

LifeAct-GFP, a fluorescent reporter for actin filaments, is used to uncover the dynamics of actin cytoskeleton remodeling in real time during TGF-β–induced EMT. Efficient actin filament remodeling and complete transition to a mesenchymal phenotype depend on an increase in expression of the ERM protein moesin.
Abstract
Remodeling of actin filaments is necessary for epithelial–mesenchymal transition (EMT); however, understanding of how this is regulated in real time is limited. We used an actin filament reporter and high-resolution live-cell imaging to analyze the regulated dynamics of actin filaments during transforming growth factor-β–induced EMT of mammary epithelial cells. Progressive changes in cell morphology were accompanied by reorganization of actin filaments from thin cortical bundles in epithelial cells to thick, parallel, contractile bundles that disassembled more slowly but remained dynamic in transdifferentiated cells. We show that efficient actin filament remodeling during EMT depends on increased expression of the ezrin/radixin/moesin (ERM) protein moesin. Cells suppressed for moesin expression by short hairpin RNA had fewer, thinner, and less stable actin bundles, incomplete morphological transition, and decreased invasive capacity. These cells also had less α-smooth muscle actin and phosphorylated myosin light chain in cortical patches, decreased abundance of the adhesion receptor CD44 at membrane protrusions, and attenuated autophosphorylation of focal adhesion kinase. Our findings suggest that increased moesin expression promotes EMT by regulating adhesion and contractile elements for changes in actin filament organization. We propose that the transciptional program driving EMT controls progressive remodeling of actin filament architectures.
INTRODUCTION
Epithelial–mesenchymal transition (EMT) is a transcriptional and morphological program that occurs during normal development and tissue remodeling and in the progression of diseases such as fibrosis and metastatic cancers. As a process of epithelial plasticity, EMT is achieved when epithelial cell–cell adhesions are dissolved, the actin cytoskeleton is reorganized, and cells acquire increased cell–matrix contacts and enhanced migratory and invasive capabilities (Xu et al., 2009; Yilmaz and Christofori, 2009). The most recognized inducers of EMT are growth factors acting through receptor tyrosine kinases, secreted signaling molecules in the Wnt and Notch families, and cytokines, such as transforming growth factor-β (TGF-β) (Moustakas and Heldin, 2007). The transcriptional program for EMT induced by TGF-β is well characterized and is coordinated primarily through Smad-dependent activation of transcription factors of the Snail, ZEB, and Twist families (Xu et al., 2009). These transcription factors drive EMT by repressing expression of epithelial genes and activating expression of mesenchymal genes. Down-regulated genes include those encoding proteins maintaining epithelial cell–cell adhesions, such as the adherens junction protein E-cadherin, and the tight junctions proteins claudins and occludin. Up-regulated genes include those encoding proteins promoting cell migration and invasion, such as the mesenchymal cell–cell adhesion protein N-cadherin, the intermediate filament protein vimentin, and the extracellular matrix proteins fibronectin and collagen.
In contrast with the transcriptional program controlling transdifferentiation and morphological changes during EMT, dynamic remodeling of the actin cytoskeleton and how this is regulated are less well understood. Actin filaments in epithelial cells are organized in cortical thin bundles. In contrast, actin filaments in transdifferentiated mesenchymal cells are bundled into thick contractile stress fibers at the ventral cell surface. For TGF-β–induced EMT, actin cytoskeleton remodeling requires activation of the guanosine triphosphatase (GTPase) RhoA, which also is necessary to disrupt localization of E-cadherin at cell–cell adhesions and to promote a mesenchymal cell morphology (Bhowmick et al., 2001; Tavares et al., 2006; Cho and Yoo, 2007). Inactivation of the RhoA effector, Rho-associated coiled-coil–containing protein kinase (ROCK), inhibits TGF-β–dependent assembly of actin filaments into stress fibers but not delocalization of E-cadherin (Bhowmick et al., 2001; Edlund et al., 2002; Masszi et al., 2003). Although a TGF-β–dependent increase in RhoA expression is reported to be necessary for EMT during embryonic chick heart development (Tavares et al., 2006), changes in RhoA expression have not been identified during EMT of cultured cells. Genome-wide expression studies of cell culture models of TGF-β–induced EMT indicate that genes encoding actin cytoskeleton–associated proteins are consistently upregulated (Zavadil et al., 2001; Xie et al., 2003; Valcourt et al., 2005; Keshamouni et al., 2006). However, the functional significance of this increased expression and whether actin cytoskeleton remodeling by proteins other than RhoA and ROCK are necessary for EMT are not known. Because remodeling of the actin cytoskeleton promotes morphological changes and cell migration during EMT and is also required for metastatic cancers to spread from primary tumors, factors controlling actin cytoskeleton remodeling are potentially key targets for therapeutics to restrict cancer progression.
We therefore asked two questions. First, how does dynamic remodeling of the actin cytoskeleton occur in real time during EMT? Second, does EMT and associated cytoskeleton remodeling depend on changes in the expression of actin regulatory proteins? In this study, we used high-resolution live-cell imaging of a fluorescent actin filament reporter to reveal regulated dynamics of filament remodeling during TGF-β–induced EMT of mouse mammary epithelial cells. We also report that increased expression of moesin, a member of the ezrin/radixin/moesin (ERM) family of actin-binding proteins, was necessary for efficient EMT. ERM proteins regulate cell morphology, migration, and adhesion by cross-linking actin filaments to plasma membrane proteins (Fievet et al., 2007; Fehon et al., 2010). Although the function of ERM proteins is often viewed as redundant, we found a distinct role for increased moesin in EMT that is not shared by ezrin or radixin. Our data show that during EMT increased moesin expression is necessary for efficient actin filament remodeling, including the stability of contractile actin filament bundles, and for cortical relocalization of adhesion and contractile elements, including CD44, α-smooth muscle actin (α-SMA), and phosphorylated myosin light chain (p-MLC). Moreover, our findings reveal a link between the transcriptional program of EMT and actin filament remodeling during transdifferentiation.
RESULTS
Dynamic changes in cell morphology and actin filament organization during TGF-β–induced EMT
To initially characterize the dynamics of cell morphological changes during EMT, we used phase contrast time-lapse microscopy over 48 h to observe mouse mammary epithelial NMuMG cells that were previously reported to undergo transdifferentiation with TGF-β treatment (Miettinen et al., 1994; Bakin et al., 2000; Bhowmick et al., 2001; Lamouille and Derynck, 2007). Untreated NMuMG epithelial cells were cuboidal shaped and organized in compact islets (Figure 1A and Supplemental Video S1). After ∼10 h with TGF-β, cells in these islets became more loosely arranged, and after ∼12 h they started to elongate. These changes progressed gradually to a spindle-shaped morphology with cells arranged in parallel, which was evident at ∼24 h with TGF-β, although cells elongated further between 24 and 48 h.
