wang et al., 2001; Raffaelli et al., 2004; Wang, 2008). The localization of the BK channel to the vicinity of voltage-gated Ca2+ channels allows it to be activated by Ca2+ microdomains, which are hemispheric sites of high [Ca2+] at the inner mouth of open Ca2+ channels (Roberts et al., 1990; Augustine et al., 2003). In mammalian striated muscle, the BK channel is enriched in the transverse tubule (t-tubule) membrane (Latorre et al., 1982; Knaus et al., 1995) with unknown function. In C. elegans body-wall muscle, which is analogous to mammalian striated muscle (Moerman and Fire, 1997), SLO-1 colocalizes with the L-type voltage-gated Ca2+ channel EGL-19. SLO-1 in muscle is potentially involved in regulating locomotion and egg-laying behaviors (Kim et al., 2009; Abraham et al., 2010; Chen et al., 2010a,b).
Through a genetic screen for suppressors of a behavioral phenotype caused by a gain-of-function (gf) isoform of SLO-1, we identified DYB-1 as a protein essential to SLO-1 function in vivo. Analyses of mutant phenotypes revealed that SLO-1 regulates Ca2+ transients in body-wall muscle cells and that DYB-1 regulates neurotransmitter release and muscle Ca2+ transients by localizing SLO-1 to presynaptic sites and muscle-dense body regions, respectively. These findings potentially help to understand the molecular mechanisms of neurological and muscular defects caused by deficiencies of the DAPC.
Materials and Methods
Growth and culture of C. elegans.
C. elegans hermaphrodites were grown on agar plates with a layer of OP50 Escherichia coli at room temperature (21–22°C) or inside an environmental chamber (21°C).
Strains.
N2 Bristol was used as the wild-type in all experiments. The other strains used in this study are as follows: ZW083 [zwIs101(Pslo-1::slo-1::GFP)]; ZW320 [zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP)]; ZW331 [zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP); dyb-1(zw11)]; ZW349 [dyb-1(zw11)]; ZW352 [zwIs101(Pslo-1::slo-1::GFP); dyb-1(zw11)]; ZW385 [zwEx132(Pdyb-1::GFP; lin-15(+)); lin-15(n765)]; ZW471 [zwEx150(Pdyb-1::αDB1; Pmyo-3::GFP); zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP); dyb-1(zw11)]; ZW495 [zwIs132(Pmyo-3::GCaMP2; lin-15(+))]; ZW527 [zwIs132(Pmyo-3::GCaMP2; lin-15(+)); slo-1(md1745)]; ZW528 [zwIs132(Pmyo-3::GCaMP2; lin-15(+)); zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP)]; ZW536 [zwIs132(Pmyo-3::GCaMP2; lin-15(+)); dyb-1(zw11)]; ZW581 [slo-1(md1745); dyb-1(zw11)]; ZW604 [zwIs101(Pslo-1::slo-1::GFP); zwIs135(Pdyb-1::dyb-1::mStrawberry; rol-6(+))]; ZW605 [zwEx164(Punc-47::slo-1::mStrawberry; Punc-25::GFP::unc-2; lin-15(+)); lin-15(n765)]; ZW608 [zwEx166(Pmyo-3::dyb-1; Pmyo-3::GFP); zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP); dyb-1(zw11)]; ZW609 [zwEx167(Prab-3::dyb-1; Prab-3::GFP); zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP); dyb-1(zw11)]; ZW610 [zwEx166(Pmyo-3::dyb-1; Pmyo-3::GFP); dyb-1(zw11)]; ZW611 [zwEx167(Prab-3::dyb-1; Prab-3::GFP); dyb-1(zw11)]; ZW612 [zwIs101(Pslo-1::slo-1::GFP); dys-1(cx18)]; ZW615 [zwIs136(Pdyb-1::dyb-1::GFP; lin-15(+)); lin-15(n765)]; ZW616 [zwIs136(Pdyb-1::dyb-1::GFP; lin-15(+)); dys-1(cx18)]; ZW622 [zwEx168(Pmyo-3::αDB1; Pmyo-3::GFP); zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP); dyb-1(zw11)]; ZW623 [zwEx169(Prab-3::αDB1; Prab-3::GFP); zwIs129(Pslo-1::slo-1(gf); Pmyo-2::YFP); dyb-1(zw11)]; ZW624 [zwEx170(Pdyb-1::αDB1; Pmyo-2::DsRed); zwIs101(Pslo-1::slo-1::GFP); dyb-1(zw11)]; ZW625 [zwEx171(Pmyo-3::slo-1; Pmyo-2::DsRed); zwIs132(Pmyo-3::GCaMP2; lin-15(+)); slo-1(md1745)]; ZW626 [zwEx172(Prab-3::slo-1; Pmyo-2::DsRed); zwIs132(Pmyo-3::GCaMP2; lin-15(+)); slo-1(md1745)].
Mutant screen.
An integrated transgenic strain expressing Pslo-1::SLO-1(E350Q) and Pmyo-2::YFP in the wild-type genetic background was used for mutant screen. The Pslo-1::SLO-1(E350Q) transgene caused a lethargic phenotype, whereas the Pmyo-2::YFP transgene was used as a genetic marker. Synchronized L4-stage worms were treated with the chemical mutagen ethyl methanesulfonate (50 mm) for 4 h at room temperature. The F2 progeny were screened for animals that moved better than the original slo-1(gf) animals.
Behavioral assay.
Locomotion velocity and the head-bending angle were quantified using an automated worm tracking and analysis system (Chen et al., 2010a). Specifically, a single adult hermaphrodite was transferred to an agar plate without food. After allowing ∼30 s for recovery from the transfer, snapshots of the worm were taken at 15 frames/s for 30 s using a VGA FireWire camera (XCD-V60; Sony) mounted on a stereomicroscope (SMZ800; Nikon). The worm was constantly kept in the center of the view field with a motorized microscope stage (OptiScan ES111; Prior Scientific). Both the camera and the motorized stage were controlled by a custom program running in MATLAB (MathWorks).
