

Abstract
Hirschsprung’s disease is a congenital disorder that occurs in 1:5000 live births. It is characterised by an absence of enteric neurons along a variable region of the gastrointestinal tract. Hirschsprung’s disease is classified as a multigenic disorder, because the same phenotype is associated with mutations in multiple distinct genes. Furthermore, the genetics of Hirschsprung’s disease are highly complex and not strictly Mendelian. The phenotypic variability and incomplete penetrance observed in Hirschsprung’s disease also suggests the involvement of modifier genes. Here, we summarise the current knowledge of the genetics underlying Hirschsprung’s disease based on human and animal studies, focusing on the principal causative genes, their interactions, and the role of modifier genes.
Keywords: Neural crest, Enteric nervous system, Hirschsprung’s disease, Aganglionosis, Modifier genes
INTRODUCTION
The enteric nervous system (ENS) comprises neurons and glial cells within the wall of the gastrointestinal tract. It is responsible for regulating intestinal motility, immune function, luminal secretions, and blood flow[1]. During development, the ENS arises from a highly migratory population of cells, called the neural crest[2-4]. The neural crest arises as a result of an epithelial to mesenchymal transition during the formation of the neural tube. Two separate populations of neural crest, arising from different axial levels, contribute to the ENS - the vagal level (defined as the post-otic hindbrain adjacent to somites 1-7) and the sacral level (caudal to somite 24 in mice and humans)[5,6]. Vagal neural crest cells migrate ventrally to the presumptive foregut, and then into and along the entire length of the gastrointestinal tract, a process that takes five days in mice (embryonic day E9.5-E14.5)[7] and three weeks in humans (during 4th-7th wk of gestation)[8].
The formation of a functional ENS requires the coordination of many processes, including survival, migration, proliferation, and differentiation of precursor cells within the gastrointestinal tract. Failure of neural crest cells to fully colonise the entire length of the gastrointestinal tract results in a region of gut that lacks enteric neurons, called an “aganglionic zone”, which affects a variable length of the distal most bowel. As enteric neurons are essential for motility, the aganglionic zone remains tonically constricted, preventing the passage of faecal material. In humans, this condition is known as Hirschsprung’s disease (HSCR) and occurs in approximately 1:5000 live births[9]. HSCR can either be familial or sporadic, and is subdivided into short or long segment HSCR (S-HSCR and L-HSCR), which refers to the extent of the aganglionic zone[10]. The less severe S-HSCR (about 80% of cases) is more common than L-HSCR (about 20% of cases) and displays a more pronounced gender bias (4:1 male:female in S-HSCR compared to 1.2:1 male:female in L-HSCR)[11]. HSCR may present as an isolated condition (about 70% of cases) or as part of a syndrome, such as Mowatt-Wilson or Waardenburg Shah type 4 (Table 1). Although HSCR is normally detected soon after birth, there have been reports of HSCR being identified in patients after childhood[12]. Failure to treat HSCR is often fatal because of malnutrition or sepsis following rupture of the bowel. Present treatment involves surgery to remove the affected portion of bowel and re-anastomosis of the remaining gut to the anus. Although refinements to surgical techniques have improved patient outcome, post-operative complications persist in a large number of patients[13,14].
Table 1.
Genes associated with Hirschsprung's disease
| Locus | Gene | Associated syndrome | Incidence | Penetrance | Inheritance | Ref. |
| 10q11 | RET | Non-syndromic HSCR | 50% familial | 70% male | Dominant | 82-84 |
| 30% sporadic | 50% female | |||||
| 5p13 | GDNF | Non-syndromic HSCR | 5 cases | Low | Dominant | 85-89 |
| 13q22 | EDNRB | Shah-Waardenburg | 5% | Low | Dominant or recessive | 44,90 |
| Non-syndromic HSCR | ||||||
| 20q13 | ET3 | Shah-Waardenburg | 1 case | N/A | Dominant or recessive | 91 |
| Non-syndromic HSCR | ||||||
| 1p36 | ECE1 | Cardiac and autonomic nervous system defects with HSCR | 1 case | N/A | Dominant | 40 |
| 22q13 | SOX10 | Shah-Waardenburg | > 5% | ~80% | Dominant | 47,49,50,92 |
| Non-syndromic HSCR | ||||||
| 2q22 | ZFHX1B | Mowat-Wilson | < 5% | 60% | Dominant | 62,93-95 |
| 4p12 | PHOX2B | CCHS–Ondines Curse | < 5% | 20% | Dominant | 96 |
| 19p13 | NTN | Non-syndromic HSCR | 1 case | Dominant | 97 | |
| 18q21 | TCF4 | Epileptic encephalopathy | 1 case | Dominant | 98 | |
| 10q21.1 | KIAA1279 | Goldberg-Shprintzen | Rare | Recessive | 21 |
HSCR: Hirschsprung's disease; CCHS: Congenital central hypoventilation syndrome.
In the majority of cases, the genetics of HSCR are complex and non-Mendelian in nature[15]. To date, more than a dozen genes have been identified as being associated with HSCR[9,16]. However, mutations in these genes account for only about 50% of all HSCR cases[17]. The phenotypic variability and incomplete penetrance observed in HSCR also suggests the involvement of modifier genes. The aim of this review is to summarise the current knowledge of the genetics underlying HSCR. We first discuss the principal causative genes and detail the interactions between these genes that alter the severity or incidence of HSCR. Finally, we discuss the accumulating evidence for the role of modifier genes in the development of HSCR.
GENES INVOLVED IN ENS DEVELOPMENT
Many of the genes associated with HSCR encode members of the Glial cell line-derived neurotrophic factor (GDNF)/RET- and ET-3/EDNRB-signalling pathways or transcription factors, such as SOX10, PHOX2B or ZFHX1B. Mutations in these genes have been shown to result in Hirschsprung’s disease in humans (Table 1) or aganglionosis in mice (Table 2).
Table 2.
