

Abstract
Glioblastoma multiforme (GBM) is the most common and aggressive primary brain tumor in adults. Despite intensive treatment, the prognosis for patients with GBM remains grim with a median survival of only 14.6 months. Immunotherapy has emerged as a promising approach for treating many cancers and affords the advantages of cellular-level specificity and the potential to generate durable immune surveillance. The complexity of the tumor microenvironment poses a significant challenge to the development of immunotherapy for GBM, as multiple signaling pathways, cytokines, and cell types are intricately coordinated to generate an immunosuppressive milieu. The development of new immunotherapy approaches frequently uncovers new mechanisms of tumor-mediated immunosuppression. In this review, we discuss many of the current approaches to immunotherapy and focus on the challenges presented by the tumor microenvironment.
1. Introduction
Glioblastoma multiforme (GBM) (WHO grade IV astrocytoma) is the most common and malignant primary brain tumor in adults. Despite aggressive, multimodal treatment with maximal surgical resection followed by temozolomide and radiation, the prognosis for patients with GBM remains grim with a median survival of 14.6 months and a 3-year survival rate of only 10% [1]. One formidable challenge in advancing GBM therapy is the complexity of the GBM microenvironment [2]. Elucidating the details of GBM resistance to traditional therapies requires consideration not only of the intrinsic properties of tumor cells, but also how these cells interact with neural precursor cells, tumor stem cells, vascular endothelial cells, stromal cells, astrocytes, microglia, lymphocytes, extracellular matrix proteins, and cytokines. It is this dynamic interplay among diverse cell populations, cytokines, and extracellular matrix proteins that coordinates GBM tumorigenesis, growth, and invasion. Effective therapies, therefore, must not only be directly cytotoxic to a molecularly diverse population of tumor cells [3], but must also overcome the protumorigenic properties of the GBM microenvironment.
Immunotherapy is a particularly attractive approach to cancer treatment as it affords the advantages of cellular level specificity and the potential for generating long-term immune surveillance against cancer cells. The notion of activating the immune system against cancer has been around for decades but has recently come to the forefront with the FDA approval of the first therapeutic cancer vaccine for the treatment of metastatic, castration-resistant prostate cancer [4]. More recently, ipilimumab, an anti-CTLA-4 antibody, was approved by the FDA for first- and second-line treatment of unresectable or metastatic melanoma [5]. Preclinical research is rapidly identifying new immunological targets leading the way for the development of powerful combination therapies [6]. In addition, several immunotherapies are currently in clinical trials and many are producing encouraging results in a variety of cancers [7].
Immunotherapy for neoplasms of the central nervous system (CNS) has been hampered by the traditional belief that the CNS is immunologically privileged [8]. This theory was based on reports of a paucity of native antigen-presenting cells (APCs) in the CNS, the lack of a traditional lymphatic system, impermeability of the blood-brain barrier (BBB) to antibodies and lymphocytes [9], low baseline levels of major histocompatibility complex (MHC) expression [10], altered expression of T cell costimulatory molecules [11], and the observation that tissues engrafted into the CNS are rejected more slowly than those grafted to other sites [12, 13]. Each of these perceived impediments to immunotherapy has subsequently undergone major revisions. Microglia [14], macrophages, and dendritic cells [15, 16] act as powerful APCs in the CNS. Antigens originating within the CNS drain in the cerebrospinal fluid through Virchow-Robin perivascular spaces to nasal and cervical lymph nodes where they can be accessed by naïve T cells [17, 18]. Subpopulations of activated T cells expressing integrins, which impart CNS tropism, such as α4β7, traverse the BBB [19] where they can act as cytotoxic or helper T cells based on CD8 or CD4 expression, respectively [20]. There is also evidence to suggest that naïve T cells traffic to the CNS, especially when inflammation locally increases the permeability of the BBB [21]. Furthermore, antibodies have been isolated from the brain, albeit in much lower concentrations than in plasma [22, 23]. Taken together, these findings represent an evolution in our understanding of the interactions between the CNS and the immune system.
This paradigm shift has generated enthusiasm for a potential role for immunotherapy in GBM. Despite encouraging results in rodent models, however, clinical trials of immunotherapy for GBM have been largely disappointing to date. One of the primary impediments to developing effective immunotherapies is the aforementioned complexity of the GBM microenvironment (Figure 1). Immunosuppressive cytokines such as prostaglandin E2 (PGE-2), TGF-β, and IL-10 are known to be highly expressed in GBMs [24, 25]. In addition, tumor-infiltrating T cells have been shown to exhibit an enriched population of CD4+, CD25+, FoxP3+ regulatory T cells (Tregs) [26]. Expression of the signal transducer and activator of transcription 3 (STAT3) is upregulated in GBM and is believed to promote immunosupression and serve as a point of convergence for several protumorigenic pathways [27]. Furthermore, tumor stem cells have been shown to be immunosuppressive in GBM [28]. Immune checkpoints, such as programmed cell death 1 (PD-1) and Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) may also be manipulated by GBM to induce T cell exhaustion [26, 29]. Finally, there is evidence to suggest that the GBM microenvironment may divert CD4+ T cell differentiation away from a tumor-directed cytotoxic Th1-mediated response and toward a Th17-mediated chronic inflammatory response [30], which has been shown to be protumorigenic in other cancers [31].
Figure 1.
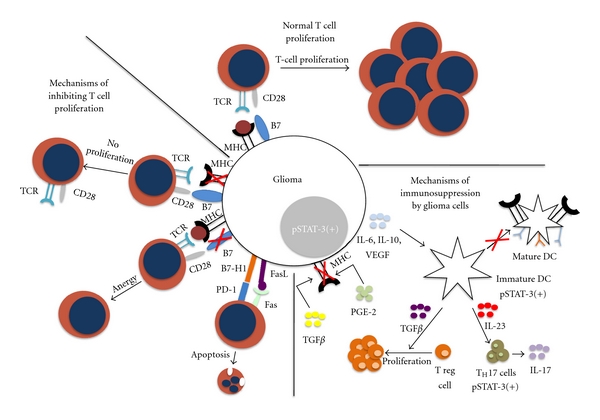
Normal T cell proliferation and mechanisms of glioma cell immunoresistance. (From top moving clockwise) Normal T cell proliferation: tumor cell antigens are presented by MHC and costimulatory molecules. Mechanisms of immunosuppression: glioma cells secrete factors leading to an immunosuppressive tumor microenvironment. TGFB and PGE-2 downregulate the expression of MHC, restricting antigen presentation and T cell proliferation. IL-6. IL-10 and VEGF are potent STAT-3 activators, leading to the proliferation of immature DCs that are not able to function as APCs. These immature DCs also secrete TGFB which aid in the proliferation of immunosuppressive Treg cells and STAT-3 positive TH17 cells. Mechanisms of inhibiting T cell proliferation: glioma cells downregulate MHC on their surface leading to the decreased antigen presentation and decreased T cell proliferation. Downregulation of B7 works via a similar mechanism in that the costimulatory signal is lost preventing T cell proliferation. Increased expression of B7-H1 and FasL act as proapoptotic signals for T cells.
Identification of appropriate tumor antigens and generation of a strong antitumor immune response against such a molecularly heterogeneous neoplasm [32] poses a considerable challenge. This challenge is amplified by the immunosuppressive tumor microenvironment. Here, we review the current approaches in immunotherapy for GBM, focusing specifically on how each approach is affected by the array of challenges presented by the tumor microenvironment.
2. Current Approaches
2.1. Cytokine Modulation
Immune responses in the CNS exhibit a distinct hierarchy skewed toward antibody responses and Th2 T cell differentiation [33–35]. It is believed that this hierarchy is maintained by the CNS cytokine milieu [35]. In the GBM microenvironment, the antitumor immune response is further suppressed by high levels of circulating immunosuppressive cytokines such as IL-10, TGF-β, and PGE2 as well as membrane-bound proteins such as FasL and B7-H1 (PD-L1) [36, 37]. The sources of these molecules and the details of their interactions are yet to be fully elucidated. It is clear, however, that the cytokine milieu plays a critical role in coordinating immunosupression in GBM. Clinical trials using cytokine modulation are summarized in Table 1.
Table 1.
Selected clinical trials using cytokine modulation.
| Reference | Patients | Cytokine | Immunologic response | Clinical response |
|---|---|---|---|---|
| [58] |
n = 145 (GBM: 103, AA: 42) |
TGF-β | — | Median survival: 39.1 mo (10 uM dose) and 35.2 mo (80 uM dose) |
| [63] |
n = 9 Recurrent GBM |
IL–2 | — | Enhancement of tumor on MRI unchanged (6/9) |
| [64] |
n = 9 (GBM: 7, AA: 2) |
IL–2 | — | PR: 1 |
| [65] |
n = 12 recurrent GBM |
IL–2 | Increased inflammatory infiltrate in biopsied tumors | PR: 2, SD: 4, Minor response: 4, Overall survival 58% (6 mo) and 25% (1 yr) |
| [69] |
n = 31 (GBM: 26, AA: 5) |
IFN-γ
(n = 14) |
— | PR: 3 (Treatment group), No difference in median survival between treatment and control groups |
| [70] |
n = 40 (GBM: 14, AA: 14, Other: 12) |
IFN-γ | — | No difference in median overall survival |
| [71] |
n = 29 (AA: 12, Other: 17) |
IFN-β | — | PR: 2, SD: 2 |
| [72] |
n = 20 (GBM/AA: 15) |
IFN-β | IFN-β treatment showed no growth suppression in ex vivo assays | SD: 3 |
| [73] |
n = 7 (GBM: 6, Recurrent AA: 1) |
IFN-β | — | No response |
| [75] |
n = 35 recurrent HGG |
IFN-α | — | Median survival: 13.3 mo |
| [76] |
n = 275 HGG |
IFN-α | — | No difference in survival |
| [77] |
n = 9 (GBM: 6, AA: 2, Other 1) |
IFN-α | — | CR: 2 |
| [80] |
n = 12 (GBM: 11, AA: 1) |
IL–4 | Positive Elispot assay | No difference in progression free survival |
| [142] |
n = 9 recurrent GBM |
IL–4 | — | Survival > 18 mo (n = 1) |
| [84] |
n = 15 (GBM: 6, AA: 7, Other: 2) |
IL–12 | — | PR: 4, Mixed response: 1 |
2.1.1. TGF-β
TGF-β is synthesized in a pre-pro-TGF-β form and undergoes homodimerization and cleavage by the convertase family of endopeptidases [38] to produce a C-terminal mature peptide and an N-terminal latency-associated peptide, which collectively form the small latency complex [39]. The small latency complex is then secreted from the cell and associates with specific binding proteins to form the large latency complex, which is bound by components of the extracellular matrix [39, 40]. TGF-β is activated when it is released from the latency-associated peptide through one of a number of context-dependent mechanisms [41]. Activated TGF-β regulates gene expression downstream via the SMAD family of transcription factors [39]. TGF-β synthesis, secretion, and signaling are reviewed in detail elsewhere [42].
TGF-β promotes immunosuppression in GBM by inhibiting T cell activation and proliferation, blocking IL-2 production, suppressing activity of NK cells, and promoting Treg activity [43, 44]. In addition, TGF-β is believed to promote tumor growth and invasion by sustaining GBM stem cells [45], promoting angiogenesis [46], and upregulating expression of molecules such as MMP-2, which are associated with tumor invasion [47]. The involvement of TGF-β in multiple tumorigenic pathways makes this cytokine an enticing target for immunotherapy.
TGF-β expression is increased by radiation both in vitro [48] and in vivo [49]. This finding is of interest because radiation therapy is a critical component of the tripartite treatment approach of resection, temozolomide, and radiation which has become standard of care for patients with GBM [1], and because there is emerging evidence to suggest that radiation therapy may alter several components of the immune microenvironment [50–52]. Radiation-induced activation of TGF-β is believed to be mediated by reactive oxygen species (ROS), which have been shown to convert latent TGF-β preferentially to the TGF-β1 isoform [53]. Although this isoform plays a more minor role in GBM pathogenesis than the TGF-β2 isoform, available evidence suggests that TGF-β1 promotes immunosuppression [54] and acts as a mediator of radiation-induced DNA damage sustained by nontargeted cell populations [55]. In addition, TGF-β2 has been shown to increase tumor invasiveness by upregulating MMP-2 expression in glioma cells [56] and evidence from other cell lines suggests that TGF-β1 may be an even more powerful inducer of MMP-2 expression [57].
The results of TGF-β blockade in preclinical models have been generally promising. The TGF-β2 antisense oligonucleotide trabedersen (AP12009) has been shown to decrease tumor cell proliferation, inhibit migration, and enhance the antitumor immune response in vitro. A randomized, phase IIb clinical trial of trabedersen reported significantly improved tumor control and a trend toward increased 2-year survival for patients with anaplastic astrocytoma as compared with standard chemotherapy (temozolomide or a combination of procarbazine, lomustine, and vincristine) [58]. This trial did not report improved survival in patients with GBM, although a subgroup analysis of young patients with good performance status indicated a trend toward improved 2- and 3-year survival rates. Of note, the reported rate of treatment-related adverse events was approximately 20% higher with standard chemotherapy than with trabedersen. Trabedersen is currently in phase III clinical trials for anaplastic astrocytoma [59]. Understanding the role of TGF-β in the tumor microenvironment may have implications for standard therapies as well. For example, given that available evidence points toward a protumorigenic role for TGF-β, the addition of TGF-β blockade to adjuvant radiation therapy may prove prudent [60].
