

Abstract
In the present study we tested the hypothesis whether hyperhomocysteinemia, an elevated homocysteine level, induces venous phenotype in artery. To test our hypothesis, we employed wild type (WT) and cystathionine β-synthase+/− (CBS+/−) mice treatment with or without folic acid (FA). Aortic blood flow and velocity were significantly lower in CBS+/− mice compared to WT. Aortic lumen diameter was significantly decreased in CBS+/− mice, whereas FA treatment normalized it. Medial thickness and collagen were significantly increased in CBS+/− aorta, whereas elastin / collagen ratio was significantly decreased. Superoxide and gelatinase activity was significantly high in CBS +/− aorta vs WT. Western blot showed significant increase in MMP-2, -9,-12, TIMP-2 and decrease in TIMP-4 in aorta. RT-PCR revealed significant increase of vena cava marker EphB4, MMP-13 and TIMP-3 in aorta. We summarize that chronic HHcy causes vascular remodeling that transduces changes in vascular wall in a way that artery expresses vein phenotype.
Introduction
Vascular remodeling is an active process of structural modification of the blood vessels in response to arterial pressure and other hemodynamic stimuli (Gibbons and Dzau, 1994), which may lead to clinical conditions, such as hypertension and myocardial infarction. Various endogenous and exogenous molecules influence hemodynamic changes and contribute to vascular remodeling. Of particular interest, homocysteine (Hcy) is one of the known endogenous molecules that potentiate vascular disease risk at a level higher than normal physiological ranges.
Hcy is sulfur containing non-protein amino acid and is formed during the metabolism of methionine. In the body, Hcy can be remethylated back to methionine or clears from the body by transsulfuration pathway. In transsulfuration pathway, the CBS (cystathionine β-synthase) enzyme breaks down the Hcy to cysteine (Giusti et al., 2004). Complete absence of CBS (CBS−/−) causes severe hyperhomocysteinemia (HHcy), an elevated plasma Hcy level, and these individuals develop complications in the cardiovascular system leading to early and aggressive vascular disease.
At pathological levels, Hcy causes remodeling of arterial wall by breaking down the elastin and accumulation of collagen in the extracellular matrix (ECM) (Giusti et al., 2004). In heart, this causes endothelial-myocyte uncoupling (Rodriguez et al., 2008). This phenomenon is one of the most important factors behind cardiac endothelial dysfunction during HHcy (Kundu et al., 2009). Accumulation of ECM between the endothelial and vascular muscle cells at elevated Hcy level, known as hyperhomocysteinemia (HHcy), may also lead to uncoupling of endothelial and vascular smooth muscle cells causing structural and functional abnormalities. The possibilities of such condition, however, not defined.
Remodeling of ECM is a dynamic process and matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) regulate this process. MMPs are the family of zinc dependent redox sensitive endopeptidases that maintains the synthesis and breakdown of the ECM protein, particularly elastin and collagen (Bobik and Tkachuk, 2003, Chun et al., 2004, Lehoux et al., 2004, Nagase et al., 2006, Nagase and Woessner, 1999, Visse and Nagase, 2003). In pathological conditions these proteinases play key role in vascular remodeling (de Kleijn et al., 2001, Dollery et al., 1995, Galli et al., 2005a, Galli et al., 2005b, Hayden et al., 2005, He et al., 2007). To date, 28 different MMPs has been discovered; of which MMP 2 (72 KD Gelatinase A) and MMP-9, (92 KD Gelatinase B), has a wide range of matrix substances. These two MMPs catalyze type IV collagen, gelatin and elastin (Visse and Nagase, 2003). Additionally, MMP-9 also breaks down Collagen I, II, and III (Patterson et al., 2001). It is interesting that MMP-2 activates MMP-9, and both these MMPs are well expressed in the vascular smooth muscle cells (VSMC) (Fridman et al., 2003). MMP-12 and 13 are also important in vascular remodeling mainly because elastin is the substrate of MMP-12, and MMP 13 breaks down collagen III (Beaudeux et al., 2004). Endogenously, MMPs activity is regulated by TIMPs. So far, four TIMPs (TIMP-1, - 2, - 3 and -4) are identified, and they inhibit all MMPs (Raffetto and Khalil, 2008b, Raffetto and Khalil, 2008a). As the activities of MMPs are balanced by the TIMPs, the remodeling of vascular structure can be demonstrated by the imbalance between the function and expression of MMPs and TIMPs (Watts et al., 2007).
Endothelial dysfunction due to reactive oxygen species (ROS) production is another important aspect of vascular remodeling (Edirimanne et al., 2007, Weiss et al., 2002). Injury or activation of the endothelial cells (EC) causes changes in the regulatory function. This dysfunction of EC is mainly defined as the impaired relaxation and contraction of the blood vessel, due to impaired nitric oxide (NO) production from the EC (De Meyer and Herman, 1997). ROS are produced by EC and their adjacent VSMC, and it is an important factor for maintaining normal vascular function. However, at higher concentration, it reduces the bioavailability of NO, and impairs the EC dependent vasodilatation (Rodford et al., 2008). The pathophysiological consequences of ROS in the vascular remodeling during HHcy are still incompletely defined.
The contributory role of intrinsic factors, such as Ephrin B2, the arterial endothelial marker and EphB4, the venous endothelial marker are well known in remodeling process during embryonic vascular development (Aitsebaomo et al., 2008, Herbert et al., 2009, Korff et al., 2006, Pacilli and Pasquinelli, 2009, Rodriguez et al., 2007, Swift and Weinstein, 2009, Taylor et al., 2007, Obi et al., 2009). However, the role of HHcy on these intrinsic factors in the developed vessels is not understood. Ephrin B2 and EphB4 are the members of the eph-ephrin subclass of receptor tyrosine kinases and complementary expression of the Ephrin B2 ligand and the EphB4 receptor in arterial and venous endothelium mediates proper remodeling of the arterial and venous vascular network, and for a distinct boundary between the two (Swift and Weinstein, 2009). Although the role of EphB4 and Ephrin B2 are well documented in embryonic development of the blood vessels, their role in the adult vasculature are still unclear (Taylor et al., 2007). Blood flow and blood pressure are two important parameters for the determination of vascular structure and function. The relationship between increased blood pressure and HHcy has long been established (Graham et al., 1997, Nygard et al., 1995, Jacques et al., 2001, Lim and Cassano, 2002, Kahleova et al., 2002). We have shown in our previous study that there is a threshold relationship between the blood flow and elastin/collagen ratio (Basu et al., 2010). However, the relationship was reverted with respect to the MMP/TIMP ratio and the vascular wall thickness. Therefore, in this study, we tested our hypothesis that HHcy causes vascular remodeling not only by ECM deposition, but also by instigating vein phenotype in artery. We used CBS+/− mice (a well established model of moderate HHcy) for our study. Folic acid (FA) treatment was given to these mice to confirm any HHcy effect, as the FA is an essential factor in remethylation pathway.
Materials and methods
Animal model
C57BL/6J (WT) and CBS+/− mice of C57BL/6J background of ages 100–130 days were used for this study. The mice were obtained from Jackson Laboratories Inc, Maine, USA, and housed in the animal care facility of the University of Louisville. The heterozygous mutants of CBS+/− mice exhibit clinically-significant HHcy and are widely used to study HHcy pathobiology in animal model. Therefore, we used these mice to study in vivo role of elevated Hcy level. Both CBS+/− and C57BL/6J animals were divided into two groups, one group were fed standard rodent chow and water ad libitum, and the other group was fed standard rodent chow supplemented with folic acid (FA, 0.03 g / L) in water for 45 days. At the end of the experiments, animals were deeply anesthetized with freshly prepared Tribromoethanol (TBE, 240 mg/Kg body weight) and sacrificed to harvest the whole aorta. All animal procedures were in accordance with the National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee of the University of Louisville.
Antibodies and reagents
MMP-2 antibody was obtained from Novas Biologicals, Littleton, CO. MMP-9, -12 antibody was purchased from Abcam Antibodies, Cambridge, MA, and TIMP-2 and -4 was obtained from Chemicon International, Tamecula, CA. GAPDH antibody and HRP conjugated secondary antibodies were bought from Santa Cruz Biotechnology, Santa Cruz, CA.
In vivo blood pressure measurement
The animals were anesthetized by injection with TBE. A small incision was made carefully in the throat area of the animal to isolate the carotid artery without damaging the attached nerve. The portion of carotid artery proximal to the brain was tied with silk thread tightly, so that no blood could circulate towards the brain. Another silk thread was used to make a knot loosely at the part of carotid artery proximal to the heart. A small incision was made between two knots in the artery. A catheter was inserted into the artery towards the heart. Extreme care was taken to minimize damage to endothelial layer of the vessels of interest. The other end of the catheter was attached to a pressure transducer. After catheterization, heparin (100 µg/ml) (USB Corporation, Cleveland, OH) flush was done. The distal part of the catheter was then attached to the PE-10 catheter cannulation instrument (CyQ, US) and blood pressure was measured. The equipment was calibrated before using a standard blood pressure meter in mmHg.
In vivo blood flow measurement
The animal was anesthetized with TBE. A window was made in the abdominal area below the kidney, to isolate abdominal aorta. A transit time perivascular flow meter (Transonic System Inc, Ithaca, NY), equipped with transonic flow probe 0.5PSB630 was used to measure blood flow in the aorta. The equipment was calibrated using a standard flow meter in ml/min. The waveform of the blood flow was recorded using DMSI-100 software.
Ultrasound Doppler study for measurement of blood flow velocity and aortic lumen diameter
Transthoracic echocardiography was performed on mice to achieve aortic blood flow from an apical view using a SONOS 1500 or 2500 Hewlett-Packard, Inc, and a 12.5 MHz transducer. The procedure was performed using TBE anesthesia to minimize cardio depressing actions produced by other anesthetics. Anesthetized mice were shaved with hair removal cream (Nair), and placed on a regulated heating pad to maintain the body temperature. The blood flow velocity was recorded as cm/sec, and the aortic lumen diameter was recorded in micron scale.
