

Abstract
Mutations in the leucine-rich repeat kinase 2 gene (LRRK2) are the most common genetic cause of Parkinson's disease (PD) and cause both autosomal dominant familial and sporadic PD. Currently, the physiological and pathogenic activities of LRRK2 are poorly understood. To decipher the biological functions of LRRK2, including the genes and pathways modulated by LRRK2 kinase activity in vivo, we assayed genome-wide mRNA expression in the brain and peripheral tissues from LRRK2 knockout (KO) and kinase hyperactive G2019S (G2019S) transgenic mice. Subtle but significant differences in mRNA expression were observed relative to wild-type (WT) controls in the cortex, striatum and kidney of KO animals, but only in the striatum in the G2019S model. In contrast, robust, consistent and highly significant differences were identified by the direct comparison of KO and G2019S profiles in the cortex, striatum, kidney and muscle, indicating opposite effects on mRNA expression by the two models relative to WT. Ribosomal and glycolytic biological functions were consistently and significantly up-regulated in LRRK2 G2019S compared with LRRK2 KO tissues. Genes involved in membrane-bound organelles, oxidative phosphorylation, mRNA processing and the endoplasmic reticulum were down-regulated in LRRK2 G2019S mice compared with KO. We confirmed the expression patterns of 35 LRRK2-regulated genes using quantitative reverse transcription polymerase chain reaction. These findings provide the first description of the transcriptional responses to genetically modified LRRK2 activity and provide preclinical target engagement and/or pharmacodynamic biomarker strategies for LRRK2 and may inform future therapeutic strategies for LRRK2-associated PD.
INTRODUCTION
Parkinson's disease (PD) is recognized as the second-most common neurodegenerative disorder after Alzheimer's disease. It affects more than 1.5% of the population over age 65 (1). Currently, no neuroprotective or neurorestorative therapies have been shown to halt or slow the progression of this disease. Delaying the progression of PD thus represents a critical unmet need. Although most PD cases appear to be sporadic, genetic forms of the disease are providing insight into this chronic, progressive neurodegenerative disease (2). Recent studies on the etiology of PD suggest a critical role for the leucine-rich repeat kinase 2 gene (LRRK2, PARK8, dardarin, OMIM 609007) in disease susceptibility (3,4). The LRRK2 gene encodes a large protein with multiple domains, including a Ras of Complex guanosine triphosphate hydrolase (GTPase) domain, a C-terminal of Roc domain, a protein kinase domain and several protein–protein interaction domains (5,6). Mutations in LRRK2 are associated with late-onset, autosomal-dominant, familial PD and also contribute to sporadic PD (3,4,7,8). The most prevalent is substitution of Ser for Gly 2019 in the kinase domain of LRRK2 (G2019S), which accounts for 5–6% of autosomal-dominant PD patients and ∼1% of PD patients with sporadic late-onset disease (1,9).
LRRK2 exhibits both GTPase and kinase activities. It is able to undergo autophosphorylation and phosphorylate numerous substrates (10–21). However, the physiologically relevant substrates of LRRK2 are unknown and under investigation. The GTPase activity of LRRK2 plays a key role in the pathogenesis of LRRK2 (22–24). Guanosine triphosphate (GTP)-binding activity of LRRK2 may be required for its kinase activity (19,25,26), but it is unclear how the GTPase and kinase activities synergize to regulate LRRK2 functions. A number of models have been developed to study the pathobiology of LRRK2, including primary neurons, budding yeast, Drosophila, Caenorhabditis elegans and mouse models. Studies in primary neuronal cultures demonstrate that the overexpression of mutant LRRK2 induces neuronal toxicity (17,18,23). In a recent yeast model, the LRRK2 GTPase domain causes more toxicity compared with other domains. The toxicity is closely associated with GTPase activity and defects in endocytic vesicular trafficking and autophagy (23). In Drosophila models, the overexpression of human LRRK2 variants induces late-onset, selective degeneration of dopaminergic neurons and L-3,4-dihydroxyphenylalanine responsive motor dysfunction (27,28). Knocking out LRRK2 in mice or its homologs in Drosophila and C. elegans reveals that LRRK2 is dispensable for the development and survival of DA neurons under physiologic conditions (15,29–34). LRRK2 R1441C knock-in, R1441G and G2019S BAC transgenic mouse models all exhibit mild impairments in nigrostriatal dopaminergic neurotransmission and mild motor dysfunction (35–37). Overexpression of LRRK2 G2019S using a cytomegalovirus (CMV)-enhanced human platelet-derived growth factor β-chain (CMVe-PDGF-β) promoter caused age-dependent dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities (38). All these models suggest a gain of function for PD causing LRRK2 mutations. However, how LRRK2 mutations cause neuronal toxicity is currently unknown and the pathogenic mechanisms accounting for LRRK2 neurotoxicity remain to be defined (39).
To further investigate the biological functions of LRRK2, including the genes and pathways modulated by LRRK2 activity in vivo, we have evaluated the gene expression patterns in LRRK2 knockout (KO) (29) and CMVe-PDGF-β LRRK2 G2019S transgenic mice (38) using microarrays. By comparing the transcriptional effects of LRRK2 KO versus G2019S overexpressing mice in multiple brain regions and peripheral tissues, we have identified a number of genes and biological processes that are regulated by LRRK2 expression and activity. Notably, ribosome and glycolysis genes were significantly up-regulated, while genes involved in membrane-bound organelles, oxidative phosphorylation, mRNA processing and endoplasmic reticulum (ER) function were down-regulated in G2019S mice compared with KO mice. These findings provide novel insights into the biological pathways regulated by LRRK2.
RESULTS
Characterization of LRRK2 KO and G2019 transgenic mice
For the expression analysis reported in this study, we used previously reported LRRK2 KO mice (29) and CMVe-PDGF-β LRRK2 G2019S transgenic mice (38). The LRRK2 KO mice lack LRRK2 protein as determined by immunoblot analysis with a LRRK2-specific monoclonal antibody (Fig. 1A). The CMVe-PDGF-β LRRK2 G2019S transgenic mice (Fig. 1B) overexpress G2019S LRRK2 3–4-fold (Fig. 1C).
Figure 1.