2.1.2. IL-2
IL-2 is a proinflammatory cytokine which promotes T cell activation and Th1 differentiation while abrogating the immunosuppressive effects of TGF-β [61]. IL-2 therapy for GBM is complicated by the fact that high systemic doses of IL-2 are required to reach therapeutic concentrations in the CNS [62]. Early trials of IL-2 alone or in combination with IFN-α [63] or lymphokine-activated killer (LAK) cells [64] attempted to obviate the severe side effects associated with systemic high-dose IL-2 therapy by delivering IL-2 intratumorally or intraventricularly; however, the patients in these trials experienced significant adverse events resulting from local edema. A more recent trial by Colombo et al. used a retroviral vector and intratumoral implantation of retroviral-producing cells to deliver combination HSV-TK/IL-2 gene therapy followed by administration of acyclovir to 12 patients with recurrent gliomas [65]. This trial reported no major adverse events and a radiographic response rate of 50%. Evidence from preclinical models additionally suggests that IL-2 therapy generates long-lasting immune surveillance, which is capable of eliminating tumor cells both inside and outside the CNS [66]. Current approaches to IL-2 therapy for GBM are focused on combination therapy and strategies for local delivery [67].
2.1.3. Interferons
Interferons are secreted by immune cells in response to viruses or other challenges and serve to coordinate the immune response. Alpha interferon (IFN-α), beta interferon (IFN-β) and gamma interferon (IFN-γ) have been extensively studied in cancer immunotherapy. These type 1 interferons have specifically been implicated in coordinating an antitumor immune response against GBM. A study by Fujita et al. demonstrated that mice deficient in type 1 interferons, and induced to develop gliomas de novo via p53 knockdown, exhibited enriched populations of tumor infiltrating myeloid-derived suppressor cells and Tregs as well as a decrease in the numbers of tumor-infiltrating CD8+ T cells [68]. Despite some preclinical evidence for efficacy against gliomas, small clinical trials using IFN-γ have been generally disappointing [69, 70]. Trials of IFN-β have produced mixed results [71–73]. The efficacy of IFN-β in combination with temozolomide is currently being investigated [74].
Of the type 1 interferons, IFN-α has been the most extensively studied in GBM. In a phase III study by Buckner et al., 214 patients were initially treated with BCNU and radiation. Patients with radiographically stable disease were subsequently randomized to treatment with a second course of BCNU or BCNU and IFN-α. This study demonstrated no difference in survival or tumor response with the addition of IFN-α [75]. Unfortunately, there was a significantly increased incidence of side effects, including fever, chills, myalgias, somnolence, confusion, and exacerbation of neurologic deficits in patients receiving IFN-α. These findings were in contrast with a prior phase II study by the same group, which reported that IFN-α was associated with radiographic evidence of tumor regression in 29% of patients and limited toxicity [76]. A more recent trial of IFN-α in combination with local BCNU delivery in patients with recurrent GBM reported 6-month progression-free survival in 2/9 patients [77]. Of interest, both patients who responded in this study were in the group receiving the lowest dose of IFN-α. Therefore, while grade 2 and grade 3 toxicities were observed somewhat frequently in the higher dose groups, only two grade 2 events and no events grades 3 or higher were observed in the treatment group containing the two responders.
2.1.4. Miscellaneous Cytokines
Many cytokines have been evaluated for their effectiveness in GBM therapy. TNF-α knockout mice implanted with GL261 glioma cells have been shown to harbor a decreased number of tumor-associated macrophages and exhibit shorter survival [78]. Knowledge of the role of TNF-α in human gliomas, however, is limited. IL-4 has been shown to increase in CD8+ tumor-infiltrating T cells in a rat model [79]. In a small clinical trial by Okada et al., patients received vaccinations of autologous glioma cells and fibroblasts retrovirally transfected with TFG-IL4-Neo-TK [80]. Treatment was well tolerated, but there was no observed progression-free survival benefit. Locally delivered IL-12 in preclinical models increases tumor-directed T cell responses [81], improves survival, and produces variable development of durable immune surveillance [82]. Tumor stem cells secreting IL-12 have also been shown to track migrating glioma cells and prolong survival [83]. Limited evaluation of IL-12 therapy in clinical trials, however, has produced mixed results [84]. Granulocyte-macrophage colony-stimulating factor (GM-CSF) promotes a CD8+ cytotoxic T cell response when combined with antitumor vaccines [85]. GM-CSF is currently being used as an adjuvant in a phase II vaccination study of patients with newly diagnosed GBM [86]. Discovery of T cell populations producing IL-17 (Th17) [87] and their association with STAT3 expression in human cancers [88] have recently generated an interest in defining the role of these cells in GBM pathogenesis. Early preclinical studies indicate IL-17 is expressed in GBM, but the significance of IL-17 expression in the tumor microenvironment is yet to be clearly defined [30].
2.2. Cellular Immunotherapy
Transfer of ex vivo matured immune cells is showing promising results as a future immunotherapeutic intervention against malignant glioma. Initially used as a treatment for melanoma, this strategy involves infusion of autologous immune cells that were matured ex vivo with activity specific for glioma cell antigens. While studies have shown lymphokine-activated killer cells cannot effectively migrate across the BBB, effector T cells are able to cross the BBB allowing for a vaccine or IV strategy to be used [89].
2.2.1. Lymphokine Activated Killer Cells
Lymphokine activated killer (LAK) cells are autologous peripheral blood lymphocytes that have been stimulated in vitro with IL-2 [90]. Results of early clinical trials infusing LAK cells directly into the surgical cavity showed promise for the use of LAK cells as an immunotherapeutic strategy [64, 91, 92]. The most encouraging of these early studies, Hayes et al. reported a median survival in 18 patients of 12.2 months compared with the control group of 6.2 months with minimal toxicity [93]. In 2004, Dillman et al. reported minimal toxicity and an increase in median survival in a trial of 31 patients. Median survival from the date of original diagnosis was 17.5 months versus 13.6 months for a control group of 41 contemporary GBM patients [94]. Of note, LAK cells must be administered directly to the tumor site since they fail to effectively migrate from the periphery into the brain [95]. Clinical trials using LAKs are summarized in Table 2.
Table 2.
Selected clinical trials using lymphokine activated killer (LAK) cells.
| Reference | Patients | Immunologic response | Clinical response |
|---|---|---|---|
| [64] |
n = 9 (GBM: 7, AA: 2) |
— | PR: 1 |
| [91] |
n = 9 HGG |
Cultured LAK cells lysed cultured glioma cells (n = 6) | Slight clinical (but not radiologic) improvement. |
| [92] |
n = 20 recurrent HGG |
— | Median survival: 63 weeks |
| [93] |
n = 19 (GBM: 5, AA:4, Other 10) |
— | CR: 1, PR: 2, median survival (GBM): 15 weeks |
| [94] |
n = 40 recurrent GBM |
— | Median survival: 17.5 months (significantly longer than contemporary patients) |
| Others: [94, 143–147] |
2.2.2. Effector T-Cells
Effector T cell therapy involves transfer of autologous cytotoxic T cells (CTLs) specific for tumor antigens, which are matured from peripheral blood mononuclear cells (PBMCs) or T cells from the tumor itself, to the host. This therapy is based on the theory that T cells can migrate to the site of a tumor by crossing the BBB, and selectively exert cytotoxic effects on tumor cells. This strategy has been studied extensively in malignant melanoma with promising results. Studies in animal models of glioma have been promising. Initial studies by Yamasaki and Kikuchi used IL-2 to activate CD8+ T cell clones with target specificity against murine malignant brain tumor cells. This strategy resulted in successful migration of T-cells to the tumor, cytotoxic activity against the tumor, and a significant increase in survival after IV infusion [96].
Early clinical studies using ex-vivo-expanded CTLs were largely disappointing for patients with GBM, however, more recent studies have shown promise. Tsurushima et al. reported that activating polyclonal T cells with IL-2 resulted in two patients with Grade III disease exhibiting complete tumor regression for at least 5 years with another patient having a partial regression [97]. A study using GM-CSF resulted in three of ten patients having at least partial tumor regression. All patients with a diagnosis of GBM survived at least one year from the time of adoptive transfer [98]. Another approach has been to genetically modify T cells to express a chimerical antigen receptor (CAR) for a known tumor antigen. Kahlon et al. genetically engineered CD8+ T cells to express CARs for IL-13Rα2 and reported regression of GBM xenografts [99]. Studies in human GBM have demonstrated that CARs can migrate to tumors in vivo [100]. Furthermore, Ahmed et al. have shown that CARs targeted to HER2 are able to eliminate CD133+ stem cells as well as bulk tumor cells in HER2+ GBMs [101]. Clinical trials using CTLs are summarized in Table 3.
Table 3.
Selected clinical trials using cytotoxic T lymphocytes (CTLs).
2.3. Antigen Identification and Targeting
Targeting of tumor-specific antigens is a promising strategy for delivering antitumor immunotherapy. The effectiveness of this approach remains controversial, however, as many vaccine trials have not demonstrated a consistent antitumor response or survival advantage despite increased tumor reactive cytotoxic T cells [102–105]. One of the challenges facing therapy directed against single antigens is the ability of a tumor to alter its antigen expression profile, resulting in immune editing. Immune editing consists of three phases: elimination, equilibrium, and escape [106]. The elimination phase is maintained by immunosurveillance of cancer cells by both the innate and adaptive immune system [107–109]. The equilibrium phase occurs when tumor cells survive the cytotoxic pressure exerted by immune cells. Finally, the escape phase results in uncontrolled tumor growth and often clinical manifestations of disease [106]. Often immune escape is preceded by mutations within cancer cells that facilitate immune evasion. For example, loss of HLA class I proteins [110, 111] and decreased response to IFN-γ [108, 112] have been described in adenomas of the lung and melanoma.
Another major challenge currently limiting antigen-targeted therapies is the inability to tailor therapy to an individual tumor's antigen expression profile. The current classification scheme for glioma does not account for the molecular diversity of GBM. A new model for classification, reported by Verhaak et al., is a molecular classification of glioblastoma consisting of four clinically relevant tumor subtypes—classical, mesenchymal, proneural, and neural [113]. A comprehensive understanding of which antigens are present on each GBM subtype would allow for better targeted immunotherapy.
2.3.1. EGFRvIII
The epidermal growth factor receptor vIII (EGFRvIII) is a truncated form of the wild-type EGF receptor [114, 115] and is an attractive antigen for immunotherapy because it is not expressed by normal brain and leads to enhanced tumorigenicity of the EGFRvIII-expressing cell [116]. This truncated protein is constitutively active despite its inability to bind extracellular ligand [117]. Efforts to target EGFRvIII, however, have been significantly hampered by immune editing [106]. For example, unpublished data from CDX-110 clinical trials reported the EGFRvIII antigen was not expressed on recurrent tumors in 20/23 patients who had been initially treated with the EGFRvIII vaccine [25].
The novel EGFRvIII epitope exists extracellulary and is a prime target for monoclonal antibody recognition [118, 119], which stimulates antitumor cytotoxic T cell maturation. EGFRvIII-specific titers are not found in normal volunteers, but are present in patients with EGFRvIII-expressing cancers, such as adenocarcinomas and gliomas [120, 121]. Early animal studies using vaccination strategies against EGFRvIII reported increased numbers of tumor-infiltrating CD4+, CD8+, natural killer (NK) cells, and macrophages as well as a dramatic increase in survival [119, 122–125].
These promising preclinical results lead to early-phase I studies looking at the use of vaccine strategies against the EGFRvIII peptide. The first study for malignant gliomas was the Vaccine for Intra-Cranial Tumors I (VICTOR1). In this study, autologous mature dendritic cells were pulsed with 500 ug of PEPvIII, which was conjugated with keyhole limpet hemocyanin (KLH). Following surgical resection and completion of radiation therapy, all patients were vaccinated three times; the first three patients were dosed with 3 × 107 mature DCs per vaccine while the remaining patients were dosed with one third of their DCs per injection. No serious adverse events were reported and immunological responses were detected ex vivo. For patients with GBM, the median time to progression (TTP) was 46.9 weeks and median survival was 110.8 weeks. These results compare favorably with patients treated with resection site carmustine wafers [126] or temozolamide [1, 121].
The follow-up phase II study, A Complementary Trial of an Immunotherapy Against Tumor Specific EGFRvIII (ACTIVATE) evaluated the efficacy of the PEPvIII-KLH and granulocyte macrophage-colony stimulating factor (GM-CSF) [127]. Patients received three vaccinations at two-week intervals. Similar to the VICTOR1 study, there were no serious adverse effects and cellular immune responses were detected ex vivo. The median TTP was 14.2 months and the median survival was 32 months. Of note, upon histological examination, recurrent tumors did not express EGFRvIII.
The currently ongoing ACTIVATE II trial was initiated to evaluate the effectiveness of adding adjuvant PEPvIII-KLH vaccination therapy to standard of care (resection, temozolomide, and radiation). Of note, temozolomide induces lymphopenia, theoretically decreasing the efficacy of an immune-based therapy. Therefore, the EGFRvIII vaccine (CDX-110) was given on day 21 of the 28 day cycle, allowing recovery of the immunosuppression caused by temozolamide [128].