FIGURE 1:

Morphological changes and actin cytoskeleton remodeling during TGF-β–induced EMT of mouse mammary epithelial NMuMG cells. (A) Images of cells treated with TGF-β for the indicated times showing morphological changes during EMT. Images were acquired by phase contrast, time-lapse microscopy using a 40× objective. Bar, 20 μm. See Supplemental Video S1. (B) Rhodamine-phalloidin labeling of F-actin in cells maintained in the absence or presence of TGF-β for 48 h. Images show F-actin staining for cells at a single Z-section (bottom, middle, or top), or a maximum-intensity projection of multiple Z-sections. Bar, 20 μm. (C) F-actin labeling of cells expressing LifeAct-GFP maintained in the absence or presence of TGF-β for 48 h and stained with rhodamine-phalloidin after fixing. Bar, 20 μm. (D–F) Images of cells expressing LifeAct-GFP and treated with TGF-β for the indicated times. Images were acquired by spinning disk confocal time-lapse microscopy using a 60× objective to show dynamic remodeling of the actin cytoskeleton during EMT. Bars, 20 μm. (D) Cells were imaged from 6 to 33 h after TGF-β treatment. Images are displayed with a γ value of 0.9 to enhance dim features. See Supplemental Video S2. (E) Cells were imaged for 1 h, beginning at 24 or 48 h after TGF-β treatment. Untreated cells were imaged after 48 h with vehicle control. See Supplemental Videos S3–S5. (F) Boxed region in E magnified and shown as a montage of images that were captured every 5 min.
Changes in cell morphology corresponded with reorganization of filamentous actin (F-actin). In NMuMG cells maintained in the absence of TGF-β, phalloidin-labeled F-actin was predominantly organized in cortical bundles tightly associated with cell–cell adhesions (Figure 1B), as previously described (Miettinen et al., 1994). In contrast, after 48 h with TGF-β, F-actin was assembled into thick parallel bundles, or actin stress fibers, traversing the ventral cell surface. To characterize the dynamics of actin filament remodeling during EMT, we transiently expressed green fluorescent protein (GFP)–tagged LifeAct in NMuMG cells. LifeAct is a yeast F-actin–binding peptide that does not interfere with actin dynamics and has been used to visualize F-actin in live cells (Riedl et al., 2008), but its use during EMT has not been reported. In NMuMG cells maintained in the absence or presence of TGF-β for 48 h, LifeAct-GFP colabeled F-actin stained with rhodamine-phalloidin (Figure 1C) and did not disrupt actin filament remodeling, which validates its use as a reporter of actin filament dynamics during EMT.
We used spinning disk confocal fluorescence time-lapse microscopy to monitor actin filament dynamics in live cells undergoing TGF-β–induced EMT. Because long-term fluorescent imaging is technically challenging, we observed a time window between 6 and 33 h after treatment with TGF-β and focused on the ventral cell surface, where stress fibers assemble and where we expected the most dramatic changes in F-actin organization to occur (Figure 1B). We did not observe a rapid switch in actin filament organization but instead found a slow and progressive increase in the number, width, and length of actin filaments that occurred in parallel with changes in cell morphology (Figure 1D and Supplemental Video S2). By ∼24 h with TGF-β, most cells had assembled thick contractile actin stress fibers. To analyze the dynamics of these actin stress fibers, we imaged cells expressing LifeAct-GFP after treatment with TGF-β for 24 or 48 h at a higher time resolution. Actin stress fibers further increased in number and size between 24 and 48 h with TGF-β (Figure 1E and Supplemental Videos S3 and S4). By 48 h with TGF-β, stress fibers appeared thicker and more bundled and remained assembled longer compared with earlier time points but they remained dynamic and contractile (Figure 1F). In contrast, untreated cells expressing LifeAct-GFP retained a randomly organized network of thin, short, noncontractile actin filaments at the basal surface (Figure 1E and Supplemental Video S5). In addition to actin filament remodeling, these time-lapse movies also indicated a decrease in the number of membrane protrusions with TGF-β treatment. Thus, the marked changes in cell morphology that occur during TGF-β–induced EMT are accompanied by a progressive and dynamic remodeling of the actin cytoskeleton that includes distinct changes in actin stress fiber bundling and contractility and fewer membrane protrusions.
ERM protein expression changes during TGF-β–induced EMT
EMT is a transcriptional program that down-regulates expression of epithelial genes and up-regulates expression of mesenchymal genes. TGF-β–induced EMT of NMuMG cells was accompanied by a gradual decrease in the abundance of the epithelial cell–cell adhesion protein E-cadherin and an increase in the abundance of the mesenchymal adhesion protein N-cadherin (Figure 2A), as previously shown (Miettinen et al., 1994; Bhowmick et al., 2001). The slow and progressive changes in cell morphology and actin cytoskeleton remodeling during EMT suggested transcriptional regulation of genes encoding proteins that control actin filament organization rather than rapid signaling events. To test this, we analyzed the expression levels of ERM proteins ezrin, radixin, and moesin, which bind actin filaments and have an established role in epithelial cell morphology (Fehon et al., 2010). Immunoblotting with specific as well as pan-ERM antibodies showed that the abundance of ezrin decreased, whereas the abundance of moesin increased, as early as 24 h after TGF-β treatment (Figure 2B). In contrast, the abundance of radixin was unchanged (Figure 2C). After 3 d with TGF-β, ezrin protein levels decreased 2.7-fold (p < 0.001; n = 3) and moesin protein levels increased 2.3-fold (p < 0.001; n = 3; Figure 2D). Increased abundance of moesin was sustained for up to 7 d with TGF-β (unpublished data). Consistent with our immunoblot data, quantitative PCR (qPCR) analysis showed that changes in ezrin and moesin protein expression were preceded by changes in gene expression (Figure 2E). After 48 h with TGF-β, ezrin mRNA levels decreased 2.8-fold (p < 0.01; n = 4) and moesin mRNA levels increased 5.2-fold (p < 0.01; n = 4). Furthermore, qPCR analysis revealed that by 48 h with TGF-β, moesin was the most abundant ERM mRNA expressed (78%), compared with untreated control cells, in which ezrin was predominant (59%). These opposing changes in expression of ezrin and moesin indicate that ERM protein switching occurs during the initial stages of TGF-β–induced EMT and suggest that ERM proteins may have nonredundant functions.
FIGURE 2:

Expression of the ERM proteins ezrin and moesin changes during TGF-β–induced EMT. (A–C) Immunoblots of lysates from NMuMG cells treated with TGF-β for the indicated times, showing changes in protein expression during EMT. Cells were maintained in the absence (−) or presence (+) of TGF-β for the indicated times. Blots were probed with E-cadherin– and N-cadherin–specific antibodies (A), ezrin- and moesin-specific and pan-ERM antibodies (B), and radixin-specific antibodies (C). Blots were also probed with antibodies for β-actin as a loading control. (D) Relative abundance of ezrin and moesin proteins in TGF-β–treated samples compared with untreated control samples was determined by semiquantitative densitometry of anti-ezrin and anti-moesin immunoblots. Data shown are means ± SEM of three independent cell preparations. (E) qPCR analysis of mRNA from NMuMG cells treated with TGF-β, showing changes in gene expression during EMT. Cells were maintained in the absence or presence of TGF-β for the indicated times. Relative mRNA expression for ezrin, moesin, and radixin in TGF-β–treated samples compared with untreated control samples was determined by qPCR and normalized to Rpl19 mRNA expression. Data shown are means ± SEM of at least three independent cell preparations. (F) Moesin immunolabeling, showing changes in localization during EMT. Cells were maintained in the absence or presence TGF-β for 48 h. Fixed cells were labeled with antibodies specific for moesin. Images show maximum-intensity projections of multiple Z-sections. Bar, 20 μm. (G) Immunoblots showing that ezrin and moesin expression is reversed during MET. Cells were maintained in the absence (−) or presence (+) of TGF-β for the indicated times to induce EMT and then cotreated with the TGF-β type I receptor inhibitor compound SB431542 in the absence or presence of TGF-β for the indicated times to induce MET. Blots were probed with E-cadherin and with ezrin- and moesin-specific antibodies. Blots were also probed with antibodies for β-actin as a loading control.