LRRK2 protein expression in LRRK2 KO and G2019S transgenic mice as determined by western blot analysis. (A) Three LRRK2 WT and three LRRK2 KO mice. (B) Schematic showing the CMVe-PDGF-β LRRK2 transgene and the position of the G2019S mutation. (C) Three LRRK2 G2019S transgenic (Tg) and three their non-transgenic (NTg) litter mate mice. (D) Fold overexpression and total LRRK2 were normalized to actin and expressed as the percentage of NTg controls with bars representing the mean ± SEM from n = 3 mice per genotype. Data were analyzed for statistical significance by the two-tailed unpaired Student's t-test against NTg controls (*P < 0.05).
LRRK2 KO and G2019S models have opposite transcriptional responses
Using Affymetrix microarrays, we assessed the genome-wide expression profiles in the striatum, cortex, kidney and muscle of the LRRK2 KO and G2019S animals. LRRK2 KOs had more pronounced transcriptional changes across all tissues than G2019S when compared with wild-type (WT) littermate controls. One-way analysis of variance (ANOVA) within each tissue identified 647, 587, 485 and 447 sequences differentially expressed (P < 0.01) for the cortex, kidney, muscle and striatum, respectively, of KO animals relative to WT controls (Fig. 2A). The kidney, which expresses high levels of LRRK2, and muscle, which is a potential clinically accessible tissue, were included in the analysis. Estimated false positive rates (FDRs) for these signatures were 30, 48, 72 and 82%, respectively. Animals overexpressing LRRK2 had smaller but significant differences in brain regions only (striatum and cortex) when compared with their non-transgenic littermates (Fig. 2B). One-way ANOVA within each tissue identified 557 and 390 sequences differentially expressed (P < 0.01) for the striatum and cortex, respectively, of G2019S animals relative to NTg controls. Estimated FDR for these signatures was above 50% for both.
Figure 2.

Transcriptional patterns in the striatum, cortex, kidney and muscle of (A) LRRK2 KO and (B) LRRK2 G2019S mice. One-way ANOVA was run in each tissue type separately using the corresponding control group (WT and NTg litter mates for KO and G2019S, respectively). Sequences expressed at P < 0.01 were used for 2D hierarchical clustering with agglomerative algorithm. The expression of genes in the striatum and cortex was significantly above the estimated 1.0 FDR (shown as yellow line) in both LRRK2 mouse models. The kidney and muscle mRNA profiles were significant in LRRK2 KO only.
We hypothesized that if LRRK2 KO and G2019S had opposite effects on gene expression, we would observe larger and more significant transcriptional differences by directly comparing KO and G2019S animals than by comparison with the respective controls. To test this hypothesis, gene expression levels in KO and G2019S animals were first normalized to expression levels in their respective NTg littermate controls to remove any potential mouse strain differences. Then, gene expression levels in KO and G2019S animals were directly compared using ANOVA to reveal robust signatures in each tissue examined (Fig. 3). This analysis identified 2986 (FDR 11.7%), 2045 (FDR 17.5%), 1646 (FDR 22%) and 761 (FDR 48%) sequences differentially expressed (P < 0.01) in the cortex, striatum, kidney and muscle between LRRK2 KO and G2019S animals (Fig. 3A).
Figure 3.

Direct comparison of LRRK2 G2019S and KO mRNA expression tested after normalization to corresponding NTg and WT controls in the striatum, cortex, kidney and muscle reveals robust differences across tissues. (A) The results of one-way ANOVA run in each tissue type separately. Sequences expressed at P < 0.01 were used for 2D hierarchical clustering with agglomerative algorithm. The estimated FDR was much lower compared with within-study comparisons of LRRK2 G2019S versus NTg and LRRK2 KO versus WT (see Results and Fig. 2 for details). 100% estimated FDR is shown as yellow line. (B) 2D agglomerative hierarchical clustering of all tissue samples using LRRK2 genotype signature (1833 transcripts; two-way ANOVA, P < 0.001). Horizontal lines on the heatmap represent biological replicates for each animal model.
In order to identify transcripts regulated similarly by genotype across tissue types, we performed two-way ANOVA using all samples controlling for tissue type and genotype. This analysis identified 1833 sequences differentially expressed (P < 0.001, FDR 1.9%) between LRRK2 KO and G2019S, which we refer to as the LRRK2 genotype signature, and these changes were generally observed in all four tissues (Fig. 3B). Moreover, we found 11 highly significant transcripts by two-way ANOVA (with factors being tissue type and genotype, P < 0.001) that were detected across all four tissues tested. Gh (growth hormone), Prl (prolactin) and Lrrk2 exhibited the highest average expression across all tissues in G2019S relative to KO animals (Table 1).
Table 1.
Common significant transcripts across all tissues with >30% change (LogR > 0.1 in any tissue) after the direct comparison between LRRK2 G2019S and KO (two-way ANOVA P < 0.001)
| Accession number | Gene symbol | Gene name | LogR LRRK2 G2019S/KO |
Cross tissue average | |||
|---|---|---|---|---|---|---|---|
| Striatum | Cortex | Kidney | Muscle | ||||
| NM_011164 | Prl | Prolactin | 1.99 | 0.94 | 0.02 | 0.00 | 0.74 |
| NM_008117 | Gh | Growth hormone | 1.9 | 1.42 | −0.01 | 0.05 | 0.84 |
| NM_008045 | Fshb | Follicle-stimulating hormone beta | 0.61 | 0.17 | −0.08 | 0.03 | 0.18 |
| AK014938 | Lrrk2 | Leucine-rich repeat kinase 2 | 0.45 | 0.49 | 0.58 | 0.71 | 0.56 |
| NM_001039677 | Slc30a2 | Solute carrier family 30 (zinc transporter), member 2 | 0.16 | 0.1 | −0.02 | 0.15 | 0.1 |
| CA751126 | Abcf2 | ATP-binding cassette, subfamily F (GCN20), member 2 | −0.12 | −0.08 | 0.01 | −0.02 | −0.05 |
| BY344923 | Tbc1d7 | TBC1 domain family, member 7 | −0.16 | −0.28 | 0.03 | 0.01 | −0.1 |
| BC117876 | Zfp287 | Zinc finger protein 287 | −0.25 | −0.29 | −0.09 | −0.16 | −0.2 |
| NM_001039094 | Negr1 | Neuronal growth regulator 1 | −0.27 | −0.17 | −0.04 | −0.24 | −0.18 |
| AK018644 | n/a | Hypotheticala | −0.33 | −0.5 | −0.14 | −0.14 | −0.28 |
| NM_198607 | 4930572J05Rik | RIKEN cDNA 4930572J05 gene | −0.35 | −0.21 | −0.13 | −0.09 | −0.2 |
aMicrobodies C-terminal targeting signal/phenylalanine-rich region containing protein.