2.3.2. IL-13 Receptor α2
The IL-13Rα2 antigen is a promising target for immunotherapy because it is highly expressed on glioma cells but not on host CNS cells [129, 130]. However, it should be noted that IL13Rα2 expression is often heterogeneous [131]. In a study by Okano et al., it was shown that a novel epitope of IL-13Rα2 induced CD8+ T cells to secrete IFNγ and lyse IL-13Rα2-expressing glioma cells in vitro. This effect was only seen in CD8+ T cells expressing the HLA-A*0201 allele [132], which 40–50% of Caucasians and Asians express [133]. To target the IL-13Rα2 in vivo, IL-13 was tagged with a mutated form of the pseudomonas exotoxin [134–138]. This fused protein (IL-13-PE38QQR), also termed Cintredekin besudotox (CB), showed promise in vivo; Kawakami et al. reported that CB injected intracranially resulted in both tumor regression and prolonged survival by 164% as compared with control animals [139].
Three phase I studies were undertaken to determine the safety of intracerebral administration of CB. Pooled results of the 51 total patients indicated a slight survival advantage as compared with BCNU wafers. Subsequently, 276 patients were enrolled in a Phase III study (PRECISE) to determine if the overall survival, safety, and quality of life differ in patients receiving the CB via local Convection-enhanced delivery (CED) compared to patients receiving BCNU wafers. There was no reported difference in median survival (36.4 weeks for the patients receiving CB compared with 35.3 weeks for the patients receiving Gliadel wafers, P = 0.476) [140, 141].
2.3.3. IL-4 Receptor
IL-4 receptor (IL-4R) is an attractive target for immunotherapy because tumor cells express a different IL-4R isoform than that which is present on circulating immune cells. This isoform of the IL-4R is commonly expressed in human gliomas and not on neural tissue [178–181]. The type 2 IL-4R signals through the Jak-STAT pathway, activating the Jak1/Jak2 tyrosine kinases, and eventually activating the STAT-6 protein, which translocates to the nucleus and regulates gene expression [182–184]. To target the IL-4R, IL-4 was fused to pseudomonas exotoxin (IL-4(38-37)-PE38KDEL) [181, 185]. Joshi et al. showed that this construct induces glioma cell death in culture [186]. In vivo studies demonstrated the same construct decreased the size of implanted human-derived glioma tumors (U251) with all treated mice showing complete regression. The tumors recurred in 50% of animals but were smaller than tumors harbored by control animals [187].
A phase I clinical trial of the IL-4-fused protein (cpIL4-PE) was performed in patients with recurrent malignant gliomas. The construct was injected intratumorally by CED. The authors concluded that direct glioma injection of cpIL4-PE was safe, had no systemic toxicity, and caused necrosis of malignant gliomas that were refractory to conventional therapy. Subsequent clinical trials using the same construct, with stereotactic injection as the delivery method, showed similar findings of safety and efficacy [142].
In addition to identifying appropriate epitopes, an effective immunotherapy strategy must be able to efficiently target these antigens in vivo. Dendritic cell, autologous tumor cell, and heat shock protein vaccines are discussed below with general principles illustrated in Figure 2.
Figure 2.
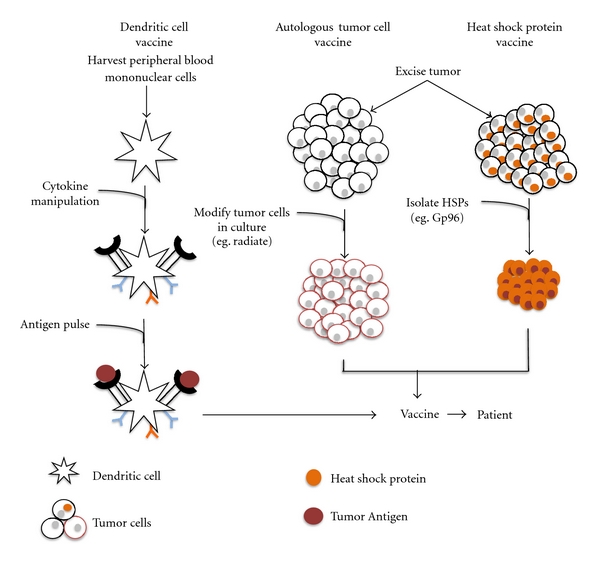
Vaccine Strategies for GBM. (From Left) Dendritic cell vaccine: peripheral blood mononuclear cells are isolated from the patient and cultured ex vivo. Cytokines are added to culture to activate the DCs. The matured DCs are pulsed with tumor antigen and then added to the vaccine preparation. Autologous tumor cell vaccine: after tumor removal, tumor cells are cultured. In some cases, these cells are modified (e.g., radiation, chemical) and then injected back into the patient. Heat shock protein vaccine: after tumor removal, tumor cells are cultured and specific heat shock proteins (e.g., Gp96) are isolated and purified. The proteins are then added to the vaccine preparation and injected into the patient.
2.3.4. Dendritic Cells
Dendritic cells (DCs) are “professional” antigen-presenting cells (APCs) that activate innate and adaptive immune responses [155]. Strategies using DCs seek to exploit this ability as GBM cells are unable to reliably present antigens to the immune system [188, 189]. DCs can be harvested from peripheral blood or bone marrow, pulsed with tumor lysate or tumor-specific peptides, and after maturation, injected back into the patient.
In a phase I trial, Yu et al. expanded peripheral blood cells ex vivo into DCs and pulsed them with peptides eluted from the surface of cultured autologous brain tumor cells. Seven patients received three biweekly intradermal vaccinations of peptide-pulsed DCs with no systemic side effects. The vaccination led to significant T-cell-specific cytotoxicity against glioma tumor cells and later biopsy showed that cytotoxic and memory T cells were able to traffic into the tumor [155]. Liau et al. reported a series of 12 patients treated with 1, 5, and 10 million autologous dendritic cells pulsed with autologous tumor peptides. Similar to the previous studies, no systemic side effects were seen and survival was improved compared to historical controls. Of note, the magnitude of the T cell infiltration was inversely correlated with TGF-β expression within the tumor microenvironment [156]. A larger trial showed 8 of 19 patients with GBM had a median survival of 33.6 months with a median time to progression of 18.1 months, surpassing that of historical controls receiving standard of care. Of note, 42% of patients have survived longer than 4 years [190].
Pulsing DCs with whole tumor lysate increases the number of targeted epitopes and prevent antigen-loss escape and immune editing [191]. Parajuli et al. reported that DCs pulsed with apoptotic tumor cells or total tumor RNA led to a more robust immune response compared to DCs pulsed with tumor cells or fused with glioma cells [192]. Clinical trials using dendritic cells are summarized in Table 4.
Table 4.
Selected clinical trials using dendritic cells (DCs).
| Reference | Patients | Immunologic response | Clinical response |
|---|---|---|---|
| [155] |
n = 7 (GBM: 6, AA: 1) |
Cytotixic and memory T cells found in recurrent tumor bulk | Median survival: 455 days (Control group: 257 days) |
| [156] |
n = 12 (GBM: 7, Recurrent GBM: 5) |
Cytotoxicity against autologous tumor cells. Cytotoxic T cells found in recurrent tumor bulk. | Median TTP: 19.9 mo (P = 0.028), Median survival: 35.8 mo (P = 0.006) |
| [157] |
n = 18 EGFRvIII expressing GBM |
82% of recurrent tumors lost EGFRvIII expression | Median survival: 26 mo (P = 0.001) |
| Others: [80, 84, 158–173] |
2.3.5. Autologous Tumor Cells
The use of autologous tumor cells (ATCs) as an immunotherapeutic approach has garnered attention due to the ability to activate the immune system with an increased number of potential glioma antigens. Several strategies for ATC vaccines have been tested including using irradiated glioma cells that were either autologous or allogenic. The autologous strategy was more beneficial in providing the most relevant antigens to the patient's tumor [193–195]. Recent clinical trials have shown this method can be used without systemic side effects. Schneider et al. reported 11 patients who received an autologous tumor vaccine with cells modified with Newcastle-Disease-Virus after surgery and radiation. Survival was no different compared to patients receiving surgery, radiation, and chemotherapy. No side effects were seen with the vaccine group [174]. A similar trial by Steiner et al. reported 23 patients who underwent surgery, radiation, and vaccination. There was a statistically significant increase in median progression-free survival (40 weeks versus 26 weeks in controls) and median overall survival of vaccinated patients (100 weeks versus 49 weeks in controls) [175]. Using an autologous formalin-fixed tumor vaccine, which is thought to preserve the antigenicity of the tumor cells, Ishikawa et al. studied 24 patients who received surgery, and radiation, showing no adverse events [176]. Selected clinical trials using ATCs are summarized in Table 5.
Table 5.
Selected clinical trials using autologous tumor cells (ATCs).
| Reference | Patients | Immunologic response | Clinical response |
|---|---|---|---|
| [174] |
n = 11 recurrent GBM |
Local skin reaction | Median survival: 46 weeks |
| [175] |
n = 23 GBM |
Delayed-type hypersensitivity, increased memory T cells, increased CD8+ T cells in recurrent tumors | Median progression free survival: 40 weeks, median survival 100 weeks |
| [176] |
n = 12 GBM |
— | CR: 1, PR: 1, minor response: 2, median survival: 10.7 mo |
2.3.6. Heat Shock Proteins
Heat shock proteins (HSPs) are chaperon proteins that aid in protein folding and are implicated in mediating adaptive and innate immune responses [195]. While there are five major families of HSPs, the HSPs Grp 96, HSP 90, HSP 70, HSP 110, and HSP 170 are considered the most immunogenic [196, 197]. HSPs aid in the folding of many proteins within the cell, and, therefore, a specific target antigen is not required, thus decreasing the potential for immune editing. Furthermore, HSPs have been shown to induce human DC maturation and to activate DCs to secrete proinflammatory cytokines making this strategy an attractive option for immunotherapy.
Clinical trials using a vaccine-based HSP strategy are currently underway. In cancers, such as metastatic melanoma, colorectal carcinoma, chronic myeloid leukemia, and renal cell carcinoma, HSP vaccines have been shown to be safe and associated with increased survival [198–201]. Parsa et al. reported a study in 12 patients with recurrent GBM, seven of the eight patients treated had a median survival time of 10.5 months compared to historical controls' median of 6.5 months [177]. Currently, two phase I/II clinical trials using the Grp 96 vaccine strategy are underway (NCT00293423, NCT00905060). Selected clinical trials using HSPs are summarized in Table 6.
Table 6.
Selected clinical trials using heat shock proteins (HSP).
| Reference | Patients | Immunologic response | Clinical response |
|---|---|---|---|
| [177] |
n = 12 recurrent GBM |
— | Median survival: 10.5 mo |
3. Challenges in the Tumor Microenvironment
3.1. Cell Populations
GBM-mediated immunosuppresion arises from coordinated interactions among the diverse cell populations, cytokines, and extracellular matrix proteins in the tumor microenvironment. The nature of these interactions is yet to be fully characterized, but is likely to be more complex than initially appreciated. For example, it has been shown that 20–90% of endothelial cells in GBM-associated vasculature harbor the same mutations as the tumor cells [202] and that a subpopulation of CD133+ tumor stem cells expresses vascular-endothelial cadherin (CD144) [203]. Taken together, these findings indicate that a significant number of GBM-associated endothelial cells may arise from tumor stem cells [204]. In addition, experiences with conventional therapies have highlighted how specific cell populations give rise to resistance. For example, tumor stem cells are largely radioresistant. A recent study by Tamura et al. found that tumors in a cohort of patients with recurrent grade III and IV gliomas following treatment with radiosurgery and external beam radiation therapy were significantly enriched for CD133+ cells [205]. Interestingly, additional cell populations have been implicated in this phenomenon as well. In vitro studies of GBM stem cell sensitivity have not clearly demonstrated that these cells are more radioresistant than CD133− tumor cells [206]. Based on these findings, Calabrese et al. have proposed that the resistance of glioma stem cells to radiotherapy may arise from interactions within the GBM microenvironment [207]. Supporting this theory is the observation that GBM stem cells tend to reside within perivascular niches, where interactions with endothelial cells appear to impart tumor stem cell radioresistance [204, 208]. Other lines of evidence indicate that extracellular matrix proteins and hypoxia within the tumor microenvironment may impart radioresistance in tumor stem cells. These two examples illustrate the fact that an effective immunotherapy must not only target tumor cells, but must also disrupt the immunosuppressive activities of a variety of cell populations in the tumor microenvironment.
3.2. Cytokines
GBM cell lines have long been known to express high levels of immunosuppressive cytokines [209]. However, our understanding of the origins of these cytokines and the roles they play in the tumor microenvironment represents one of the most significant challenges to cytokine-based therapies for GBM. A recent study by Rodriques et al. demonstrated that expression of IL-10, TGF-β, and B7-H1 is induced in normal human monocytes after exposure to GBM cells [37]. TGF-β has also been implicated in the transformation of vascular endothelial cells to a proangiogenic phenotype characteristically associated with GBM [46]. Other studies indicate that TGF-β and IL-10 are more highly expressed in CD133+ than in CD133− glioma cells and that elevated expression of these cytokines specifically within tumor stem cell population correlates with a poorer prognosis [45, 210]. In order to fully understand the relationship between specific cytokines and the variety of cell populations present in the GBM microenvironment, subclassification of these cell populations may be necessary. For example, it has been suggested that the level of TGF-β expression as well as the effects of TGF-β signaling may vary among cancer stem cell subtypes [211]. Another recent study has shown that exposing GBM cells to IFN-γ decreased TGF-β but increased expression of PD-1 ligand and Indoleamine-2,3-Dioxygenase (IDO) [212]. It is reasonable to speculate that other immunosuppressive cytokines exhibit comparably complex interactions.