Cloning of dyb-1.
dyb-1(zw11) was used for single nucleotide polymorphism-based genetic mapping (Davis et al., 2005). After mapping the mutation to a small interval, the candidate gene was identified by testing whether mutants of candidate genes would complement with zw11 in suppressing the lethargic phenotype of slo-1(gf). Full-length dyb-1 cDNA was obtained by RT-PCR. Molecular lesions of dyb-1 mutants were identified by sequencing the cDNA or genomic DNA of dyb-1 gene prepared from three randomly picked mutant alleles.
Analysis of expression pattern and subcellular localization.
The expression pattern of dyb-1 was determined by expressing GFP under the control of a 3.1-kb dyb-1 promoter (Pdyb-1::GFP, wp747). The plasmid was injected into the lin-15(n765) strain using a lin-15 rescue plasmid as a transformation marker. The transgenic animals were photographed using a Carl Zeiss Axiovert 200M fluorescence microscope (40× objective) with an apotome device (Carl Zeiss) for optical sectioning.
Subcellular localization of DYB-1 was determined by expressing DYB-1 with GFP tagged to its C terminus under the control of Pdyb-1 (Pdyb-1::DYB-1::GFP, wp1132). SLO-1 subcellular localization was examined with an integrated transgenic strain expressing Pslo-1::SLO-1::GFP (wp5) (Chen et al., 2010a). To determine whether DYB-1 and SLO-1 are colocalized, mStrawberry-tagged DYB-1 (Pdyb::DYB-1::mStrawberry, wp1130) was expressed in the transgenic strain expressing Pslo-1::SLO-1::GFP. To determine whether SLO-1 colocalized with UNC-2 in neurons, SLO-1::mStrawberry and UNC-2::GFP (Saheki and Bargmann, 2009) fusion proteins were expressed under the control of Punc-47 and Punc-25, respectively. Epifluorescence of the fusion proteins in transgenic animals was visualized and photographed with a Nikon TE2000-U inverted microscope (100× objective) and a Peltier cooled digital camera (F-view II; Olympus).
Recording of postsynaptic currents.
Evoked postsynaptic currents (ePSCs) and miniature postsynaptic currents (mPSCs) were recorded from the C. elegans neuromuscular junction (NMJ) using an established technique (Richmond et al., 1999; Liu et al., 2007). The recording pipette solution contained the following (in mm): 120 KCl, 20 KOH, 5 Tris, 0.25 CaCl2, 4 MgCl2, 36 sucrose, 5 EGTA, and 4 Na2ATP, pH 7.2. Two external solutions with different [Ca2+] (5 and 0.5 mm) were used. The external solution with the higher [Ca2+] contained the following (in mm): 140 NaCl, 5 KCl, 5 CaCl2, 5 MgCl2, 11 dextrose, and 5 HEPES, pH 7.2. This solution was modified by reducing CaCl2 to 0.5 mm and increasing NaCl to 145 mm to make the external solution with the lower [Ca2+].
Ca2+ imaging.
An integrated Pmyo-3::GCaMP2 transgene was crossed into different mutant backgrounds. Ca2+ transients were monitored by imaging GCaMP2 fluorescence in body-wall muscle cells of dissected worms as described previously (Liu et al., 2011).
Western blot.
To examine SLO-1::GFP protein level expressed in worms, worms of mixed stages were homogenized in a lysis buffer containing 2% SDS/100 mm NaCl/10% glycerol/50 mm Tris, pH 6.8. Soluble protein extracts were separated on 4–12% SDS-PAGE gels and probed with GFP (Invitrogen) and α-tubulin (Santa Cruz Biotechnology) antibodies. Anti-mouse IgG HRP (Santa Cruz Biotechnology) was used as the secondary antibody for detection by enhanced chemiluminescence (Pierce).
Data analysis.
The averaged locomotion speed and head-bending angle were determined using an automated worm tracking and analysis system (Chen et al., 2010a,b). Briefly, 13 marker points were placed at equal intervals along the spline of each worm, which was imaged at 15 frames/s, and the angle supplementary to the angle formed by the two straight lines connecting the marker points 1 and 2 and the marker points 2 and 3 was determined. Both the full-amplitude dorsal/ventral head-bending angle and the root mean square of head-bending angles were determined. The mean locomotion speed was calculated based on the distance traveled over time.
Amplitude and frequency of mPSCs were analyzed using MiniAnalysis (Synaptosoft). A detection threshold of 10 pA was used in initial automatic analysis, followed by visual inspections to include missed smaller events (5 pA or larger) and to exclude false events resulting from baseline fluctuations. For each recording, we adjusted the position of the stimulating electrode to obtain the largest ePSCs and used an average of the two largest values to derive an amplitude measurement for that recording.
For Ca2+ imaging data, all the muscle cells within the camera imaging field were chosen as separate regions of interest for quantification of fluorescence intensity changes over successive frames. Fluorescence intensity was first plotted as absolute signal intensity over time using the NIS-Elements software (Nikon) and then converted to F/F0 using MATLAB (MathWorks) running a custom module. The frequency of Ca2+ transient peaks (defined as F/F0 ≥ 0.05) was quantified for each cell. The mean frequency of all cells in the imaging field per preparation except for the slo-1(gf) group was treated as one sample. For the slo-1(gf) group, only data from cells with Ca2+ transients were included in the analysis.
Data graphing and statistical analyses.
Graphing and statistical analyses were performed with Origin (version 8.5; OriginLab). Either unpaired t test or one-way ANOVA (followed by Bonferroni's post hoc test) was used for statistical comparisons. p < 0.05 is considered statistically significant. All values are expressed as mean ± SEM.
Results
dyb-1 mutants suppressed the lethargic phenotype caused by slo-1(gf)
To identify proteins important to BK channel function in vivo, we performed a genetic screen for mutants that suppressed the lethargic phenotype of a worm strain expressing SLO-1(gf) under the control of slo-1 promoter (Pslo-1) (Chen et al., 2010b). SLO-1(gf) was created by mutating glutamate 350 to glutamine, which causes a 40 mV shift toward more hyperpolarized potentials in the SLO-1 conductance–voltage relationship (Chen et al., 2010b). Among 25 mutants isolated from a screen of 24,000 haploid genomes, seven mutants belonged to the dystrobrevin gene dyb-1 (locus F47G6.1, www.wormbase.org, WS225) based on complementation tests and rescue experiments. Although there are two dystrobrevin genes (α and β) with multiple splice variants in human (Peters et al., 1998; Holzfeind et al., 1999), the C. elegans genome has only one dystrobrevin gene with probably a single isoform (www.wormbase.org, WS225). DYB-1 shares 45.5 and 46.9% sequence identity with human αDB1 and βDB, respectively (Fig. 1). Three mutants were randomly chosen for sequencing, and all of them were found to be putative nulls (Fig. 1). Subsequent analyses were performed with the dyb-1(zw11) allele.