Conversely, hypothetical (microbodies C-terminal targeting signal/phenylalanine-rich region containing protein), zinc finger protein 287 (Znf287), RIKEN cDNA 4930572J05 and neuronal growth regulator 1 (Ngr1, particularly in the striatum) genes exhibited the lowest average expression across all tissues in G2019S relative to KO animals.
To identify what tissue(s) had the biggest changes in mRNA expression, we ran correlation analyses between cross-tissue (combined expression) and each of the tissue LRRK2 G2019S and LRRK2 KO sets. Out of four tissues studied, we found that the cortex and striatum mRNA expressions contributed most to detected changes by the Pearson correlation (r2 = 0.98 and 0.95, respectively).
Validation of the transcriptional responses in LRRK2 KO and G2019S models
To validate these findings, we used TaqMan to assay 80 named genes that met the following criteria in the striatum: (i) more than 60% expression difference between LRRK2 G2019S and KO; (ii) more than 20% expression difference between the corresponding model (LRRK2 KO or G2019S) and control (WT or non-transgenic litter mates, NTg); (iii) dynamics of expression was opposite (up-down and down-up) in LRRK2 G2019S and LRRK2 KO (Fig. 4). It should be noted that LRRK2 did not meet this criteria, as the array probes do not detect the G2019S transgene.
Figure 4.

TaqMan validated 22 genes selected by the direct comparison between LRRK2 KO and G2019S expression profiles in the striatum (see Results for all selection criteria). (A) Gene expression in the striatum (primary selection tissue). (B) Gene expression in the cortex (secondary tissue). Log-transformed mean values of fold change and standard errors represent WT or NTg normalized values. Note that asterisks indicate significance between each model and corresponding control (WT or NTg, data not shown), not LRRK2 KO versus LRRK2 G2019S. Student's t-test: n = 3–5 and *P < 0.05.
In the striatum, 22 of the genes were confirmed to be significantly (P < 0.05) different between LRRK2 G2019S versus KO by TaqMan with direction in agreement with the array data (Fig. 4). Forty-four of the other 58 assayed genes exhibited trends in differential expression between GS and KO consistent with the array data. Importantly, for all 22 significant genes, LRRK2 KO and LRRK2 G2019S had significant or trending opposite effects on expression relative to their respective WT controls. Nine of the 22 genes were confirmed to be increased in LRRK2 G2019S versus KO. Genes that were increased >10-fold in 2019S relative to KO include S100a8, S100a9, Gabra6, Pcp2 and Crtam, 5–10-fold are Cbln3 and Cdkn1a and <5-fold are Abcb11 and Spef2. Six of the nine increased transcripts (S100a8, S100a9, Gabra6, Crtam, Cbln3 and Cdkn1a) were also significantly different when compared with non-transgenic littermate controls (Fig. 4A). Thirteen of 22 transcripts were decreased in the striatum of LRRK2 G2019S versus KO mice. Four of these were increased within the 5–10-fold range (Cdh1, Prg4, Slc22a6 and Prdm6), while five were significant against non-transgenic controls. Cdkn1a (increased in G2019S) and Slc22a6 (decreased in G2019S) were confirmed to be significant against both the matching genotype controls and after direct LRRK2 G2019S versus KO comparison in the striatum.
We also assessed the expression of these genes in the cortex by TaqMan. Although this gene set was not selected based on microarray evidence for changes in the cortex, nine genes showed expression changes in the overall same direction as the striatum. Those genes that showed obvious similar direction of changes in both the striatum and cortex included S100a8, S100a9, Abcb1, Cdkn1a, Pcp2 and Mid1 (Fig. 4B). Interestingly, five genes in the cortex changed in a direction opposite to the striatum (Tnfrsf11b, 4Cytl1, Fmod, Prdm6 and Prg4). S100a8, S100a9 and Slc22a6 were found to be significantly altered in the cortex, when compared with NTg controls (LRRK2 G2019S), and Cdkn1a, when compared with WT (P < 0.05, LRRK2 KO).
To help prioritize these genes for follow-up, we used IPA and DAVID to assess whether the human homologs were targets of existing drugs or biomarkers. Only Gabra6 was found to be targeted by existing drugs according to the Ingenuity database, and six of the 22 genes have been proposed as diagnostic/efficacy biomarkers according to IPA. For detailed annotations of the 22 genes, see Table 2.
Table 2.