4. Therapies Directed at the Immune Microenvironment
4.1. STAT3 Blockade
STAT3 is a member of the signal transducer and activator of transcription (STAT) family of transcription factors. The detailed activities of STAT3 in cancer are reviewed elsewhere [213]. In brief, STAT3 is activated when Janus kinases (JAKs) phosphorylate the cytoplasmic tail of activated IL-6 family cytokine receptors [214]. The phosphorylated receptor then recruits STAT1 and STAT3 via the Src homology 2 (SH2) domain of the STAT protein [214, 215]. JAK tyrosine kinase activity subsequently phosphorylates STAT3 on Tyr 705, leading to formation of a phosphorylated-STAT3 (p-STAT3) homodimer which translocates to the nucleus and binds several promoters which regulate cytokine expression, cell differentiation, proliferation, apoptosis, and angiogenesis [216–219]. Constitutive activation of STAT3 has been implicated in the tumorigenesis of many cancers both inside and outside of the CNS and has been shown to be sufficient to transform cells to a malignant phenotype in vitro [220].
Some authors have reported that p-STAT3 is present in high levels in GBM cell lines [221] and in greater than 75% of tumor tissue samples [222]; however, other authors have failed to corroborate these findings [27]. In tumors exhibiting high levels of STAT3 activity, this transcription factor has emerged as a critical convergence point for many pathways known to be associated with GBM growth and invasion. In addition, increased STAT3 activation has been correlated with shorter overall survival in a cohort of patients with GBM [222].
Numerous lines of evidence indicate a protumorigenic role for STAT3 in the GBM microenvironment. STAT3 activation has been shown to be increased in GBM under hypoxic conditions, leading to elevated expression of proangiogenic factors such as vascular endothelial growth factor (VEGF) and hypoxic inducible factor-1 (HIF-1) [223]. Furthermore, STAT3 inhibition results in a reduction in endothelial cell tube formation in vitro [216, 223]. STAT3 has also been implicated in tumor invasion and suppression of apoptosis. For example, Chen et al. recently demonstrated that STAT3 inhibition reduces expression of the proinvasive factor matrix metalloprotease-2 (MMP-2) and the antiapoptotic factors Bcl-xL and survivin [224]. STAT3 is also critical for maintaining tumor stem cells [225]. A recent study by Villalva et al. demonstrated that siRNA knockdown or inhibition of STAT3 with the small molecule inhibitor Stattic led to decreased GBM stem cell proliferation and inhibited neurosphere formation [226]. In addition to its roles in angiogenesis, tumor invasion, apoptosis, and maintenance of tumor stem cells, STAT3 is known to act as a potent inhibitor of both innate [227] and adaptive [228] immune responses. STAT3 also induces tolerance via Treg activity, potentially through an HIF-1-mediated mechanism [229].
Although STAT3 has been most extensively studied as a tumor-promoting factor in GBM, evidence has recently emerged to suggest that it may act alternately as a protumorigenic factor or a tumor suppressor based on the genetic background of the tumor [230]. The theory that STAT3 may exert tumor-suppressing effects in GBM originated from the observation that STAT3 plays a prominent role in astrocyte differentiation [231, 232]. Studies of STAT3−/− astrocytes have demonstrated that these cells exhibit increased proliferation and invasion, although this mutation is not sufficient to produce malignancy [230]. In addition, STAT3 suppresses malignant transformation of astrocytes deficient in PTEN in an orthotopic transplant model in SCID mice [230] and a correlation between PTEN mutation and low levels of STAT3 activity has also been reported in human GBMs [233]. Conversely, STAT3 appears to be protumorigenic in EGFRvIII-expressing tumors [230]. The details of STAT3's interaction with EGFRvIII are currently unknown; however, evidence from breast cancer cell lines suggests that EGFRvIII may translocate to the nucleus and alter the binding of STAT3 to DNA [234].
The multiplicity of pro-oncogenic effects ascribed to STAT3 makes this transcription factor an attractive target for immunotherapy. Strategies to block STAT3 in GBM have focused primarily on direct inhibition using RNA interference and small molecule inhibitors or indirect inhibition by targeting upstream kinases or regulatory SOCS proteins [221, 235–237]. Although STAT3 inhibition has yielded promising results in vitro, applying this approach to animal models of GBM has produced mixed outcomes. In light of the finding that STAT3 may be alternately protumorigenic or suppressive to tumor growth, additional research is needed to elucidate the role of STAT3 in a variety of genetic contexts, including the background genotype of the host.
Even if the correct patients are identified, the tumor microenvironment may pose a number of additional challenges to effective GBM therapy with STAT3 blockade. For example, although inhibiting STAT3 may overcome some of the immunosuppressive mechanisms employed by GBM, immune cells must still efficiently identify appropriate tumor-specific antigens in order to avoid immune editing. In addition, evidence has already emerged to suggest that cancer stem cells express a different immunosuppressive cytokine profile in response to STAT3 blockade than bulk tumor cells [238]. This finding highlights the principle that it will be critical to consider the effects of STAT3 inhibition on cytokine expression and signaling in the variety of cell populations present in the GBM microenvironment individually as well as in aggregate. Even if STAT3 inhibition results in generation of an antitumor immune response, this activity may be thwarted by activation of immune checkpoints such as PD-1 [29] and CTLA-4 [26]. Other barriers to STAT3 inhibition in the treatment of brain tumors include identifying small molecule inhibitors that can either cross the blood-brain barrier or be delivered locally. Nevertheless, STAT3 remains one of the most promising targets in immunotherapy for GBM and at least one small molecule inhibitor, WP1066, is currently in preclinical development.
4.2. Regulatory T Cell Depletion
Tregs are a CD25+, FoxP3+ subset of CD4+ helper T cells which suppress immune activation through interactions with T cells, B cells, NK cells, DCs, and macrophages [239–243]. Tregs have been shown to express CTLA-4, to decrease the secretion of IL-2 and IFN-γ [244], and to skew the immune response away from a cytotoxic Th1-mediated response in favor of a Th2 response [245]. Studies of human GBM tissue samples have reported tumor-infiltrating lymphocyte populations significantly enriched for Tregs [26]. GBM cells also appear to secrete high levels of CCL22 and CCL2, which facilitates Treg trafficking to the tumor [246]. In addition, high-grade gliomas have been reported to exhibit a higher density of Tregs than low-grade tumors [247]. These observations have led to interest in developing immunotherapies for GBM that target Tregs.
Tregs have been shown to be associated with a number of other known immunomodulatory pathways [248]. For example, the STAT3 inhibitor WP1066 has been shown to decrease Treg proliferation. In addition, CTLA-4 blockade may abrogate the immunosuppressive effects of Tregs in the tumor microenvironment without directly inhibiting their immunosuppressive properties [249–251]. Direct inhibition of Tregs is also possible with anti-CD25 antibodies and has been shown to improve survival in mouse glioma models [252]. A number of other approaches have also been proposed to inhibit Tregs in gliomas. These approaches are reviewed in detail elsewhere [253].
Indirect evidence for the efficacy of Treg depletion in human glioma comes from combining immunotherapy with cyclophosphamide, which preferentially inhibits Treg activity at low doses [254]. Clinical trials combining cyclophosphamide with a dendritic cell vaccine for renal cell carcinoma [255] or with a protein antigen vaccine for breast cancer [256] have demonstrated that the addition of cyclophosphamide augmented the antitumor effect. Blocking antibodies against CTLA-4 [249] and CD25 [252] have been shown to be effective against gliomas in mice; however, neither of these approaches has been evaluated in clinical trials.
One of the primary challenges impeding the development and implementation of Treg depletion for treatment of GBM is precisely delineating how these cells interact with the other immunosuppressive factors in the tumor environment. Despite numerous lines of evidence implicating a protumorigenic role for Tregs, and the theoretical appeal of these cells as targets for immunotherapy, fundamental questions about the role of Tregs in GBM tumorigenesis remain unanswered. For example, several studies have failed to convincingly correlate the density of tumor-infiltrating lymphocytes with prognosis in human gliomas [257–259]. Because these studies did not account for lymphocyte activity, it has been proposed that local immunosuppression in GBMs results from inhibition of T cell function secondary to an enriched population of Tregs [247]. Studies directly evaluating the relationship between Treg fractions and survival in patients with GBM, however, have not demonstrated a reliable correlation [260].
Tregs have been implicated in association with many other known immunosuppressive factors in the GBM microenvironment, such as CTLA-4 and STAT3. The lack of a clearly defined mechanism underlying the interactions between Tregs and CTLA-4, however, precludes the development of maximally effective combination therapies. The finding that STAT3 blockade inhibits Treg function is intriguing and deserves further exploration. In particular, STAT3 signaling may coordinate the activities of Tregs with other cell populations in the tumor microenvironment, including tumor stem cells [238]. Ultimately, defining the roles of Tregs in GBM represents a critical step toward understanding the mechanisms underlying the immunosuppressive tumor microenvironment and may serve as a valuable target for intervention.
5. Conclusion
We have reviewed challenges presented by the tumor microenvironment and many of the current approaches to immunotherapy for GBM. It is becoming increasingly clear that GBM-mediated immunosuppression arises not only from the intrinsic properties of tumor cells, but from the ability of these cells to coordinate the activities of a diverse set of cell types and signaling pathways in the tumor microenvironment. Therefore, the development of effective immunotherapies will require careful study of how intervening at any point in this system alters the dynamics of these interactions. For example, the finding that treatment with IFN-γ increases expression of PD-L1 demonstrates potentially redundant immunosuppressive mechanisms. The differential effects of STAT3 blockade based on tumor genetics highlights the importance of developing molecular classification schemes that reflect responsiveness to various immunotherapy approaches. Furthermore, the finding that tumor stem cells may differentiate into vascular endothelial cells suggests potential interactions between tumor endothelial cells and immune cells that have not yet been elucidated. With these challenges, however, comes enormous potential to precisely target the defense mechanisms in GBM and tip the balance back in favor of the immune system.
Authors' Contribution
C. Jackson and J. Ruzevick contributed equally to this work.
Abbreviations
- CR:
Complete response
- PR:
Partial response
- SD:
Stable disease
- TTP:
Time to progression
- GBM:
Glioblastoma multiforme
- AA:
Anaplastic astrocytoma.