Figure 1.

dyb-1 encodes a homolog of mammalian dystrobrevins. Shown is the alignment of predicted amino acid sequences of DYB-1, human αDB1 (accession number NM_001390), and human βDB (accession number AF022728). Positions of molecular lesions of three dyb-1 alleles are indicated. Two alleles have mutations leading to premature stop codon (marked by *) and one allele (marked by an arrow) disrupts the splice acceptor site of the fourth intron, leading to the removal of exon 5 and a frame shift after Y171, which was shortly followed by a stop codon.
DYB-1 was coexpressed and colocalized with SLO-1 in both neurons and muscle cells
The isolation of dyb-1 mutants as suppressors of the slo-1(gf) lethargic phenotype suggests that DYB-1 might be required for SLO-1 function in vivo and have similar expression and subcellular localization patterns as SLO-1. Expression of GFP under the control of the dyb-1 promoter (Pdyb-1) resulted in GFP epifluorescence in many neurons and several muscles, including body-wall muscle (Fig. 2A). This expression pattern of dyb-1 is indistinguishable from that of slo-1 (Chen et al., 2010a,b). When mStrawberry-tagged full-length DYB-1 (DYB-1::mStrawberry) was coexpressed with GFP-tagged full-length SLO-1 (SLO-1::GFP) under the controls of their respective promoters, the two fusion proteins appeared as puncta with colocalization at muscle dense body regions and along the dorsal nerve cord (Fig. 2B,C). The two fusion proteins also showed overlapping expression in the nerve ring (Fig. 2D).
Figure 2.
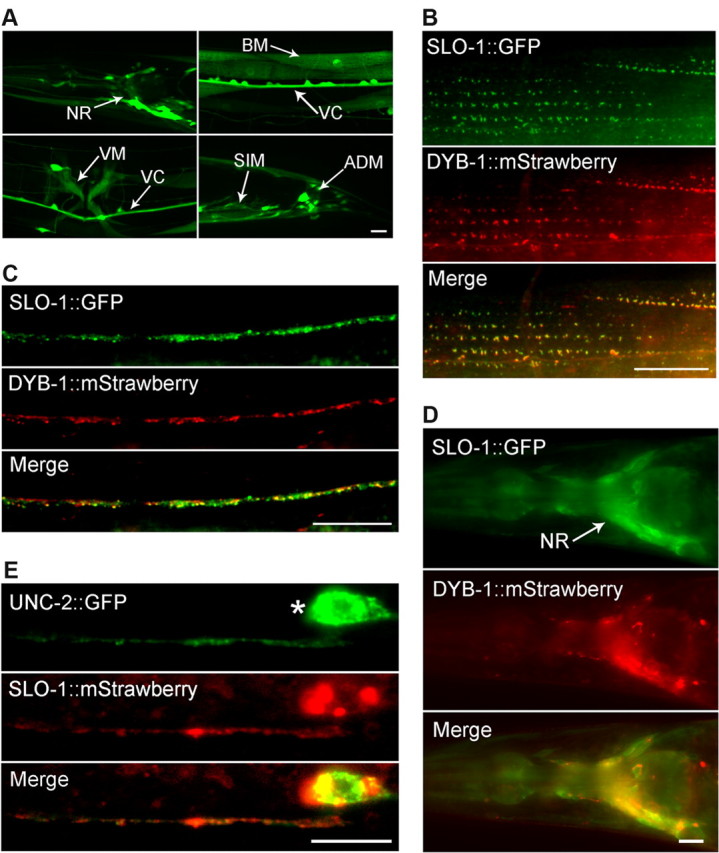
DYB-1 and SLO-1 were coexpressed and colocalized in neurons and muscle cells. A, Expression of GFP under the control of Pdyb-1 resulted in GFP epifluorescence in many neurons in the nerve ring (NR), ventral cord (VC), and tail (not labeled), as well as several types of muscles, including body-wall muscle (BM), vulval muscle (VM), stomatointestinal muscle (SIM), and anal depressor muscle (ADM). B–D, SLO-1::GFP and DYB-1::mStrawberry colocalized in body-wall muscle cells (B), the dorsal nerve cord (C), and the nerve ring (D) when they were expressed under the control of their native promoters. E, SLO-1::mStrawberry and UNC-2::GFP, which were expressed in GABAergic neurons, colocalized in the ventral nerve cord. * indicates the soma of a GABAergic motoneuron. Scale bars, 10 μm.
To determine whether the SLO-1::GFP puncta in the dorsal nerve cord corresponded to synaptic sites, we coexpressed mStrawberry-tagged SLO-1 (SLO-1::mStrawberry) and GFP-tagged UNC-2 (UNC-2::GFP) in GABAergic neurons under the controls of the unc-47 (vesicular GABA transporter) and unc-25 (glutamic acid decarboxylase) promoters, respectively, and analyzed their localization in the ventral nerve cord (SLO-1::mStrawberry signal in the dorsal cord was too weak to be imaged). UNC-2 is an N/P/Q-type (CaV2) voltage-gated Ca2+ channel (Bargmann, 1998) and is localized to presynaptic sites in motoneurons (Saheki and Bargmann, 2009). We found that the two fusion proteins colocalized in the ventral nerve cord (Fig. 2E). The separate observations of SLO-1 colocalization with DYB-1 and UNC-2 at the nerve cords collectively suggest that SLO-1 and DYB-1 colocalized at presynaptic sites in motoneurons.