In addition to increased expression, changes in moesin localization were observed during EMT. In NMuMG cells maintained in the absence of TGF-β, moesin immunolabeling was localized at the apical membrane, associated with microvilli at the apical surface and concentrated at cell–cell adhesions (Figure 2F). In contrast, after 48 h with TGF-β, immunolabeling was predominantly localized at distinct large membrane protrusions on the dorsal cell surface and was also found at filopodia extending from the ventral cell surface. Consistent with its known role as a membrane–cytoskeleton linker, moesin colocalized with the plasma membrane and membrane-associated F-actin, as indicated by wheat germ agglutinin and phalloidin labeling, respectively (Supplemental Figure S1).
We also confirmed that changes in moesin and ezrin protein expression during EMT were reversible, by treating transdifferentiated NMuMG cells with the TGF-β type I receptor inhibitor SB431542, which induces mesenchymal–epithelial transition (MET; Halder et al., 2005). We confirmed MET of transdifferentiated cells treated with SB431542 for 2–3 d, as indicated by morphological changes from mesenchymal to epithelial (unpublished data) and increased abundance of E-cadherin protein (Figure 2G). In the presence of SB431542, the abundance of ezrin increased and the abundance of moesin decreased. These data show that ezrin and moesin expression in NMuMG cells is dynamically and reversibly regulated during transdifferentiation.
We next tested whether changes in ezrin and moesin expression are conserved during EMT in other cell types. Human mammary epithelial MCF-10A cells undergo EMT in 2–6 d when treated with TGF-β (Brown et al., 2004; Maeda et al., 2005). As expected, this was accompanied by morphological changes from epithelial to mesenchymal (unpublished data) and by increased abundance of the extracellular matrix protein fibronectin, a mesenchymal marker (Figure 3A). The abundance of moesin also increased, similar to what we observed during EMT of NMuMG cells. In contrast to NMuMG cells, however, there was no change in the abundance of ezrin and E-cadherin. During TGF-β–induced EMT of human lung adenocarcinoma A549 cells, which down-regulate E-cadherin expression (Kasai et al., 2005), the abundance of moesin and fibronectin increased, similar to MCF-10A cells. However, although the abundance of E-cadherin decreased, the abundance of ezrin was unchanged. These data suggest that increased expression of moesin is a conserved feature of TGF-β–induced EMT. Whether decreased expression of ezrin observed in NMuMG cells occurs in cell types other than MCF-10A or A549 cells remains to be determined.
FIGURE 3:
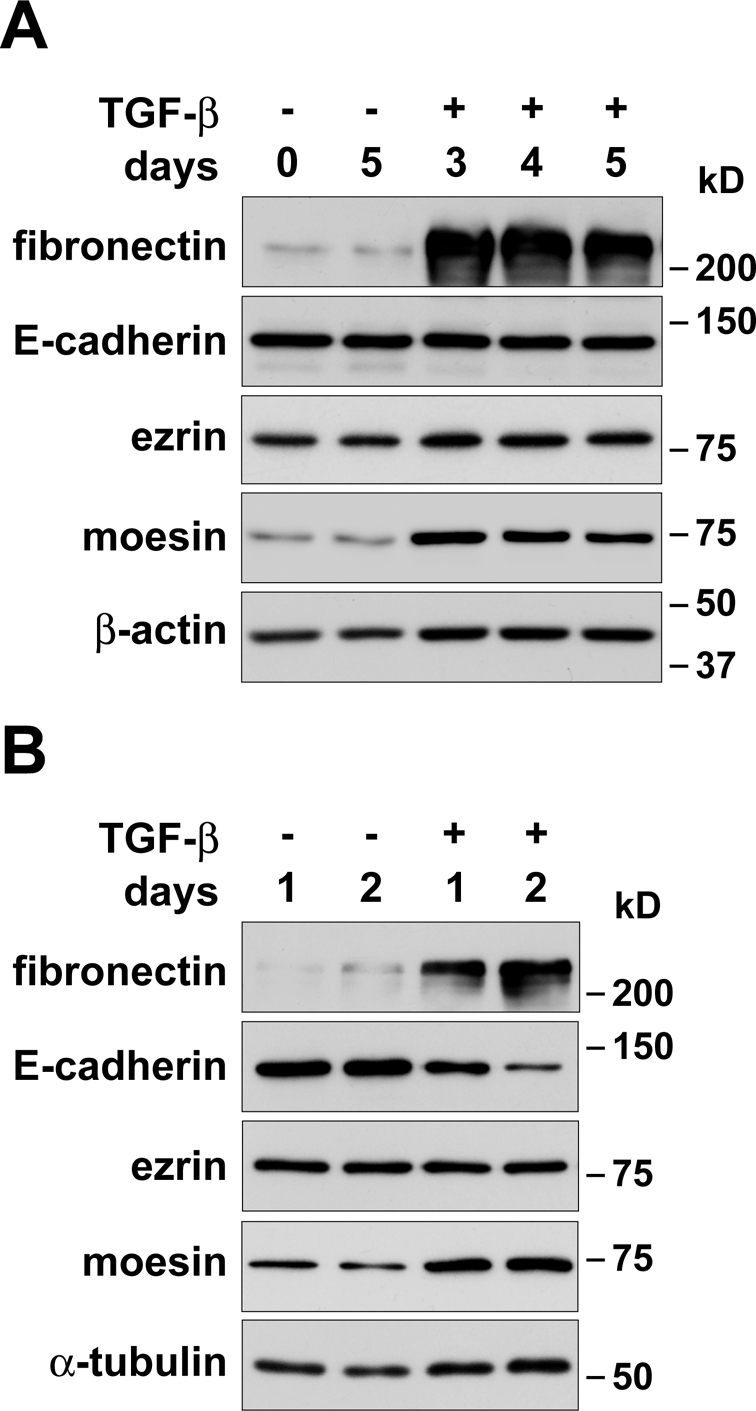
Increased expression of moesin during TGF-β–induced EMT is conserved in human cell lines. Immunoblots of lysates from cells treated with TGF-β for the indicated times, showing changes in protein expression during EMT. (A) MCF-10A cells were maintained in the absence (−) or presence (+) of TGF-β for 3–5 d. Blots were probed with antibodies for fibronectin, E-cadherin, ezrin, and moesin. Blots were also probed with antibodies for β-actin as a loading control. (B) A549 cells were maintained in the absence (−) or presence (+) of TGF-β for 1–2 d. Blots were probed with antibodies for fibronectin, E-cadherin, ezrin, and moesin. Blots were also probed with antibodies for α-tubulin as a loading control.