Histological tissue sectioning and staining
The aorta was isolated from each group of animal. Extreme care was taken at the time of isolation to minimize tissue damage. The tissue was cleaned with phosphate-buffered saline (PBS) containing 20 unit/ml heparin to get rid of any unwanted blood and viscera. The tissue was then cut into approximately 2 mm pieces, placed vertically in tissue freezing media (Triangle Biomedical Sciences, Inc., Durham, NC)] and was frozen in liquid nitrogen. Frozen blocks with the molds were placed in a −70°C freezer. After one hour, a series of 7µm sections were cut and put on glass slides. Before staining, the slides were allowed to come to RT. Frozen tissue sections were stained with one of the following staining kits according to supplied protocol: Masson Trichrome, van Gieson’s or Hematoxylin and Eosin (Richard Allen Scientific, Kalamazoo, MI).
Measurement of elastin and collagen intensity and blood vessel wall width
Cryosections of blood vessels were stained with Masson Trichrome, van Gieson or Hematoxylin Eosin. All pictures were taken under light microscope attached with a digital camera. In order to measure intensity of collagen and elastin staining of blood vessels, densitometric data were collected by using Un-ScanIt software on scanned staining pictures. From densitometric data, elastin: collagen ratio was calculated. Blood vessel widths were also measured of hematoxylin eosin-stained vessels.
Western blotting technique
Samples of isolated aorta were taken from anesthetized mice. RIPA buffer (Thermo Scientific Inc., Rockford, IL), supplemented with protease inhibitor and PMSF was used for protein extraction. Protein was estimated by BCA assay, and 30 µg of total protein was loaded in each well of 10% SDS-PAGE gels for detecting protein of interests. After overnight transfer, membranes were blocked in 5% milk and incubated overnight with primary antibody followed by secondary antibody for 2 hrs. An ECL plus Western blotting detection system (GE Health Care, Little Chalfont, Buckinghamshire) was used to detect bands. The membranes were stripped with membrane-stripping buffer (Boston BioProducts, Worcester, MA) and reprobed with GAPDH antibody (Chemicon International) for loading control. Intensity of bands was detected by Gel –Doc software, and was normalized with their corresponding GAPDH control.
Concentration response and Passive stretch-tension relationship study
The aorta was isolated from the animal to measure passive stretch–tension relationship and placed in ice cold physiological salt solution (PSS, 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2 PO4, 1.2 mM MgSO4, 12.5 mM NaHCO3 and 11.1 mM glucose, pH 7.4). The solution was continuously aerated with 20% O2, 5% CO2, and 75% N2. Fat tissues and connective tissues were removed, and the vessels were cut into 2.5–3 mm pieces. The vessels rings were mounted with two tungsten wires of same diameter attached to the myobath and were placed in a 25 ml organ bath filled with PSS at 37°C. The PSS in the myobath was constantly aerated with O2:CO2, 95%:5%, respectively. Rings were stretched gradually to obtain optimal resting tensions, which were 1 g for the aorta and were equilibrated for an hour. After equilibration, 25 µl phenylephrine (Phe) of 10−6 to 10−2 M was added in the organ bath to make a final concentration of 10−9 to 10−5 M, respectively. Acetylcholine (Ach) was added to the organ bath in similar manner as described for Phe. Then the tissues were treated with sodium nitropruside (SNP) after a brief treatment of Phe (10−5 M). The tissue responses were recorded graphically using mp100 software for 10 minutes of each for each drug concentration. The passive-stretch tension relationship was obtained by increasing stretch 1mm at each time. The resulting increase in tension (g) was recorded by using same software as described above. Tissue responses by Phe concentrations and passive stretch-tension responses were normalized by respective tissue weight, and the Ach and SNP responses was graphed in a % scale, assuming the basal tissue condition as 100%.
Dihydroethidium staining for determination of ROS
The oxidative fluorescent dye, dihydroethidium (DHE; Invitrogen, Carlsbad, CA), was used in frozen, 7µm aorta sections (40µmol, 30 mins), and the fluorescence intensity was measured by a laser scanning confocal microscopy (Fluo View 1000, Olympus). The method was adopted from Dayal et al (Dayal et al., 2006), and previously used by our group (Sen et al., 2009). Negative control sections were incubated for 25 min with 250 U/ml polyethylene glycol-superoxide dismutase (PEG-SOD; Sigma-Aldrich), before incubation with DHE. Fluorescent images were analyzed with ImagePro software (Media Cybernetics, Bethesda, MD).
In situ Zymography for determination of MMP activity
Seven µm frozen sections were used for this purpose. Tissue sections were incubated with 40 µmol DQ gelatin (Invitrogen) for 2 hours and fluorescence were measured by laser scanning confocal microscopy without washing. Fluorescent images were analyzed with ImagePro software.
RNA isolation and reverse transcriptase polymerase chain reaction
Total RNA was isolated from the tissues by using Trizol reagent (Invitrogen) according to the protocol provided with reagent. Complementary total DNA was made by incubation of RNA with oligo dT at 70°C for 6.00 min. The RT cycle was 25°C for 2.00 min, 42°C for 50.00 min, 75°C for 5.00 min, 4°C forever as described previously (Mishra et al., 2009). All the primers were bought from Invitrogen. The primer sequences were:
| EphrinB2 | Forward, 5′TCCAGGAGGGACTCTGTGTGGAAG3′ |
| Reverse,5′CGGGGTATTCTCCTTCTTAATTGT3′ | |
| EphB4 | Forward, 5′CCCAAATAGGAGACGAGTCC3′ |
| Reverse, 5′CGGGGTATTCTCCTTCTTAATTGT3′ | |
| TIMP-3 | Forward, 5′GCCCTCCCATATGTATACCC3′ |
| Reverse, 5′TAGGCCTCACCTCAAGTCTG3′ | |
| TIMP-1 | Forward, 5′TCATCGGGCCCCAAGGGATCT3′ |
| Reverse, 5′GCAGTGAAGAGTTTCTCATC3′ | |
| MMP-13 | Forward, 5′AGGCCTTCAGAAAAGCCTTC3′ |
| Reverse, 5′CCCACCATAGTTTGGTCCAG3′ | |
| GAPDH | Forward, 5′ATGGGAAGCTGGTCATCAAC3′ |
| reverse,5′TGTGAGGGAGATGCTCAGTG3′ |
Statistical analysis
Each experiment was carried out using 4–6 specimens in each group. Data were analyzed by ANOVA followed by Scheffe’s post-hoc test. Comparison between groups was made according to Student’s independent “t’ test. Significance between groups was accepted at p < 0.05. Values are means ± standard deviation (SD) of measurement.
Results
Blood pressure was increased in CBS+/− mice and was mitigated by folic acid treatment
Mean arterial blood pressure (MAP) as measured from the carotid artery was 104 ± 8 mmHg in wild type (WT) mice (Figure 1). MAP in WT mice treated with folic acid (FA) did not change (Figure 1, 103 ± 10 mmHg). Average MAP in CBS+/− mice was 119 ± 12 mmHg, and it was significant increase compared to WT. Interestingly, CBS+/− mice treated with FA showed normalized blood pressure (Figure 1). There was no difference in heart rate among the groups (data not shown).
Figure 1. Mean blood pressure.
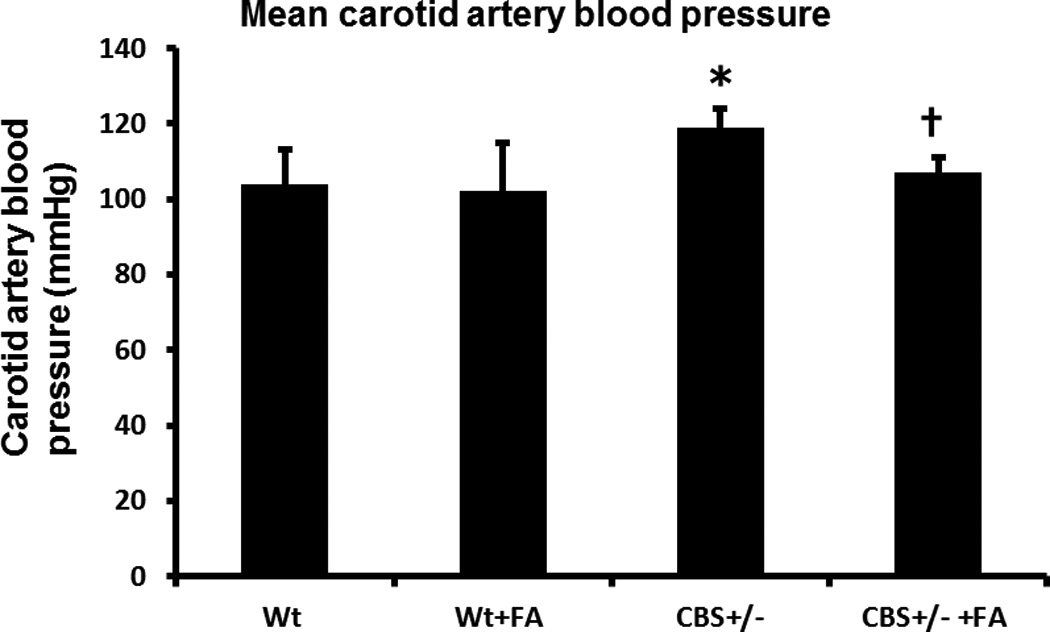
Bar graph showed mean blood pressure in different study groups. The mean carotid blood pressure in CBS+/− mice significantly increased compared to WT, and was mitigated by FA (folic acid) treatment. Data represents mean ± SD, n = 5. The * represents p< 0.01 versus WT, and † represent p<0 .01 versus CBS+/− mice.
Decreased blood flow, blood flow velocity and arterial lumen diameter in CBS+/− mice were mitigated by folic acid treatment
The blood flow (ml/min) in WT and WT + FA aorta was similar, which was 1.38 ± 0.07 ml/min in WT vs 1.44 ± 0.07 ml/min in WT + FA (Figures 2A & B). The CBS+/− mice showed significant decrease in blood flow (0.80 ± 0.2 ml/min), whereas CBS +/− mice treated with FA ameliorated blood flow towards normal (0.99 ± 0.14 ml/min) (Figures 2A & B).
Figure 2. Arterial blood flow.