DYB-1 was required for SLO-1 function in both neurons and muscle cells
To determine whether DYB-1 contributes to SLO-1 function in neurons, muscles, or both, we expressed wild-type DYB-1 in the slo-1(gf);dyb-1 double mutant using either the muscle-specific myo-3 promoter (Pmyo-3) or the pan-neuronal rab-3 promoter (Prab-3) and analyzed its effects on animal behaviors. Expression of wild-type DYB-1 in either neurons or muscle cells primarily reinstated the locomotion and egg-laying defects of slo-1(gf) (Fig. 3A,B), suggesting that DYB-1 is required for SLO-1 function in both neurons and muscle cells. In addition, we analyzed a head-bending phenotype associated with slo-1(lf) mutant (Kim et al., 2009; Chen et al., 2010a,b). The head-bending angle was increased to a similar degree in the dyb-1 mutant, slo-1(lf), and the double-mutant, slo-1(lf);dyb-1 (Fig. 3C,D). The head-bending phenotype of the dyb-1 mutant could be rescued by expressing wild-type DYB-1 in muscle cells but not neurons (Fig. 3C,D), suggesting that DYB-1 functions together with SLO-1 in muscle to regulate the head-bending angle.
Figure 3.

dyb-1 mutant suppressed locomotion and egg-laying phenotypes of slo-1(gf) and phenocopied slo-1(lf) in head-bending behavior. A, B, dyb-1 mutant suppressed the locomotion (A) and egg-laying (B) defects caused by slo-1(gf), which could be reversed by expressing wild-type DYB-1 in either neurons (Prab-3) or body-wall muscle cells (Pmyo-3). Twenty to 30 worms per group in A and 10 worms per group in B. C, D, Comparisons of full-amplitude head-bending angle and root mean square (RMS) head-bending angle. The head-bending angle was similarly increased in dyb-1 mutant, slo-1(lf), and the double mutant. This phenotype of dyb-1 mutant could be rescued by expressing wild-type DYB-1 in muscle but not neurons. Ten worms per group. *p < 0.01, significantly different between the indicated groups (A, B) or from wild type (C, D) (one-way ANOVA with Bonferroni's post hoc tests).
DYB-1 was required by SLO-1 to regulate neurotransmitter release
SLO-1 is a potent negative regulator of neurotransmitter release at the C. elegans NMJ (Wang et al., 2001; Liu et al., 2007). To determine whether DYB-1 is required for this function of SLO-1, we analyzed the effect of dyb-1 on ePSCs at the NMJ at two different extracellular Ca2+ concentrations (5 and 0.5 mm). The higher [Ca2+] is more suitable for determining whether dyb-1 may reverse an inhibitory effect of slo-1(gf) on neurotransmitter release, whereas the lower [Ca2+] is more suitable for testing whether dyb-1 could increase ePSC amplitude as slo-1(lf) does (Liu et al., 2007; Chen et al., 2010b). At 5 mm [Ca2+]o, the inhibitory effect of slo-1(gf) on the ePSC amplitude was reversed by dyb-1, whereas dyb-1 alone did not increase ePSC amplitude (Fig. 4A), which resembles slo-1(lf) and is probably attributable to the limited capacity of the readily releasable pool of synaptic vesicles (Wang et al., 2001). At 0.5 mm [Ca2+]o, ePSC amplitude was increased to a similar degree in dyb-1 and slo-1(lf) as well as in the slo-1(lf);dyb-1 double mutant (Fig. 4B). These observations suggest that DYB-1 is required by SLO-1 to control neurotransmitter release.
Figure 4.

DYB-1 was required by SLO-1 to control neurotransmitter release at NMJs. A, dyb-1 mutant counteracted the inhibitory effect of slo-1(gf) on ePSCs at 5 mm [Ca2+]o. B, The ePSC amplitude of dyb-1 mutant was significantly larger than that of the wild type but similar to that of slo-1(lf) or the slo-1(lf);dyb-1 double mutant. C, The frequency of mPSCs was increased, whereas the mean amplitude did not change in slo-1(lf), dyb-1, and slo-1(lf);dyb-1 compared with the wild type. D, Responses of body-wall muscle cells to exogenous ACh (100 μm) and GABA (100 μm) were unaltered in the mutants compared with the wild type. The holding potential for all recordings was −60 mV. *p < 0.05, significantly different from the wild type (one-way ANOVA with Bonferroni's post hoc test). The number of samples analyzed is indicated inside each column.
To test whether the abnormal ePSCs in slo-1 and dyb-1 mutants resulted from altered sensitivity of postsynaptic receptors, we recorded the mPSCs at 0.5 mm [Ca2+]o and compared the frequency and amplitude of mPSCs between the wild-type and the mutants. We found that the frequency of mPSCs was increased, whereas the amplitude of mPSCs did not change in slo-1(lf), dyb-1, and slo-1(lf);dyb-1 (Fig. 4C). We also examined the sensitivities of postsynaptic acetylcholine (ACh) and GABA receptors by puffing exogenous ACh and GABA directly onto the muscle cells. We found that the amplitude of ACh- and GABA-induced currents was similar between wild-type worms and the mutants (Fig. 4D). These observations suggest that SLO-1 and DYB-1 regulated neuromuscular transmission through a presynaptic mechanism.
SLO-1 and DYB-1 controlled Ca2+ transients in body-wall muscle cells
Because SLO-1 colocalizes with the L-type voltage-gated Ca2+ channel EGL-19 (Kim et al., 2009) and EGL-19 is the predominant voltage-gated Ca2+ channel in body-wall muscle (Jospin et al., 2002), SLO-1 could potentially be activated by Ca2+ entering through EGL-19 and, in turn, regulate intracellular [Ca2+]. To examine this possibility, we expressed the calcium indicator GCaMP2 in body-wall muscle under the control of Pmyo-3 and imaged Ca2+ transients as described previously (Liu et al., 2011). Ca2+ transients were observed in all muscle cells examined in wild-type and slo-1(lf) worms, with the frequency being much higher in the slo-1(lf) mutant than the wild type (Fig. 5). In slo-1(gf) worms, however, Ca2+ transients were detected in only approximately one-third of the preps with reduced frequency (Fig. 5). These observations suggest that a physiological function of SLO-1 is to inhibit muscle Ca2+ transients. Expression of wild-type SLO-1 in either muscle cells or neurons reversed the effect of slo-1(lf) on Ca2+ transients (Fig. 5), suggesting that SLO-1 regulates muscle Ca2+ transients through both presynaptic and postsynaptic effects.