Phenotypes of mouse models of enteric nervous system defects
| Wild-type | Colonic aganglionosis | Total intestinal aganglionosis | Hypoganglionosis | |
| Ret | +/- | 51/51 | -/- | C620R/+ |
| 9/9 | S697A/S697A | Y1062F/Y1062F | ||
| Y162F/+ | C620R/C620R | |||
| Ednb | sl/+ | sl/sl | ||
| Ednrbtm1Ywa/Ednrbtm1Ywa | ||||
| Et3 | ls/+ | ls/ls | ||
| Et3tm1Ywa/ Et3tm1Ywa | ||||
| Sox10 | Dom/+ (~80%) | Dom/+ (~20%) | Dom/Dom | |
| LacZ/+ (~80%) | LacZ/+ (~20%) | LacZ/LacZ | ||
| Interactions | Ret+/-; Ednrbs/s | Ret51/51; Et3ls/ls | ||
| Ret51/+; Et3ls/ls | Sox10Dom/+; Et3ls/ls | |||
| Sox10Dom/+; Edrnbsl/sl | ||||
| Other genotypes | Sall4-/- | Gdnf-/- | Gdnf+/- | |
| B1Intergrin-/- | Gfra1-/- | Hlx1-/- | ||
| Ece1-/- | Phox2b-/- | |||
| Pax3-/- |
GDNF/RET-GFRα1
GDNF is a secreted protein and a distant member of the TGF-β superfamily[18]. GDNF binds to the glycosylphosphatidylinsoitol-linked receptor, GFRα1. The GDNF-GFRα1 complex then binds to and activates the transmembrane receptor tyrosine kinase, RET[19]. Mutations in genes encoding members of the GDNF/RET-GFRα1 signalling pathway account for about 50% of familial cases and around 30% of sporadic cases of HSCR[17]. Non-coding mutations in RET have also been proposed to increase susceptibility to HSCR[20-23]. In mice, Gdnf is expressed by the gut mesenchyme prior to the entry of neural crest cells[24]. Ret is expressed exclusively by neural crest-derived cells and Gfrα1 is expressed by both crest-derived cells and the gut mesenchyme[25]. Gdnf-, Gfrα1- or Ret-null mice die within 24 hours of birth, and lack enteric neurons along the entire length of the gastrointestinal tract caudal to the stomach[25-27]. Gdnf+/- and Ret+/- mice are viable and do not exhibit aganglionosis[28].
RET is subject to alternative splicing and translated into two functional isoforms, RET51 and RET9, which differ in the number of amino acids at their C terminal end[29]. These isoforms are highly conserved between human and mouse[30]. Mice lacking the Ret51 isoform (Ret9/9 mice) have enteric neurons along the entire length of the gastrointestinal tract, while mice lacking the Ret9 isoform (Ret51/51 mice) suffer colonic aganglionosis and kidney hypodysplasia[31]. The phenotype of the Ret51/51 mice is highly reminiscent of the colonic aganglionosis observed in patients with HSCR. Interestingly, the developing ENS in humans appears to be more sensitive to reduced RET signalling than that of the ENS in mice. RET mutations in humans act dominantly to give rise to HSCR, whereas ENS development is normal in Ret heterozygous mice[28]. In fact, it has recently been shown that a loss of around 60%-70% of Ret expression in mice is required to mimic the aganglionic phenotype observed in humans[32].
Targeted mutations in RET have identified signalling sites that are required for ENS development. Mutation of a putative protein kinase A phosphorylation site, which changes serine to alanine (RetS697A), results in aganglionosis of the distal colon[33]. Mutation of an intracellular docking site, which converts tyrosine to phenylalanine (RetY1062F), induces total intestinal aganglionosis[34]. The mutation of cysteine to arginine (RetC620R), which is observed in some MEN2A/HSCR patients, has also been shown to result in total intestinal aganglionosis[35].
ENDOTHELIN SIGNALLING PATHWAY
Endothelin 3 (ET-3) is a secreted peptide, which is expressed by the gut mesenchyme[36]. ET-3 is initially expressed in an immature form before being processed to an active peptide by the enzyme, endothelin converting enzyme 1 (ECE1)[37,38]. ET-3 signals through the receptor Endothelin receptor B (EDNRB), which is expressed on migrating enteric neural crest cells[39].
Mutations in ET3 and EDNRB account for around 5% of HSCR cases, whilst only a single case of ECE1-associated HSCR has been reported[40]. ET3- and EDNRB-associated HSCR can present as both syndromic (such as Wardenburg-Shah syndrome) and non-syndromic forms of HSCR. In mice, lethal spotted (ls) and piebald lethal (sl) are naturally occurring mutants of Et-3 and Ednrb respectively, and lack enteric neurons in the distal bowel[37,41]. Although enteric neurons are absent only from the distal colon of Et-3 and Ednrb-null mice, the migration of neural crest cells through the small intestine is also delayed[42,43]. As with RET, the human ENS appears to be more sensitive to reduced EDNRB signalling than that in mice. Around 21% of patients heterozygous for the W276C mutation in EDNRB develop HSCR[44], while heterozygous piebald lethal (sl) mice do not develop any form of aganglionosis[45].
SOX10
SRY (Sex determining region Y)-box 10 (SOX10) is a high mobility group transcription factor of the SRY (sex determining factor) family. Mutations in SOX10 account for around 5% of HSCR cases[46-48], and comprise both syndromic [Waardenburg-Shah types 4 (WS4)] and non-syndromic forms[49]. Some WS4 patients with SOX10 mutations also suffer dysmyelination of the central and peripheral nervous systems[47]. Sox10 is expressed by migrating enteric neural crest cells[50]. Dom is a naturally occurring mouse mutant of Sox10, which carries a single base insertion in the Sox10 locus that prematurely truncates the transcription factor downstream of the DNA binding domain, producing a dominant negative form of the protein[50]. Mice lacking Sox10 are devoid of enteric neurons throughout the entire gastrointestinal tract[50,51]. Around 20% of Sox10+/- mice suffer colonic aganglionosis, although the incidence varies depending on the background strain[45,50,52,53]. These mice also exhibit coat colour defects analogous to the pigmentation defects seen in WS4 patients. Sox10 has been shown to regulate the expression of both Ret and Ednrb[54-56].
ADDITIONAL TRANSCRIPTION FACTORS ASSOCIATED WITH HSCR
PHOX2B
Paired-like homeobox 2b (PHOX2B) is a transcription factor that is expressed by enteric neural crest cells[57]. Human studies have linked mutations in PHOX2B with HSCR associated with congenital central hypoventilation syndrome (CCHS)/Ondines curse in two thirds of patients[58]. The causative mutations in these patients are most commonly polyalanine expansions[59]. Phox2b null mice lack enteric neurons along the entire length of the gastrointestinal tract[60]. Phox2b has been shown to regulate the expression of Ret[60-61].