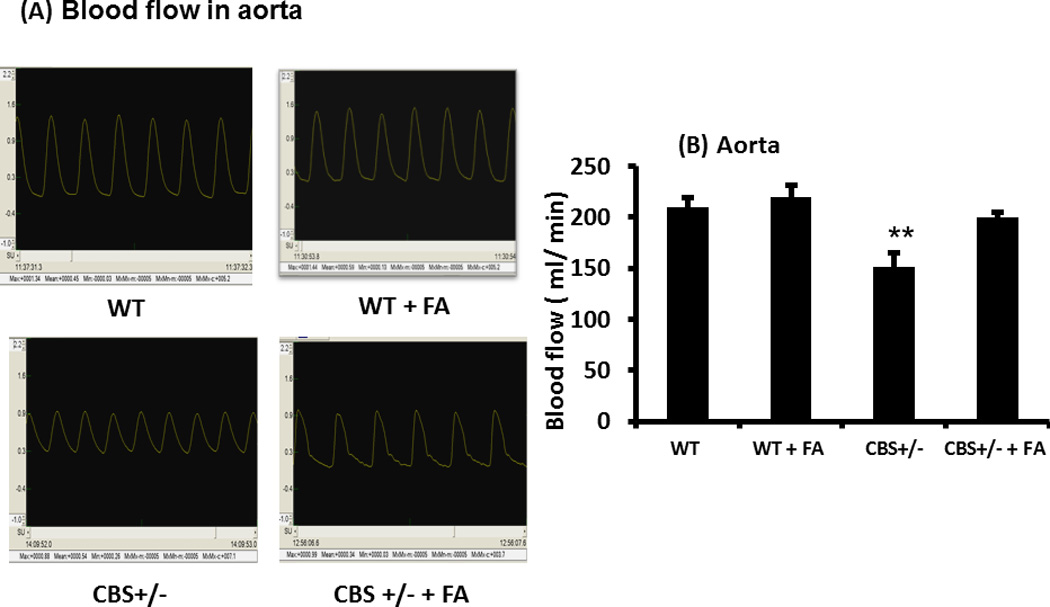
(A) Typical arterial blood flow waves from different experimental groups as measured in the Transonic perivascular flow meter. (B) Graphical representation of the blood flow in aorta of the same study groups. Blood flow in the aorta was significantly lower in the CBS+/− mice compared to WT, and this was ameliorated by FA treatment. Data represents mean ± SD, n = 6. The ** represent p<0.01 versus WT.
The aortic blood flow velocity in WT mice and WT mice treated with FA did not change significantly (Figures 3A & B). In CBS+/− mice the aortic blood flow velocity was significantly decreased compared to WT (Figure 3B). Folic acid treatment in the CBS+/− group normalized the blood flow velocity (Figure 3B). The aortic lumen diameter was significantly decreased in CBS+/− mice compared to WT, which was significantly increased after FA treatment (Figure 3C). Contrary to this there was no significant difference of aortic lumen diameter between WT and WT mice treated with FA (Figure 3C).
Figure 3. Aortic blood flow velocity and lumen diameter.
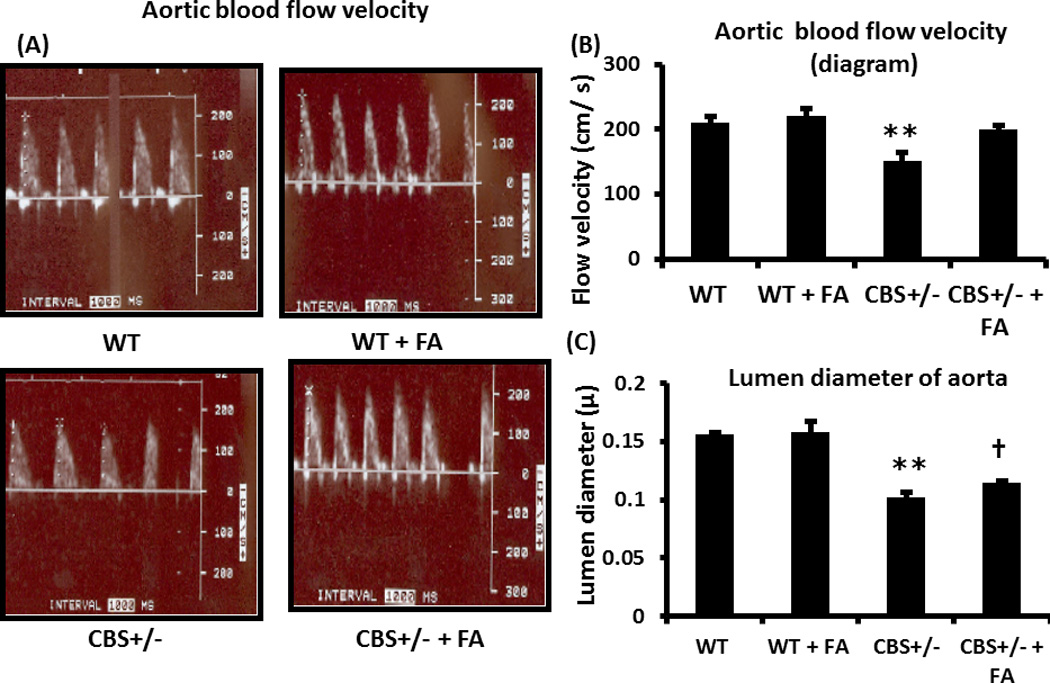
(A) Typical arterial blood velocity peaks are shown from different experimental groups as measured in the ultrasound Doppler. (B) Graphical representation of the blood velocity in different study groups. Blood flow velocity showed significant decrease in the CBS+/− mice compared to WT, and ameliorated with FA. (C). Lumen diameter showed significant decrease in the CBS+/− mice, compared to WT, and lumen diameter was increased with FA treatment. Data represents mean ± SD, n = 6. The ** represents p< 0.01 versus WT, and † represents p<0.01 versus CBS+/− mice.
Increased aortic medial thickness in CBS+/− mice was mitigated by the folic acid treatment
The aortic medial thickness was 1.5 µm×10−2 ± 0.2 in WT mice. This thickness was significantly increased (3 µm×10−2 ± 0.5) in CBS+/− mice. Folic acid treatment almost normalized medial thickness in CBS+/− mice (Figure 4).
Figure 4. H & E staining and medial thickness.
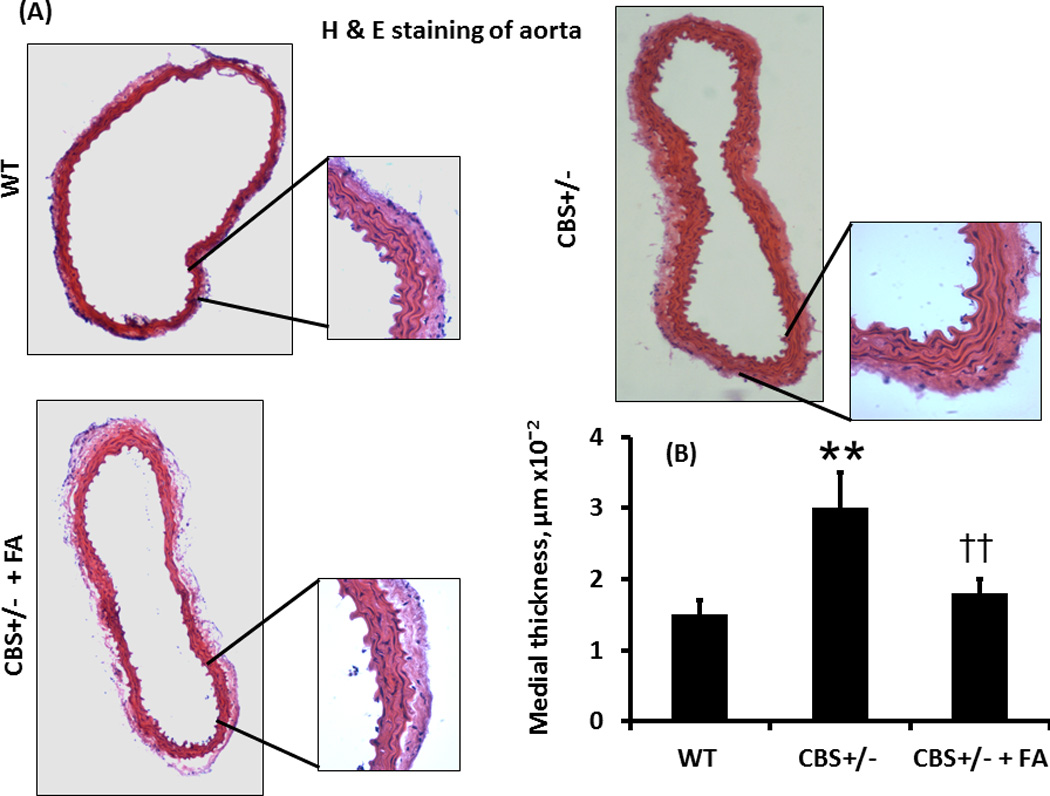
(A) H & E staining of the aorta showed gross morphological difference of the tissue in different study groups. The whole picture of the aortas was taken in 4×1.6X; magnification and the inset pictures were taken in 20×1.6X magnification. (B) Bar graphs showed significant increase in the medial area in CBS +/− mice compared to WT, and this was mitigated by FA treatment. Data represent mean ± SD, n = 5; and ** indicates p< 0.01 versus WT, and †† represents p<0.01 versus CBS+/− mice.
Folic acid mitigated increased collagen/elastin ratio in CBS+/− aorta
The elastin content in WT aorta was 153±5 AU (Arbitrary Unit) (Figures 5A & B). In CBS+/− mice without or with FA treatment elastin content was 150±4 AU and 151±5 AU, respectively. There were virtually no changes in elastin content between the groups (Figures 5A & B). The collagen content in the WT aorta, CBS+/− aorta and in the FA treated CBS+/− aorta was 109.9±5 AU, 141.05±11 AU and 134.9±8 AU, respectively (Figures 6A & B). Thus, CBS+/− aorta showed a significant increase in the collagen content. The FA treatment however did not mitigate the collagen content in CBS+/− aorta (Figures 6A & B).
Figure 5. van Gieson staining and elastin content.