Figure 5.

The frequency of Ca2+ transients in body-wall muscle cells was altered by mutations of slo-1 and dyb-1. A, Representative traces of Ca2+ transients from wild-type worms and various mutants. B, Quantification of the frequency of Ca2+ transients. The frequency of muscle Ca2+ transients was increased in slo-1(lf), which was restored by expressing wild-type SLO-1 in either muscles (Pmyo-3::SLO-1) or neurons (Prab-3::SLO-1). The frequency of Ca2+ transients was decreased in slo-1(gf), which was reversed by dyb-1 mutant. dyb-1 mutant showed a similar increase in the frequency of Ca2+ transients as slo-1(lf) did. *p < 0.05, significantly different from the wild type (one-way ANOVA with Bonferroni's post hoc test). The number of samples analyzed is indicated inside each column.
To determine whether DYB-1 also regulates muscle Ca2+ transients by affecting SLO-1, we analyzed Ca2+ transients in the dyb-1 mutant and slo-1(gf);dyb-1 double mutant. The frequency of muscle Ca2+ transients was increased to a similar degree in the dyb-1 mutant as in the slo-1(lf) mutant; furthermore, the dyb-1 mutant counteracted the effect of slo-1(gf) on muscle Ca2+ transients (Fig. 5). These observations suggest that DYB-1 is required by SLO-1 to regulate Ca2+ transients.
DYB-1 was required for SLO-1 subcellular localization in neurons and body-wall muscle cells
DYB-1 could contribute to SLO-1 function through several potential mechanisms. We found that DYB-1 did not affect channel functional properties when it was coexpressed with SLO-1 in Xenopus oocytes (data not shown). In contrast, SLO-1::GFP puncta appeared diffuse and dim in the ventral and dorsal nerve cords (Fig. 6A,E) and were essentially absent in body-wall muscle cells (Fig. 6B) of the dyb-1 mutant. The apparent SLO-1::GFP mislocalization did not result from a gross destabilization of presynaptic sites or muscle-dense bodies because the presynaptic marker RIM (Koushika et al., 2001) (Fig. 6C,E) and the dense body marker vinculin (Francis and Waterston, 1985) (Fig. 6D) were normally localized in the dyb-1 mutant, nor did it result from a decrease of SLO-1::GFP protein level, which was similar between the wild-type and dyb-1 mutant (Fig. 6F). These observations suggest that DYB-1 is required for proper SLO-1 subcellular localization in neurons and muscle cells and that the observed behavioral, synaptic, and muscle Ca2+ transient phenotypes of the dyb-1 mutant were likely caused by SLO-1 mislocalization in these cells.
Figure 6.

SLO-1 was mislocalized in both body-wall muscle cells and neurons of dyb-1 mutant. A, SLO-1::GFP in the dorsal cord and ventral cord appeared as puncta in the wild type but was diffusely distributed in dyb-1 mutant. B, SLO-1::GFP in muscle was localized to dense body regions in the wild type but undetectable in dyb-1 mutant. C, D, The presynaptic marker RIM (C) and dense body marker vinculin (D) were normally localized in dyb-1 mutant. E, Quantification of the density of dorsal cord puncta shown in A and C. *p < 0.0001, significantly different from the wild type (unpaired t test). F, Western blot shows that the level of SLO-1::GFP total protein was comparable between the wild-type and dyb-1 mutant. α-Tubulin was blotted to show equal loading of the protein samples. Scale bars, 10 μm.
DYB-1 subcellular localization did not depend on dystrophin
Dystrophin has been implicated in controlling dystrobrevin expression in mouse (Ohlendieck and Campbell, 1991) and SLO-1 subcellular localization in C. elegans (Kim et al., 2009; Chen et al., 2010a). We found that SLO-1::GFP was mislocalized in body-wall muscle but normally localized in the dorsal nerve cord in the dystrophin null mutant dys-1(cx18) (Fig. 7A,B,E). However, DYB-1 expression and subcellular localization were unaltered in both muscle cells and neurons of the dys-1 mutant (Fig. 7C–E). These observations suggest that in neurons DYB-1 localizes SLO-1 through a mechanism that is independent of DYS-1.
Figure 7.

Effects of dys-1 mutant on SLO-1 and DYB-1 subcellular localization. A, B, SLO-1::GFP was mislocalized in body-wall muscle but not neurons of the dystrophin mutant dys-1(cx18). C, D, DYB-1::GFP was normally localized in both muscle cells and neurons of dys-1 mutant. E, Quantification of SLO-1::GFP and DYB-1::GFP puncta density in the dorsal nerve cord. Scale bars, 10 μm.
Mouse αDB1 partially rescued dyb-1 mutant phenotype
The relatively high level of sequence homology between DYB-1 and mammalian dystrobrevins (Fig. 1) raised the possibility that a mammalian dystrobrevin may be able to substitute DYB-1 with respect to SLO-1 function in vivo. Therefore, we analyzed the effect of mouse αDB1 on various phenotypes of the dyb-1 mutant. Targeted expression of αDB1 in neurons but not muscle cells of the slo-1(gf);dyb-1 double mutant essentially reinstated behavioral phenotypes of slo-1(gf), including reduced locomotion speed and increased egg retention in the uterus (Fig. 8A,B). Expression of αDB1 in slo-1(gf);dyb-1 double mutant also essentially restored the small ePSC amplitude of slo-1(gf) (Fig. 8C). Furthermore, expression of αDB1 under the control of Pdyb-1 essentially rescued dyb-1-induced SLO-1::GFP mislocalization in the dorsal cord but had no effect in body-wall muscle (Fig. 8D,E). The differential rescuing effects of mouse αDB1 in neurons and muscle cells suggest that DYB-1 likely mediates SLO-1 localization by interacting with different proteins in neurons and muscle cells and that αDB1 may interact with the related protein(s) in neurons but not muscle cells.
Figure 8.