ZFHX1B
ZFHX1B, also known as SMAD interacting protein 1 (SMADIP1/SIP1), is a zinc finger homeodomain transcription factor. Mutations in ZFHX1B are associated with Mowat-Wilson syndrome, and have been shown to result in HSCR with a varying degree of penetrance[62]. In mice, Zfhx1b is expressed by vagal neural crest cells, which are absent in Zfhx1b null mutant mice[63].
INTERACTIONS BETWEEN KNOWN HSCR ASSOCIATED GENES
Interactions between known HSCR associated genes significantly influence the incidence and severity of intestinal aganglionosis (Table 2) (Figure 1).
Figure 1.
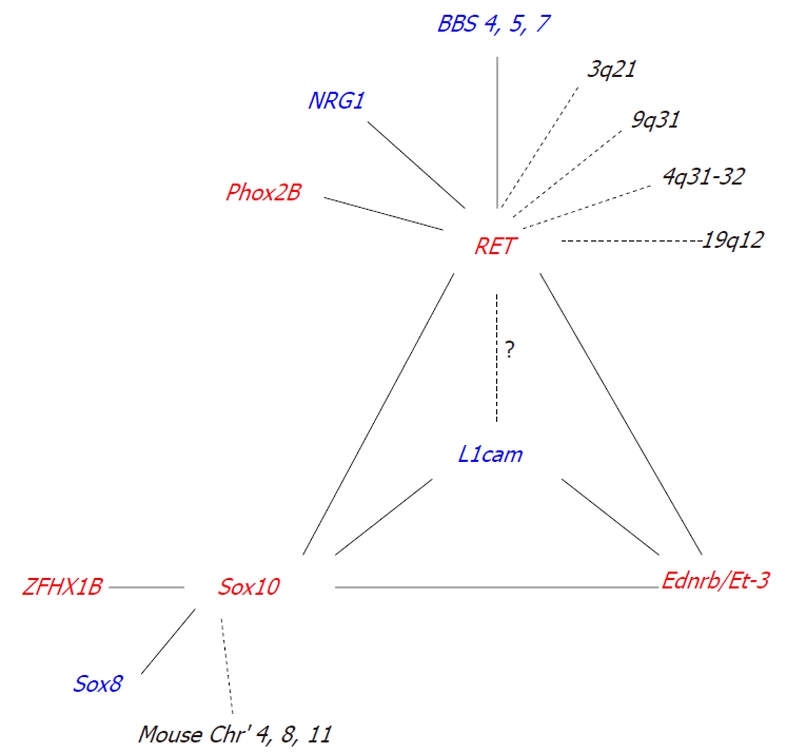
Interactions between known Hirschsprung's disease associated genes and their modifiers. Confirmed and putative interactions are shown by the solid and dotted lines, respectively. Red: HSCR associated gene; Blue: Known HSCR modifier gene; Black: Modifier loci with unknown gene. The question mark represents a disparity in the existing data obtained from human clinical and animal studies. HSCR: Hirschsprung's disease; BBS: Bardet-Biedel syndrome.
Gdnf/Ret and Et-3/Ednrb signalling pathways
Genetic interactions were first proposed based on a human study of the genetically isolated Mennonite population, suggesting that the RET and EDNRB loci may interact to govern the susceptibility to Hirschsprung’s disease[64]. Studies in mice, using a two-locus complementation approach, confirmed a genetic interaction between the Ret and Ednrb loci by showing that the generation of Ret+/-; Ednrbs/s mice resulted in colonic aganglionosis; a phenotype not observed in Ret+/- or Ednrbs/s mice alone[64,65]. A similar genetic interaction was also reported using Ret51 and Et-3ls mice[42]. A significant increase in aganglionosis, extending all the way to the stomach, was observed in Ret51/51; Et-3ls/ls mice compared to the colonic aganglionosis normally observed in Ret51/51 or Et-3ls/ls mice alone[42]. The mechanism underlying these interactions is not yet known; however, it has been proposed that Ret and Ednrb may interact by activating common downstream signalling molecules, such as PKA[42].
Gdnf/Ret signalling pathway and the transcription factors Sox10 and Phox2b
Although genome-wide linkage studies failed to detect any genetic interaction between the Sox10 and Ret loci[66], Sox10 has been shown to form a transcriptional complex with the transcription factor, Pax3, to directly regulate the expression of RET[67]. In addition to Sox10, Phox2B has also been shown to bind the RET promoter and regulate transcription[61]. Although no genetic interactions were observed in double heterozygotic mice (Ret+/-; Phox2b+/- mice), human clinical studies have reported interactions between RET and PHOX2B in CCHS patients[68].
Sox10 and Zfhx1b
Sox10 has been shown to interact with the transcription factor, Zfhx1b, in mice[69]. The generation of double heterozygotic progeny (Sox10LacZ/+; Zfhx1bΔ/+ mice) resulted in a significant increase in the severity of aganglionosis compared to a mutation in Sox10LacZ/+ or Zfhx1bΔ/+ mice alone[69]. The mechanism underlying this interaction is not known, but is likely to be mediated by the modulation of Bmp expression[69].
Et-3/Ednrb signalling pathway and Sox10
In mice, interactions between Sox10 and members of the endothelin signalling pathway (Et-3 and Ednrb) have been reported[45,52]. Using a two-locus complementation approach, mice carrying mutations in Sox10 and Et-3 (Sox10Dom/+; Et-3ls/ls) or Ednrb (Sox10Dom/+;Ednrbs/s and Sox10Dom/+; Ednrbsl/sl) exhibited a significant increase in the severity of intestinal aganglionosis compared to mutations in Sox10, Et-3, or Ednrb alone[45,52]. In addition, the expression of Ednrb has been shown to be significantly reduced in Sox10Dom/+ mice[50]. The mechanism underlying this interaction can be explained, at least in part, by the presence of SOX10 binding sites within a conserved enhancer region of the Ednrb promoter, which are required for the spatiotemporal expression of Ednrb in the ENS[56].