Top differentially expressed genes between LRRK2 G2019S and KO mouse models selected using the striatum and relevant to human
| Symbol | Entrez gene name | Location | Family | Entrez gene ID |
|
|---|---|---|---|---|---|
| Human | Mouse | ||||
| Abcb11 | ATP-binding cassette, subfamily B (MDR/TAP), member 11 | EC | Transporter | 8647 | 27413 |
| Cbln3 | Cerebellin 3 precursor | EC | Other | 643866 | 56410 |
| Cdh1 | Cadherin 1, type 1, E-cadherin (epithelial) | PM | Other | 999 | 12550 |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | NC | Other | 1026 | 12575 |
| Crtam | Cytotoxic and regulatory T cell molecule | PM | Other | 56253 | 54698 |
| Cytl1 | Cytokine-like 1 | EC | Other | 54360 | 231162 |
| Fmod | Fibromodulin | EC | Other | 2331 | 14264 |
| Gabra6a | Gamma-aminobutyric acid (GABA) A receptor, alpha 6 | PM | Ion channel | 2559 | 14399 |
| Gpr182 | G protein-coupled receptor 182 | PM | G-protein-coupled receptor | 11318 | 11536 |
| Igfn1 | Immunoglobulin-like and fibronectin type III domain containing 1 | CP | Other | 91156 | 226438 |
| Mid1 | Midline 1 (Opitz/BBB syndrome) | NC | Other | 4281 | 17318 |
| Pcp2 | Purkinje cell protein 2 | CP | Other | 126006 | 18545 |
| Prdm6 | PR domain containing 6 | NC | Other | 93166 | 225518 |
| Prg4 | Proteoglycan 4 | EC | Other | 10216 | 96875 |
| Qrfpr | Pyroglutamylated RFamide peptide receptor | PM | G-protein-coupled receptor | 84109 | 229214 |
| Ranbp3l | RAN-binding protein 3-like | UK | Other | 202151 | 223332 |
| S100a8 | S100 calcium-binding protein A8 | CP | Other | 6279 | 20201 |
| S100a9 | S100 calcium-binding protein A9 | CP | Other | 6280 | 20202 |
| Slc22a6 | Solute carrier family 22 (organic anion transporter), member 6 | PM | Transporter | 9356 | 18399 |
| Spef2 | Sperm flagellar 2 | UK | Other | 79925 | 320277 |
| Tnfrsf11b | tumor necrosis factor receptor superfamily, member 11b | PM | Transmembrane receptor | 4982 | 18383 |
| Gm889b | Predicted gene 889 | UK | 380755 | ||
Molecules with diagnostic/efficacy biomarker applications (IPA) are shown in bold.aKnown drugs from Ingenuity (IPA) database: methohexital, aspirin/butalbital/caffeine, aspirin/butalbital/caffeine/codeine, pagoclone, alphadolone, SEP174559, acetaminophen/butalbital/caffeine, sevoflurane, isoflurane, gaboxadol, isoniazid, felbamate, etomidate, muscimol, halothane, fluoxetine/olanzapine, amobarbital, atropine/hyoscyamine/phenobarbital/scopolamine, acetaminophen/butalbital, eszopiclone, mephobarbital, hyoscyamine/phenobarbital, acetaminophen/butalbital/caffeine/codeine, butabarbital, diazepam, temazepam, zolpidem, lorazepam, olanzapine, clonazepam, zaleplon, secobarbital, butalbital, phenobarbital, pentobarbital, thiopental, D23129, desflurane, methoxyflurane, enflurane, pregnenolone.
bFrom DAVID; EC, extracellular space; PM, plasma membrane; NC, nucleus; CP, cytoplasm; UK, unknown.
LRRK2 regulates ribosomal function and glycolysis pathways
To identify the biological pathways that may underlie the transcriptional responses to the loss or the gain of LRRK2 function, we ran gene set enrichment analyses using multiple public ontological databases (see Methods for details). It was strikingly evident that animals overexpressing LRRK2 had an overall up-regulation, while LRRK2 KOs an overall down-regulation in genes involved in the ribosomal function. The kidney (E-value 0.003), cortex (E-value 0.001) and striatum (E-value 0.026) showed highly significant enrichment for genes involved in ribosomal function/translation function when analyses were run using gene signatures (P < 0.01) from each tissue separately or from the cross-tissue genotype signature (P < 0.001; E-value 4.8E–10; Table 3).
Table 3.
Biological pathways significantly enriched and up-regulated in LRRK2 G2019S animals compared with LRRK2 KO
| Tissue | Similar set | Bonferroni P-value (E-value) | Source | Sequence number |
|||
|---|---|---|---|---|---|---|---|
| Input | Overlap | Set | Background | ||||
| Kidney | Cytosolic ribosome | 0.003 | GO | 462 | 13 | 70 | 14359 |
| Ribosomal subunit | 0.003 | GO | 462 | 19 | 151 | 14359 | |
| Cortex | Cytosolic part | 0.001 | GO | 778 | 23 | 123 | 14359 |
| Cytosolic ribosome | 0.007 | GO | 778 | 16 | 70 | 14359 | |
| Ribosome | 0.009 | KEGG | 281 | 17 | 74 | 4722 | |
| Translation | 0.09 | GeneGo | 1005 | 16 | 97 | 21511 | |
| Striatum | Glycolysis | 0.009 | Panther | 704 | 11 | 44 | 17413 |
| Ribosome | 0.026 | KEGG | 208 | 14 | 74 | 4722 | |
| Cross-tissue | Cytosolic ribosome | 4.81E−10 | GO | 473 | 20 | 70 | 14359 |
| Ribosome | 3.99E−08 | KEGG | 191 | 20 | 74 | 4722 | |
| Cytosolic part | 7.14E−07 | GO | 473 | 22 | 123 | 14359 | |
| Translation | 1.53E−06 | GeneGo | 599 | 18 | 97 | 21511 | |
| Ribosomal subunit | 4.08E−05 | GO | 473 | 22 | 151 | 14359 | |
| Cytosolic large ribosomal subunit | 8.07E−05 | GO | 473 | 11 | 35 | 14359 | |
| Ribosome | 0.0001 | GO | 473 | 24 | 190 | 14359 | |
| Ribonucleoprotein complex | 0.002 | GO | 473 | 33 | 374 | 14359 | |
| Large ribosomal subunit | 0.01 | GO | 473 | 14 | 90 | 14359 | |
| Protein biosynthesis | 0.06 | Panther | 547 | 28 | 351 | 17413 | |
| Cytosolic small ribosomal subunit | 0.06 | GO | 473 | 8 | 32 | 14359 | |
E-value, Bonferroni–Hochberg P-value corrected for multiple comparison (<0.1 significant); GO, gene ontology; ‘Background’ represents the total number of genes in collection (source); ‘Set’ represents the total number of genes per biological function in collection; ‘Input’ column contains the number of ANOVA significant genes downloaded for gene set enrichment analysis; ‘Overlap’ is matched intersection genes between downloaded and annotated in database. ‘Ribosome’ function is consistently present across tissues and databases.
Interestingly, another highly significant pathway expressed higher in the striatum of G2019S versus KO was glycolysis (E-value 0.009, Table 3). Only two other pathways were borderline significant in this comparison, and both of these were enriched in the muscle signature: clathrin-mediated endocytosis (E-value 0.1; 11 overlapping genes) and cell adhesion_alpha 4 integrin (E-value 0.26; 6 overlapping genes), identified by Ingenuity and GeneGo, respectively (not shown).