Increased moesin expression contributes to morphological changes and actin filament remodeling during EMT
To determine the functional significance of increased moesin during EMT, we suppressed moesin expression by infecting NMuMG cells with lentivirus expressing moesin-specific short hairpin RNA (shRNA) sequences. We selected stable clones having the greatest and most homogeneous knockdown of moesin, as determined by immunoblotting and immunolabeling, respectively (unpublished data). Control cells expressing nonsilencing shRNA sequences (control shRNA) showed changes in protein expression during EMT similar to those seen in wild-type cells, including decreased expression of E-cadherin and ezrin, and increased expression of N-cadherin and moesin (Figure 4A). Two clones of epithelial cells expressing moesin-specific shRNAs (moesin shRNA) had ∼80% less moesin but no change in the abundance of ezrin (Figure 4A). After 48 h with TGF-β, these cells had decreased abundance of E-cadherin and ezrin and increased abundance of N-cadherin, similar to wild-type and control shRNA cells. The abundance of moesin increased slightly, although total protein expression was still markedly less than with control cells.
FIGURE 4:

Moesin shRNA knockdown attenuates actin stress fiber assembly and full morphological transition during TGF-β–induced EMT. (A) Immunoblots of lysates from NMuMG cells treated with TGF-β, showing changes in protein expression during EMT. Wild-type (WT), control shRNA, and moesin shRNA cells were maintained in the absence (–) or presence (+) of TGF-β for 48 h. Blots were probed with antibodies for E-cadherin, N-cadherin, moesin, and ezrin. Blots were also probed with antibodies for β-actin and α-tubulin as loading controls. (B) E-cadherin immunolabeling and F-actin staining of cells, showing changes in organization of cell–cell adhesions and actin cytoskeleton during EMT. Wild-type, control shRNA, and moesin shRNA cells were maintained in the absence (–) or presence (+) of TGF-β (5 ng/ml) for 48 h. Also shown are wild-type cells cotreated with the ROCK inhibitor Y-27632. Fixed cells were labeled with antibodies for E-cadherin and with rhodamine-phalloidin. Arrows indicate cells having fewer, thinner, or absent actin stress fibers. Bar, 20 μm. (C) Quantitative analysis of cell morphology of TGF-β–treated cells in B, showing that the degree of elongated cell morphology, or morphological index, is reduced in moesin shRNA knockdown cells. Horizontal lines indicate the mean value for each cell type. Representative data shown are from a single experiment, for which n was between 30 and 40 for each cell type. Similar data were obtained from two independent cell preparations. **p < 0.01 by one-way ANOVA followed by Dunnett's post test. (D) F-actin staining, showing impaired assembly of actin stress fibers in moesin shRNA knockdown cells. Control shRNA and moesin shRNA cells were maintained in the absence (−) or presence (+) of a low concentration of TGF-β (1.25 ng/ml) for 24 h. Fixed cells were stained with rhodamine-phalloidin. Bar, 20 μm.
Moesin shRNA cells treated with TGF-β had distinct differences in cell morphology and actin filament organization compared with wild-type and control shRNA cells. Although E-cadherin was down-regulated (Figure 4A) and delocalized from cell–cell adhesions (Figure 4B), quantitative morphometric analysis showed that moesin shRNA cells did not reach a full morphological transition and were significantly less elongated than control shRNA cells (Figure 4C; p < 0.01). In addition, moesin shRNA cells had markedly fewer actin stress fibers, and bundled filaments were thinner, shorter, and less uniformly aligned along the major cell axes (Figure 4B). However, abundant thick and parallel stress fibers were observed in moesin shRNA cells transiently expressing moesin-GFP that is not targeted by moesin shRNA sequences (Supplemental Figure S2). These cells were also more elongated, but no differences in actin filaments or cell morphology occurred with expression of GFP alone (unpublished data). Moreover, when treated with a fourfold lower concentration of TGF-β for 24 h, moesin shRNA cells had no actin stress fibers, although short, bundled fibers were present in control shRNA cells (Figure 4D). To compare these data with the established regulation of actin cytoskeleton organization by ROCK during EMT, we treated cells with Y-27632, a pharmacological inhibitor of ROCK activity. Actin stress fibers were absent in wild-type cells treated with both TGF-β and Y-27632, although E-cadherin was delocalized from cell–cell adhesions as in control cells (Figure 4B). This is consistent with previous reports that inhibiting ROCK activity specifically blocks actin stress fiber formation without affecting dissolution of cell–cell adhesions during EMT (Bhowmick et al., 2001; Masszi et al., 2003; Hutchison et al., 2009). Our data indicate that increased moesin expression during EMT promotes the acquisition of a mesenchymal morphology and increased number and size of actin stress fibers.
Transdifferentiated cells with suppressed moesin expression also had impaired actin stress fiber dynamics. After treatment with TGF-β for 48 h, actin filaments in cells transiently expressing LifeAct-GFP assembled into stress fibers with varying degrees of thickness, stability, and movement. Approximately 40% of wild-type and control shRNA cells contained mainly thick, bundled actin stress fibers, and only ∼10% of cells had mostly thin fibers (Figure 5, A–C, and Supplemental Video S6). In contrast, only 5% of moesin shRNA cells had mainly thick fibers, whereas 55% of cells had mostly thin or no fibers (Figure 5, A–C, and Supplemental Video S7). The thick stress fiber bundles were generally aligned along the major cell axis, as seen with phalloidin labeling, and often appeared by lateral fusion of thinner fibers. Conversely, thick bundles often dissolved by spreading into a less tightly bundled array of thin fibers. This complexity of stress fiber dynamics made it difficult to quantitatively compare control and moesin shRNA cells. Qualitatively, however, actin stress fiber bundles appeared more stable in control cells, and although these bundles changed structure over time, they often remained visible for the duration of the movie (60 min). In contrast, the thin stress fiber bundles observed in moesin shRNA cells were shorter lived and were also less uniformly aligned compared with the thick stress fibers in control cells. Kymograph analysis of time-lapse sequences perpendicular to the stress fibers indicated that thin stress fiber bundles in moesin shRNA cells displayed increased lateral movement compared with thick stress fiber bundles in control cells, as indicated by continuous, relatively horizontal lines across the kymographs (Figure 5D). These data indicate that moesin promotes the assembly, organization, and stability of thick, bundled actin stress fibers in transdifferentiated cells.
FIGURE 5:

Moesin shRNA knockdown impairs actin stress fiber dynamics during TGF-β–induced EMT. (A) Images of WT, control shRNA, and moesin shRNA NMuMG cells expressing LifeAct-GFP and treated with TGF-β for 48 h. Images were acquired every 1 min for 1 h by spinning disk confocal time-lapse microscopy using a 60× objective (see Supplemental Videos S6 and S7). Bar, 20 μm. (B) Boxed regions in A magnified and shown as montages of images that were captured every 12 min. Bar, 20 μm. (C) Quantitative analysis of actin stress fiber thickness. The number of cells having mostly thick, intermediate or mixed, or mostly thin stress fibers were counted and expressed as percentage of the total number of cells counted. Data shown are from two independent experiments, for which n was between 46 and 51 for each cell type. (D) Kymographs of regions in A marked with yellow or cyan arrows, showing the stability of stress fibers over time.