References
- 1.Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England Journal of Medicine. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59(8):1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 3.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 4.Gulley JL, Drake CG. Immunotherapy for prostate cancer: recent advances, lessons learned, and areas for further research. Clinical Cancer Research. 2011;17(12):3884–3891. doi: 10.1158/1078-0432.CCR-10-2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cameron F, Whiteside G, Perry C. Ipilimumab: first global approval. Drugs. 2011;71(8):1093–1104. doi: 10.2165/11594010-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 6.Karan D, Dubey S, Van Veldhuizen P, Holzbeierlein JM, Tawfik O, Thrasher JB. Dual antigen target-based immunotherapy for prostate cancer eliminates the growth of established tumors in mice. Immunotherapy. 2011;3(6):735–746. doi: 10.2217/imt.11.59. [DOI] [PubMed] [Google Scholar]
- 7.Disis ML. Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer Immunology, Immunotherapy. 2011;60(3):433–442. doi: 10.1007/s00262-010-0960-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pachter JS, De Vries HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. Journal of Neuropathology and Experimental Neurology. 2003;62(6):593–604. doi: 10.1093/jnen/62.6.593. [DOI] [PubMed] [Google Scholar]
- 9.Fabry Z, Raine CS, Hart MN. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunology Today. 1994;15(5):218–224. doi: 10.1016/0167-5699(94)90247-X. [DOI] [PubMed] [Google Scholar]
- 10.Lampson LA, Hickey WF. Monoclonal antibody analysis of MHC expression in human brain biopsies: tissue ranging from ’histologically normal’ to that showing different levels of glial tumor involvement. Journal of Immunology. 1986;136(11):4054–4062. [PubMed] [Google Scholar]
- 11.Wintterle S, Schreiner B, Mitsdoerffer M, et al. Expression of the B7-Related Molecule B7-H1 by Glioma Cells: A Potential Mechanism of Immune Paralysis. Cancer Research. 2003;63(21):7462–7467. [PubMed] [Google Scholar]
- 12.Sloan DJ, Wood MJ, Charlton HM. The immune response to intracerebral neural grafts. Trends in Neurosciences. 1991;14(8):341–346. doi: 10.1016/0166-2236(91)90159-r. [DOI] [PubMed] [Google Scholar]
- 13.Tambur AR. Transplantation immunology and the central nervous system. Neurological Research. 2004;26(3):243–255. doi: 10.1179/016164104225013932. [DOI] [PubMed] [Google Scholar]
- 14.Yang I, Han SJ, Kaur G, Crane C, Parsa AT. The role of microglia in central nervous system immunity and glioma immunology. Journal of Clinical Neuroscience. 2010;17(1):6–10. doi: 10.1016/j.jocn.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussain SF, Heimberger AB. Immunotherapy for human glioma: Innovative approaches and recent results. Expert Review of Anticancer Therapy. 2005;5(5):777–790. doi: 10.1586/14737140.5.5.777. [DOI] [PubMed] [Google Scholar]
- 16.Serot JM, Foliguet B, Béné MC, Faure GC. Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. NeuroReport. 1997;8(8):1995–1998. doi: 10.1097/00001756-199705260-00039. [DOI] [PubMed] [Google Scholar]
- 17.Goldmann J, Kwidzinski E, Brandt C, Mahlo J, Richter D, Bechmann I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. Journal of Leukocyte Biology. 2006;80(4):797–801. doi: 10.1189/jlb.0306176. [DOI] [PubMed] [Google Scholar]
- 18.Weller RO, Engelhardt B, Phillips MJ. Lymphocyte targeting of the central nervous system: A review of afferent and efferent CNS-immune pathways. Brain Pathology. 1996;6(3):275–288. doi: 10.1111/j.1750-3639.1996.tb00855.x. [DOI] [PubMed] [Google Scholar]
- 19.Calzascia T, Masson F, Di Berardino-Besson W, et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity. 2005;22(2):175–184. doi: 10.1016/j.immuni.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Quattrocchi KB, Miller CH, Cush S, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. Journal of Neuro-Oncology. 1999;45(2):141–157. doi: 10.1023/a:1006293606710. [DOI] [PubMed] [Google Scholar]
- 21.Brabb T, Von Dassow P, Ordonez N, Schnabel B, Duke B, Goverman J. In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. Journal of Experimental Medicine. 2000;192(6):871–880. doi: 10.1084/jem.192.6.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gigliotti F, Lee D, Insel RA, Scheld WM. IgG penetration into the cerebrospinal fluid in a rabbit model of meningitis. Journal of Infectious Diseases. 1987;156(2):394–398. doi: 10.1093/infdis/156.2.394. [DOI] [PubMed] [Google Scholar]
- 23.Stemmler HJ, Schmitt M, Willems A, Bernhard H, Harbeck N, Heinemann V. Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2-positive breast cancer patients with brain metastases and impairment of blood-brain barrier. Anti-Cancer Drugs. 2007;18(1):23–28. doi: 10.1097/01.cad.0000236313.50833.ee. [DOI] [PubMed] [Google Scholar]
- 24.Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro-Oncology. 2011;13(1):3–13. doi: 10.1093/neuonc/noq169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rolle CE, Sengupta S, Lesniak MS. Challenges in clinical design of immunotherapy trials for malignant glioma. Neurosurgery Clinics of North America. 2010;21(1):201–214. doi: 10.1016/j.nec.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro-Oncology. 2006;8(3):234–243. doi: 10.1215/15228517-2006-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Molecular Cancer Research. 2008;6(5):675–684. doi: 10.1158/1541-7786.MCR-07-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Tomaso T, Mazzoleni S, Wang E, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clinical Cancer Research. 2010;16(3):800–813. doi: 10.1158/1078-0432.CCR-09-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobs JFM, Idema AJ, Bol KF, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro-Oncology. 2009;11(4):394–402. doi: 10.1215/15228517-2008-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wainwright DA, Sengupta S, Han Y, Ulasov IV, Lesniak MS. The presence of IL-17A and T helper 17 cells in experimental mouse brain tumors and human gliom. PLoS ONE. 2010;5(10, article e15390) doi: 10.1371/journal.pone.0015390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 32.Brennan C, Momota H, Hambardzumyan D, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE. 2009;4(11, article e7752) doi: 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harling-Berg C, Knopf PM, Merriam J, Cserr HF. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. Journal of Neuroimmunology. 1989;25(2-3):185–193. doi: 10.1016/0165-5728(89)90136-7. [DOI] [PubMed] [Google Scholar]
- 34.Harling-Berg CJ, Hallett JJ, Park JT, Knopf PM. Hierarchy of immune responses to antigen in the normal brain. Current Topics in Microbiology and Immunology. 2002;265:1–22. doi: 10.1007/978-3-662-09525-6_1. [DOI] [PubMed] [Google Scholar]
- 35.Harling-Berg CJ, Park JT, Knopf PM. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. Journal of Neuroimmunology. 1999;101(2):111–127. doi: 10.1016/s0165-5728(99)00130-7. [DOI] [PubMed] [Google Scholar]
- 36.Jansen T, Tyler B, Mankowski JL, et al. FasL gene knock-down therapy enhances the antiglioma immune response. Neuro-Oncology. 2010;12(5):482–489. doi: 10.1093/neuonc/nop052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodrigues JC, Gonzalez GC, Zhang L, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro-Oncology. 2010;12(4):351–365. doi: 10.1093/neuonc/nop023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubois CM, Laprise MH, Blanchette F, Gentry LE, Leduc R. Processing of transforming growth factor β1 precursor by human furin convertase. Journal of Biological Chemistry. 1995;270(18):10618–10624. doi: 10.1074/jbc.270.18.10618. [DOI] [PubMed] [Google Scholar]
- 39.Tritschler I, Gramatzki D, Capper D, et al. Modulation of TGF-β activity by latent TGF-β-binding protein 1 in human malignant glioma cells. International Journal of Cancer. 2009;125(3):530–540. doi: 10.1002/ijc.24443. [DOI] [PubMed] [Google Scholar]
- 40.Kanzaki T, Olofsson A, Moren A, et al. TGF-β1 binding protein: A component of the large latent complex of TGF-β1 with multiple repeat sequences. Cell. 1990;61(6):1051–1061. doi: 10.1016/0092-8674(90)90069-q. [DOI] [PubMed] [Google Scholar]
- 41.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFβ activation. Journal of Cell Science. 2003;116, part 2:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 42.Kirkbride KC, Blobe GC. Inhibiting the TGF-β signalling pathway as a means of cancer immunotherapy. Expert Opinion on Biological Therapy. 2003;3(2):251–261. doi: 10.1517/14712598.3.2.251. [DOI] [PubMed] [Google Scholar]
- 43.de Martin R, Haendler B, Hofer-Warbinek R, et al. Complementary DNA for human glioblastoma-derived T cell suppressor factor, a novel member of the transforming growth factor-beta gene family. EMBO Journal. 1987;6(12):3673–3677. doi: 10.1002/j.1460-2075.1987.tb02700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wrann M, Bodmer S, de Martin R, et al. T cell suppressor factor from human glioblastoma cells is a 12.5-kd protein closely related to transforming growth factor-beta. EMBO Journal. 1987;6(6):1633–1636. doi: 10.1002/j.1460-2075.1987.tb02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, et al. TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010;18(6):655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 46.Pen A, Moreno MJ, Durocher Y, Deb-Rinker P, Stanimirovic DB. Glioblastoma-secreted factors induce IGFBP7 and angiogenesis by modulating Smad-2-dependent TGF-β signaling. Oncogene. 2008;27(54):6834–6844. doi: 10.1038/onc.2008.287. [DOI] [PubMed] [Google Scholar]
- 47.Arslan F, Bosserhoff AK, Nickl-Jockschat T, Doerfelt A, Bogdahn U, Hau P. The role of versican isoforms V0/V1 in glioma migration mediated by transforming growth factor-β2. British Journal of Cancer. 2007;96(10):1560–1568. doi: 10.1038/sj.bjc.6603766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tabatabai G, Frank B, Möhle R, Weller M, Wick W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-β-dependent HIF-1α-mediated induction of CXCL12. Brain. 2006;129, part 9:2426–2435. doi: 10.1093/brain/awl173. [DOI] [PubMed] [Google Scholar]
- 49.Ehrhart EJ, Segarini P, Tsang MLS, Carroll AG, Barcellos-Hoff MH. Latent transforming growth factor β1 activation in situ: quantitative and functional evidence after low-dose γ-irradiation. FASEB Journal. 1997;11(12):991–1002. doi: 10.1096/fasebj.11.12.9337152. [DOI] [PubMed] [Google Scholar]
- 50.Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Research. 2004;64(21):7985–7994. doi: 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 51.Klein B, Loven D, Lurie H, et al. The effect of irradiation on expression of HLA class I antigens in human brain tumors in culture. Journal of Neurosurgery. 1994;80(6):1074–1077. doi: 10.3171/jns.1994.80.6.1074. [DOI] [PubMed] [Google Scholar]
- 52.Olschowka JA, Kyrkanides S, Harvey BK, et al. ICAM-1 induction in the mouse CNS following irradiation. Brain, Behavior, and Immunity. 1997;11(4):273–285. doi: 10.1006/brbi.1997.0506. [DOI] [PubMed] [Google Scholar]
- 53.Jobling MF, Mott JD, Finnegan MT, et al. Isoform-specific activation of latent transforming growth factor β (LTGF-β) by reactive oxygen species. Radiation Research. 2006;166(6):839–848. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 54.Wu A, Wei J, Kong LY, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology. 2010;12(11):1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shao C, Folkard M, Prise KM. Role of TGF-β1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene. 2008;27(4):434–440. doi: 10.1038/sj.onc.1210653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wick W, Platten M, Weller M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-β . Journal of Neuro-Oncology. 2001;53(2):177–185. doi: 10.1023/a:1012209518843. [DOI] [PubMed] [Google Scholar]
- 57.Kim GH, Kang SY, Kwak HJ, et al. Transforming growth factor-β1 bioassay involving matrix metalloproteinase-2 induction. Journal of Interferon and Cytokine Research. 2010;30(9):667–672. doi: 10.1089/jir.2009.0058. [DOI] [PubMed] [Google Scholar]
- 58.Bogdahn U, Hau P, Stockhammer G, et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro-Oncology. 2011;13(1):132–142. doi: 10.1093/neuonc/noq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaschinski F, Rothhammer T, Jachimczak P, Seitz C, Schneider A, Schlingensiepen KH. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-beta2. doi: 10.2174/138920111798808266. Current Pharmaceutical Biotechnology. In press. [DOI] [PubMed] [Google Scholar]
- 60.Barcellos-Hoff MH, Newcomb EW, Zagzag D, Narayana A. Therapeutic targets in malignant glioblastoma microenvironment. Seminars in Radiation Oncology. 2009;19(3):163–170. doi: 10.1016/j.semradonc.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gansbacher B, Zier K, Daniels B, Cronin K, Bannerji R, Gilboa E. Interleukin 2 gene transfer into tumor cells abrogates tumorigenicity and induces protective immunity. Journal of Experimental Medicine. 1990;172(4):1217–1224. doi: 10.1084/jem.172.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saris SC, Rosenberg SA, Friedman RB, Rubin JT, Barba D, Oldfield EH. Penetration of recombinant interleukin-2 across the blood-cerebrospinal fluid barrier. Journal of Neurosurgery. 1988;69(1):29–34. doi: 10.3171/jns.1988.69.1.0029. [DOI] [PubMed] [Google Scholar]
- 63.Merchant RE, McVicar DW, Merchant LH, Young HF. Treatment of recurrent malignant glioma by repeated intracerebral injections of human recombinant interleukin-2 alone or in combination with systemic interferon-α. Results of a phase I clinical trial. Journal of Neuro-Oncology. 1992;12(1):75–83. doi: 10.1007/BF00172459. [DOI] [PubMed] [Google Scholar]
- 64.Barba D, Saris SC, Holder C, Rosenberg SA, Oldfield EH. Intratumoral LAK cell and interleukin-2 therapy of human gliomas. Journal of Neurosurgery. 1989;70(2):175–182. doi: 10.3171/jns.1989.70.2.0175. [DOI] [PubMed] [Google Scholar]
- 65.Colombo F, Barzon L, Franchin E, et al. Combined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical results. Cancer Gene Therapy. 2005;12(10):835–848. doi: 10.1038/sj.cgt.7700851. [DOI] [PubMed] [Google Scholar]