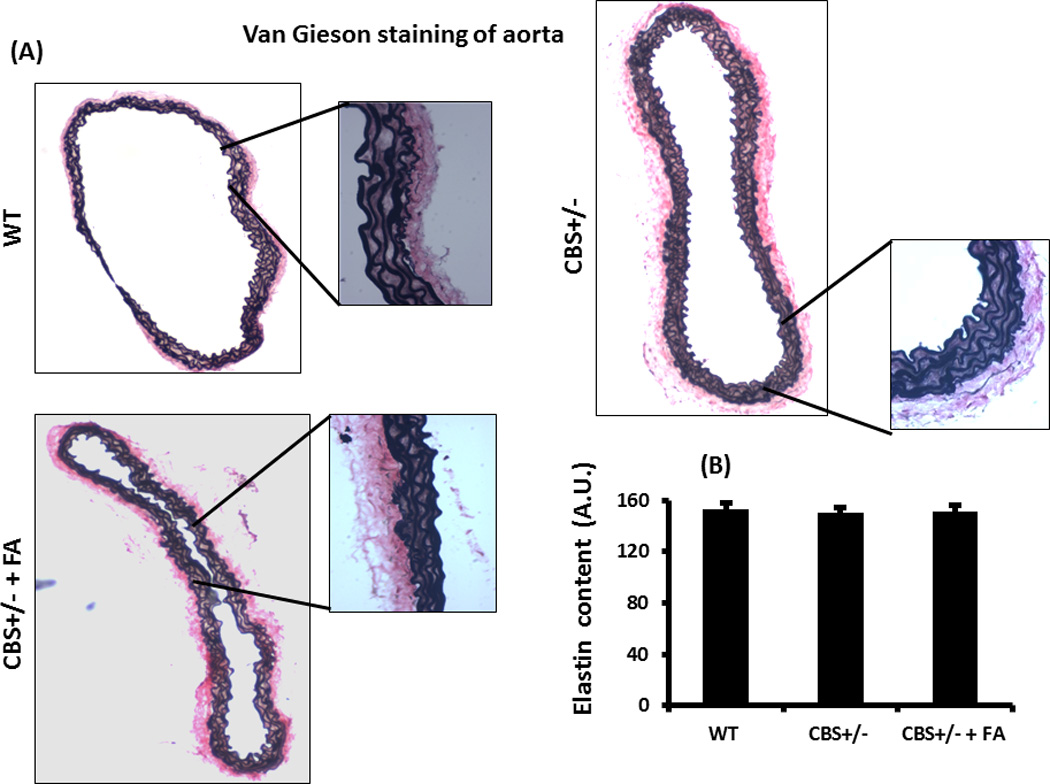
(A) The van Gieson staining of aorta showing elastin content of the tissue in different study groups. The whole picture of the cross sectional aorta was taken in 4×1.6X magnification and the inset pictures were taken in 20×1.6X magnification. The elastin content of CBS+/− aorta showed no change. (B) The bar graph representing elastin content (Arbitrary Unit, A.U.), and no difference was observed among the groups. Data represent mean ± SD, n = 5.
Figure 6. Masson Trichrome staining and collagen content.
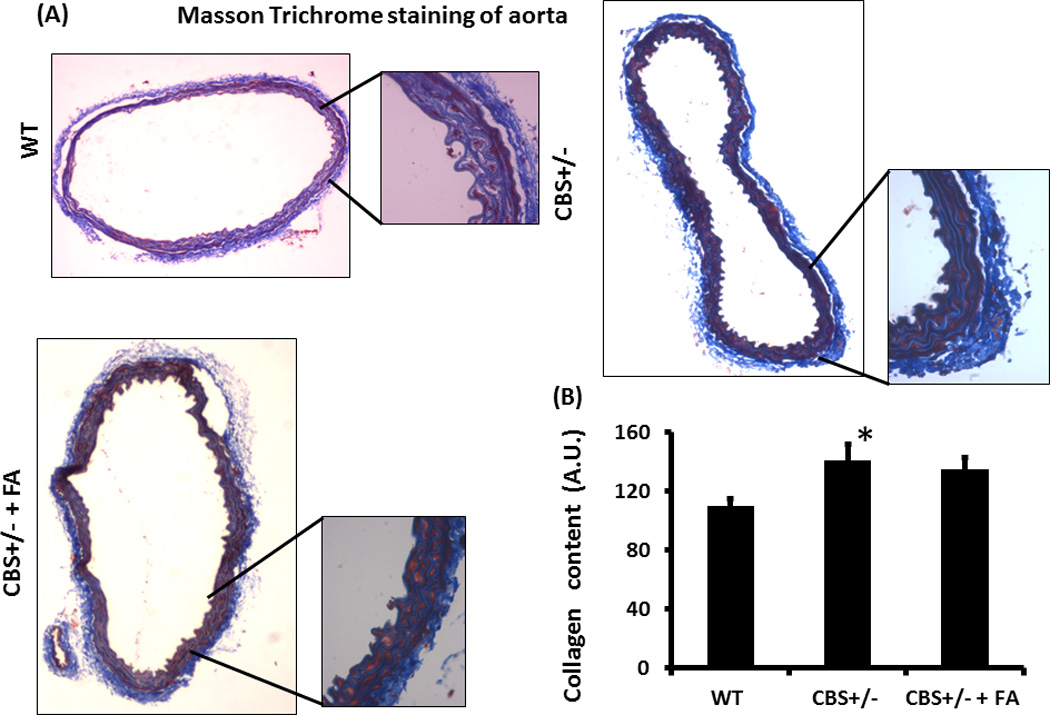
(A) The Masson Trichrome staining of the aorta showing collagen in blue color. The whole cross sectional picture of the aorta was taken in 4×1.6X magnification and the inset pictures were taken in 20×1.6X magnification. (B) The bar diagram showing graphical presentation of the collagen content. There was a significant increase of collagen deposition in the CBS+/− aorta. Data represent mean ± SD, n = 5; *represent p<0.05 versus WT.
Interestingly, although there was not much difference of elastin content in the aorta among the groups, the collagen/elastin ratio was significantly higher in the CBS+/− mice (0.95±0.1 AU) compared to WT (0.81±2.2 AU) (Figure 7A). FA treatment normalized this ratio in CBS+/− aorta (0.81±0.2 AU) (Figure 7A). No difference was observed between WT and WT with FA (data not shown).
Figure 7. Collagen/elastin ratio in aorta.
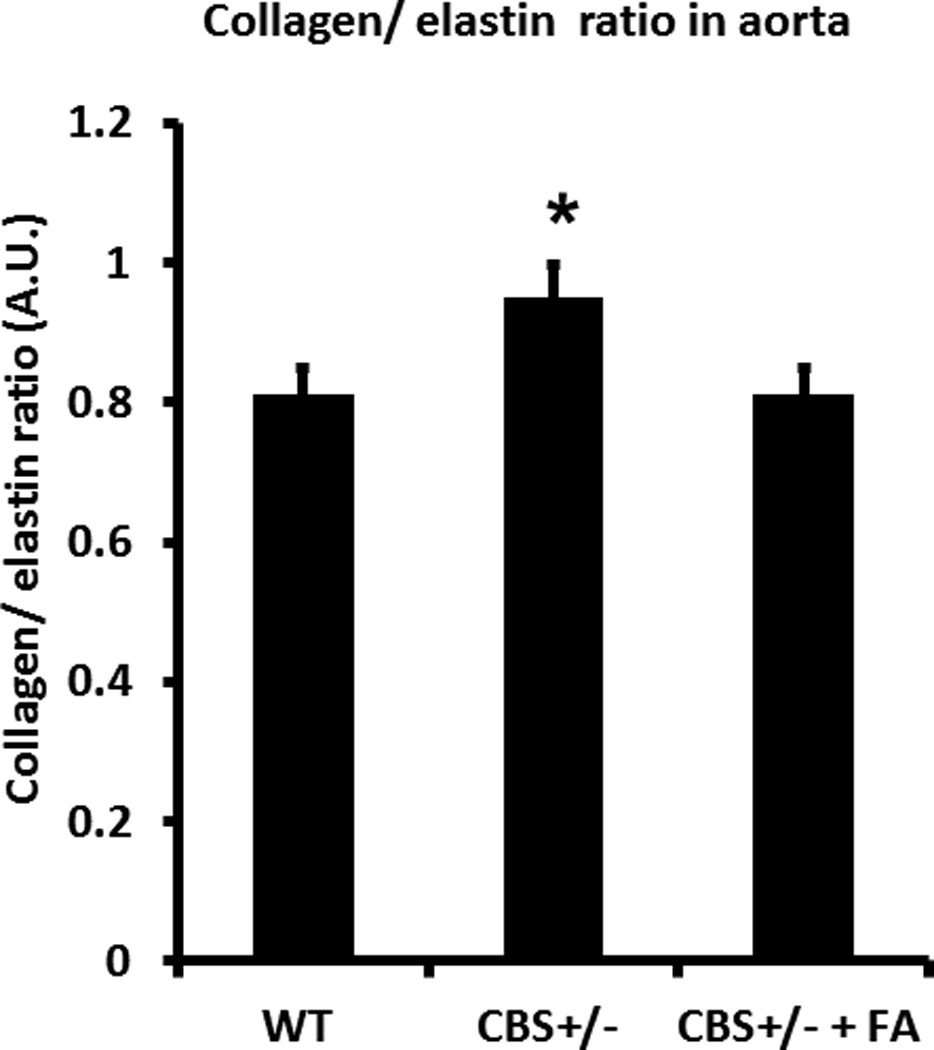
Bar graph showing collagen/elastin ratio, and was significantly high in CBS+/− aorta compared to WT. Data represent mean ± SD, n = 5; and * indicates p<0.05 versus WT.
Increased super oxide in the CBS+/− aorta was mitigated by FA treatment
The level of ROS, particularly super oxide in CBS+/− aorta showed significant increase compared to WT and was normalized by FA treatment (Figures 8A & B). In WT aorta, fluorescence intensity of DHE was 0.98±0.2 AU, in CBS+/− mice it was 2.33±0.4 AU, and in CBS+/− + FA mice it was 0.96±0.1 AU.
Figure 8. Dihydroethidium (DHE) staining of aorta to detect super oxide.