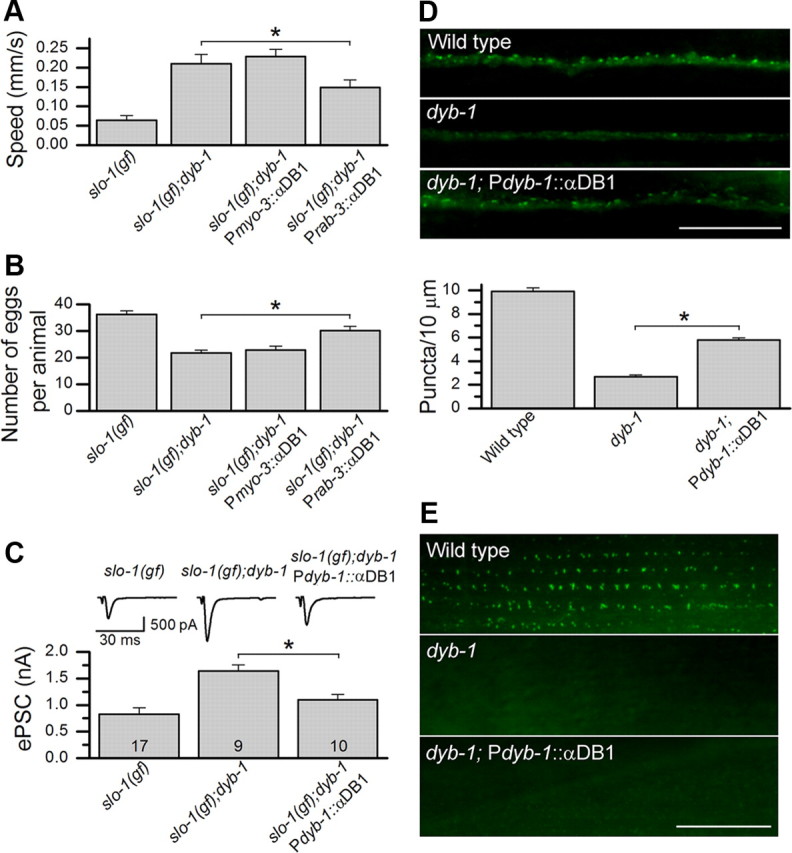
Mouse αDB1 partially rescued the neuronal phenotypes of dyb-1 mutant. A, B, Expression of αDB1 in neurons (Prab-3::αDB1) but not muscles (Pmyo-3::αDB1) partially reinstated the defective locomotion (A) and egg-laying (B) phenotypes of slo-1(gf) in slo-1(gf);dyb-1 mutant. The datasets for slo-1(gf) and slo-1(gf);dyb-1 in Figures 3 were replotted here for comparison. C, Expression of αDB1 in slo-1(gf);dyb-1 mutant under the control of Pdyb-1 essentially restored the small ePSC amplitude of slo-1(gf). The datasets for slo-1(gf) and slo-1(gf);dyb-1 in Figure 4 were replotted here for comparison. The holding potential used for recording ePSCs was −60 mV. D, E, Expression of αDB1 under the control of Pdyb-1 partially rescued SLO-1::GFP localization in the dorsal nerve cord but did not have an effect in body-wall muscle. *p < 0.05, significantly different between the indicated groups (one-way ANOVA with Bonferroni's post hoc test). Scale bars, 10 μm.
Discussion
The present study shows that DYB-1 plays pivotal roles in controlling muscle Ca2+ transients and synaptic exocytosis in C. elegans by localizing the BK channel to subcellular domains containing a high density of voltage-gated Ca2+ channels. These observations are consistent with the previous reports that dystrobrevin is localized to axon terminals (Ueda et al., 2000) and that mutations of either dystrobrevin or the BK channel may contribute to synaptic defects and muscular dystrophy (Grady et al., 1999; Wang et al., 2001; Carre-Pierrat et al., 2006; Grady et al., 2006).
Ca2+ mishandling has been suggested as the basis of muscle degeneration in Duchenne muscular dystrophy (DMD) patients (Duncan, 1978). It is thought that sarcolemma damage or enhanced activity of Ca2+ leak channels leads to elevation of intracellular [Ca2+], which activates Ca2+-dependent proteases such as calpains and eventually leads to cell death (Deconinck and Dan, 2007; Allen et al., 2010). However, measurements of Ca2+ concentration in skeletal muscle from DMD patients and mdx mice have produced controversial results (Constantin et al., 2006). Through Ca2+ imaging in live worms, we found that SLO-1 is a potent regulator of muscle Ca2+ transients in C. elegans body-wall muscle and that its mislocalization in dyb-1 mutant led to a large increase in Ca2+ transient frequency. Given that SLO-1 colocalizes with the L-type voltage-gated Ca2+ channel EGL-19 at dense body regions in body-wall muscle (Kim et al., 2009), Ca2+ microdomains formed at the inner mouth of EGL-19 in response to action potentials likely activate colocalized SLO-1, which in turn terminates further Ca2+ entry by facilitating membrane repolarization. This function of SLO-1 in regulating muscle Ca2+ transients might be an important contributing factor to the muscular degenerative changes related to slo-1, dyb-1, and dys-1 mutants.
Contraction of mammalian skeletal muscle requires the functions of both L-type voltage-gated Ca2+ channels in the sarcolemma and ryanodine receptors (RyRs) in the sarcoplasmic reticulum (SR) membrane (Rios and Brum, 1987; Takeshima et al., 1994), which is similar to C. elegans body-wall muscle (Liu et al., 2011). Although RyR-mediated Ca2+ release from the SR serves as the primary source of Ca2+ for skeletal muscle contraction, the activation of RyRs is coupled to depolarization-induced conformational changes of voltage-gated Ca2+ channels through protein/protein interactions occurring in t-tubules (Ríos et al., 1992; Schneider, 1994). Because the BK channel is also enriched or localized in t-tubules (Latorre et al., 1982; Knaus et al., 1995), whether or not it colocalizes with voltage-gated Ca2+ channels to regulate Ca2+ concentrations in mammalian skeletal muscle is worthy of investigation.
The similar locomotion and muscle Ca2+ transient phenotypes of dyb-1 and slo-1 mutants suggest that DYB-1 likely regulates muscle Ca2+ transients mainly through SLO-1. Nevertheless, we cannot exclude the potential involvement of other mechanisms. For example, Ca2+ mishandling in dystrophic mammalian skeletal muscle has been attributed to weakened membrane integrity and increased opening of other ion channels (e.g., store-operated cation channels and inositol triphosphate receptors) (Constantin et al., 2006). It would be interesting to determine whether these kinds of mechanisms are also implicated in the regulatory effect of DYB-1 on worm muscle Ca2+ transients.