We selected 13 ribosomal genes identified by gene set enrichment analysis that were significantly modulated in the cross-tissue genotype signature (P < 0.001) for TaqMan validation based on the assay availability. The results of expression of these 13 ribosomal transcripts in each tissue tested can be seen in Figure 5.
Figure 5.

TaqMan validation of ribosomal genes in the striatum (A), cortex (B) and kidney (C). The majority of ribosomal genes expressed lower in LRRK2 KO than in LRRK2 G2019S. Log-transformed mean values of fold change and standard errors represent WT or NTg normalized values. Note that asterisks indicate significance between each model and corresponding control (WT or NTg, data not shown), not LRRK2 KO versus LRRK2 G2019S. Student's t-test: n = 3–5 and *P < 0.05 and **P < 0.01.
Confirming the array findings, the majority of ribosomal genes were expressed significantly lower in LRRK2 KO than in LRRK2 G2019S in the kidney, cortex and striatum. Only Rps2 was significantly higher in the LRRK2 KO kidney when compared with both G2019S and WT, while no change was observed in the striatum and somewhat down-regulation in the LRRK2 KO cortex. Rpl15 and Rpl27a were consistently significantly regulated (P < 0.05) compared with matching controls (WT and NTg) in all tissues tested.
LRRK2 modulates the expression of membrane-bound organelle, protein metabolism and mitochondrial genes
We ran similar gene set enrichment analyses on the gene set that were down-regulated in LRRK2 G2019S mice compared with KO. We found that genes associated with membrane-bound organelles were highly enriched in both brain regions (cortex, E-value 8.39E–07, 680 transcripts; striatum, E-value 1.82E–124, 410 transcripts). In the cortex, biological sets comprising ubiquitination processes were also highly significantly down-regulated in LRRK2 G2019 animals compared with KO, while in the striatum ER, oxidative phosphorylation, RNA processing and mitochondrial function were down-regulated. The kidney had only one pathway of borderline significance (sensory perception of smell; E-value 0.2; 34 overlapping genes, not shown) and muscle had none (Table 4).
Table 4.
Significant down-regulated biological pathways identified in the striatum (A) and cortex (B) of LRRK2 G2019S animals compared with KO
| Tissue | Similar set | Bonferroni, P-value (E-value) | Source | Sequence number |
|||
|---|---|---|---|---|---|---|---|
| Input | Overlap | Set | Background | ||||
| Striatum | Membrane-bounded organelle | 1.82E−124 | GO | 415 | 410 | 6749 | 14359 |
| Oxidative phosphorylation | 0.00184 | Ingenuity | 418 | 14 | 128 | 21511 | |
| mRNA processing | 0.00222 | GO | 387 | 23 | 251 | 14575 | |
| Endoplasmic reticulum | 0.0174 | GO | 415 | 54 | 963 | 14359 | |
| Oxidative phosphorylation | 0.0221 | KEGG | 125 | 14 | 120 | 4722 | |
| RNA processing | 0.0225 | GO | 387 | 34 | 537 | 14575 | |
| RNA metabolic process | 0.024 | GO | 387 | 117 | 2984 | 14575 | |
| Mitochondrial dysfunction | 0.04 | Ingenuity | 418 | 12 | 122 | 21511 | |
| mRNA metabolic process | 0.05 | GO | 387 | 24 | 323 | 14575 | |
| Electron transport | 0.06 | Panther | 392 | 18 | 237 | 17413 | |
| Oxidative phosphorylation | 0.07 | GeneGo | 418 | 10 | 88 | 21511 | |
| Cortex | Intracellular membrane-bounded organelle | 2.48E−05 | GO | 1096 | 608 | 6745 | 14359 |
| Protein modification | 0.0036 | GO | 1131 | 211 | 1980 | 14575 | |
| Protein deubiquitination | 0.0068 | GO | 1131 | 15 | 45 | 14575 | |
| Protein ubiquitination pathway | 0.017 | Ingenuity | 1445 | 31 | 184 | 21511 | |
| Ubiquitin-mediated proteolysis | 0.026 | KEGG | 357 | 26 | 130 | 4722 | |
E-value, Bonferroni–Hochberg P-value corrected for multiple comparison (<0.1 significant); GO, gene ontology; ‘Background’ represents the total number of genes in collection (source); ‘Set’ represents the total number of genes per biological function in collection; ‘Input’ column contains the number of ANOVA significant genes downloaded for gene set enrichment analysis; ‘Overlap’ is matched intersection genes between downloaded and annotated in database.
Additionally, using public brain expression data, Ingenuity and Metabase sources from targeted geoscience initiative, we found that in the striatum 116 of 410 transcripts identified for membrane-bound organelle (∼30%) were interconnected. Two of 116 genes—Ctcf (CCCTC-binding factor known to promote histone methylation) and Zfx (zinc-finger protein X linked)—had most connections with other genes and thus were overrepresented (Supplementary Material). Similar patterns (interconnectivity and overrepresentation) were observed in the mouse cortex for ‘membrane-bound organelle’ function (not shown).
DISCUSSION
Numerous in vitro studies suggest that LRRK2 is involved in diverse cell biological functions, including translational control, mitogen-activated protein kinase, tumor necrosis factor α/Fas ligand and Wnt signaling pathways (40). To decipher the biological functions of LRRK2 in vivo, including the targets and pathways modulated by LRRK2 activity, we examined and compared gene expression in LRRK2 KO and G2019S transgenic mice. Instead of testing the expression of one gene at a time, microarrays can examine the expression levels of thousands of genes simultaneously (41). This allows for both identifying candidate genes and discovering networks and pathways that may play a role in LRRK2 pathogenesis. In this study, we profiled mRNA from brain and peripheral tissues of LRRK2 KO and LRRK2 G2019S transgenic mice using custom Affymetrix arrays. Our results provide the first evidence of transcriptional responses to genetically modified LRRK2 in mammalian models. Interestingly, LRRK2 KO and G2019S overexpression have opposite transcriptional responses. Pathway analysis revealed that ribosomal/translational functions are significantly up-regulated in LRRK2 G2019S model compared with LRRK2 KO and WT animals, indicating that LRRK2 regulates translation biological pathways. Furthermore, gene sets associated with membrane-bound organelles, oxidative phosphorylation, mRNA processing and ER are down-regulated in the G2019S model, suggesting that LRRK2 is also involved in these processes.