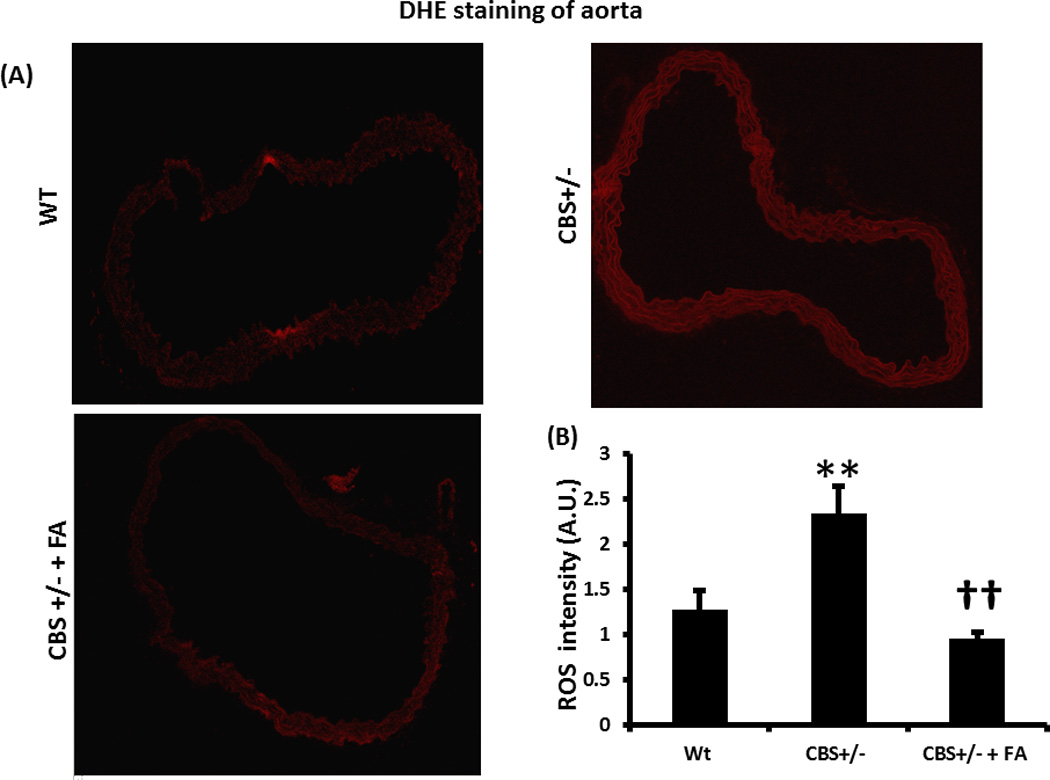
(A) CBS+/− aorta showing higher intensity of red fluorescence, indicating more super oxide, compared to WT. (B) The bar graph showing densitometric analysis of red fluorescence. Data represent mean ± SD, n = 5; and ** indicates p<0.01 versus WT, and †† represents p<0.01 versus CBS+/−.
Differential expression of MMP/TIMP regulated vascular remodeling in CBS+/− aorta
The expressions of MMP-2, -9 and -12 in the CBS +/− aorta were significantly increased compared to WT, and these increases were mitigated by FA treatment (Figure 9). Similarly, TIMP-2 expression significantly increased in CBS+/− aorta compared to the WT, and this increase expression was mitigated by FA treatment (Figure 9). Unlike TIMP-2, TIMP-4 was significantly decreased in the CBS+/− aorta compared to WT, and this FA was not able to normalize this change (Figure 9).
Figure 9. Western blot analysis of MMPs/TIMPs in the aorta.
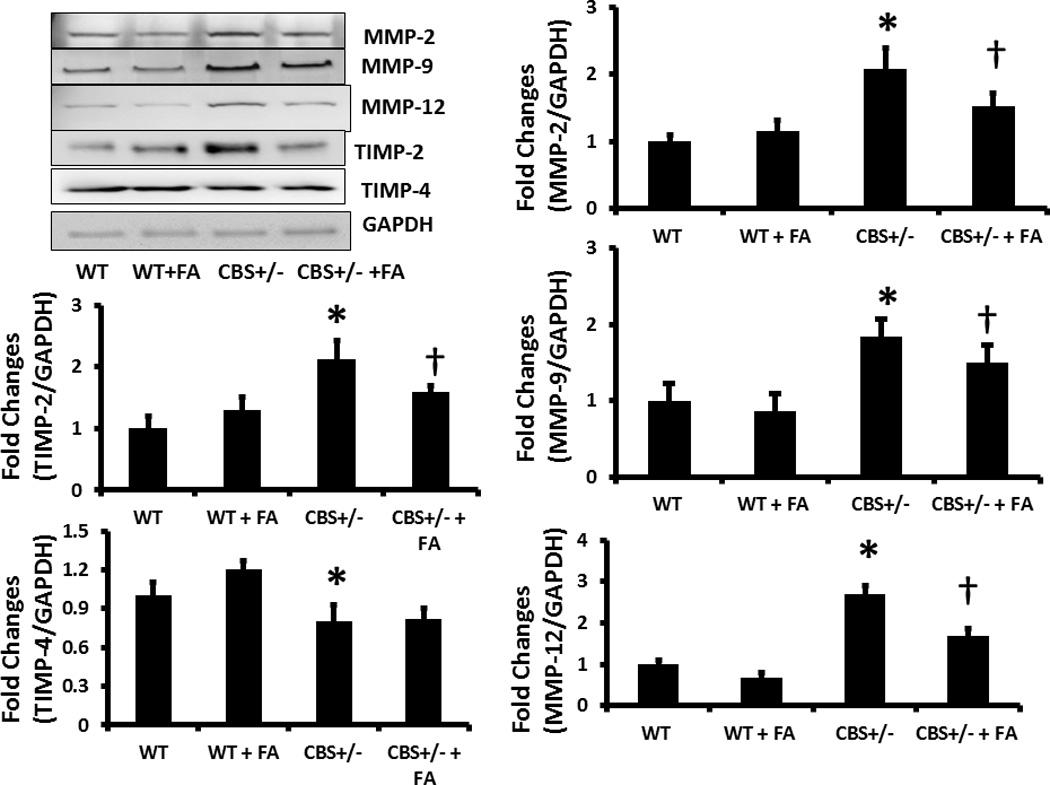
(A) Protein was extracted from aorta and equal amount of protein was resolved by SDS-PAGE gel electrophoresis. Protein was transferred to PVDF membrane, and different MMPs and TIMPs, as indicated in the figure, were detected using specific antibodies. Membrane reprobed with GAPDH for loading control. (B) Bar graphs showing densitometric analyses of protein expression and represented as fold changes over control (WT). Data represents mean ± SD, n = 5. The * represent p<0.05 versus WT and † represent P<0.05 versus CBS+/−.
In CBS +/− mice, increased vascular MMP activity was mitigated by FA treatment
The MMP activity level as measured in in situ Zymography, showed significant increase in the CBS +/− aorta compared to WT (Figure 10). This was attenuated by FA treatment (Figure 10).
Figure 10. In situ zymography.
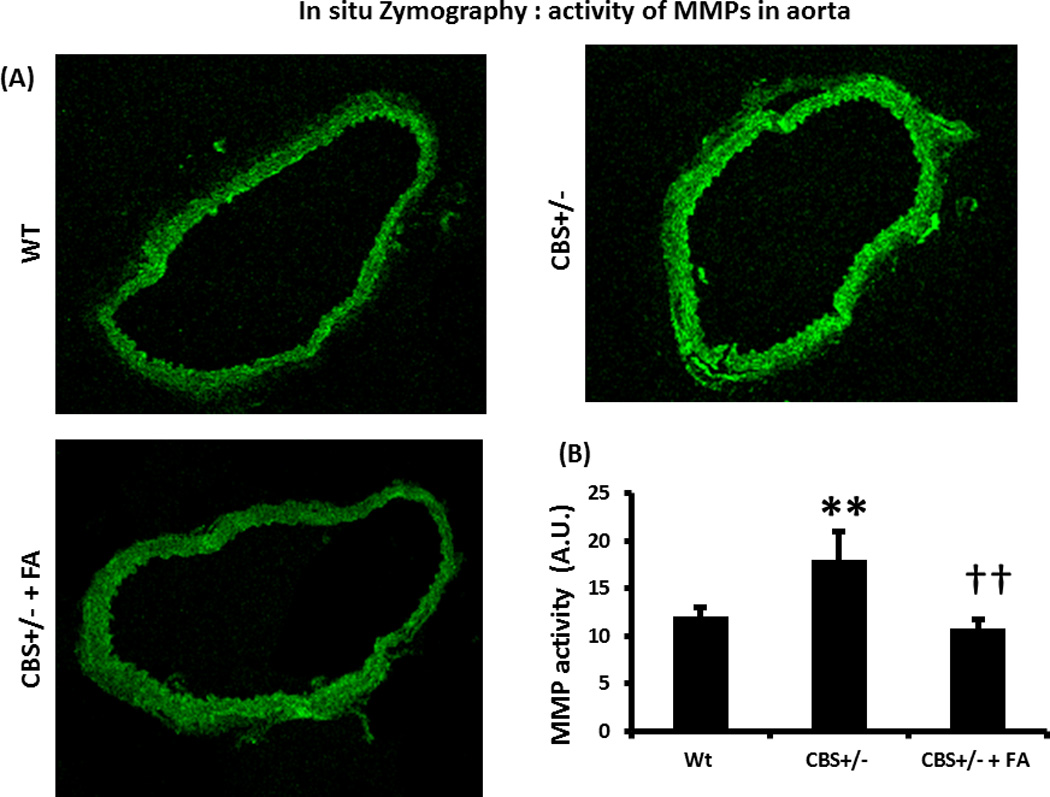
(A) In situ zymography of aorta showing increase MMP activity in the CBS+/− aorta, which was mitigated by FA treatment. Green fluorescence indicates active MMP. (B) Graphical presentation of MMP activity. Data represent mean ± SD, n = 4; ** indicates p<0.01 versus WT, and †† represents p<0.01 versus CBS+/−.
Expression of MMP/TIMP mRNA and vascular markers in CBS+/−
TIMP-3 and MMP-13 RNA expression showed significant increase in the CBS+/− aorta compared to WT (Figure 11), whereas TIMP-1 expression did not change significantly (Figure 11). Treatment with FA mitigated TIMP-3 and MMP-13 expressions in CBS+/− aorta (Figures 11).