The DAPC has also been implicated in synaptic transmission. For example, αDB1 is required for ACh receptor clustering at postsynaptic sites in mouse skeletal muscle (Pawlikowski and Maimone, 2008). In C. elegans, mutations of a DAPC component (DYS-1, DYB-1, or STN-1/syntrophin) result in increased cholinergic transmission as suggested by behavioral and pharmacological assays (Gieseler et al., 1999; Giugia et al., 1999). The present study shows that dystrobrevin controls neurotransmitter release by localizing SLO-1 to presynaptic sites, which potentially allows SLO-1 to be functionally coupled with UNC-2, the predominant voltage-gated Ca2+ channel at the C. elegans NMJ (Richmond et al., 2001; Saheki and Bargmann, 2009). Given that mouse αDB1 is also localized to axon terminals in mammals (Ueda et al., 2000) and may partially substitute DYB-1 in neurons with respect to SLO-1 function in C. elegans, it would be interesting to see whether dystrobrevins play a similar role in mammalian brain.
Dystrobrevins are mainly known to function as a component in the DAPC. However, there is evidence that dystrobrevins may also function independently of dystrophin. In mice lacking dystrophin, the assembly of the DAPC is disrupted in skeletal muscle but the dystrobrevin–syntrophin complexes are still formed in the brains (Blake et al., 1999). Although DYB-1 was required for SLO-1 localization in neurons as well as body-wall muscle cells, DYS-1 was dispensable for normal SLO-1 localization in neurons, suggesting that, at least in neurons, DYB-1 localizes SLO-1 through a mechanism independent of DYS-1. This conclusion is consistent with the data showing that mouse αDB1 was able to partially rescue SLO-1 mislocalization in neurons but not muscle cells in dyb-1 mutant. It remains to be determined what differences exist in the protein partners interacting with DYB-1 to mediate SLO-1 localization in neurons and body-wall muscle cells.
αDB and βDB are widely expressed in mammalian tissues. It has been suggested that mutations of dystrobrevins may underlie some behavioral and cognitive defects (Rees et al., 2007). However, relatively little is known about the functions of dystrobrevins. Patients with αDB deficiency may display severe congenital muscular dystrophy with ophthalmoplegia (Jones et al., 2003). Studies with mice have shown that knock-out of αDB causes mild muscular dystrophy and cardiomyopathy, abnormal distribution and reduced level of ACh receptors at the NMJs, and reduced level of neuronal nitric oxide synthase at the sarcolemma, and knock-out of βDB causes reduction in the number of GABAAα1 receptors in cerebellar Purkinje cells (Rees et al., 2007). Our analyses with C. elegans revealed new physiological functions of dystrobrevins and raised an interesting question as to whether dystrobrevin-dependent BK channel localization is involved in DAPC-associated diseases.
Footnotes
This work was supported by National Institute of Health Grants 1R01MH085927 and 5R01GM083049 (Z.-W.W.). We thank Margaret M. Maimone for mouse α-dystrobrevin cDNA plasmids, Cori Bargmann for an unc-2::gfp plasmid, and Michael Nonet for the RIM antibody. The vinculin antibody (MH24) developed by Robert H. Waterson was obtained from the Developmental Studies Hybridoma Bank under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa.
References
- Abraham LS, Oh HJ, Sancar F, Richmond JE, Kim H. An alpha-catulin homologue controls neuromuscular function through localization of the dystrophin complex and BK channels in Caenorhabditis elegans. PLoS Genet. 2010;6:e1001077. doi: 10.1371/journal.pgen.1001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG, Gervasio OL, Yeung EW, Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol. 2010;88:83–91. doi: 10.1139/Y09-058. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Hawkes R, Benson MA, Beesley PW. Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol. 1999;147:645–658. doi: 10.1083/jcb.147.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatz AL, Magleby KL. Ion conductance and selectivity of single calcium-activated potassium channels in cultured rat muscle. J Gen Physiol. 1984;84:1–23. doi: 10.1085/jgp.84.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carre-Pierrat M, Grisoni K, Gieseler K, Mariol MC, Martin E, Jospin M, Allard B, Ségalat L. The SLO-1 BK channel of Caenorhabditis elegans is critical for muscle function and is involved in dystrophin-dependent muscle dystrophy. J Mol Biol. 2006;358:387–395. doi: 10.1016/j.jmb.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Chen B, Liu P, Wang SJ, Ge Q, Zhan H, Mohler WA, Wang ZW. α-Catulin CTN-1 is required for BK channel subcellular localization in C. elegans body-wall muscle cells. EMBO J. 2010a;29:3184–3195. doi: 10.1038/emboj.2010.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Ge Q, Xia XM, Liu P, Wang SJ, Zhan H, Eipper BA, Wang ZW. A novel auxiliary subunit critical to BK channel function in Caenorhabditis elegans. J Neurosci. 2010b;30:16651–16661. doi: 10.1523/JNEUROSCI.3211-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin B, Sebille S, Cognard C. New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: implications in DMD. J Muscle Res Cell Motil. 2006;27:375–386. doi: 10.1007/s10974-006-9085-2. [DOI] [PubMed] [Google Scholar]
- Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7:762–773. doi: 10.1038/nrm2024. [DOI] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics. 2005;6:118. doi: 10.1186/1471-2164-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck N, Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Duncan CJ. Role of intracellular calcium in promoting muscle damage: a strategy for controlling the dystrophic condition. Experientia. 1978;34:1531–1535. doi: 10.1007/BF02034655. [DOI] [PubMed] [Google Scholar]
- Francis GR, Waterston RH. Muscle organization in Caenorhabditis elegans: localization of proteins implicated in thin filament attachment and I-band organization. J Cell Biol. 1985;101:1532–1549. doi: 10.1083/jcb.101.4.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieseler K, Bessou C, Ségalat L. Dystrobrevin- and dystrophin-like mutants display similar phenotypes in the nematode Caenorhabditis elegans. Neurogenetics. 1999;2:87–90. doi: 10.1007/s100480050057. [DOI] [PubMed] [Google Scholar]