MODIFIER GENES
The incomplete penetrance and interfamilial variation commonly observed in HSCR strongly suggests the involvement of modifier genes. We define a modifier gene as a gene that, when mutated, is insufficient on its own to produce an effect, but, when coupled with another genetic mutation, it produces or enhances an effect[70]. To date, only a handful of modifier genes have been identified for HSCR (Figure 1).
Modifiers for RET
Linkage studies and genome-wide screens have identified a number of putative modifying loci for RET, such as 3q21, 4q31-32, 8p12, 9q31, and 19q12[71,72]. However, many of the genes responsible for interacting with RET at these loci are yet to be identified. One gene that has been identified is neuregulin 1 (NRG1)[72]. Association studies have shown that individuals that possess a specific NRG1 haplotype have an increased risk of HSCR conferred by RET[72]. NRG1 signals through ErbB2 and ErbB3 receptors to regulate neural crest cell development and in turn, ErbB3 is regulated by the HSCR associated gene Sox10[73].
Although not detected in any of the genome-wide screens, three further modifier genes for RET were identified through the Bardet-Biedel syndrome (BBS). Subsets of patients with BBS, a genetically heterogeneous disorder with 14 identified causative loci, also present with HSCR. BBS patients with HSCR are more frequent carriers of a common RET intronic hypomorphic allele than the general population[74]. In zebrafish, suppression of Ret in conjunction with a loss of either Bbs 4, 5, or 7 has been shown to significantly increase the severity of ENS defects compared to loss of these genes independently[75].
Human clinical studies have also suggested that the X-linked gene L1CAM, may act as a modifier gene for RET. Some individuals with L1CAM mutations who have HSCR, also possess a common RET polymorphism that is over-represented in HSCR populations[76]. However, animal model studies using a two-locus complementation approach failed to detect any genetic interaction between L1cam and Ret[70]. One reason for this discrepancy could be that humans are more sensitive to a reduction in RET levels than mice[28,32]. It is not yet known whether interactions with L1cam can be detected in Ret51/51 and RetS697A/S697A mice that exhibit colonic aganglionosis and more closely resemble human HSCR[31,33].
Modifiers for Sox10
A genome-wide screen in mice has identified five putative modifying loci for Sox10 on chromosomes 3, 5, 8, 11, and 14[66]. Two of these loci have been identified as Ednrb and Phox2b[66], while the other three loci, on chromosomes 3, 8, and 11, are yet to be determined.
Although not identified in the Sox10 genome-wide screen, one modifier gene that has been shown to significantly increase the penetrance and extent of aganglionosis in Sox10 heterozygous mice, is Sox8[53]. Sox8 is a transcription factor that is closely related to Sox10, and is expressed by all enteric neural crest cells[53]. Sox8-/- mice are viable and fertile and show no ENS phenotype[53]. Using a two-locus complementation approach, double heterozygotic progeny (Sox8+/-; Sox10+/- mice) were shown to have a significant increase in the incidence and severity of aganglionosis compared to a mutation in Sox8 or Sox10 alone[53]. The most likely mechanism underlying this interaction is genetic redundancy, as Sox8 has been shown to have DNA binding and subcellular redistribution properties similar to that of Sox10[77-80] and is capable of activating Sox10 target genes[78].
The X-linked gene, L1cam, can also act as a modifier gene for Sox10 in mice[70]. Loss or haploinsufficiency of L1cam in conjunction with a heterozygous loss of Sox10 significantly increases the incidence of intestinal aganglionosis compared to a mutation in Sox10 alone[70]. Sox10 has been shown to directly regulate the expression of endogenous L1cam[70].
Modifiers of Et3/Ednrb
To date, only one modifier gene has been identified for members of the endothelin signalling pathway. Loss or haploinsufficiency of L1cam in conjunction with a null mutation in Et-3 or Ednrb significantly increases the severity of intestinal aganglionosis compared to a loss of Et-3 or Ednrb alone[81]. Although the mechanism underlying these interactions is not yet known, it is most likely mediated through the activation of common downstream targets, such as PI3K[81].
CONCLUSION
HSCR research has now entered a second phase. Having identified many of the key genes capable of independently inducing HSCR, we are now undertaking the difficult task of identifying the interactions that modulate the severity and penetrance of this disease. By combining human genetic data from patients, family pedigrees, and genome wide association screens with animal studies, we are beginning to assemble the pieces of the HSCR puzzle into a coherent picture of multigenetic inheritance and interactions. To further aid this goal, as the cost of genome sequencing becomes more affordable, the potential to sequence the entire genome of individual HSCR patients becomes viable, which is likely to provide significant advances into our understanding of the genetic basis of HSCR.