Figure 11. Expressions of MMP, TIMP, Ephrin B2 and EphB4 mRNA in aorta.
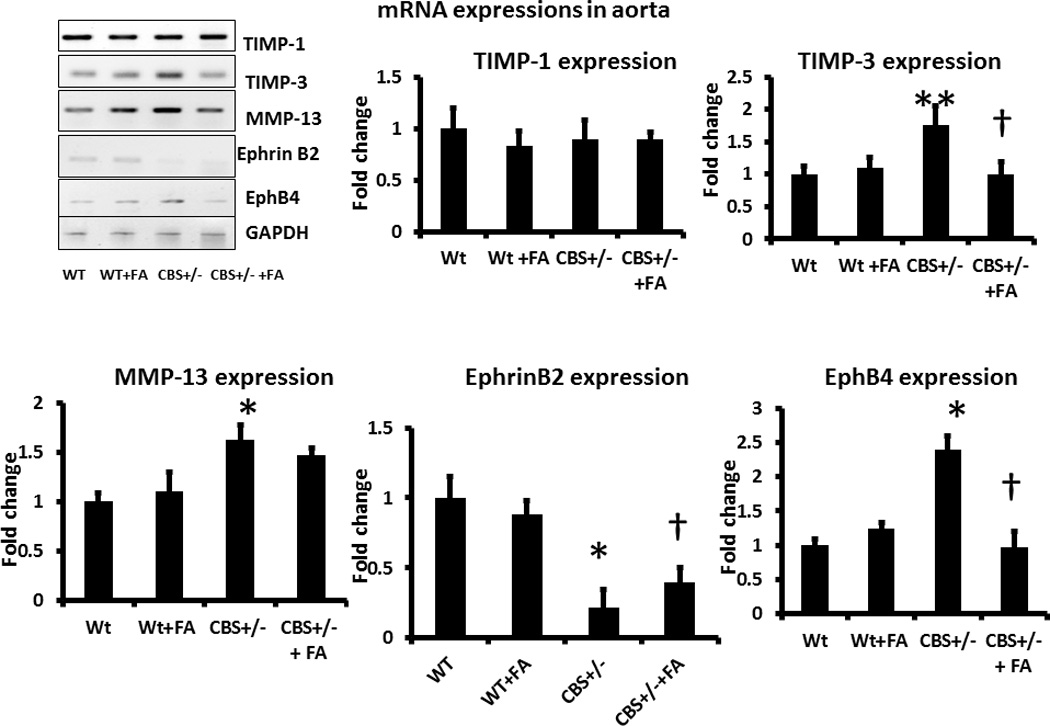
Total RNA was isolated and expression of specific mRNA was detected as described in the Methods. Bar diagram indicating densitometric analyses of expressed mRNA over control (WT). Data represents mean ± SD, n = 6. The *p<0.01 and ** p<0.05 versus WT, and † represent p<0.05 versus CBS+/−.
The expression of aorta marker EphrinB2 in the aorta was significantly repressed in CBS+/− aorta, whereas the expression of vena cava marker EphB4 showed significant increase (Figure 11). FA treatment normalized the expressions of EphrinB2 and EphB4 in CBS+/− aorta (Figure 11).
Endothelial dependent attenuated relaxation of CBS+/− aorta was ameliorated by FA treatment
The concentration response of Phe (phenylephrine) showed similar response in aorta among the groups as shown in the figures 12A. However, the CBS+/− aorta showed a slightly increased, although not significant, at a Phe contraction of 10−5 mol compared to WT (Figure 12A).The concentration response of Ach (acetylcholine) showed significant decreased relaxation in CBS+/− aorta compared to WT. This decreased relaxation was ameliorated by FA treatment (Figures 12B). Tissue relaxation in response to SNP did not show significant differences among the groups (Figure 12C). Passive stretch-tension relationship of aorta was impaired in CBS+/− animals compared to WT, and FA supplement did not change this relationship in CBS+/− animals (Figure 12D).
Figure 12. Vascular reactivity and passive stretch-tension in aorta.
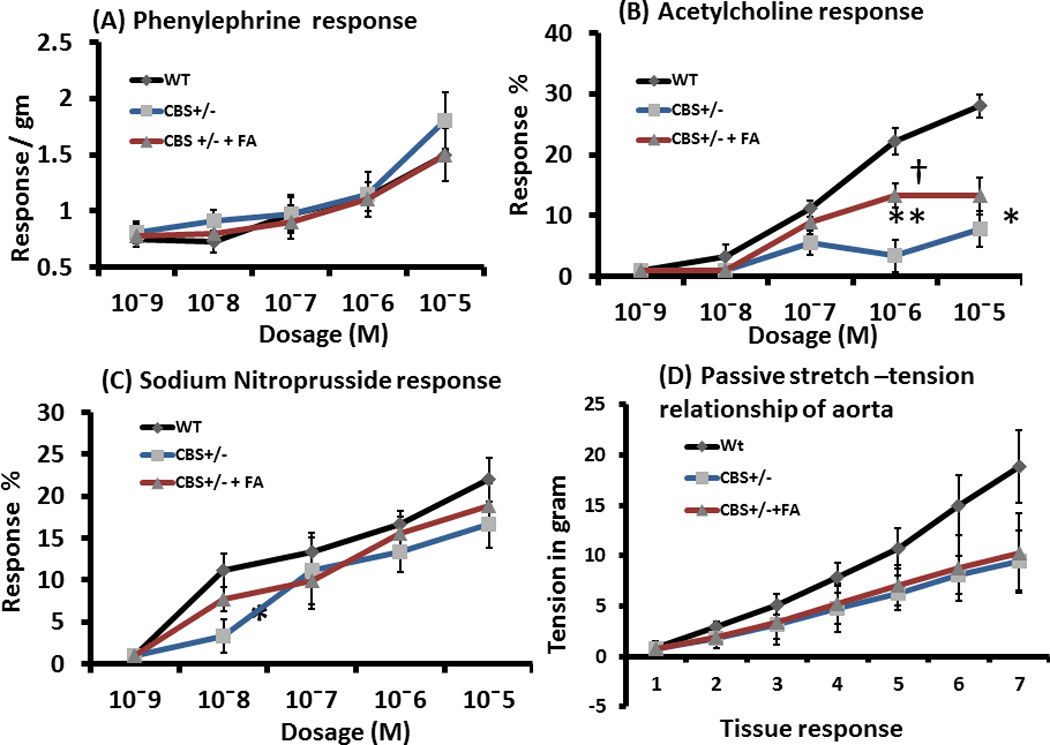
(A) Concentration dependent contraction of aortic rings by phenylephrine (Phe). (B) The rings were pre-contracted by 10−5 M Phe, and then gradient dosages of acetylcholine (Ach) applied to the myobath to relax the vessel in endothelial-dependent manner. (C) Pre-contracted (by 10−5 M Phe) aortic rings were allowed to relax endothelial-independent way by gradient concentrations of sodium nitropruside (SNP). : (D) The passive stretch-tension relationship applied to the rings with gentle increase of tension manually in the myobath and recorded. All data represents mean ± SD, n = 4–5. The ** and* represent p<0.01 and p<0.05, respectively versus same concentration in WT, and † represent p<0.05 versus CBS+/−.
Discussion
Hyperhomocysteinemia (HHcy), an elevated homocysteine (Hcy) level, is a known condition that causes vascular remodeling (Giusti et al., 2004), which includes hypertrophy (Rodionov et al., 2010), collagen addition (Sen et al., 2010), matrix remodeling (Steed et al., 2010), elevated reactive oxygen species (ROS) (Bao et al., 2010) and potential endothelial dysfunction (Bhalodia et al., 2011). Many of these findings have previously been reported in murine model of HHcy and summarized in the review article by Dayal et al (Dayal and Lentz, 2008). Therefore, the purpose of our study was not to confirm these vascular changes during HHcy, but was to determine any switch of venous characteristics in HHcy mediated aortic remodeling. To our knowledge vascular remodeling and phenotypic change in the artery was not studied before under HHcy condition. Our study is therefore novel in the sense that this is the first attempt to address a phenotypic shift of aorta to vein during HHcy.
Artery by its phenotype has relatively more elastic tissue and therefore compliance with vascular elasticity than vein. With the decreasing elasticity by either smooth muscle cell proliferation and/or apoptosis, matrix remodeling by increased elastin degradation and collagen deposition, artery may adopt physiochemical properties of vein. In this study, we have demonstrated increased ratio of collagen/elastin and TMIP-3 during hyperhomocysteinemia (HHcy). TIMP-3 is known to induce cell apoptosis (Bond et al., 2002) and thus increased expression and activity contributes to vascular rigidity during pathological condition. Here, we report that in addition to remodeling, which is related to MMP/TIMP imbalance, increased matrix accumulation and apoptotic TIMP-3 expression, aorta also exhibited increased expression of vein marker, EphB4 in HHcy. Folic acid (FA) supplementation normalized this phenotypic shift of aorta in HHcy, suggesting that Hcy is playing a role in this process.
All the measurements we have performed in the aorta support active vascular remodeling in HHcy mice. Active remodeling was observed as an increase in the medial thickness, increased collagen/elastin ratio, increased ROS production as evident by the DHE staining, decreased lumen diameter and by the altered expression of MMPs and TIMPs than normal (Bobik and Tkachuk, 2003). Increased MMP-2 level in the HHcy mice aorta suggests the vascular remodeling, and the increased TIMP-2 expression that was observed in the HHcy mice aorta is presumably an adaptive increase to mitigate the higher level of MMP-2 found in CBS+/− mice. MMP-9 expression was also increased in the HHcy mice aorta. A similar finding was observed by Watts et al (Watts et al., 2007), where an increase in MMP-9 level suggests remodeling in the DOCA-salt sensitive hypertensive rats. Higher MMP-12 activity in HHcy mice aorta suggests higher elastinase activity, although, the elastin content of the HHcy mice did not show any difference than that of WT mice. It is possible that other factors, such as different elastinases or TIMPs might have played a role in normalizing elastin content in HHcy mice. Additionally, MMP-13 RNA, as determined from the PCR study showed an increase in expression, which is presumably an adaptive increase to mitigate the higher level of collagen content in the HHcy mice. Not only had that, in situ zymography showed an increased expression of activated MMPs in the HHcy mice, which is also in accordance to the vascular remodeling observed in the HHcy mice aorta tissue.