Suppressing moesin expression during EMT limits relocalization of CD44, α-SMA, and p-MLC and the autophosphorylation of focal adhesion kinase
Additional cytoskeleton-associated changes that occur during TGF-β–induced EMT include increased expression of extracellular matrix proteins and acquisition of cell–substrate adhesions and cell contractility (Maeda et al., 2005). CD44, a cell surface receptor for extracellular matrix components that regulates cell adhesion and migration and binds to ERM proteins (Fehon et al., 2010), had increased abundance in wild-type and control shRNA cells treated with TGF-β (Figure 6A), consistent with recent findings that increased CD44 is a marker for EMT (Mani et al., 2008). In addition, CD44 relocalized from cell–cell adhesions in the absence of TGF-β to large dorsal membrane protrusions and numerous smaller membrane microextensions after 48 h with TGF-β. As expected, CD44 showed a high degree of colocalization with moesin in both the absence and presence of TGF-β (Figure 6B). Suppressing moesin expression slightly attenuated the increase in CD44 expression during EMT (Figure 6A); however, more markedly, it reduced the abundance of CD44 in dorsal protrusions compared with wild-type and control cells (Figure 6C), although CD44 remained localized to plasma membrane microextensions. Consistent with moesin regulating a cell–substrate adhesion protein, the increased abundance of autophosphorylated focal adhesion kinase (p-FAK) seen in wild-type and control shRNA cells, and previously reported for TGF-β-induced EMT (Nakamura et al., 2001), was markedly reduced in moesin shRNA cells (Figure 6D). The abundance of total FAK was unchanged during EMT in wild-type and moesin shRNA cells. Suppressing moesin expression had no effect on the increased abundance of fibronectin during EMT (Figure 6D) and it did not alter the size and number of paxillin-labeled focal adhesions compared with controls (Supplemental Figure S3), although our data do not rule out possible dual effects of moesin on focal adhesion assembly and turnover. However, clear effects of moesin on CD44 localization and p-FAK suggest that its increased expression contributes to cell–substrate adhesions during EMT.
FIGURE 6:

Moesin shRNA knockdown impairs relocalization of CD44 and α-SMA and attenuates increased phosphorylation of FAK during TGF-β–induced EMT. (A) Immunoblots of lysates from NMuMG cells treated with TGF-β, showing changes in expression of CD44 during EMT. Wild-type, control shRNA, and moesin shRNA cells were maintained in the absence (−) or presence (+) of TGF-β for 48 h. Blots were probed with antibodies for E-cadherin, CD44, and moesin. Blots were also probed with antibodies for β-actin as a loading control. (B, C) CD44 immunolabeling showing relocalization during EMT. Cells were maintained in the absence (−) or presence (+) of TGF-β for 48 h. (B) Wild-type cells were fixed and immunolabeled with antibodies for CD44 and moesin. Bar, 20 μm. (C) Control shRNA and moesin shRNA cells were fixed and immunolabeled with antibodies for CD44. The projections show total CD44 immunolabeling for TGF-β–treated cells that were imaged at multiple Z-sections. Bar, 20 μm. (D) Immunoblots of lysates from cells treated with TGF-β, showing changes in expression of cytoskeleton-associated proteins during EMT. Wild-type, control shRNA, and moesin shRNA cells were maintained in the absence (−) or presence (+) of TGF-β for 24 or 48 h. Also shown are wild-type cells cotreated with the ROCK inhibitor Y-27632. Blots were probed with antibodies for E-cadherin, fibronectin, p-FAK, FAK, and a-SMA and with antibodies for p-ERM or pan-ERM (ERM). Blots were also probed with antibodies for β-actin and α-tubulin as loading controls. (E) Immunolabeling of cells showing relocalization of α-SMA during EMT. Wild-type, control shRNA, and moesin shRNA cells were maintained in the absence (−) or presence (+) of TGF-β for 48 h. Fixed cells were immunolabeled with antibodies for α-SMA. The projections show total α-SMA immunolabeling for TGF-β–treated cells that were imaged at multiple Z-sections. Bar, 20 μm.
To compare our findings with established effects of ROCK activity on cell–substrate adhesions, we confirmed that cotreating wild-type cells with Y-27632 blocked TGF-β–induced increases in p-FAK and focal adhesion size and abundance but not fibronectin expression (Figure 6D and Supplemental Figure S3). Y-27632 also blocked an increase in the abundance of phosphorylated moesin. In wild-type cells treated with TGF-β, there was a time-dependent increase in phosphorylated moesin, with a 5.0-fold increase after 48 h, compared with a 2.0-fold increase in total moesin protein (Figure 6D; phosphorylated moesin is the lower band recognized by phosphorylated ERM [p-ERM] antibodies). Phosphorylation of moesin increases its actin cross-linking ability, which these data suggest may function in promoting EMT. Although Y-27632 prevented the increase in phosphorylated moesin, consistent with ERM proteins being substrates for ROCK (Matsui et al., 1998; Oshiro et al., 1998; Shaw et al., 1998), it had no effect on the increased abundance of total moesin protein. Despite the view that Rho, ROCK, and ERM proteins function in the same pathways regulating actin cytoskeleton organization (Mackay et al., 1997; Takahashi et al., 1997; Shaw et al., 1998; Speck et al., 2003; Lee et al., 2004), our data suggest that a transcriptional program for increased moesin expression during EMT is independent of ROCK activity.
Another notable cytoskeleton-associated change that occurs during TGF-β–induced EMT is increased expression of α-SMA (Masszi et al., 2003; Cho and Yoo, 2007). Immunoblot analysis confirmed a modest increase in the abundance of α-SMA in wild-type and control shRNA cells treated with TGF-β (Figure 6D), as previously described for NMuMG cells (Valcourt et al., 2005). The increase in α-SMA expression was blocked in wild-type cells cotreated with Y-27632, similar to previous findings (Cho and Yoo, 2007), but not in moesin shRNA cells (Figure 6D). A more distinct change in α-SMA during EMT of NMuMG cells was its relocalization from a diffuse distribution in the cytoplasm to prominent patches at the cell cortex (Figure 6E). In moesin shRNA cells, however, the abundance of α-SMA in cortical patches was markedly reduced compared with wild-type and control cells (Figure 6E), indicating that relocalization was incomplete. To our knowledge, cortical clustering of α-SMA has not previously been reported during EMT, but it may be a conserved feature because we also noted relocalization of α-SMA to cortical patches during EMT of A549 cells (Supplemental Figure S4A).
We further characterized these cortical α-SMA patches in transdifferentiated NMuMG cells by showing that although they did not localize at actin stress fibers or label with phalloidin (Figure 7A), they were still present after Triton extraction to remove soluble proteins prior to fixation and immunolabeling, which indicates cytoskeleton association (Supplemental Figure S4B). Moreover, a subset colocalized with moesin, as indicated by immunolabeling for moesin and for phosphorylated ERM proteins (Figure 7B). Also colocalizing with a subset of α-SMA patches were the p34Arc subunit of the Arp2/3 complex that binds and nucleates actin filaments and p-MLC (Figure 7B). Association with p34Arc and p-MLC suggested that cortical α-SMA patches could be regulated by actomyosin contractility. To confirm this, we treated transdifferentiated cells with Y-27632 or with blebbistatin, a myosin II inhibitor, which disassembled actin stress fibers and completely abolished cortical α-SMA localization (Figure 7A). In addition, treating transdifferentiated cells with the microtubule-depolymerizing agent nocodazole, which stimulates contractility, increased the number and thickness of actin stress fibers and the number of cortical α-SMA patches (Figure 7A). Together, these findings indicate that moesin regulates a contractility-dependent clustering of α-SMA at the cell cortex that we predict is necessary for a complete EMT.