Footnotes
Supported by The National Health and Medical Research Council of Australia to Anderson RB: Project grant, No. 509219; and a CDA Fellowship, No. 454773
Peer reviewer: Nageshwar D Reddy, Professor, Asian Institute of Gastroenterology, 6-3-652, Somajiguda, Hyderabad-500 082, India
S- Editor Sun H L- Editor Stewart GJ E- Editor Zhang DN
References
- 1.Furness JB. The Enteric Nervous System. Oxford: Blackwell Publishing; 2006. [Google Scholar]
- 2.Anderson RB, Newgreen DF, Young HM. Neural crest and the development of the enteric nervous system. Adv Exp Med Biol. 2006;589:181–196. doi: 10.1007/978-0-387-46954-6_11. [DOI] [PubMed] [Google Scholar]
- 3.Burns AJ, Champeval D, Le Douarin NM. Sacral neural crest cells colonise aganglionic hindgut in vivo but fail to compensate for lack of enteric ganglia. Dev Biol. 2000;219:30–43. doi: 10.1006/dbio.1999.9592. [DOI] [PubMed] [Google Scholar]
- 4.Yntema CL, Hammond WS. The origin of intrinsic ganglia of trunk viscera from vagal neural crest in the chick embryo. J Comp Neurol. 1954;101:515–541. doi: 10.1002/cne.901010212. [DOI] [PubMed] [Google Scholar]
- 5.Young HM, Newgreen D. Enteric neural crest-derived cells: origin, identification, migration, and differentiation. Anat Rec. 2001;262:1–15. doi: 10.1002/1097-0185(20010101)262:1<1::AID-AR1006>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 6.Wallace AS, Burns AJ. Development of the enteric nervous system, smooth muscle and interstitial cells of Cajal in the human gastrointestinal tract. Cell Tissue Res. 2005;319:367–382. doi: 10.1007/s00441-004-1023-2. [DOI] [PubMed] [Google Scholar]
- 7.Burns AJ, Le Douarin NM. Enteric nervous system development: analysis of the selective developmental potentialities of vagal and sacral neural crest cells using quail-chick chimeras. Anat Rec. 2001;262:16–28. doi: 10.1002/1097-0185(20010101)262:1<16::AID-AR1007>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 8.Anderson RB, Stewart AL, Young HM. Phenotypes of neural-crest-derived cells in vagal and sacral pathways. Cell Tissue Res. 2006;323:11–25. doi: 10.1007/s00441-005-0047-6. [DOI] [PubMed] [Google Scholar]
- 9.Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, Brooks AS, Antinolo G, de Pontual L, Clement-Ziza M, Munnich A, Kashuk C, West K, Wong KK, Lyonnet S, Chakravarti A, Tam PK, Ceccherini I, Hofstra RM, Fernandez R. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- 10.Tam PK, Garcia-Barceló M. Genetic basis of Hirschsprung’s disease. Pediatr Surg Int. 2009;25:543–558. doi: 10.1007/s00383-009-2402-2. [DOI] [PubMed] [Google Scholar]
- 11.Amiel J, Lyonnet S. Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet. 2001;38:729–739. doi: 10.1136/jmg.38.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doodnath R, Puri P. A systematic review and meta-analysis of Hirschsprung’s disease presenting after childhood. Pediatr Surg Int. 2010;26:1107–1110. doi: 10.1007/s00383-010-2694-2. [DOI] [PubMed] [Google Scholar]
- 13.Catto-Smith AG, Trajanovska M, Taylor RG. Long-term continence after surgery for Hirschsprung’s disease. J Gastroenterol Hepatol. 2007;22:2273–2282. doi: 10.1111/j.1440-1746.2006.04750.x. [DOI] [PubMed] [Google Scholar]
- 14.Tannuri AC, Tannuri U, Romão RL. Transanal endorectal pull-through in children with Hirschsprung’s disease--technical refinements and comparison of results with the Duhamel procedure. J Pediatr Surg. 2009;44:767–772. doi: 10.1016/j.jpedsurg.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Belknap WM. The pathogenesis of Hirschsprung disease. Curr Opin Gastroenterol. 2002;18:74–81. doi: 10.1097/00001574-200201000-00013. [DOI] [PubMed] [Google Scholar]
- 16.Gariepy CE. Genetic basis of Hirschsprung disease: implications in clinical practice. Mol Genet Metab. 2003;80:66–73. doi: 10.1016/j.ymgme.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Parisi MA, Kapur RP. Genetics of Hirschsprung disease. Curr Opin Pediatr. 2000;12:610–617. doi: 10.1097/00008480-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 18.Schindelhauer D, Schuffenhauer S, Gasser T, Steinkasserer A, Meitinger T. The gene coding for glial cell line derived neurotrophic factor (GDNF) maps to chromosome 5p12-p13.1. Genomics. 1995;28:605–607. doi: 10.1006/geno.1995.1202. [DOI] [PubMed] [Google Scholar]
- 19.Cik M, Masure S, Lesage AS, Van Der Linden I, Van Gompel P, Pangalos MN, Gordon RD, Leysen JE. Binding of GDNF and neurturin to human GDNF family receptor alpha 1 and 2. Influence of cRET and cooperative interactions. J Biol Chem. 2000;275:27505–27512. doi: 10.1074/jbc.M000306200. [DOI] [PubMed] [Google Scholar]
- 20.Burzynski GM, Nolte IM, Osinga J, Ceccherini I, Twigt B, Maas S, Brooks A, Verheij J, Plaza Menacho I, Buys CH, Hofstra RM. Localizing a putative mutation as the major contributor to the development of sporadic Hirschsprung disease to the RET genomic sequence between the promoter region and exon 2. Eur J Hum Genet. 2004;12:604–612. doi: 10.1038/sj.ejhg.5201199. [DOI] [PubMed] [Google Scholar]
- 21.Brooks AS, Bertoli-Avella AM, Burzynski GM, Breedveld GJ, Osinga J, Boven LG, Hurst JA, Mancini GM, Lequin MH, de Coo RF, Matera I, de Graaff E, Meijers C, Willems PJ, Tibboel D, Oostra BA, Hofstra RM. Homozygous nonsense mutations in KIAA1279 are associated with malformations of the central and enteric nervous systems. Am J Hum Genet. 2005;77:120–126. doi: 10.1086/431244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griseri P, Lantieri F, Puppo F, Bachetti T, Di Duca M, Ravazzolo R, Ceccherini I. A common variant located in the 3’UTR of the RET gene is associated with protection from Hirschsprung disease. Hum Mutat. 2007;28:168–176. doi: 10.1002/humu.20397. [DOI] [PubMed] [Google Scholar]
- 23.Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, Portnoy ME, Cutler DJ, Green ED, Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- 24.Natarajan D, Marcos-Gutierrez C, Pachnis V, de Graaff E. Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development. 2002;129:5151–5160. doi: 10.1242/dev.129.22.5151. [DOI] [PubMed] [Google Scholar]