It is plausible that the effect of LRRK2 on translational processes identified in this study may underlie LRRK2′s association with PD pathogenesis. Consistent with our observations, in Drosophila LRRK2 (dLRRK) was shown to regulate protein translational pathways. Imai et al. (15) found that LRRK2 phosphorylates eukaryotic initiation factor 4E (eIF4E)-binding protein (4E-BP), a negative regulator of eIF4E-mediated protein translation and a key mediator of various stress responses. Overexpression of 4E-BP rescues the Parkinsonian phenotypes in Drosophila models of PD by inhibiting cap-dependent translation and inducing the expression of genes involved in the stress response (15,42). 4E-BP is not directly phosphorylated by LRRK2 in vitro, suggesting that 4E-BP may be indirectly phosphorylated by LRRK2 (43). A link between dLRRK and protein synthesis was further strengthened by the observation that dLRRK interacts with the microRNA pathway to regulate protein synthesis (44). Taken together with our results, there is strong convergent evidence that LRRK2 regulates the cell protein translation machinery in diverse species and tissues. The Drosophila studies need to be extended to mammalian systems and the significant up-regulation of genes in ribosomal/translational pathways should serve as a strong starting point for future studies to elucidate and clarify the underlying mechanisms of potential regulation of protein translation by LRRK2.
In this study, we also found that LRRK2 regulates gene sets associated with membrane-bound organelles, ER genes and oxidative phosphorylation genes. Consistent with our results, it is known that LRRK2 localizes to a wide range of vesicular and membranous structures in neurons including multivesicular bodies, microtubule transport vesicles, endosomes, lysosomes, the ER, Golgi and mitochondria (45,46). LRRK2 is also present in the synaptosomal compartment of cortical neurons in which it interacts with several proteins involved in vesicular recycling (47). Furthermore, it has been reported that LRRK2 causes defects in synaptic vesicle endocytosis and exocytosis in neurons (23,48). Our findings and previous studies suggest that LRRK2 may be involved in vesicular trafficking by regulating membrane-bound proteins. In addition, ablating LRK-1 in C. elegans causes an enhanced vulnerability of dopaminergic neurons to rotenone, the mitochondrial toxin (49). Dopaminergic neurons in Drosophila with overexpression of mutant LRRK2 are more vulnerable to rotenone (28). These studies, together with our findings suggest that LRRK2 may regulate the oxidative phosphorylation.
In summary, our genome-wide mRNA expression profiling of the loss or the gain of function of LRRK2 mammalian models uncover biological pathways regulated by LRRK2 which could underlie its association with PD. They are summarized in Table 5. Importantly, these findings could serve as the preclinical biomarkers of LRRK2 activity in the absence of known physiological LRRK2 substrates. In addition, there is utility in these for drug development even if they are not directly translatable to humans such as assessing pharmacodynamics across lead compounds. Further, determining the biological pathways altered by LRRK2 hyperactivity that contribute to PD onset will provide additional therapeutic opportunities in addition to direct LRRK2 inhibition.
Table 5.
Summary of all significant biological processes identified in the cortex and striatum of LRRK2 G2019S and KO animals after the direct comparison (E-value <0.1)
| Biological function: LRRK2 G2019S/KO | Cortex | Striatum |
|---|---|---|
| Ribosome | ↑ | ↑ |
| Mitochondria: glycolysis | – | ↑ |
| Membrane-bound organelle | ↓ | ↓ |
| Ubiquitination | ↓ | – |
| Mitochondria: oxidative phosphorylation | – | ↓ |
| Cytosol: endoplasmic reticulum | – | ↓ |
MATERIALS AND METHODS
Animals
Mice were housed and treated strictly in accordance with the NIH ‘Guide for the Care and Use of Laboratory Animals’. The Johns Hopkins University Animal Care and Use Committees (ACUC) approved all procedures.
LRRK2 KO mice utilized in this study were previously generated by targeting the kinase domain of LRRK2 (29). Heterozygous LRRK2 mutant mice were backcrossed to C57BL/6J for over 10 generations. LRRK2 KO and littermate WT mice were obtained from heterozygous LRRK2 mutant male and female breeding. LRRK2 KO mice were maintained on a C57BL/6J background. LRRK2 transgenic (G2019S) mice overexpress LRRK2 under the direction of a CMVe-PDGF-β promoter as described previously (38,50). LRRK2 G2019S transgenic mice were backcrossed to C57BL/6J for 3–4 generations and age-matched littermate controls from heterozygous LRRK2 G2019S and C57BL/6J WT matings were used.
LRRK2 KO, LRRK2 G2019S transgenic and C57BL/6J animals were sacrificed by decapitation, and four tissues were preserved in RNAlater for RNA isolation and included two brain regions (cortex and striatum), kidney (highest LRRK2 expression) and muscle. For the LRRK2 KO experiment, we had six KO and six WT animals combined within each tissue in twos (n = 2 per amplification, overall n = 3 biological replicates per group). For LRRK2 G2019S, we had six biological replicates per group (transgenic, NTg).
mRNA isolation and microarray profiling
All steps including RNA isolation, quality control, amplification, labeling and microarray processing were performed by the Rosetta Inpharmatics Gene Expression Laboratory (Seattle, WA) using standard protocols. Briefly, tissue was pulverized prior to homogenization using the Covaris cryo-prep. Tissue homogenization was performed using the Covaris S2 instrument in a solution of GITC/BME (1:50 ratio), followed by the addition of a TRIzol water solution (4:1 ratio). 100% chloroform was added to the TRIzol/GITC lysate (1:5 ratio) to facilitate the separation of the organic and aqueous components using the phaselock (Eppendorf) system. The aqueous supernatant was further purified using the Promega SV-96 Total RNA Kit, incorporating a DNase treatment during the procedure. Isolated total RNA samples were then assayed for quality (Agilent Bioanalyzer) and yield (Ribogreen) metrics prior to amplification. About 50 ng of total RNA samples were amplified and labeled using a custom-automated version of the NuGEN Ovation WB protocol before hybridization to custom mouse Affymetrix GeneChip® microarrays containing 38,385 probe sets designed to monitor additional genes and poly-A sites than commercially available microarrays. Hybridization, labeling and scanning were conducted using Affymetrix ovens, fluidics stations and scanners followed the recommended protocols (NuGEN, San Carlos, CA).