FIGURE 7:

Cortical α-SMA localization is regulated by actomyosin contractility and is associated with moesin. (A) F-actin staining and α-SMA immunolabeling, showing α-SMA localization in the presence of pharmacological inhibitors that regulate actomyosin contractility. Wild-type cells were maintained in the presence of TGF-β for 48 h and then cotreated for 1 h with the ROCK inhibitor Y-27632, the myosin II inhibitor blebbistatin, or the microtubule-depolymerizing agent nocodazole. Fixed cells were labeled with rhodamine-phalloidin and with antibodies for α-SMA. The projections show total α-SMA immunolabeling for cells that were imaged at multiple Z-sections. Bar, 20 μm. (B) Wild-type NMuMG cells treated with TGF-β for 48 h immunolabeled for α-SMA and colabeled with antibodies for moesin, p-ERM, p-MLC, and the p34 subunit of the Arp2/3 complex (p34Arc). Bars, 20 μm.
To further test a role for moesin in contractility-dependent cortical clustering, we recorded time-lapse movies of wild-type cells transiently expressing moesin-GFP. In transdifferentiated cells, we also observed clusters of moesin-GFP enriched at membrane protrusions that clearly formed as a result of contractile intracellular movements and that were reminiscent of α-SMA patches (Supplemental Video S8, right, arrows). In contrast, contractile moesin clusters were not evident in cells maintained in the absence of TGF-β, in which moesin-GFP localized to highly dynamic membrane patches and filamentous structures (Supplemental Video S8, left). We also asked whether the localization of p-MLC changes during transdifferentiation and whether this is dependent on increased moesin expression. In wild-type and control shRNA cells maintained in the absence of TGF-β, p-MLC was distributed diffusely in the cytoplasm and enriched at cell–cell adhesions. After 48 h with TGF-β, p-MLC was predominantly localized along actin stress fibers and in small cortical aggregates near the dorsal cell surface (Figure 8A). Suppressing moesin expression during EMT had no obvious effect on p-MLC localized at actin stress fibers; however, it markedly reduced the abundance of cortical p-MLC aggregates. Furthermore, p-MLC colocalized with moesin at a subset of membrane protrusions in transdifferentiated wild-type cells (Figure 8B). Control cells treated with TGF-β also had increased abundance of p-MLC, as indicated by immunoblotting, which was not different in cells with suppressed moesin expression (unpublished data). These data confirm that increased moesin expression during EMT is necessary for the cortical localization of p-MLC and α-SMA, which is associated with the cytoskeleton and regulated by actomyosin contractility.
FIGURE 8:

Moesin shRNA knockdown impairs relocalization of phosphorylated MLC to cortical patches during TGF-β–induced EMT. (A) Immunolabeling of NMuMG cells showing relocalization of p-MLC during EMT. Wild-type, control shRNA, and moesin shRNA cells were maintained in the absence or presence of TGF-β for 48 h. Fixed cells were immunolabeled with antibodies for MLC-pS19. Bar, 20 μm. (B) Colocalization p-MLC and moesin at cortical patches. Wild-type cells treated with TGF-β for 48 h immunolabeled for p-MLC and colabeled with antibodies for moesin. Bar, 20 μm.
Suppressing moesin expression during EMT increases cell migration in monolayer wound healing but decreases cell invasion
In addition to inducing changes in cell morphology, actin cytoskeleton organization, and adhesions, TGF-β promotes increased cell migration and invasion, which contribute to the progression of metastatic cancers (Yilmaz and Christofori, 2009). To determine whether moesin regulates the migration of transdifferentiated cells, we wounded a monolayer of cells treated with TGF-β for 48 h and monitored wound closure by time-lapse microscopy (Figure 9, A and B, and Supplemental Videos S9 and S10). Wild-type and control shRNA cells migrated at similar rates of 10.39 ± 0.84 and 12.09 ± 0.95 μm/h, respectively (mean ± SEM over 20 h; n = 4), consistent with previous reports (Lamouille and Derynck, 2007). In contrast, moesin-shRNA cells migrated significantly faster, at a rate of 16.50 ± 1.77 μm/h (n = 4; p < 0.05), which was a 1.4-fold increase compared with control shRNA cells.
FIGURE 9:

Moesin shRNA knockdown increases cell migration but decreases cell invasion during EMT. (A) Images of WT, control shRNA, and moesin shRNA NMuMG cells treated with TGF-β for 48 h and migrating at the indicated times after monolayer wounding. Images were acquired by time-lapse microscopy for 20 h, using a 10× objective. Bar, 200 μm. See Supplemental Videos S9 and S10. (B) Quantitative analysis of wound-healing migration of cells in A. Wound areas were measured every 4 h for a total of 20 h. Migratory rates were determined from the total decrease in wound area after 20 h. Data shown are means ± SEM of four independent experiments. Asterisks indicate significant difference between moesin shRNA and control shRNA cells. *p < 0.05 by one-way ANOVA followed by Newman–Keuls multiple comparisons post test. (C) Quantification of WT, control shRNA, and moesin shRNA cells treated for 48 h with TGF-β and invading a Matrigel matrix for 21 h. Data shown are means ± SEM of at least five membranes from three independent cell preparations. **p < 0.01; ***p < 0.001; n.s., not significant by one-way ANOVA followed by Newman–Keuls multiple comparisons post test.
In contrast to increased migration with monolayer wounding, suppressing moesin expression decreased invasion of transdifferentiated cells. Wild-type, control shRNA, and moesin shRNA cells were treated with TGF-β for 48 h and then seeded onto Matrigel basement membrane matrix–coated filters, after which cell invasion was determined at 21 h. Wild-type and control shRNA cells invaded the matrix and migrated through the filters at similar numbers (Figure 9C). However, moesin shRNA cells had a significant 1.8-fold decrease in invasion compared with control shRNA cells (p < 0.001). Thus, although transdifferentiated cells with suppressed moesin expression had increased wound-healing migration, their ability to invade a basement membrane matrix was significantly impaired. These differences may reflect reduced tensional force from thinner, less stable actin stress fibers in moesin shRNA cells compared with force generated from thicker, more stable fibers in control cells. Taken together, our findings indicate that moesin regulates actin cytoskeleton remodeling and morphological changes for TGF-β–induced EMT of NMuMG cells, which in turn modulates cell migration and invasion.
DISCUSSION
EMT is driven by changes in gene expression and cell morphology that promote migration and invasion during normal development and the progression of diseases such as metastatic cancer and fibrosis. Despite notable changes in actin cytoskeleton architecture during EMT (Miettinen et al., 1994; Zavadil et al., 2001; Masszi et al., 2003; Brown et al., 2004), how this occurs in real time, how it contributes to morphological changes, and whether it is regulated by changes in gene expression remain relatively unknown. Actin regulatory genes are among the most highly up-regulated groups during TGF-β-induced EMT (Zavadil et al., 2001; Xie et al., 2003; Valcourt et al., 2005; Keshamouni et al., 2006; Safina et al., 2009; Lenferink et al., 2010); however, the functional significance of this regulation is largely unknown (Mori et al., 2009). We used LifeAct-GFP, a recently developed fluorescent reporter for F-actin (Riedl et al., 2008), to reveal in real time the progressive changes in actin filament organization and properties that are consistent with transcriptional regulation rather than rapid signaling events.