- 25.Tomac AC, Grinberg A, Huang SP, Nosrat C, Wang Y, Borlongan C, Lin SZ, Chiang YH, Olson L, Westphal H, Hoffer BJ. Glial cell line-derived neurotrophic factor receptor alpha1 availability regulates glial cell line-derived neurotrophic factor signaling: evidence from mice carrying one or two mutated alleles. Neuroscience. 2000;95:1011–1023. doi: 10.1016/s0306-4522(99)00503-5. [DOI] [PubMed] [Google Scholar]
- 26.Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 27.Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 28.Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130:2187–2198. doi: 10.1242/dev.00433. [DOI] [PubMed] [Google Scholar]
- 29.Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M. Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene. 1990;5:97–102. [PubMed] [Google Scholar]
- 30.Carter MT, Yome JL, Marcil MN, Martin CA, Vanhorne JB, Mulligan LM. Conservation of RET proto-oncogene splicing variants and implications for RET isoform function. Cytogenet Cell Genet. 2001;95:169–176. doi: 10.1159/000059341. [DOI] [PubMed] [Google Scholar]
- 31.de Graaff E, Srinivas S, Kilkenny C, D’Agati V, Mankoo BS, Costantini F, Pachnis V. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev. 2001;15:2433–2444. doi: 10.1101/gad.205001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uesaka T, Nagashimada M, Yonemura S, Enomoto H. Diminished Ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in mice. J Clin Invest. 2008;118:1890–1898. doi: 10.1172/JCI34425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asai N, Fukuda T, Wu Z, Enomoto A, Pachnis V, Takahashi M, Costantini F. Targeted mutation of serine 697 in the Ret tyrosine kinase causes migration defect of enteric neural crest cells. Development. 2006;133:4507–4516. doi: 10.1242/dev.02616. [DOI] [PubMed] [Google Scholar]
- 34.Jain S, Knoten A, Hoshi M, Wang H, Vohra B, Heuckeroth RO, Milbrandt J. Organotypic specificity of key RET adaptor-docking sites in the pathogenesis of neurocristopathies and renal malformations in mice. J Clin Invest. 2010;120:778–790. doi: 10.1172/JCI41619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carniti C, Belluco S, Riccardi E, Cranston AN, Mondellini P, Ponder BA, Scanziani E, Pierotti MA, Bongarzone I. The Ret(C620R) mutation affects renal and enteric development in a mouse model of Hirschsprung’s disease. Am J Pathol. 2006;168:1262–1275. doi: 10.2353/ajpath.2006.050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nataf V, Amemiya A, Yanagisawa M, Le Douarin NM. The expression pattern of endothelin 3 in the avian embryo. Mech Dev. 1998;73:217–220. doi: 10.1016/s0925-4773(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 37.Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–1285. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 38.Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M. ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78:473–485. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- 39.Nataf V, Lecoin L, Eichmann A, Le Douarin NM. Endothelin-B receptor is expressed by neural crest cells in the avian embryo. Proc Natl Acad Sci USA. 1996;93:9645–9650. doi: 10.1073/pnas.93.18.9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofstra RM, Valdenaire O, Arch E, Osinga J, Kroes H, Löffler BM, Hamosh A, Meijers C, Buys CH. A loss-of-function mutation in the endothelin-converting enzyme 1 (ECE-1) associated with Hirschsprung disease, cardiac defects, and autonomic dysfunction. Am J Hum Genet. 1999;64:304–308. doi: 10.1086/302184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, Yanagisawa M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell. 1994;79:1267–1276. doi: 10.1016/0092-8674(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 42.Barlow A, de Graaff E, Pachnis V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron. 2003;40:905–916. doi: 10.1016/s0896-6273(03)00730-x. [DOI] [PubMed] [Google Scholar]
- 43.Druckenbrod NR, Epstein ML. Age-dependent changes in the gut environment restrict the invasion of the hindgut by enteric neural progenitors. Development. 2009;136:3195–3203. doi: 10.1242/dev.031302. [DOI] [PubMed] [Google Scholar]
- 44.Puffenberger EG, Hosoda K, Washington SS, Nakao K, deWit D, Yanagisawa M, Chakravart A. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell. 1994;79:1257–1266. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 45.Cantrell VA, Owens SE, Chandler RL, Airey DC, Bradley KM, Smith JR, Southard-Smith EM. Interactions between Sox10 and EdnrB modulate penetrance and severity of aganglionosis in the Sox10Dom mouse model of Hirschsprung disease. Hum Mol Genet. 2004;13:2289–2301. doi: 10.1093/hmg/ddh243. [DOI] [PubMed] [Google Scholar]
- 46.Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Préhu MO, Puliti A, Herbarth B, Hermans-Borgmeyer I, Legius E, Matthijs G, Amiel J, Lyonnet S, Ceccherini I, Romeo G, Smith JC, Read AP, Wegner M, Goossens M. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18:171–173. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 47.Touraine RL, Attié-Bitach T, Manceau E, Korsch E, Sarda P, Pingault V, Encha-Razavi F, Pelet A, Augé J, Nivelon-Chevallier A, Holschneider AM, Munnes M, Doerfler W, Goossens M, Munnich A, Vekemans M, Lyonnet S. Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. Am J Hum Genet. 2000;66:1496–1503. doi: 10.1086/302895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Southard-Smith EM, Angrist M, Ellison JS, Agarwala R, Baxevanis AD, Chakravarti A, Pavan WJ. The Sox10(Dom) mouse: modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome Res. 1999;9:215–225. [PubMed] [Google Scholar]
- 49.Sánchez-Mejías A, Watanabe Y, M Fernández R, López-Alonso M, Antiñolo G, Bondurand N, Borrego S. Involvement of SOX10 in the pathogenesis of Hirschsprung disease: report of a truncating mutation in an isolated patient. J Mol Med (Berl) 2010;88:507–514. doi: 10.1007/s00109-010-0592-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Southard-Smith EM, Kos L, Pavan WJ. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat Genet. 1998;18:60–64. doi: 10.1038/ng0198-60. [DOI] [PubMed] [Google Scholar]