- 66.Ewend MG, Thompson RC, Anderson R, et al. Intracranial paracrine interleukin-2 therapy stimulates prolonged antitumor immunity that extends outside the Central Nervous System. Journal of Immunotherapy. 2000;23(4):438–448. doi: 10.1097/00002371-200007000-00007. [DOI] [PubMed] [Google Scholar]
- 67.Pedretti M, Verpelli C, Mårlind J, et al. Combination of temozolomide with immunocytokine F16-IL2 for the treatment of glioblastoma. British Journal of Cancer. 2010;103(6):827–836. doi: 10.1038/sj.bjc.6605832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fujita M, Scheurer ME, Decker SA, et al. Role of type 1 IFNs in antiglioma immunosurveillance - Using mouse studies to guide examination of novel prognostic markers in humans. Clinical Cancer Research. 2010;16(13):3409–3419. doi: 10.1158/1078-0432.CCR-10-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farkkila M, Jaaskelainen J, Kallio M, et al. Randomised, controlled study of intratumoral recombinant γ-interferon treatment in newly diagnosed glioblastoma. British Journal of Cancer. 1994;70(1):138–141. doi: 10.1038/bjc.1994.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolff JEA, Wagner S, Reinert C, et al. Maintenance treatment with interferon-gamma and low-dose cyclophosphamide for pediatric high-grade glioma. Journal of Neuro-Oncology. 2006;79(3):315–321. doi: 10.1007/s11060-006-9147-8. [DOI] [PubMed] [Google Scholar]
- 71.Allen J, Packer R, Bleyer A, Zeltzer P, Prados M, Nirenberg A. Recombinant interferon beta: a phase I-II trial in children with recurrent brain tumors. Journal of Clinical Oncology. 1991;9(5):783–788. doi: 10.1200/JCO.1991.9.5.783. [DOI] [PubMed] [Google Scholar]
- 72.Fetell MR, Housepian EM, Oster MW, et al. Intratumor administration of beta-interferon in recurrent malignant gliomas. A Phase I clinical and laboratory study. Cancer. 1990;65(1):78–83. doi: 10.1002/1097-0142(19900101)65:1<78::aid-cncr2820650117>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 73.Mahaley MS, Dropcho EJ, Bertsch L, Tirey T, Gillespie GY. Systemic beta-interferon therapy for recurrent gliomas: a brief report. Journal of Neurosurgery. 1989;71(5, part 1):639–641. doi: 10.3171/jns.1989.71.5.0639. [DOI] [PubMed] [Google Scholar]
- 74.Wakabayashi T, Kayama T, Nishikawa R, et al. A multicenter phase I trial of interferon-β and temozolomide combination therapy for high-grade gliomas (INTEGRA Study) Japanese Journal of Clinical Oncology. 2008;38(10):715–718. doi: 10.1093/jjco/hyn095. [DOI] [PubMed] [Google Scholar]
- 75.Buckner JC, Brown LD, Kugler JW, et al. Phase II evaluation of recombinant interferon alpha and BCNU in recurrent glioma. Journal of Neurosurgery. 1995;82(3):430–435. doi: 10.3171/jns.1995.82.3.0430. [DOI] [PubMed] [Google Scholar]
- 76.Buckner JC, Schomberg PJ, McGinnis WL, et al. A Phase III study of radiation therapy plus carmustine with or without recombinant interferon-α in the treatment of patients with newly diagnosed high-grade glioma. Cancer. 2001;92(2):420–433. doi: 10.1002/1097-0142(20010715)92:2<420::aid-cncr1338>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 77.Olson JJ, McKenzie E, Skurski-Martin M, Zhang Z, Brat D, Phuphanich S. Phase I analysis of BCNU-impregnated biodegradable polymer wafers followed by systemic interferon alfa-2b in adults with recurrent glioblastoma multiforme. Journal of Neuro-Oncology. 2008;90(3):293–299. doi: 10.1007/s11060-008-9660-z. [DOI] [PubMed] [Google Scholar]
- 78.Villeneuve J, Tremblay P, Vallières L. Tumor necrosis factor reduces brain tumor growth by enhancing macrophage recruitment and microcyst formation. Cancer Research. 2005;65(9):3928–3936. doi: 10.1158/0008-5472.CAN-04-3612. [DOI] [PubMed] [Google Scholar]
- 79.Benedetti S, Bruzzone MG, Pollo B, et al. Eradication of rat malignant gliomas by retroviral-mediated, in vivo delivery of the interleukin 4 gene. Cancer Research. 1999;59(3):645–652. [PubMed] [Google Scholar]
- 80.Okada H, Lieberman FS, Walter KA, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. Journal of Translational Medicine. 2007;5, article 67 doi: 10.1186/1479-5876-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu Y, Ehtesham M, Samoto K, et al. In situ adenoviral interleukin 12 gene transfer confers potent and long-lasting cytotoxic immunity in glioma. Cancer Gene Therapy. 2002;9(1):9–15. doi: 10.1038/sj.cgt.7700399. [DOI] [PubMed] [Google Scholar]
- 82.Dimeco F, Rhines LD, Hanes J, et al. Paracrine delivery of IL-12 against intracranial 9L gliosarcoma in rats. Journal of Neurosurgery. 2000;92(3):419–427. doi: 10.3171/jns.2000.92.3.0419. [DOI] [PubMed] [Google Scholar]
- 83.Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, Yu JS. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Research. 2002;62(20):5657–5663. [PubMed] [Google Scholar]
- 84.Kikuchi T, Akasaki Y, Abe T, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. Journal of Immunotherapy. 2004;27(6):452–459. doi: 10.1097/00002371-200411000-00005. [DOI] [PubMed] [Google Scholar]
- 85.Herrlinger U, Kramm CM, Johnston KM, et al. Vaccination for experimental gliomas using GM-CSF-transduced glioma cells. Cancer Gene Therapy. 1997;4(6):345–352. [PubMed] [Google Scholar]
- 86.Butowski N. Immunostimulants for Malignant Gliomas. Neurosurgery Clinics of North America. 2010;21(1):53–65. doi: 10.1016/j.nec.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 87.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. Journal of Clinical Investigation. 2006;116(5):1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15(2):114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yong VW, Marks S. The interplay between the immune and central nervous systems in neuronal injury. Neurology. 2010;74(supplement 1):S9–S16. doi: 10.1212/WNL.0b013e3181c97d04. [DOI] [PubMed] [Google Scholar]
- 90.Dillman RO, Duma CM, Ellis RA, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. Journal of Immunotherapy. 2009;32(9):914–919. doi: 10.1097/CJI.0b013e3181b2910f. [DOI] [PubMed] [Google Scholar]
- 91.Jacobs SK, Wilson DJ, Kornblith PL, Grimm EA. Interleukin-2 or autologous lymphokine-activated killer cell treatment of malignant glioma: phase I trial. Cancer Research. 1986;46(4, part 2):2101–2104. [PubMed] [Google Scholar]
- 92.Lillehei KO, Mitchell DH, Johnson SD, McCleary EL, Kruse CA. Long-term follow-up of patients with recurrent malignant gliomas treated with adjuvant adoptive immunotherapy. Neurosurgery. 1991;28(1):16–23. doi: 10.1097/00006123-199101000-00003. [DOI] [PubMed] [Google Scholar]
- 93.Hayes RL, Koslow M, Hiesiger EM, et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. 1995;76(5):840–852. doi: 10.1002/1097-0142(19950901)76:5<840::aid-cncr2820760519>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 94.Dillman RO, Duma CM, Schiltz PM, et al. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. Journal of Immunotherapy. 2004;27(5):398–404. doi: 10.1097/00002371-200409000-00009. [DOI] [PubMed] [Google Scholar]
- 95.Itoh K, Sawamura Y, Hosokawa M, Kobayashi H. Scintigraphy with In-111 labeled lymphokine-activated killer cells of malignant brain tumor. Radiation Medicine—Medical Imaging and Radiation Oncology. 1988;6(6):276–281. [PubMed] [Google Scholar]
- 96.Yamasaki T, Kikuchi H. An experimental approach to specific adoptive immunotherapy for malignant brain tumors. Archiv fur Japanische Chirurgie. 1989;58(6):485–492. [PubMed] [Google Scholar]
- 97.Tsurushima H, Liu SQ, Tuboi K, et al. Reduction of end-stage malignant glioma by injection with autologous cytotoxic T lymphocytes. Japanese Journal of Cancer Research. 1999;90(5):536–545. doi: 10.1111/j.1349-7006.1999.tb00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Plautz GE, Miller DW, Barnett GH, et al. T cell adoptive immunotherapy of newly diagnosed gliomas. Clinical Cancer Research. 2000;6(6):2209–2218. [PubMed] [Google Scholar]
- 99.Kahlon KS, Brown C, Cooper LJN, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Research. 2004;64(24):9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 100.Yaghoubi SS, Jensen MC, Satyamurthy N, et al. Noninvasive detection of therapeutic cytolytic T cells with 18 F-FHBG PET in a patient with glioma. Nature Clinical Practice Oncology. 2009;6(1):53–58. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ahmed N, Salsman VS, Kew Y, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clinical Cancer Research. 2010;16(2):474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hsu FJ, Benike C, Fagnoni F, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nature Medicine. 1996;2(1):52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 103.Kugler A, Stuhler G, Walden P, et al. Regression of human metastatic renal cell carcinoma after vaccination with tumor cell-dendritic cell hybrids. Nature Medicine. 2000;6(3):332–336. doi: 10.1038/73193. [DOI] [PubMed] [Google Scholar]
- 104.Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nature Medicine. 1998;4(3):328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 105.Tjoa BA, Simmons SJ, Elgamal A, et al. Follow-up evaluation of a phase II prostate cancer vaccine trial. Prostate. 1999;40(2):125–129. doi: 10.1002/(sici)1097-0045(19990701)40:2<125::aid-pros8>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 106.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 107.Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFNγ receptors. Immunity. 1994;1(6):447–456. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 108.Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(13):7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shankaran V, Ikeda H, Bruce AT, et al. IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 110.Algarra I, Cabrera T, Garrido F. The HLA crossroad in tumor immunology. Human Immunology. 2000;61(1):65–73. doi: 10.1016/s0198-8859(99)00156-1. [DOI] [PubMed] [Google Scholar]
- 111.Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Advances in Immunology. 2000;74:181–273. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- 112.Wong LH, Krauer KG, Hatzinisiriou I, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3γ . Journal of Biological Chemistry. 1997;272(45):28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 113.Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bigner SH, Humphrey PA, Wong AJ, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Research. 1990;50(24):8017–8022. [PubMed] [Google Scholar]
- 115.Libermann TA, Nusbaum HR, Razon N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313(5998):144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 116.Li G, Mitra S, Wong AJ. The epidermal growth factor variant III peptide vaccine for treatment of malignant gsliomas. Neurosurgery Clinics of North America. 2010;21(1):87–93. doi: 10.1016/j.nec.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 117.Fernandes H, Cohen S, Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/ΔEGFR) expressed in cancer cells. Journal of Biological Chemistry. 2001;276(7):5375–5383. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- 118.Humphrey PA, Wong AJ, Vogelstein B, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(11):4207–4211. doi: 10.1073/pnas.87.11.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Research. 1995;55(14):3140–3148. [PubMed] [Google Scholar]
- 120.Purev E, Cai D, Miller E, et al. Immune responses of breast cancer patients to mutated epidermal growth factor receptor (EGF-RvIII, Delta EGF-R, and de2-7 EGF-R) The Journal of Immunology. 2004;173(10):6472–6480. doi: 10.4049/jimmunol.173.10.6472. [DOI] [PubMed] [Google Scholar]
- 121.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Seminars in Immunology. 2008;20(5):267–275. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ciesielski MJ, Kazim AL, Barth RF, Fenstermaker RA. Cellular antitumor immune response to a branched lysine multiple antigenic peptide containing epitopes of a common tumor-specific antigen in a rat glioma model. Cancer Immunology, Immunotherapy. 2005;54(2):107–119. doi: 10.1007/s00262-004-0576-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harris JR, Markl J. Keyhole limpet hemocyanin (KLH): a biomedical review. Micron. 1999;30(6):597–623. doi: 10.1016/s0968-4328(99)00036-0. [DOI] [PubMed] [Google Scholar]
- 124.Heimberger AB, Crotty LE, Archer GE, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clinical Cancer Research. 2003;9(11):4247–4254. [PubMed] [Google Scholar]
- 125.Moscatello DK, Ramirez G, Wong AJ. A naturally occurring mutant human epidermal growth factor receptor as a target for peptide vaccine immunotherapy of tumors. Cancer Research. 1997;57(8):1419–1424. [PubMed] [Google Scholar]
- 126.Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-Oncology. 2003;5(2):79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Heimberger A. Tumor-specific peptide vaccination in newly-diagnosed patients with GBM. Journal of Clinical Oncology. 2006;24:2275–2282. [Google Scholar]
- 128.Emens LA, Reilly RT, Jaffee EM. Cancer vaccines in combination with multimodality therapy. Cancer Treatment and Research. 2005;123:227–245. doi: 10.1007/0-387-27545-2_10. [DOI] [PubMed] [Google Scholar]
- 129.Debinski W, Gibo DM. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor-associated cancer/testis antigen. Molecular Medicine. 2000;6(5):440–449. [PMC free article] [PubMed] [Google Scholar]
- 130.Eguchi J, Hatano M, Nishimura F, et al. Identification of interleukin-13 receptor α2 peptide analogues capable of inducing improved antiglioma CTL responses. Cancer Research. 2006;66(11):5883–5891. doi: 10.1158/0008-5472.CAN-06-0363. [DOI] [PubMed] [Google Scholar]
- 131.Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor α2 in glioblastoma multiforme: implications for targeted therapies. Cancer Research. 2007;67(17):7983–7986. doi: 10.1158/0008-5472.CAN-07-1493. [DOI] [PubMed] [Google Scholar]
- 132.Okano F, Storkus WJ, Chambers WH, Pollack IF, Okada H. Identification of a novel HLA-A*0201-restricted, cytotoxic T lymphocyte epitope in a human glioma-associated antigen, interleukin 13 receptor α2 chain. Clinical Cancer Research. 2002;8(9):2851–2855. [PubMed] [Google Scholar]