Our findings with three distinct epithelial cell types suggest a conserved and significant increase in moesin expression during EMT. Moesin expression also increases during TGF-β–induced EMT of keratinocytes (Zavadil et al., 2001; Valcourt et al., 2005), mammary epithelial cells (Valcourt et al., 2005), and lung carcinoma cells (Keshamouni et al., 2006), further suggesting a conserved event. However, the functional significance of increased moesin expression during EMT has not been reported. Moesin and the other ERM proteins ezrin and radixin regulate actin cytoskeleton remodeling for dynamic cellular processes, including cell morphogenesis, adhesion, and migration (Fievet et al., 2007; Fehon et al., 2010). ERM proteins also regulate epithelial cell integrity and formation of the apical membrane domain (Gautreau et al., 2000; Speck et al., 2003; Saotome et al., 2004). Although ERM proteins are known to promote epithelial plasticity for morphogenesis and migration, their role in EMT is not clearly established. Binding of moesin and ezrin to the small, mucin-like transmembrane glycoprotein podoplanin was shown to be necessary for EMT of MDCK cells by inducing activation of RhoA (Martin-Villar et al., 2006), although this effect was not noted to be dependent on changes in ERM protein expression. In addition, recent work shows that moesin promotes actin remodeling during tumor necrosis factor-α–induced EMT of retinal pigment epithelial cells (Takahashi et al., 2010). Analyses of our LifeAct-GFP time-lapse movies indicate that increased moesin expression is necessary for dynamic actin filament remodeling during EMT, including filament bundling, organization, and stability.
We also found a moesin-dependent relocalization of CD44, α-SMA, and p-MLC, and increased autophosphorylation of FAK during EMT. High expression of CD44 is emerging as a marker of TGF-β–induced EMT and a feature shared by epithelial stem cells (Mani et al., 2008), and repressed CD44 expression is associated with tumor suppression (Godar et al., 2008). In addition, recent findings suggest that a CD44–ERM linkage at the cell cortex might be an important step in reorganization of the actin cytoskeleton during cytokine-induced EMT of human lung carcinoma cells (Buckley et al., 2011). Our data indicate that increased moesin expression is necessary for the relocalization of CD44 at dorsal membrane protrusions in transdifferentiated cells. Increased moesin expression is also necessary for relocalization of α-SMA during EMT to cortical patches that contain moesin, p34Arc, and p-MLC but not F-actin. Moreover, cortical α-SMA patches are dependent on actomyosin contractility, as indicated by their decreased abundance after inhibiting myosin activity. In addition, dynamic clustering of moesin-GFP–enriched membrane protrusions occurs as a result of contractile intracellular movements in transdifferentiated cells. Together, our data suggest of model of moesin-dependent assembly of contractile elements at cortical adhesion sites as a required mechanism for actin filament remodeling, actin stress fiber stability, and a complete morphological transition during EMT.
Our data also reveal new insights on the regulation and function of moesin and differences compared with other ERM proteins. First, increased moesin expression during EMT is independent of ROCK activity, despite ROCK-dependent increases in phosphorylated moesin abundance and shared effects of ROCK and moesin in promoting actin filament remodeling. Second, the robust increase in moesin expression during EMT is not seen for ezrin or radixin. Moreover, ezrin abundance is not altered in moesin shRNA cells, which indicates the phenotype of disrupted actin filament remodeling, decreased α-SMA cortical patches, and attenuated cell invasion of these cells is selectively moesin dependent. A previous view was that ERM proteins have redundant functions, based on their high sequence homology and tissue-specific expression, and the lack of morphological or functional effects when a single ERM protein is inactivated in cells expressing two or all three ERM proteins (Doi et al., 1999). However, a revised view of nonredundant functions is supported by mouse models of gene inactivation revealing essential functions for ezrin and radixin in tissues expressing only a single ERM protein (Kikuchi et al., 2002; Kitajiri et al., 2004; Saotome et al., 2004; Tamura et al., 2005). In addition, recent studies show distinct functions for ezrin and moesin for leukocyte immunological synapse formation (Ilani et al., 2007) and in melanoma cells during invasion and lung colonization (Estecha et al., 2009). Our findings that suppressing moesin expression has functional effects during EMT of NMuMG cells that also express ezrin and radixin further support the revised view that ERM proteins have nonredundant functions.
The significance of increased ezrin expression we observed during EMT of NMuMG cells is unclear. Although this was not seen with EMT of MCF-10A or A549 cells, decreased expression of ezrin occurs during EMT of peritoneal mesothelial cells (Vargha et al., 2006) and retinal pigment epithelial cells (Takahashi et al., 2010). In addition, fibroblasts deficient for the EMT-inducing transcription factor Snail1 have increased expression of ezrin and decreased expression of moesin and are unable to invade a three-dimensional extracellular matrix (Rowe et al., 2009). Hence, ERM protein switching with opposing changes in ezrin and moesin may be a feature of EMT of some cell types. Decreased ezrin expression during EMT could reflect a distinct transdifferentiation program and perhaps a requirement to achieve epithelial plasticity. This would be analogous to inactivation of moesin being necessary for cytoskeletal remodeling during immunological synapse formation in leukocytes (Faure et al., 2004; Ilani et al., 2007).
The ERM protein switching we report during TGF-β–induced EMT may also be a feature of the early stages of breast cancer progression, consistent with the relative abundance of ezrin being higher in noninvasive, more epithelial-like breast cancer cells, whereas the relative abundance of moesin is higher in invasive, more mesenchymal-like breast cancer cells (Ostapkowicz et al., 2006). Changes in expression of ERM proteins may also contribute to the progression of fibrosis. Fibrotic pulmonary fibroblasts have increased expression of ezrin and moesin compared with normal pulmonary fibroblasts (Shi-Wen et al., 2004), and after acute liver injury, fibrosis of hepatic stellate cells is decreased in moesin-null mice (Okayama et al., 2008). The critical role of actin cytoskeleton remodeling in the progression of diseases such as metastatic cancer and fibrosis underscores the importance of understanding its complex regulation, which we show for EMT is determined by increased moesin expression and a moesin-dependent assembly of contractile elements at the cell cortex.