- 51.Kapur RP. Early death of neural crest cells is responsible for total enteric aganglionosis in Sox10(Dom)/Sox10(Dom) mouse embryos. Pediatr Dev Pathol. 1999;2:559–569. doi: 10.1007/s100249900162. [DOI] [PubMed] [Google Scholar]
- 52.Stanchina L, Baral V, Robert F, Pingault V, Lemort N, Pachnis V, Goossens M, Bondurand N. Interactions between Sox10, Edn3 and Ednrb during enteric nervous system and melanocyte development. Dev Biol. 2006;295:232–249. doi: 10.1016/j.ydbio.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 53.Maka M, Stolt CC, Wegner M. Identification of Sox8 as a modifier gene in a mouse model of Hirschsprung disease reveals underlying molecular defect. Dev Biol. 2005;277:155–169. doi: 10.1016/j.ydbio.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 54.Lang D, Chen F, Milewski R, Li J, Lu MM, Epstein JA. Pax3 is required for enteric ganglia formation and functions with Sox10 to modulate expression of c-ret. J Clin Invest. 2000;106:963–971. doi: 10.1172/JCI10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lang D, Epstein JA. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum Mol Genet. 2003;12:937–945. doi: 10.1093/hmg/ddg107. [DOI] [PubMed] [Google Scholar]
- 56.Zhu L, Lee HO, Jordan CS, Cantrell VA, Southard-Smith EM, Shin MK. Spatiotemporal regulation of endothelin receptor-B by SOX10 in neural crest-derived enteric neuron precursors. Nat Genet. 2004;36:732–737. doi: 10.1038/ng1371. [DOI] [PubMed] [Google Scholar]
- 57.Young HM, Ciampoli D, Hsuan J, Canty AJ. Expression of Ret-, p75(NTR)-, Phox2a-, Phox2b-, and tyrosine hydroxylase-immunoreactivity by undifferentiated neural crest-derived cells and different classes of enteric neurons in the embryonic mouse gut. Dev Dyn. 1999;216:137–152. doi: 10.1002/(SICI)1097-0177(199910)216:2<137::AID-DVDY5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 58.Garcia-Barceló M, Sham MH, Lui VC, Chen BL, Ott J, Tam PK. Association study of PHOX2B as a candidate gene for Hirschsprung’s disease. Gut. 2003;52:563–567. doi: 10.1136/gut.52.4.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, Vekemans M, Munnich A, Gaultier C, Lyonnet S. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 60.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399:366–370. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- 61.Leon TY, Ngan ES, Poon HC, So MT, Lui VC, Tam PK, Garcia-Barcelo MM. Transcriptional regulation of RET by Nkx2-1, Phox2b, Sox10, and Pax3. J Pediatr Surg. 2009;44:1904–1912. doi: 10.1016/j.jpedsurg.2008.11.055. [DOI] [PubMed] [Google Scholar]
- 62.Yamada K, Yamada Y, Nomura N, Miura K, Wakako R, Hayakawa C, Matsumoto A, Kumagai T, Yoshimura I, Miyazaki S, Kato K, Sonta S, Ono H, Yamanaka T, Nagaya M, Wakamatsu N. Nonsense and frameshift mutations in ZFHX1B, encoding Smad-interacting protein 1, cause a complex developmental disorder with a great variety of clinical features. Am J Hum Genet. 2001;69:1178–1185. doi: 10.1086/324343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van de Putte T, Maruhashi M, Francis A, Nelles L, Kondoh H, Huylebroeck D, Higashi Y. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am J Hum Genet. 2003;72:465–470. doi: 10.1086/346092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carrasquillo MM, McCallion AS, Puffenberger EG, Kashuk CS, Nouri N, Chakravarti A. Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet. 2002;32:237–244. doi: 10.1038/ng998. [DOI] [PubMed] [Google Scholar]
- 65.McCallion AS, Stames E, Conlon RA, Chakravarti A. Phenotype variation in two-locus mouse models of Hirschsprung disease: tissue-specific interaction between Ret and Ednrb. Proc Natl Acad Sci USA. 2003;100:1826–1831. doi: 10.1073/pnas.0337540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Owens SE, Broman KW, Wiltshire T, Elmore JB, Bradley KM, Smith JR, Southard-Smith EM. Genome-wide linkage identifies novel modifier loci of aganglionosis in the Sox10Dom model of Hirschsprung disease. Hum Mol Genet. 2005;14:1549–1558. doi: 10.1093/hmg/ddi163. [DOI] [PubMed] [Google Scholar]
- 67.Lang D, Epstein JA. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum Mol Genet. 2003;12:937–945. doi: 10.1093/hmg/ddg107. [DOI] [PubMed] [Google Scholar]
- 68.de Pontual L, Pelet A, Trochet D, Jaubert F, Espinosa-Parrilla Y, Munnich A, Brunet JF, Goridis C, Feingold J, Lyonnet S, Amiel J. Mutations of the RET gene in isolated and syndromic Hirschsprung’s disease in human disclose major and modifier alleles at a single locus. J Med Genet. 2006;43:419–423. doi: 10.1136/jmg.2005.040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stanchina L, Van de Putte T, Goossens M, Huylebroeck D, Bondurand N. Genetic interaction between Sox10 and Zfhx1b during enteric nervous system development. Dev Biol. 2010;341:416–428. doi: 10.1016/j.ydbio.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 70.Wallace AS, Schmidt C, Schachner M, Wegner M, Anderson RB. L1cam acts as a modifier gene during enteric nervous system development. Neurobiol Dis. 2010;40:622–633. doi: 10.1016/j.nbd.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 71.Bolk S, Pelet A, Hofstra RM, Angrist M, Salomon R, Croaker D, Buys CH, Lyonnet S, Chakravarti A. A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc Natl Acad Sci. 2000;97:268–273. doi: 10.1073/pnas.97.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garcia-Barcelo MM, Tang CS, Ngan ES, Lui VC, Chen Y, So MT, Leon TY, Miao XP, Shum CK, Liu FQ, et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung’s disease. Proc Natl Acad Sci USA. 2009;106:2694–2699. doi: 10.1073/pnas.0809630105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Pontual L, Pelet A, Clement-Ziza M, Trochet D, Antonarakis SE, Attie-Bitach T, Beales PL, Blouin JL, Dastot-Le Moal F, Dollfus H, et al. Epistatic interactions with a common hypomorphic RET allele in syndromic Hirschsprung disease. Hum Mutat. 2007;28:790–796. doi: 10.1002/humu.20517. [DOI] [PubMed] [Google Scholar]