Microarray data pre-processing and statistics
Data quality was assessed using the standard Affymetrix metrics (image reconstruction and histograms of raw signal intensities). Normalization was performed using the robust multi-array average method (41,51), which calculates the final expression measures using the Tukey median polish algorithm. Principal component analysis was used to identify outliers and clustering of normalized data. An FDR approach was used for initial microarray analysis to assess the robustness of data.
A series of one-way ANOVAs were run within each mouse line (LRRK2 KO or LRRK2 G2019S versus littermate controls) for all tissues tested. To directly compare KO and G2019S, each tissue for each genotype was first normalized to the average expression in its corresponding controls. A series of one-way ANOVAs comparing G2019S and KO were then performed for each tissue as well as two-way ANOVA across all tissues accounting for both tissue and genotype.
Biological function enrichment analyses
The gene sets of interest were cross-referenced for enrichment of biological processes and pathways against multiple public databases (GeneGo, Gene Ontology, KEGG, Ingenuity, Panther, SuperArray). Hypergeometric P-value (probability distribution after sampling without replacement) was calculated for the overlap between each input gene set and those found in the public databases. Bonferroni-corrected P-values (expectation, or E-values) were calculated to control for multiple comparisons (gene sets) across all databases. E-value of <0.1 was considered significant. Additional biological annotations were obtained using Ingenuity (IPA 9.0) and DAVID (the Database for Annotation, Visualization and Integrated Discovery).
Quantitative RT–PCR
Four micrograms of total RNA was converted to cDNA using Applied Biosystems High Capacity RNA-to-cDNA (ABI). Primers for 22 top selected (S100a8, S100a9, Gabra6, Pcp2, Crtam, Cbln3, Abcb11, Spef2, Cdkn1a, Mid1, Gpr182, Gm889, Igfn1, Ranbp3L, Qrfpr, Tnfrsf11b, 4Cytl1, Fmod, Prdm6, Slc22a6, Prg4 and Cdh1) and 13 ribosomal (Rpl10a, Rpl11, Rpl12, Rpl15, Rpl21, Rpl26, Rpl27a, Rpl28, Rpl35a, Rps2, Rps15, Rps28 and Mark1) genes were designed to cover the exon–exon junction for every gene (ABI, see full list of the assays in Supplementary Material). 100–200 ng total cDNA was used as templates in a standard 40 cycle, 25 µl of polymerase chain reaction. Each sample was run in duplicate on a Stratagene Mx 3000P or Mx 3005P real-time PCR instrument, using ABI's TaqMan Universal PCR Master Mix. Endogenous 18S was used for normalization in a separate reaction (not multiplexed). Relative expression of target gene against WT in respective LRRK2 KO or G2019S overexpressing samples was calculated by the comparative Ct method with the 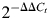 equation (41,52). Two-tailed Student's t-test was run to identify significance in PCR data (with P-value threshold <0.05).
equation (41,52). Two-tailed Student's t-test was run to identify significance in PCR data (with P-value threshold <0.05).
Western blotting
Soluble brain extracts were prepared from brains of 2-month-old LRRK2 WT and KO mice, and LRRK2 G2019S and their NTg litter mate mice by homogenization in lysis buffer [1× phosphate-buffered saline, 1% Triton X-100, 1× Complete protease inhibitor cocktail (Roche), 1× phosphatase inhibitor cocktail I and II (Sigma)]. Protein concentration was determined by BCA method (Pierce Biotech). About 100 µg of protein was resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membrane and probed with MJFF2 (clone c41–2) anti-LRRK2 rabbit monoclonal antibody recognizing both mouse and human LRRK2 (The Michael J. FOX foundation for Parkinson's Research), or with anti-actin rabbit polyclonal antibody for loading control (Sigma, 1:10 000 dilution). Densitometric analysis was conducted to quantify the fold overexpression of LRRK2 relative to endogenous mouse LRRK2. Total LRRK2 protein levels were normalized to actin and expressed as the percent of NTg controls. Mean values from three mice per genotype/control were analyzed for statistical significance by two-tailed unpaired Student's t-test compared with NTg controls (*P < 0.05).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This study was supported by the National Institute of Health (NIH) (NINDS NS38377 to T.M.D.) and Merck & Co., Inc.
Supplementary Material
ACKNOWLEDGEMENTS
T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
REFERENCES
- 1.Lees A.J., Hardy J., Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 2.Moore D.J., West A.B., Dawson V.L., Dawson T.M. Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 3.Paisan-Ruiz C., Jain S., Evans E.W., Gilks W.P., Simon J., van der Brug M., Lopez de Munain A., Aparicio S., Gil A.M., Khan N., et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 4.Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B., et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Mata I.F., Wedemeyer W.J., Farrer M.J., Taylor J.P., Gallo K.A. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Cookson M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat. Rev. Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giasson B.I., Covy J.P., Bonini N.M., Hurtig H.I., Farrer M.J., Trojanowski J.Q., Van Deerlin V.M. Biochemical and pathological characterization of Lrrk2. Ann. Neurol. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- 8.Gilks W.P., Abou-Sleiman P.M., Gandhi S., Jain S., Singleton A., Lees A.J., Shaw K., Bhatia K.P., Bonifati V., Quinn N.P., et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 9.Biskup S., West A.B. Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson's disease. Biochim. Biophys. Acta. 2009;1792:625–633. doi: 10.1016/j.bbadis.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamikawaji S., Ito G., Iwatsubo T. Identification of the autophosphorylation sites of LRRK2. Biochemistry. 2009;48:10963–10975. doi: 10.1021/bi9011379. [DOI] [PubMed] [Google Scholar]