Increased production of ROS is known to cause endothelial injury and inflammation leading to organ damage (Doughan et al., 2008, Mariappan et al., 2010). In the present study, ROS production in the aorta was increased many folds in the HHcy mice, and was ameliorated by folic acid (FA) treatment, suggesting ROS may play a role in Hcy mediated vascular remodeling in the aorta tissue. Moreover, the increased collagen content and increased collagen / elastin ratio in HHcy mice suggest a stiffer and less compliant vessel wall, characteristic of a remodeled vessel. The decreased lumen diameter and increased medial thickness also imply on the vascular remodeling. In addition, elevated blood pressure, as determined in our study in HHcy animal group, may also be an underlying cause for the remodeling process. Similarly, elevation of MMP-9 expression also supports these observations, and in accordance with the previous report (Rodriguez et al., 2007). Our findings also suggest endothelial dependent attenuated vasorelaxation in HHcy mice group, which is in accordance with the previous investigations (Stanger and Weger, 2003). The elevated level of ROS in the HHcy mice group may partly explains that the impaired vasorelaxation not only due to endothelial injury but also due to deactivation of NO by ROS (Stanger and Weger, 2003). Although we did not measure inflammatory markers and / or molecules in this study, our previous studies demonstrated that inflammatory molecules play a significant role in the process to vascular remodeling in HHcy (Sen et al., 2009, Sen et al., 2010, Sen et al., 2011). We also previously reported that HHcy induces TGF-β1 induction (Sen et al., 2006) and is involved in cytoskeleton remodeling in cyclic stretch (Sen et al., 2007). Whether a similar mechanism also contributes to the present remodeling process needs to be thoroughly investigated. Furthermore, in stretch tension relationship the wild type aorta showed highest response. This was presumably due to its low content of collagen compared to HHcy mice.
The aortic endothelial marker EphrinB2 and the vena cava endothelial marker EphB4 expressions were changed in HHcy condition. EphrinB2 expression was decreased and the EphB4 expression was increased in the aorta in HHcy condition indicating the phenotypic change in the tissue and FA mitigated this phenotypic shift. In previous studies, change in EphB4 and Ephrin B2 were reported in endothelial progenitor cells (EPC), with change in sheer stress (Obi et al., 2009), which is somewhat related to our findings, as the high blood pressure in the HHcy animals also causes high sheer stress. To our knowledge, not many studies were done in adult animals. Our study is novel in studying the expression of these two markers in the adult HHcy animals. Although we hypothesized that the HHcy condition causes vascular remodeling by instigating vein phenotype in aorta, we cannot conclude that only HHcy condition itself is instigating these changes in EphB4 and EphrinB2 expression in the aorta. Rather, vascular remodeling caused by the HHcy condition, such as increase in collagen/elastin ratio, smooth muscle proliferation and cell apoptosis may also be instigating changes in the expression in these two markers and contributing to phenotypic shift. Detailed studies on the upstream and downstream pathways can only explain the mechanism(s) of this change.
Two observations are of enormously important in our study. First, FA corrected most of the outcomes, and second, FA lowered the blood pressure to normal levels. One might argue that the out comes, a purported venous phenotype, cannot be attributed to HHcy when blood pressure is the more likely culprit. Using similar set up of experiments for coronary vasospasm study, we previously reported that FA lowered Hcy level in CBS+/− animals (21 µmole/L in CBS+/− vs 12 µmole/L in CBS+/− + FA) (Qipshidze et al., 2010). Others have also shown that in patients of HHcy FA reduced Hcy level (Toole et al., 2004, Armitage et al., 2010, Ebbing et al., 2008). Since, it is known phenomena of FA, in this particular study we did not measured homocysteine level. By FA administration to CBS+/− animals we also observed normalized blood pressure. It is possible that by lowering Hcy, FA mitigated hypertension. Measurement of Hcy level, and use of blood pressure lowering agent that does not lower homocysteine level would have further reinforced our hypothesis, and need to be addressed in future.
Acknowledgements
This study was supported, in part, by NIH grants HL-71010 and HL-88012 (to SCT) and HL-104103 (to US).
Footnotes
A part of this work was presented at High Blood Pressure Research 2010 Scientific Sessions, Washington DC.
References
- Aitsebaomo J, Portbury AL, Schisler JC, Patterson C. Brothers and sisters: molecular insights into arterial-venous heterogeneity. Circ Res. 2008;103:929–939. doi: 10.1161/CIRCRESAHA.108.184937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage JM, Bowman L, Clarke RJ, Wallendszus K, Bulbulia R, Rahimi K, Haynes R, Parish S, Sleight P, Peto R, Collins R. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA. 2010;303:2486–2494. doi: 10.1001/jama.2010.840. [DOI] [PubMed] [Google Scholar]
- Bao XM, Wu CF, Lu GP. Atorvastatin inhibits homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4 and p38MAPK. Atherosclerosis. 2010;210:114–121. doi: 10.1016/j.atherosclerosis.2009.11.032. [DOI] [PubMed] [Google Scholar]
- Basu P, Sen U, Tyagi N, Tyagi SC. Blood flow interplays with elastin: collagen and MMP: TIMP ratios to maintain healthy vascular structure and function. Vasc Health Risk Manag. 2010;6:215–228. doi: 10.2147/vhrm.s9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudeux JL, Giral P, Bruckert E, Foglietti MJ, Chapman MJ. Matrix metalloproteinases, inflammation and atherosclerosis: therapeutic perspectives. Clin Chem Lab Med. 2004;42:121–131. doi: 10.1515/CCLM.2004.024. [DOI] [PubMed] [Google Scholar]
- Bhalodia YS, Sheth NR, Vaghasiya JD, Jivani NP. Homocysteine-dependent endothelial dysfunction induced by renal ischemia/reperfusion injury. J Nephrol. 2011 doi: 10.5301/JN.2011.6245. [DOI] [PubMed] [Google Scholar]
- Bobik A, Tkachuk V. Metalloproteinases and plasminogen activators in vessel remodeling. Curr Hypertens Rep. 2003;5:466–472. doi: 10.1007/s11906-003-0054-5. [DOI] [PubMed] [Google Scholar]
- Bond M, Murphy G, Bennett MR, Newby AC, Baker AH. Tissue inhibitor of metalloproteinase-3 induces a Fas-associated death domain-dependent type II apoptotic pathway. J Biol Chem. 2002;277:13787–13795. doi: 10.1074/jbc.M111507200. [DOI] [PubMed] [Google Scholar]
- Chun TH, Sabeh F, Ota I, Murphy H, Mcdonagh KT, Holmbeck K, Birkedal-Hansen H, Allen ED, Weiss SJ. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004;167:757–767. doi: 10.1083/jcb.200405001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal S, Lentz SR. Murine models of hyperhomocysteinemia and their vascular phenotypes. Arterioscler Thromb Vasc Biol. 2008;28:1596–1605. doi: 10.1161/ATVBAHA.108.166421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal S, Wilson KM, Leo L, Arning E, Bottiglieri T, Lentz SR. Enhanced susceptibility to arterial thrombosis in a murine model of hyperhomocysteinemia. Blood. 2006;108:2237–2243. doi: 10.1182/blood-2006-02-005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kleijn DP, Sluijter JP, Smit J, Velemas E, Richard W, Schoneveld AH, Pasterkamp G, Borst C. Furin and membrane type-1 metalloproteinase mRNA levels and activation of metalloproteinase-2 are associated with arterial remodeling. FEBS Lett. 2001;501:37–41. doi: 10.1016/s0014-5793(01)02622-9. [DOI] [PubMed] [Google Scholar]
- De Meyer GR, Herman AG. Vascular endothelial dysfunction. Prog Cardiovasc Dis. 1997;39:325–342. doi: 10.1016/s0033-0620(97)80031-x. [DOI] [PubMed] [Google Scholar]
- Dollery CM, Mcewan JR, Henney AM. Matrix metalloproteinases and cardiovascular disease. Circ Res. 1995;77:863–868. doi: 10.1161/01.res.77.5.863. [DOI] [PubMed] [Google Scholar]
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction - Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circulation Research. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- Ebbing M, Bleie O, Ueland PM, Nordrehaug JE, Nilsen DW, Vollset SE, Refsum H, Pedersen EK, Nygard O. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA. 2008;300:795–804. doi: 10.1001/jama.300.7.795. [DOI] [PubMed] [Google Scholar]
- Edirimanne VE, Woo CW, Siow YL, Pierce GN, Xie JY, O K. Homocysteine stimulates NADPH oxidase-mediated superoxide production leading to endothelial dysfunction in rats. Can J Physiol Pharmacol. 2007;85:1236–1247. doi: 10.1139/Y07-112. [DOI] [PubMed] [Google Scholar]
- Fridman R, Toth M, Chvyrkova I, Meroueh SO, Mobashery S. Cell surface association of matrix metalloproteinase-9 (gelatinase B) Cancer Metastasis Rev. 2003;22:153–166. doi: 10.1023/a:1023091214123. [DOI] [PubMed] [Google Scholar]
- Galli A, Svegliati-baroni G, Ceni E, Milani S, Ridolfi F, Salzano R, Tarocchi M, Grappone C, Pellegrini G, Benedetti A, Surrenti C, Casini A. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology. 2005a;41:1074–1084. doi: 10.1002/hep.20683. [DOI] [PubMed] [Google Scholar]
- Galli F, Piroddi M, Annetti C, Aisa C, Floridi E, Floridi A. Oxidative stress and reactive oxygen species. Contrib Nephrol. 2005b;149:240–260. doi: 10.1159/000085686. [DOI] [PubMed] [Google Scholar]
- Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330:1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- Giusti B, Marcucci R, Lapini I, Sestini I, Lenti M, Yacoub M, Pepe G. Role of hyperhomocysteinemia in aortic disease. Cell Mol Biol (Noisy-le-grand) 2004;50:945–952. [PubMed] [Google Scholar]
- Graham IM, Daly LE, Refsum HM, Robinson K, Brattstrom LE, Ueland PM, Palmareis RJ, Boers GH, Sheahan RG, Israelsson B, Uiterwaal CS, Meleady R, Mcmaster D, Verhoef P, Witteman J, Rubba P, Bellet H, Wautrecht JC, De Valk HW, Sales Luis AC, Parrot-Rouland FM, Tan KS, Higgins I, Garcon D, Andria G, et al. Plasma homocysteine as a risk factor for vascular disease. The European Concerted Action Project. JAMA. 1997;277:1775–1781. doi: 10.1001/jama.1997.03540460039030. [DOI] [PubMed] [Google Scholar]
- Hayden MR, Sowers JR, Tyagi SC. The central role of vascular extracellular matrix and basement membrane remodeling in metabolic syndrome and type 2 diabetes: the matrix preloaded. Cardiovasc Diabetol. 2005;4:9. doi: 10.1186/1475-2840-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He JZ, Quan A, Xu Y, Teoh H, Wang G, Fish JE, Steer BM, Itohara S, Marsden PA, Davidge ST, Ward ME. Induction of matrix metalloproteinase-2 enhances systemic arterial contraction after hypoxia. Am J Physiol Heart Circ Physiol. 2007;292:H684–H693. doi: 10.1152/ajpheart.00538.2006. [DOI] [PubMed] [Google Scholar]
- Herbert SP, Huisken J, Kim TN, Feldman ME, Houseman BT, Wang RA, Shokat KM, Stainier DY. Arterial-venous segregation by selective cell sprouting: an alternative mode of blood vessel formation. Science. 2009;326:294–298. doi: 10.1126/science.1178577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques PF, Bostom AG, Wilson PW, Rich S, Rosenberg IH, Selhub J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am J Clin Nutr. 2001;73:613–621. doi: 10.1093/ajcn/73.3.613. [DOI] [PubMed] [Google Scholar]
- Kahleova R, Palyzova D, Zvara K, Zvarova J, Hrach K, Novakova I, Hyanek J, Bendlova B, Kozich V. Essential hypertension in adolescents: association with insulin resistance and with metabolism of homocysteine and vitamins. Am J Hypertens. 2002;15:857–864. doi: 10.1016/s0895-7061(02)02984-9. [DOI] [PubMed] [Google Scholar]
- Korff T, Dandekar G, Pfaff D, Fuller T, Goettsch W, Morawietz H, Schaffner F, Augustin HG. Endothelial ephrinB2 is controlled by microenvironmental determinants and associates context-dependently with CD31. Arterioscler Thromb Vasc Biol. 2006;26:468–474. doi: 10.1161/01.ATV.0000200081.42064.e7. [DOI] [PubMed] [Google Scholar]
- Kundu S, Kumar M, Sen U, Mishra PK, Tyagi N, Metreveli N, Lominadze D, Rodriguez W, Tyagi SC. Nitrotyrosinylation, remodeling and endothelial-myocyte uncoupling in iNOS, cystathionine beta synthase (CBS) knockouts and iNOS/CBS double knockout mice. J Cell Biochem. 2009;106:119–126. doi: 10.1002/jcb.21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoux S, Lemarie CA, Esposito B, Lijnen HR, Tedgui A. Pressure-induced matrix metalloproteinase-9 contributes to early hypertensive remodeling. Circulation. 2004;109:1041–1047. doi: 10.1161/01.CIR.0000115521.95662.7A. [DOI] [PubMed] [Google Scholar]
- Lim U, Cassano PA. Homocysteine and blood pressure in the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol. 2002;156:1105–1113. doi: 10.1093/aje/kwf157. [DOI] [PubMed] [Google Scholar]
- Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, Francis J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res. 2010;85:473–483. doi: 10.1093/cvr/cvp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra PK, Tyagi N, Kundu S, Tyagi SC. MicroRNAs Are Involved in Homocysteine-Induced Cardiac Remodeling. Cell Biochemistry and Biophysics. 2009;55:153–162. doi: 10.1007/s12013-009-9063-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nagase H, Woessner JF., Jr Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- Nygard O, Vollset SE, Refsum H, Stensvold I, Tverdal A, Nordrehaug JE, Ueland PM, Kvale G. Total Plasma Homocysteine and Cardiovascular Risk Profile - the Hordaland Homocysteine Study. Jama-Journal of the American Medical Association. 1995;274:1526–1533. doi: 10.1001/jama.1995.03530190040032. [DOI] [PubMed] [Google Scholar]
- Obi S, Yamamoto K, Shimizu N, Kumagaya S, Masumura T, Sokabe T, Asahara T, Ando J. Fluid shear stress induces arterial differentiation of endothelial progenitor cells. J Appl Physiol. 2009;106:203–211. doi: 10.1152/japplphysiol.00197.2008. [DOI] [PubMed] [Google Scholar]
- Pacilli A, Pasquinelli G. Vascular wall resident progenitor cells: a review. Exp Cell Res. 2009;315:901–914. doi: 10.1016/j.yexcr.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Patterson ML, Atkinson SJ, Knauper V, Murphy G. Specific collagenolysis by gelatinase A, MMP-2, is determined by the hemopexin domain and not the fibronectin-like domain. FEBS Lett. 2001;503:158–162. doi: 10.1016/s0014-5793(01)02723-5. [DOI] [PubMed] [Google Scholar]
- Qipshidze N, Metreveli N, Lominadze D, Tyagi SC. Folic acid improves acetylcholine-induced vasoconstriction of coronary vessels isolated from hyperhomocysteinemic mice: An implication to coronary vasospasm. J Cell Physiol. 2010 doi: 10.1002/jcp.22621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008a;75:346–359. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffetto JD, Khalil RA. Matrix metalloproteinases in venous tissue remodeling and varicose vein formation. Curr Vasc Pharmacol. 2008b;6:158–172. doi: 10.2174/157016108784911957. [DOI] [PubMed] [Google Scholar]
- Rodford JL, Torrens C, Siow RC, Mann GE, Hanson MA, Clough GF. Endothelial dysfunction and reduced antioxidant protection in an animal model of the developmental origins of cardiovascular disease. J Physiol. 2008;586:4709–4720. doi: 10.1113/jphysiol.2008.156976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov RN, Dayoub H, Lynch CM, Wilson KM, Stevens JW, Murry DJ, Kimoto M, Arning E, Bottiglieri T, Cooke JP, Baumbach GL, Faraci FM, Lentz SR. Overexpression of dimethylarginine dimethylaminohydrolase protects against cerebral vascular effects of hyperhomocysteinemia. Circ Res. 2010;106:551–558. doi: 10.1161/CIRCRESAHA.109.200360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JA, Orbe J, Paramo JA. Metalloproteases, vascular remodeling and atherothrombotic syndromes. Rev Esp Cardiol. 2007;60:959–967. doi: 10.1157/13109649. [DOI] [PubMed] [Google Scholar]
- Rodriguez WE, Sen U, Tyagi N, Kumar M, Carneal G, Aggrawal D, Newsome J, Tyagi SC. PPAR gamma agonist normalizes glomerular filtration rate, tissue levels of homocysteine, and attenuates endothelial-myocyte uncoupling in alloxan induced diabetic mice. Int J Biol Sci. 2008;4:236–244. doi: 10.7150/ijbs.4.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen U, Basu P, Abe OA, Givvimani S, Tyagi N, Metreveli N, Shah KS, Passmore JC, Tyagi SC. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am J Physiol Renal Physiol. 2009;297:F410–F419. doi: 10.1152/ajprenal.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen U, Givvimani S, Abe OA, Lederer ED, Tyagi SC. Cystathionine beta-synthase and cystathionine gamma-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. American Journal of Physiology-Cell Physiology. 2011;300:C155–C163. doi: 10.1152/ajpcell.00143.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen U, Moshal KS, Singh M, Tyagi N, Tyagi SC. Homocysteine-induced biochemical stress predisposes to cytoskeletal remodeling in stretched endothelial cells. Mol Cell Biochem. 2007;302:133–143. doi: 10.1007/s11010-007-9435-4. [DOI] [PubMed] [Google Scholar]
- Sen U, Moshal KS, Tyagi N, Kartha GK, Tyagi SC. Homocysteine-induced myofibroblast differentiation in mouse aortic endothelial cells. J Cell Physiol. 2006;209:767–774. doi: 10.1002/jcp.20752. [DOI] [PubMed] [Google Scholar]
- Sen U, Munjal C, Qipshidze N, Abe O, Gargoum R, Tyagi SC. Hydrogen sulfide regulates homocysteine-mediated glomerulosclerosis. Am J Nephrol. 2010;31:442–455. doi: 10.1159/000296717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger O, Weger M. Interactions of homocysteine, nitric oxide, folate and radicals in the progressively damaged endothelium. Clin Chem Lab Med. 2003;41:1444–1454. doi: 10.1515/CCLM.2003.222. [DOI] [PubMed] [Google Scholar]
- Steed MM, Tyagi N, Sen U, Schuschke DA, Joshua IG, Tyagi SC. Functional consequences of the collagen/elastin switch in vascular remodeling in hyperhomocysteinemic wild-type, eNOS−/−, and iNOS−/− mice. Am J Physiol Lung Cell Mol Physiol. 2010;299:L301–L311. doi: 10.1152/ajplung.00065.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res. 2009;104:576–588. doi: 10.1161/CIRCRESAHA.108.188805. [DOI] [PubMed] [Google Scholar]
- Taylor AC, Murfee WL, Peirce SM. EphB4 expression along adult rat microvascular networks: EphB4 is more than a venous specific marker. Microcirculation. 2007;14:253–267. doi: 10.1080/10739680601141829. [DOI] [PubMed] [Google Scholar]
- Toole JF, Malinow MR, Chambless LE, Spence JD, Pettigrew LC, Howard VJ, Sides EG, Wang CH, Stampfer M. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004;291:565–575. doi: 10.1001/jama.291.5.565. [DOI] [PubMed] [Google Scholar]
- Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- Watts SW, Rondelli C, Thakali K, Li X, Uhal B, Pervaiz MH, Watson RE, Fink GD. Morphological and biochemical characterization of remodeling in aorta and vena cava of DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292:H2438–H2448. doi: 10.1152/ajpheart.00900.2006. [DOI] [PubMed] [Google Scholar]
- Weiss N, Heydrick S, Zhang YY, Bierl C, Cap A, Loscalzo J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22:34–41. doi: 10.1161/hq1201.100456. [DOI] [PubMed] [Google Scholar]