- 75.de Pontual L, Zaghloul NA, Thomas S, Davis EE, McGaughey DM, Dollfus H, Baumann C, Bessling SL, Babarit C, Pelet A, et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc Natl Acad Sci USA. 2009;106:13921–13926. doi: 10.1073/pnas.0901219106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parisi MA, Kapur RP, Neilson I, Hofstra RM, Holloway LW, Michaelis RC, Leppig KA. Hydrocephalus and intestinal aganglionosis: is L1CAM a modifier gene in Hirschsprung disease? Am J Med Genet. 2002;108:51–56. doi: 10.1002/ajmg.10185. [DOI] [PubMed] [Google Scholar]
- 77.Rehberg S, Lischka P, Glaser G, Stamminger T, Wegner M, Rosorius O. Sox10 is an active nucleocytoplasmic shuttle protein, and shuttling is crucial for Sox10-mediated transactivation. Mol Cell Biol. 2002;22:5826–5834. doi: 10.1128/MCB.22.16.5826-5834.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stolt CC, Lommes P, Friedrich RP, Wegner M. Transcription factors Sox8 and Sox10 perform non-equivalent roles during oligodendrocyte development despite functional redundancy. Development. 2004;131:2349–2358. doi: 10.1242/dev.01114. [DOI] [PubMed] [Google Scholar]
- 79.Schepers G, Wilson M, Wilhelm D, Koopman P. SOX8 is expressed during testis differentiation in mice and synergizes with SF1 to activate the Amh promoter in vitro. J Biol Chem. 2003;278:28101–28108. doi: 10.1074/jbc.M304067200. [DOI] [PubMed] [Google Scholar]
- 80.Schmidt K, Glaser G, Wernig A, Wegner M, Rosorius O. Sox8 is a specific marker for muscle satellite cells and inhibits myogenesis. J Biol Chem. 2003;278:29769–29775. doi: 10.1074/jbc.M301539200. [DOI] [PubMed] [Google Scholar]
- 81.Wallace AS, Tan MX, Schachner M, Anderson RB. L1cam acts as a modifier gene for members of the endothelin signalling pathway during enteric nervous system development. Neurogastroenterol Motil. 2011;23:e510–e522. doi: 10.1111/j.1365-2982.2011.01692.x. [DOI] [PubMed] [Google Scholar]
- 82.Attié T, Pelet A, Edery P, Eng C, Mulligan LM, Amiel J, Boutrand L, Beldjord C, Nihoul-Fékété C, Munnich A. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet. 1995;4:1381–1386. doi: 10.1093/hmg/4.8.1381. [DOI] [PubMed] [Google Scholar]
- 83.Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, Pasini B, Höppener JW, van Amstel HK, Romeo G. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367:375–376. doi: 10.1038/367375a0. [DOI] [PubMed] [Google Scholar]
- 84.Hofstra RM, Wu Y, Stulp RP, Elfferich P, Osinga J, Maas SM, Siderius L, Brooks AS, vd Ende JJ, Heydendael VM, et al. RET and GDNF gene scanning in Hirschsprung patients using two dual denaturing gel systems. Hum Mutat. 2000;15:418–429. doi: 10.1002/(SICI)1098-1004(200005)15:5<418::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 85.Angrist M, Bolk S, Halushka M, Lapchak PA, Chakravarti A. Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet. 1996;14:341–344. doi: 10.1038/ng1196-341. [DOI] [PubMed] [Google Scholar]
- 86.Hofstra RM, Osinga J, Buys CH. Mutations in Hirschsprung disease: when does a mutation contribute to the phenotype. Eur J Hum Genet. 1997;5:180–185. [PubMed] [Google Scholar]
- 87.Ivanchuk SM, Myers SM, Eng C, Mulligan LM. De novo mutation of GDNF, ligand for the RET/GDNFR-alpha receptor complex, in Hirschsprung disease. Hum Mol Genet. 1996;5:2023–2026. doi: 10.1093/hmg/5.12.2023. [DOI] [PubMed] [Google Scholar]
- 88.Martucciello G, Ceccherini I, Lerone M, Jasonni V. Pathogenesis of Hirschsprung’s disease. J Pediatr Surg. 2000;35:1017–1025. doi: 10.1053/jpsu.2000.7763. [DOI] [PubMed] [Google Scholar]
- 89.Salomon R, Attié T, Pelet A, Bidaud C, Eng C, Amiel J, Sarnacki S, Goulet O, Ricour C, Nihoul-Fékété C, et al. Germline mutations of the RET ligand GDNF are not sufficient to cause Hirschsprung disease. Nat Genet. 1996;14:345–347. doi: 10.1038/ng1196-345. [DOI] [PubMed] [Google Scholar]
- 90.Chakravarti A. Endothelin receptor-mediated signaling in hirschsprung disease. Hum Mol Genet. 1996;5:303–307. [PubMed] [Google Scholar]
- 91.Bidaud C, Salomon R, Van Camp G, Pelet A, Attié T, Eng C, Bonduelle M, Amiel J, Nihoul-Fékété C, Willems PJ, et al. Endothelin-3 gene mutations in isolated and syndromic Hirschsprung disease. Eur J Hum Genet. 1997;5:247–251. [PubMed] [Google Scholar]
- 92.Pingault V, Puliti A, Préhu MO, Samadi A, Bondurand N, Goossens M. Human homology and candidate genes for the Dominant megacolon locus, a mouse model of Hirschsprung disease. Genomics. 1997;39:86–89. doi: 10.1006/geno.1996.4476. [DOI] [PubMed] [Google Scholar]
- 93.Lurie IW, Supovitz KR, Rosenblum-Vos LS, Wulfsberg EA. Phenotypic variability of del(2) (q22-q23): report of a case with a review of the literature. Genet Couns. 1994;5:11–14. [PubMed] [Google Scholar]
- 94.Mowat DR, Croaker GD, Cass DT, Kerr BA, Chaitow J, Adès LC, Chia NL, Wilson MJ. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet. 1998;35:617–623. doi: 10.1136/jmg.35.8.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wakamatsu N, Yamada Y, Yamada K, Ono T, Nomura N, Taniguchi H, Kitoh H, Mutoh N, Yamanaka T, Mushiake K, et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat Genet. 2001;27:369–370. doi: 10.1038/86860. [DOI] [PubMed] [Google Scholar]
- 96.Trochet D, O’Brien LM, Gozal D, Trang H, Nordenskjöld A, Laudier B, Svensson PJ, Uhrig S, Cole T, Niemann S, et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet. 2005;76:421–426. doi: 10.1086/428366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Doray B, Salomon R, Amiel J, Pelet A, Touraine R, Billaud M, Attié T, Bachy B, Munnich A, Lyonnet S. Mutation of the RET ligand, neurturin, supports multigenic inheritance in Hirschsprung disease. Hum Mol Genet. 1998;7:1449–1452. doi: 10.1093/hmg/7.9.1449. [DOI] [PubMed] [Google Scholar]
- 98.Rosenfeld JA, Leppig K, Ballif BC, Thiese H, Erdie-Lalena C, Bawle E, Sastry S, Spence JE, Bandholz A, Surti U, et al. Genotype-phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med. 2009;11:797–805. doi: 10.1097/GIM.0b013e3181bd38a9. [DOI] [PubMed] [Google Scholar]