- 133.Kawakami M, Leland P, Kawakami K, Puri RK. Mutation and functional analysis of IL-13 receptors in human malignant glioma cells. Oncology Research. 2001;12(11-12):459–467. doi: 10.3727/096504001108747468. [DOI] [PubMed] [Google Scholar]
- 134.Debinski W, Obiri NI, Pastan I, Puri RK. A novel chimeric protein composed of interleukin 13 and Pseudomonas exotoxin is highly cytotoxic to human carcinoma cells expressing receptors for interleukin 13 and interleukin 4. Journal of Biological Chemistry. 1995;270(28):16775–16780. doi: 10.1074/jbc.270.28.16775. [DOI] [PubMed] [Google Scholar]
- 135.Husain SR, Joshi BH, Puri RK. Interleukin-13 receptor as a unique target for anti-glioblastoma therapy. International Journal of Cancer. 2001;92(2):168–175. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1182>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 136.Husain SR, Puri RK. Interleukin-13 fusion cytotoxin as a potent targeted agent for AIDS- Kaposi’s sarcoma xenograft. Blood. 2000;95(11):3506–3513. [PubMed] [Google Scholar]
- 137.Kawakami K, Kawakami M, Joshi BH, Puri RK. Interleukin-13 receptor-targeted cancer therapy in an immunodeficient animal model of human head and neck cancer. Cancer Research. 2001;61(16):6194–6200. [PubMed] [Google Scholar]
- 138.Maini A, Hillman G, Haas GP, et al. Interleukin-13 receptors on human prostate carcinoma cell lines represent a novel target for a chimeric protein composed of IL-13 and a mutated form of pseudomonas exotoxin. Journal of Urology. 1997;158(3, part 1):948–953. doi: 10.1097/00005392-199709000-00077. [DOI] [PubMed] [Google Scholar]
- 139.Kawakami K, Kioi M, Liu Q, Kawakami M, Puri RK. Evidence that IL-13R alpha2 chain in human glioma cells is responsible for the antitumor activity mediated by receptor-directed cytotoxin therapy. Journal of Immunotherapy. 2005;28(3):193–202. doi: 10.1097/01.cji.0000161393.04207.e1. [DOI] [PubMed] [Google Scholar]
- 140.Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995;345(8956):1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 141.Kunwar S, Chang SM, Prados MD, et al. Safety of intraparenchymal convection-enhanced delivery of cintredekin besudotox in early-phase studies. Neurosurgical Focus. 2006;20(4):p. E15. [PubMed] [Google Scholar]
- 142.Rand RW, Kreitman RJ, Patronas N, Varricchio F, Pastan I, Puri RK. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clinical Cancer Research. 2000;6(6):2157–2165. [PubMed] [Google Scholar]
- 143.Blancher A, Roubinet F, Grancher AS, et al. Local immunotherapy of recurrent glioblastoma multiforme by intracerebral perfusion of interleukin-2 and LAK cells. European Cytokine Network. 1993;4(5):331–341. [PubMed] [Google Scholar]
- 144.Boiardi A, Silvani A, Ruffini PA, et al. Loco-regional immunotherapy with recombinant interleukin-2 and adherent lymphokine-activated killer cells (A-LAK) in recurrent glioblastoma patients. Cancer Immunology Immunotherapy. 1994;39(3):193–197. doi: 10.1007/BF01533386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hayes RL, Arbit E, Odaimi M, et al. Adoptive cellular immunotherapy for the treatment of malignant gliomas. Critical Reviews in Oncology/Hematology. 2001;39(1-2):31–42. doi: 10.1016/s1040-8428(01)00122-6. [DOI] [PubMed] [Google Scholar]
- 146.Jeffes EWB, Beamer YB, Jacques S, et al. Therapy of recurrent high grade gliomas with surgery, and autologous mitogen activated IL-2 stimulated killer (MAK) lymphocytes: I. Enhancement of MAK lytic activity and cytokine production by PHA and clinical use of PHA. Journal of Neuro-Oncology. 1993;15(2):141–155. doi: 10.1007/BF01053935. [DOI] [PubMed] [Google Scholar]
- 147.Sankhla SK, Nadkarni JS, Bhagwati SN. Adoptive immunotherapy using lymphokine-activated killer (LAK) cells and interleukin-2 for recurrent malignant primary brain tumors. Journal of Neuro-Oncology. 1996;27(2):133–140. doi: 10.1007/BF00177476. [DOI] [PubMed] [Google Scholar]
- 148.Holladay FP, Heitz-Turner T, Bayer WL, Wood GW. Autologous tumor cell vaccination combined with adoptive cellular immunotherapy in patients with grade III/IV astrocytoma. Journal of Neuro-Oncology. 1996;27(2):179–189. doi: 10.1007/BF00177482. [DOI] [PubMed] [Google Scholar]
- 149.Kitahara T, Watanabe O, Yamaura A, et al. Establishment of interleukin 2 dependent cytotoxic T lymphocyte cell line specific for autologous brain tumor and its intracranial administration for therapy of the tumor. Journal of Neuro-Oncology. 1987;4(4):329–336. doi: 10.1007/BF00195603. [DOI] [PubMed] [Google Scholar]
- 150.Kruse CA, Cepeda L, Owens B, Johnson SD, Stears J, Lillehei KO. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic T lymphocytes and interleukin-2. Cancer Immunology Immunotherapy. 1997;45(2):77–87. doi: 10.1007/s002620050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Plautz GE, Barnett GH, Miller DW, et al. Systemic T cell adoptive immunotherapy of malignant gliomas. Journal of Neurosurgery. 1998;89(1):42–51. doi: 10.3171/jns.1998.89.1.0042. [DOI] [PubMed] [Google Scholar]
- 152.Sloan AE, Dansey R, Zamorano L, et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. Neurosurgical Focus. 2000;9(6, article e9) doi: 10.3171/foc.2000.9.6.10. [DOI] [PubMed] [Google Scholar]
- 153.Tsuboi K, Saijo K, Ishikawa E, et al. Effects of local injection of ex vivo expanded autologous tumor-specific T lymphocytes in cases with recurrent malignant gliomas. Clinical Cancer Research. 2003;9(9):3294–3302. [PubMed] [Google Scholar]
- 154.Wood GW, Holladay FP, Turner T, Wang YY, Chiga M. A pilot study of autologous cancer cell vaccination and cellular immunotherapy using anti-CD3 stimulated lymphocytes in patients with recurrent grade III/IV astrocytoma. Journal of Neuro-Oncology. 2000;48(2):113–120. doi: 10.1023/a:1006456421177. [DOI] [PubMed] [Google Scholar]
- 155.Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Research. 2001;61(3):842–847. [PubMed] [Google Scholar]
- 156.Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clinical Cancer Research. 2005;11(15):5515–5525. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 157.Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of Clinical Oncology. 2010;28(31):4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ardon H, De Vleeschouwer S, Van Calenbergh F, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatric Blood and Cancer. 2010;54(4):519–525. doi: 10.1002/pbc.22319. [DOI] [PubMed] [Google Scholar]
- 159.Caruso DA, Orme LM, Amor GM, et al. Results of a phase I study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children with stage 4 neuroblastoma. Cancer. 2005;103(6):1280–1291. doi: 10.1002/cncr.20911. [DOI] [PubMed] [Google Scholar]
- 160.De Vleeschouwer S, Fieuws S, Rutkowski S, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clinical Cancer Research. 2008;14(10):3098–3104. doi: 10.1158/1078-0432.CCR-07-4875. [DOI] [PubMed] [Google Scholar]
- 161.De Vleeschouwer S, Van Calenbergh F, Demaerel P, et al. Transient local response and persistent tumor control in a child with recurrent malignant glioma: treatment with combination therapy including dendritic cell therapy. Case report. Journal of Neurosurgery. 2004;100(supplement 5):492–497. doi: 10.3171/ped.2004.100.5.0492. [DOI] [PubMed] [Google Scholar]
- 162.Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunology, Immunotherapy. 2001;50(7):337–344. doi: 10.1007/s002620100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liau LM, Black KL, Martin NA, et al. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Case Report. Neurosurgical Focus. 2000;9(6, article e8) doi: 10.3171/foc.2000.9.6.9. [DOI] [PubMed] [Google Scholar]
- 164.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature Medicine. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rutkowski S, De Vleeschouwer S, Kaempgen E, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. British Journal of Cancer. 2004;91(9):1656–1662. doi: 10.1038/sj.bjc.6602195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Molecular Cancer Therapeutics. 2009;8(10):2773–2779. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. Journal of Clinical Neuroscience. 2008;15(2):114–121. doi: 10.1016/j.jocn.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 168.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. Journal of Clinical Oncology. 2010;28(11):1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 169.Wheeler CJ, Black KL, Liu G, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Research. 2008;68(14):5955–5964. doi: 10.1158/0008-5472.CAN-07-5973. [DOI] [PubMed] [Google Scholar]
- 170.Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clinical Cancer Research. 2004;10(16):5316–5326. doi: 10.1158/1078-0432.CCR-04-0497. [DOI] [PubMed] [Google Scholar]
- 171.Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. British Journal of Cancer. 2003;89(7):1172–1179. doi: 10.1038/sj.bjc.6601268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yamanaka R, Homma J, Yajima N, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clinical Cancer Research. 2005;11(11):4160–4167. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 173.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Research. 2004;64(14):4973–4979. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 174.Schneider T, Gerhards R, Kirches E, Firsching R. Preliminary results of active specific immunization with modified tumor cell vaccine in glioblastoma multiforme. Journal of Neuro-Oncology. 2001;53(1):39–46. doi: 10.1023/a:1011856406683. [DOI] [PubMed] [Google Scholar]
- 175.Steiner HH, Bonsanto MM, Beckhove P, et al. Antitumor vaccination of patients with glioblastoma multiforme: a pilot study to assess feasibility, safety, and clinical benefit. Journal of Clinical Oncology. 2004;22(21):4272–4281. doi: 10.1200/JCO.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 176.Ishikawa E, Tsuboi K, Yamamoto T, et al. Clinical trial of autologous formalin-fixed tumor vaccine for glioblastoma multiforme patients. Cancer Science. 2007;98(8):1226–1233. doi: 10.1111/j.1349-7006.2007.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Parsa A, Crane C, Wilson S, et al. Autologous tumor derived gp96 evokes a tumor specific immune response in recurrent glioma patients that correlates with clinical response to therapy. In: Proceedings of the AACE-NCI-EORTC International Conference Molecular Targets and Cancer Therapeutics; 2007. [Google Scholar]
- 178.Husain SR, Behari N, Kreitman RJ, Pastan I, Puri RK. Complete regression of established human glioblastoma tumor xenograft by interleukin-4 toxin therapy. Cancer Research. 1998;58(16):3649–3653. [PubMed] [Google Scholar]
- 179.Joshi BH, Leland P, Asher A, Prayson RA, Varricchio F, Puri RK. In situ expression of interleukin-4 (IL-4) receptors in human brain tumors and cytotoxicity of a recombinant IL-4 cytotoxin in primary glioblastoma cell cultures. Cancer Research. 2001;61(22):8058–8061. [PubMed] [Google Scholar]
- 180.Kawakami K, Leland P, Puri RK. Structure, function, and targeting of interleukin 4 receptors on human head and neck cancer cells. Cancer Research. 2000;60(11):2981–2987. [PubMed] [Google Scholar]
- 181.Puri RK, Leland P, Kreitman RJ, Pastan I. Human neurological cancer cells express interleukin-4 (IL-4) receptors which are targets for the toxic effects of IL4-Pseudomonas exotoxin chimeric protein. International Journal of Cancer. 1994;58(4):574–581. doi: 10.1002/ijc.2910580421. [DOI] [PubMed] [Google Scholar]
- 182.Murata T, Noguchi PD, Puri RK. Receptors for interleukin (IL)-4 do not associate with the common γ chain, and IL-4 induces the phosphorylation of JAK2 tyrosine kinase in human colon carcinoma cells. Journal of Biological Chemistry. 1995;270(51):30829–30836. doi: 10.1074/jbc.270.51.30829. [DOI] [PubMed] [Google Scholar]
- 183.Murata T, Obiri NI, Puri RK. Structure of and signal transduction through interleukin-4 and interleukin-13 receptors (review) International Journal of Molecular Medicine. 1998;1(3):551–557. doi: 10.3892/ijmm.1.3.551. [DOI] [PubMed] [Google Scholar]
- 184.Murata T, Taguchi J, Puri RK. Interleukin-13 receptor α’ but not α chain: A functional component of interleukin-4 receptors. Blood. 1998;91(10):3884–3891. [PubMed] [Google Scholar]
- 185.Debinski W, Puri RK, Kreitman RJ, Pastan I. A wide range of human cancers express interleukin 4 (IL4) receptors that can be targeted with chimeric toxin composed of IL4 and Pseudomonas exotoxin. Journal of Biological Chemistry. 1993;268(19):14065–14070. [PubMed] [Google Scholar]
- 186.Joshi BH, Leland P, Silber J, et al. IL-4 receptors on human medulloblastoma tumours serve as a sensitive target for a circular permuted IL-4-Pseudomonas exotoxin fusion protein. British Journal of Cancer. 2002;86(2):285–291. doi: 10.1038/sj.bjc.6600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kawakami M, Kawakami K, Puri RK. Interleukin-4-Pseudomonas exotoxin chimeric fusion protein for malignant glioma therapy. Journal of Neuro-Oncology. 2003;65(1):15–25. doi: 10.1023/a:1026294416718. [DOI] [PubMed] [Google Scholar]
- 188.Lampson LA, Wen P, Roman VA, Morris JH, Sarid JA. Disseminating tumor cells and their interactions with leukocytes visualized in the brain. Cancer Research. 1992;52(4):1018–1025. [PubMed] [Google Scholar]
- 189.Satoh J, Lee YB, Kim SU. T-cell costimulatory molecules B7-1 (CD80) and B7-2 (CD86) are expressed in human microglia but not in astrocytes in culture. Brain Research. 1995;704(1):92–96. doi: 10.1016/0006-8993(95)01177-3. [DOI] [PubMed] [Google Scholar]
- 190.Wheeler CJ, Black KL. DCVax-Brain and DC vaccines in the treatment of GBM. Expert Opinion on Investigational Drugs. 2009;18(4):509–519. doi: 10.1517/13543780902841951. [DOI] [PubMed] [Google Scholar]
- 191.Jäger E, Ringhoffer M, Karbach J, Arand M, Oesch F, Knuth A. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic-T-cell responses: evidence for immunoselection of antigen-loss variants in vivo. International Journal of Cancer. 1996;66(4):470–476. doi: 10.1002/(SICI)1097-0215(19960516)66:4<470::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 192.Parajuli P, Mathupala S, Sloan AE. Systematic comparison of dendritic cell-based immunotherapeutic strategies for malignant gliomas: In vitro induction of cytolytic and natural killer-like T cells. Neurosurgery. 2004;55(5):1194–1203. doi: 10.1227/01.neu.0000141082.20865.48. [DOI] [PubMed] [Google Scholar]