- Giugia J, Gieseler K, Arpagaus M, Ségalat L. Mutations in the dystrophin-like dys-1 gene of Caenorhabditis elegans result in reduced acetylcholinesterase activity. FEBS Lett. 1999;463:270–272. doi: 10.1016/s0014-5793(99)01651-8. [DOI] [PubMed] [Google Scholar]
- Grady RM, Grange RW, Lau KS, Maimone MM, Nichol MC, Stull JT, Sanes JR. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol. 1999;1:215–220. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]
- Grady RM, Wozniak DF, Ohlemiller KK, Sanes JR. Cerebellar synaptic defects and abnormal motor behavior in mice lacking α- and β-dystrobrevin. J Neurosci. 2006;26:2841–2851. doi: 10.1523/JNEUROSCI.4823-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzfeind PJ, Ambrose HJ, Newey SE, Nawrotzki RA, Blake DJ, Davies KE. Tissue-selective expression of alpha-dystrobrevin is determined by multiple promoters. J Biol Chem. 1999;274:6250–6258. doi: 10.1074/jbc.274.10.6250. [DOI] [PubMed] [Google Scholar]
- Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, Storm JF. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21:9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KJ, Compton AG, Yang N, Mills MA, Peters MF, Mowat D, Kunkel LM, Froehner SC, North KN. Deficiency of the syntrophins and alpha-dystrobrevin in patients with inherited myopathy. Neuromuscul Disord. 2003;13:456–467. doi: 10.1016/s0960-8966(03)00066-x. [DOI] [PubMed] [Google Scholar]
- Jospin M, Jacquemond V, Mariol MC, Ségalat L, Allard B. The L-type voltage-dependent Ca2+ channel EGL-19 controls body wall muscle function in Caenorhabditis elegans. J Cell Biol. 2002;159:337–348. doi: 10.1083/jcb.200203055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Pierce-Shimomura JT, Oh HJ, Johnson BE, Goodman MB, McIntire SL. The dystrophin complex controls bk channel localization and muscle activity in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000780. doi: 10.1371/journal.pgen.1000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus HG, Eberhart A, Koch RO, Munujos P, Schmalhofer WA, Warmke JW, Kaczorowski GJ, Garcia ML. Characterization of tissue-expressed alpha subunits of the high conductance Ca2+-activated K+ channel. J Biol Chem. 1995;270:22434–22439. doi: 10.1074/jbc.270.38.22434. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post-docking role for active zone protein Rim. Nat Neurosci. 2001;4:997–1005. doi: 10.1038/nn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre R, Vergara C, Hidalgo C. Reconstitution in planar lipid bilayers of a Ca2+-dependent K+ channel from transverse tubule membranes isolated from rabbit skeletal muscle. Proc Natl Acad Sci U S A. 1982;79:805–809. doi: 10.1073/pnas.79.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Ge Q, Chen B, Salkoff L, Kotlikoff MI, Wang ZW. Genetic dissection of ion currents underlying all-or-none action potentials in C. elegans body-wall muscle cells. J Physiol. 2011;589:101–117. doi: 10.1113/jphysiol.2010.200683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Ge Q, Wang ZW. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J Neurosci. 2007;27:10404–10413. doi: 10.1523/JNEUROSCI.5634-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman DG, Fire A. Muscle: structure, function, and development. In: Riddle RL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Plainview, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 417–470. [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowski BT, Maimone MM. alpha-Dystrobrevin isoforms differ in their colocalization with and stabilization of agrin-induced acetylcholine receptor clusters. Neuroscience. 2008;154:582–594. doi: 10.1016/j.neuroscience.2008.01.052. [DOI] [PubMed] [Google Scholar]
- Peters MF, Sadoulet-Puccio HM, Grady MR, Kramarcy NR, Kunkel LM, Sanes JR, Sealock R, Froehner SC. Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J Cell Biol. 1998;142:1269–1278. doi: 10.1083/jcb.142.5.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J Physiol. 2004;557:147–157. doi: 10.1113/jphysiol.2004.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees ML, Lien CF, Górecki DC. Dystrobrevins in muscle and non-muscle tissues. Neuromuscul Disord. 2007;17:123–134. doi: 10.1016/j.nmd.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 2001;412:338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios E, Brum G. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. 1987;325:717–720. doi: 10.1038/325717a0. [DOI] [PubMed] [Google Scholar]
- Ríos E, Pizarro G, Stefani E. Charge movement and the nature of signal transduction in skeletal muscle excitation-contraction coupling. Annu Rev Physiol. 1992;54:109–133. doi: 10.1146/annurev.ph.54.030192.000545. [DOI] [PubMed] [Google Scholar]
- Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Saheki Y, Bargmann CI. Presynaptic CaV2 calcium channel traffic requires CALF-1 and the alpha(2)delta subunit UNC-36. Nat Neurosci. 2009;12:1257–1265. doi: 10.1038/nn.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MF. Control of calcium release in functioning skeletal muscle fibers. Annu Rev Physiol. 1994;56:463–484. doi: 10.1146/annurev.ph.56.030194.002335. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Iino M, Takekura H, Nishi M, Kuno J, Minowa O, Takano H, Noda T. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 1994;369:556–559. doi: 10.1038/369556a0. [DOI] [PubMed] [Google Scholar]
- Ueda H, Baba T, Kashiwagi K, Iijima H, Ohno S. Dystrobrevin localization in photoreceptor axon terminals and at blood-ocular barrier sites. Invest Ophthalmol Vis Sci. 2000;41:3908–3914. [PubMed] [Google Scholar]
- Wang ZW. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol Neurobiol. 2008;38:153–166. doi: 10.1007/s12035-008-8039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZW, Saifee O, Nonet ML, Salkoff L. SLO-1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction. Neuron. 2001;32:867–881. doi: 10.1016/s0896-6273(01)00522-0. [DOI] [PubMed] [Google Scholar]
- Yazejian B, Sun XP, Grinnell AD. Tracking presynaptic Ca2+ dynamics during neurotransmitter release with Ca2+-activated K+ channels. Nat Neurosci. 2000;3:566–571. doi: 10.1038/75737. [DOI] [PubMed] [Google Scholar]