- 11.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Greggio E., Taymans J.M., Zhen E.Y., Ryder J., Vancraenenbroeck R., Beilina A., Sun P., Deng J., Jaffe H., Baekelandt V., et al. The Parkinson's disease kinase LRRK2 autophosphorylates its GTPase domain at multiple sites. Biochem. Biophys. Res. Commun. 2009;389:449–454. doi: 10.1016/j.bbrc.2009.08.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pungaliya P.P., Bai Y., Lipinski K., Anand V.S., Sen S., Brown E.L., Bates B., Reinhart P.H., West A.B., Hirst W.D., et al. Identification and characterization of a leucine-rich repeat kinase 2 (LRRK2) consensus phosphorylation motif. PLoS One. 2010;5:e13672. doi: 10.1371/journal.pone.0013672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greggio E., Zambrano I., Kaganovich A., Beilina A., Taymans J.M., Daniels V., Lewis P., Jain S., Ding J., Syed A., et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imai Y., Gehrke S., Wang H.Q., Takahashi R., Hasegawa K., Oota E., Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaleel M., Nichols R.J., Deak M., Campbell D.G., Gillardon F., Knebel A., Alessi D.R. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem. J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith W.W., Pei Z., Jiang H., Dawson V.L., Dawson T.M., Ross C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 18.West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl Acad. Sci. USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West A.B., Moore D.J., Choi C., Andrabi S.A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K.L., Dawson V.L., et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 20.Li X., Moore D.J., Xiong Y., Dawson T.M., Dawson V.L. Reevaluation of phosphorylation sites in the Parkinson disease-associated leucine-rich repeat kinase 2. J. Biol. Chem. 2010;285:29569–29576. doi: 10.1074/jbc.M110.127639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen S., Webber P.J., West A.B. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 2009;284:36346–36356. doi: 10.1074/jbc.M109.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis P.A., Greggio E., Beilina A., Jain S., Baker A., Cookson M.R. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem. Biophys. Res. Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong Y., Coombes C.E., Kilaru A., Li X., Gitler A.D., Bowers W.J., Dawson V.L., Dawson T.M., Moore D.J. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6:e1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X., Tan Y.C., Poulose S., Olanow C.W., Huang X.Y., Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J. Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito G., Okai T., Fujino G., Takeda K., Ichijo H., Katada T., Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- 26.Liu M., Dobson B., Glicksman M.A., Yue Z., Stein R.L. Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2: characterization of the kinase and GTPase activities. Biochemistry. 2010;49:2008–2017. doi: 10.1021/bi901851y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z., Wang X., Yu Y., Li X., Wang T., Jiang H., Ren Q., Jiao Y., Sawa A., Moran T., et al. A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl Acad. Sci. USA. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng C.H., Mok S.Z., Koh C., Ouyang X., Fivaz M.L., Tan E.K., Dawson V.L., Dawson T.M., Yu F., Lim K.L. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci. 2009;29:11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andres-Mateos E., Mejias R., Sasaki M., Li X., Lin B.M., Biskup S., Zhang L., Banerjee R., Thomas B., Yang L., et al. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) J. Neurosci. 2009;29:15846–15850. doi: 10.1523/JNEUROSCI.4357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S.B., Kim W., Lee S., Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- 31.Wang D., Tang B., Zhao G., Pan Q., Xia K., Bodmer R., Zhang Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol. Neurodegener. 2008;3:3. doi: 10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakaguchi-Nakashima A., Meir J.Y., Jin Y., Matsumoto K., Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 33.Yao C., El Khoury R., Wang W., Byrd T.A., Pehek E.A., Thacker C., Zhu X., Smith M.A., Wilson-Delfosse A.L., Chen S.G. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson's disease. Neurobiol. Dis. 2010;40:73–81. doi: 10.1016/j.nbd.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R.J., 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl Acad. Sci. USA. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y., Liu W., Oo T.F., Wang L., Tang Y., Jackson-Lewis V., Zhou C., Geghman K., Bogdanov M., Przedborski S., et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat. Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong Y., Pisani A., Martella G., Karouani M., Yamaguchi H., Pothos E.N., Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl Acad. Sci. USA. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X., Patel J.C., Wang J., Avshalumov M.V., Nicholson C., Buxbaum J.D., Elder G.A., Rice M.E., Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J. Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramonet D., Daher J.P., Lin B.M., Stafa K., Kim J., Banerjee R., Westerlund M., Pletnikova O., Glauser L., Yang L., et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dawson T.M., Ko H.S., Dawson V.L. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berwick D.C., Harvey K. LRRK2 signaling pathways: the key to unlocking neurodegeneration? Trends Cell. Biol. 2011;21:257–265. doi: 10.1016/j.tcb.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Winrow C.J., Tanis K.Q., Reiss D.R., Rigby A.M., Uslaner J.M., Uebele V.N., Doran S.M., Fox S.V., Garson S.L., Gotter A.L., et al. Orexin receptor antagonism prevents transcriptional and behavioral plasticity resulting from stimulant exposure. Neuropharmacology. 2010;58:185–194. doi: 10.1016/j.neuropharm.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 42.Tain L.S., Mortiboys H., Tao R.N., Ziviani E., Bandmann O., Whitworth A.J. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar A., Greggio E., Beilina A., Kaganovich A., Chan D., Taymans J.M., Wolozin B., Cookson M.R. The Parkinson's disease associated LRRK2 exhibits weaker in vitro phosphorylation of 4E-BP compared to autophosphorylation. PLoS One. 2010;5:e8730. doi: 10.1371/journal.pone.0008730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gehrke S., Imai Y., Sokol N., Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biskup S., Moore D.J., Celsi F., Higashi S., West A.B., Andrabi S.A., Kurkinen K., Yu S.W., Savitt J.M., Waldvogel H.J., et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 46.Hatano T., Kubo S., Imai S., Maeda M., Ishikawa K., Mizuno Y., Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum. Mol. Genet. 2007;16:678–690. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- 47.Piccoli G., Condliffe S.B., Bauer M., Giesert F., Boldt K., De Astis S., Meixner A., Sarioglu H., Vogt-Weisenhorn D.M., Wurst W., et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin N., Jeong H., Kwon J., Heo H.Y., Kwon J.J., Yun H.J., Kim C.H., Han B.S., Tong Y., Shen J., et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell. Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 49.Saha S., Guillily M.D., Ferree A., Lanceta J., Chan D., Ghosh J., Hsu C.H., Segal L., Raghavan K., Matsumoto K., et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci. 2009;29:9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu B.H., Wang X., Ma Y.X., Wang S. CMV enhancer/human PDGF-beta promoter for neuron-specific transgene expression. Gene Ther. 2004;11:52–60. doi: 10.1038/sj.gt.3302126. [DOI] [PubMed] [Google Scholar]
- 51.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 52.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-▵▵C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.