- 193.Mahaley MS, Bigner DD, Dudka LF, et al. Immunobiology of primary intracranial tumors—part 7: active immunization of patients with anaplastic human glioma cells: a pilot study. Journal of Neurosurgery. 1983;59(2):201–207. doi: 10.3171/jns.1983.59.2.0201. [DOI] [PubMed] [Google Scholar]
- 194.Wikstrand CJ, Bigner DD. Immunobiologic aspects of the brain and human gliomas. A review. American Journal of Pathology. 1980;98(2):515–567. [PMC free article] [PubMed] [Google Scholar]
- 195.Yang I, Han S, Parsa AT. Heat-shock protein vaccines as active immunotherapy against human gliomas. Expert Review of Anticancer Therapy. 2009;9(11):1577–1582. doi: 10.1586/era.09.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Binder RJ. Heat shock protein vaccines: From bench to bedside. International Reviews of Immunology. 2006;25(5-6):353–375. doi: 10.1080/08830180600992480. [DOI] [PubMed] [Google Scholar]
- 197.Srivastava PK, Amato RJ. Heat shock proteins: the ØSwiss Army KnifeÙ vaccines against cancers and infectious agents. Vaccine. 2001;19(17–19):2590–2597. doi: 10.1016/s0264-410x(00)00492-8. [DOI] [PubMed] [Google Scholar]
- 198.Belli F, Testori A, Rivoltini L, et al. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: clinical and immunologic findings. Journal of Clinical Oncology. 2002;20(20):4169–4180. doi: 10.1200/JCO.2002.09.134. [DOI] [PubMed] [Google Scholar]
- 199.Li Z, Qiao Y, Liu B, et al. Combination of imatinib mesylate with autologous leukocyte-derived heat shock protein and chronic myelogenous leukemia. Clinical Cancer Research. 2005;11(12):4460–4468. doi: 10.1158/1078-0432.CCR-05-0250. [DOI] [PubMed] [Google Scholar]
- 200.Mazzaferro V, Coppa J, Carrabba MG, et al. Vaccination with autologous tumor-derived heat-shock protein Gp96 after liver resection for metastatic colorectal cancer. Clinical Cancer Research. 2003;9(9):3235–3245. [PubMed] [Google Scholar]
- 201.Rivoltini L, Castelli C, Carrabba M, et al. Human tumor-derived heat shock protein 96 mediates in vitro activation and in vivo expansion of melanoma- and colon carcinoma-specific T cells. Journal of Immunology. 2003;171(7):3467–3474. doi: 10.4049/jimmunol.171.7.3467. [DOI] [PubMed] [Google Scholar]
- 202.Ricci-Vitiani L, Pallini R, Biffoni M, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468(7325):824–830. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 203.Wang R, Chadalavada K, Wilshire J, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468(7325):829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 204.Mannino M, Chalmers AJ. Radioresistance of glioma stem cells: intrinsic characteristic or property of the ‘microenvironment-stem cell unit’? Molecular Oncology. 2011;5(4):374–386. doi: 10.1016/j.molonc.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tamura K, Aoyagi M, Wakimoto H, et al. Accumulation of CD133-positive glioma cells after high-dose irradiation by gamma knife surgery plus external beam radiation: clinical article. Journal of Neurosurgery. 2010;113(2):310–318. doi: 10.3171/2010.2.JNS091607. [DOI] [PubMed] [Google Scholar]
- 206.McCord AM, Jamal M, Williams ES, Camphausen K, Tofilon PJ. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clinical Cancer Research. 2009;15(16):5145–5153. doi: 10.1158/1078-0432.CCR-09-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 208.Soeda A, Park M, Lee D, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α . Oncogene. 2009;28(45):3949–3959. doi: 10.1038/onc.2009.252. [DOI] [PubMed] [Google Scholar]
- 209.Bodmer S, Strommer K, Frei K, et al. Immunosuppression and transforming growth factor-β in glioblastoma. Preferential production of transforming growth factor-β2. Journal of Immunology. 1989;143(10):3222–3229. [PubMed] [Google Scholar]
- 210.Qiu B, Zhang D, Wang C, et al. IL-10 and TGF-β2 are overexpressed in tumor spheres cultured from human gliomas. Molecular Biology Reports. 2011;38(5):3585–3591. doi: 10.1007/s11033-010-0469-4. [DOI] [PubMed] [Google Scholar]
- 211.Lottaz C, Beier D, Meyer K, et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Research. 2010;70(5):2030–2040. doi: 10.1158/0008-5472.CAN-09-1707. [DOI] [PubMed] [Google Scholar]
- 212.Avril T, Saikali S, Vauleon E, et al. Distinct effects of human glioblastoma immunoregulatory molecules programmed cell death ligand-1 (PDL-1) and indoleamine 2,3-dioxygenase (IDO) on tumour-specific T cell functions. Journal of Neuroimmunology. 2010;225(1-2):22–33. doi: 10.1016/j.jneuroim.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 213.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature Reviews Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 215.Darnell JE., Jr. STATs and gene regulation. Science. 1997;277(5332):1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 216.Gariboldi MB, Ravizza R, Monti E. The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochemical Pharmacology. 2010;80(4):455–462. doi: 10.1016/j.bcp.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 217.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr., Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267(5202):1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 218.Wen Z, Zhong Z, Darnell JE., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82(2):241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 219.Zhong Z, Wen Z, Darnell JE., Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 220.Turkson J, Jove R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene. 2000;19(56):6613–6626. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- 221.Iwamaru A, Szymanski S, Iwado E, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26(17):2435–2444. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- 222.Birner P, Toumangelova-Uzeir K, Natchev S, Guentchev M. STAT3 tyrosine phosphorylation influences survival in glioblastoma. Journal of Neuro-Oncology. 2010;100(3):339–343. doi: 10.1007/s11060-010-0195-8. [DOI] [PubMed] [Google Scholar]
- 223.Kang SH, Yu MO, Park KJ, Chi SG, Park DH, Chung YG. Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery. 2010;67(5):1386–1395. doi: 10.1227/NEU.0b013e3181f1c0cd. [DOI] [PubMed] [Google Scholar]
- 224.Chen F, Xu Y, Luo Y, et al. Down-regulation of stat3 decreases invasion activity and induces apoptosis of human glioma cells. Journal of Molecular Neuroscience. 2010;40(3):353–359. doi: 10.1007/s12031-009-9323-3. [DOI] [PubMed] [Google Scholar]
- 225.Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009;27(10):2383–2392. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Villalva C, Martin-Lannerée S, Cortes U, et al. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? International Journal of Cancer. 2011;128(4):826–838. doi: 10.1002/ijc.25416. [DOI] [PubMed] [Google Scholar]
- 227.Zhang L, Alizadeh D, van Handel M, Kortylewski M, Yu H, Badie B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia. 2009;57(13):1458–1467. doi: 10.1002/glia.20863. [DOI] [PubMed] [Google Scholar]
- 228.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nature Medicine. 2005;11(12):1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 229.Wei J, Wu A, Kong LY, et al. Hypoxia potentiates glioma-mediated immunosuppression. PLoS One. 2011;6(1, article e16195) doi: 10.1371/journal.pone.0016195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.De La Iglesia N, Konopka G, Puram SV, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes and Development. 2008;22(4):449–462. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Aberg MA, Ryttsen F, Hellgren G, et al. Selective introduction of antisense oligonucleotides into single adult CNS progenitor cells using electroporation demonstrates the requirement of STAT3 activation for CNTF-induced gliogenesis. Molecular and Cellular Neuroscience. 2001;17(3):426–443. doi: 10.1006/mcne.2000.0947. [DOI] [PubMed] [Google Scholar]
- 232.Bonni A, Sun Y, Nadal-Vicens M, et al. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278(5337):477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 233.De La Iglesia N, Puram SV, Bonni A. STAT3 regulation of glioblastoma pathogenesis. Current Molecular Medicine. 2009;9(5):580–590. doi: 10.2174/156652409788488739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Lo HW, Hsu SC, Ali-Seyed M, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7(6):575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 235.Gao L, Li F, Dong B, et al. Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. International Journal of Radiation Oncology Biology Physics. 2010;77(4):1223–1231. doi: 10.1016/j.ijrobp.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 236.Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21(55):8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 237.Xiong H, Du W, Zhang YJ, et al. Trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing the promoter-associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells. doi: 10.1002/mc.20777. Molecular Carcinogenesis. In press. [DOI] [PubMed] [Google Scholar]
- 238.Wei J, Barr J, Kong LY, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Molecular Cancer Therapeutics. 2010;9(1):67–78. doi: 10.1158/1535-7163.MCT-09-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Dittmer U, He H, Messer RJ, et al. Functional impairment of CD8+ T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20(3):293–303. doi: 10.1016/s1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- 240.Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. Journal of Immunology. 2004;172(8):4676–4680. doi: 10.4049/jimmunol.172.8.4676. [DOI] [PubMed] [Google Scholar]
- 241.Ralainirina N, Poli A, Michel T, et al. Control of NK cell functions by CD4+CD25+ regulatory T cells. Journal of Leukocyte Biology. 2007;81(1):144–153. doi: 10.1189/jlb.0606409. [DOI] [PubMed] [Google Scholar]
- 242.Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood. 2006;107(10):3925–3932. doi: 10.1182/blood-2005-11-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Taams LS, Van Amelsfort JMR, Tiemessen MM, et al. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Human Immunology. 2005;66(3):222–230. doi: 10.1016/j.humimm.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4+CD25+ T cells with regulatory properties isolated from peripheral blood. Journal of Experimental Medicine. 2001;193(11):1285–1294. doi: 10.1084/jem.193.11.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cosmi L, Liotta F, Angeli R, et al. Th2 cells are less susceptible than Th1 cells to the suppressive activity of CD25+ regulatory thymocytes because of their responsiveness to different cytokines. Blood. 2004;103(8):3117–3121. doi: 10.1182/blood-2003-09-3302. [DOI] [PubMed] [Google Scholar]
- 246.Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunology, Immunotherapy. 2008;57(1):123–131. doi: 10.1007/s00262-007-0336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clinical Cancer Research. 2008;14(16):5166–5172. doi: 10.1158/1078-0432.CCR-08-0320. [DOI] [PubMed] [Google Scholar]
- 248.Kong LY, Abou-Ghazal MK, Wei J, et al. A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clinical Cancer Research. 2008;14(18):5759–5768. doi: 10.1158/1078-0432.CCR-08-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4 + T cell compartment without affecting regulatory T-cell function. Clinical Cancer Research. 2007;13(7):2158–2167. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- 250.Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. Journal of Experimental Medicine. 2000;192(2):295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sutmuller RPM, Van Duivenvoorde LM, Van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. Journal of Experimental Medicine. 2001;194(6):823–832. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fecci PE, Sweeney AE, Grossi PM, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clinical Cancer Research. 2006;12(14, part 1):4294–4305. doi: 10.1158/1078-0432.CCR-06-0053. [DOI] [PubMed] [Google Scholar]
- 253.Humphries W, Wei J, Sampson JH, Heimberger AB. The role of tregs in glioma-mediated immunosuppression: potential target for intervention. Neurosurgery Clinics of North America. 2010;21(1):125–137. doi: 10.1016/j.nec.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Taieb J, Chaput N, Schartz N, et al. Chemoimmunotherapy of tumors: cyclophosphamide synergizes with exosome based vaccines. Journal of Immunology. 2006;176(5):2722–2729. doi: 10.4049/jimmunol.176.5.2722. [DOI] [PubMed] [Google Scholar]
- 255.Holtl L, Ramoner R, Zelle-Rieser C, et al. Allogeneic dendritic cell vaccination against metastatic renal cell carcinoma with or without cyclophosphamide. Cancer Immunology, Immunotherapy. 2005;54(7):663–670. doi: 10.1007/s00262-004-0629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.MacLean GD, Miles DW, Rubens RD, Reddish MA, Longenecker BM. Enhancing the effect of THERATOPE STn-KLH cancer vaccine in patients with metastatic breast cancer by pretreatment with low-dose intravenous cyclophosphamide. Journal of Immunotherapy With Emphasis on Tumor Immunology. 1996;19(4):309–316. doi: 10.1097/00002371-199607000-00006. [DOI] [PubMed] [Google Scholar]
- 257.Brooks WH, Markesbery WR, Gupta GD, Roszman TL. Relationship of lymphocyte invasion and survival of brain tumor patients. Annals of Neurology. 1978;4(3):219–224. doi: 10.1002/ana.410040305. [DOI] [PubMed] [Google Scholar]
- 258.Safdari Gh. H. H, Hochberg FH, Richardson EP., Jr. Prognostic value of round cell (lymphocyte) infiltration in malignant gliomas. Surgical Neurology. 1985;23(3):221–226. doi: 10.1016/0090-3019(85)90086-2. [DOI] [PubMed] [Google Scholar]
- 259.Strik HM, Stoll M, Meyermann R. Immune cell infiltration of intrinsic and metastatic intracranial tumours. Anticancer Research. 2004;24(1):37–42. [PubMed] [Google Scholar]
- 260.Jacobs JFM, Idema AJ, Bol KF, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. Journal of Neuroimmunology. 2010;225(1-2):195–199. doi: 10.1016/j.jneuroim.2010.05.020. [DOI] [PubMed] [Google Scholar]