MATERIALS AND METHODS
Antibodies
Primary antibodies to E-cadherin (clone 36) and FAK (clone 77) were purchased from BD Transduction Laboratories (Lexington, KY). Primary antibodies to N-cadherin, ezrin, moesin (Q480), pan-ERM, phospho-ERM, and phospho-MLC (Ser19; mouse monoclonal) were purchased from Cell Signaling Technology (Beverly, MA). Primary antibodies to radixin, fibronectin, α-SMA (clone 1A4), and α-tubulin (clone DM1A) were purchased from Sigma-Aldrich (St. Louis, MO). Primary antibody to CD44 (clone A020) was purchased from Calbiochem (La Jolla, CA). Primary antibody to phospho-MLC (S19/S20; rabbit polyclonal) was purchased from Rockland Immunochemicals (Gilbertsville, PA). Primary antibodies to p34-Arc/ARPC2 and β-actin (clone C4) were purchased from Millipore (Billerica, MA). Primary antibodies to FAK-pY397 and secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 568 were purchased from Invitrogen (Carlsbad, CA). Secondary antibodies conjugated to peroxidase were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
Cell culture, treatments, and transfections
NMuMG normal mouse mammary gland epithelial cells (obtained from R. Derynck, University of California, San Francisco) and A549 human lung adenocarcinoma cells (obtained from H. Chapman, University of California, San Francisco) were maintained in DME medium (4.5 g/l glucose) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin. Growth medium for NMuMG cells was also supplemented with 10 μg/ml insulin (Sigma-Aldrich). MCF-10A human mammary epithelial cells (obtained from J. Debnath, University of California, San Francisco) were maintained in DME/F-12 medium (4.5 g/l glucose) supplemented with 5% horse serum (Invitrogen), 10 μg/ml insulin, 20 ng/ml epidermal growth factor (PeproTech, Rocky Hill, NJ), 0.5 μg/ml hydrocortisone (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich), 100 U/ml penicillin, and 100 μg/ml streptomycin. 293TA human embryonic kidney cells were maintained in DME medium (4.5 g/l glucose) supplemented with 10% tetracycline-free FBS (Clontech, Mountain View, CA) and 110 mg/l sodium pyruvate. All cell lines were maintained at 37°C in 5% CO2.
Unless otherwise indicated, NMuMG cells were treated with 5 ng/ml recombinant human TGF-β (in 10 mM citric acid, pH 3; PeproTech) for 48 h to induce EMT. MCF-10A and A549 cells were treated with 10 ng/ml TGF-β for 3–5 d or for 1–2 d in serum-free small airway basal medium (Lonza, Basel, Switzerland), respectively. To inhibit TGF-β type I receptor signaling, cells were treated with 5 μM SB431542 (in dimethyl sulfoxide [DMSO]; Sigma-Aldrich). To inhibit Rho kinase (ROCK), cells were treated with 5 μM Y-27632 (in dH2O; Calbiochem) for 45 min before TGF-β treatment. For short-term treatments with pharmacological inhibitors, cells were incubated with 10 μM Y-27632, 5 μM blebbistatin (in DMSO; Calbiochem), or 5 μM nocodazole (in DMSO; Calbiochem) for 1 h after 48 h with TGF-β. Cells were transfected using Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. Transfected cells were seeded on glass coverslips and cultured for 2–3 d before experimental analysis.
DNA constructs, lentivirus production, and generation of stable cell lines
The plasmid containing mEGFP-N1-LifeAct sequence was kindly provided by Roland Wedlich-Sölder (Max Planck Institute of Biochemistry, Martinsried, Germany). The moesin-GFP construct was kindly provided by Francisco Sanchez-Madrid (Universidad Autonoma de Madrid, Madrid, Spain). Lentiviral plasmids (pLKO.1-puro) containing shRNA sequences to mouse moesin were obtained from Sigma-Aldrich (NM_010833, MISSION shRNA). MISSION Non-Target shRNA Control Vector (Sigma-Aldrich) was used as a control. Lentiviruses were produced in 293TA packaging cells using the Lenti-X HT Packaging System (Clontech), according to the manufacturer's protocol. For lentiviral transduction, NMuMG cells were infected with lentivirus expressing control or moesin shRNA in growth medium supplemented with 4 μg/ml Polybrene (Sigma-Aldrich). Stable clonal cell lines were selected with 10 μg/ml puromycin (Cellgro; Mediatech, Manassas, VA) and were maintained in 2.5 μg/ml puromycin.
Immunoblot analysis
Immunoblot analyses were performed using lysates from cells lysed in ice-cold RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.5% NaDOC, 0.1% SDS, and protease inhibitors) containing phosphatase inhibitors (50 mM NaF, 1 mM NaVO4, 1 mM ethylene glycol tetraacetic acid, 10 mM sodium pyrophosphate, 1 mM glycerol phosphate, and 10 nM calyculin A). Protein concentrations of clarified cell lysates were determined using a bicinchoninic acid protein assay kit (Pierce, Thermo Scientific, Rockford, IL). Proteins (5–10 μg) were separated by SDS–PAGE and transferred to polyvinylidene fluoride membranes. Membranes were blocked in 5% milk or 3% BSA, incubated with primary antibodies for 1 h or overnight, and incubated with peroxidase-conjugated secondary antibodies for 45 min. Bound antibodies were detected using enhanced chemiluminescence (PerkinElmer, Waltham, MA). Semiquantitative densitometric analysis of anti-ezrin and anti-moesin immunoblots from three independent experiments was performed using ImageJ software (National Institutes of Health, Bethesda, MD). The values for ezrin and moesin protein levels were normalized using β-actin as a loading control.
qPCR
RNA was extracted from NMuMG cells using the RNeasy Mini Kit (Qiagen, Valencia, CA), and first-strand cDNA was synthesized from total RNA using iScript reverse transcriptase (Bio-Rad Laboratories, Hercules, CA). cDNA was amplified using iQ SYBR Green Supermix (Bio-Rad Laboratories) and detected on a CFX96 Real-Time PCR detection system (Bio-Rad Laboratories). Quantitative analysis of ezrin, moesin, and radixin gene expression from at least three independent experiments was performed using CFX Manager software (Bio-Rad Laboratories) and the ribosomal protein gene Rpl19 for normalization. Primers specific for mouse ezrin, moesin, and radixin cDNA were obtained from Qiagen (NM_009510, NM_010833, and NM_009041, respectively). The data were statistically analyzed using one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparison post test (Prism 4; GraphPad Software, La Jolla, CA).
Immunolabeling and image acquisition
NMuMG cells grown on glass coverslips were washed three times with PBS at room temperature, fixed with 4% formaldehyde in PBS for 12 min, permeabilized with 0.5% Triton X-100 in PBS for 10 min, and then blocked with 3% BSA in PBS for 30 min or overnight. Fixed cells were incubated with primary antibodies for 1 or 2 h, washed with PBS, and incubated with fluorophore-conjugated secondary antibodies for 45 min. Fixed cells were also incubated with rhodamine-conjugated phalloidin (Invitrogen) for 10 min to stain F-actin and with Hoechst 33342 (Invitrogen) for 10 min to stain nuclei. For plasma membrane labeling, cells were incubated with 4 μg/ml Oregon Green 488–conjugated wheat germ agglutinin (Invitrogen) in PBS for 10 min at 37°C prior to fixation. Coverslips were mounted on slides with ProLong Gold antifade reagent (Invitrogen). Cells were imaged using a 63× Plan-Apochromat/1.40 or a 40× EC Plan-Neofluar/1.30 oil immersion objective on an inverted laser-scanning confocal microscope (LSM510 META; Carl Zeiss, Thornwood, NY), and images were captured using Zeiss software (LSM 510 Meta 4.2 Software). Z-Series projections represent confocal images combined from 16 optical sections acquired at 0.3-μm intervals.
Quantification of elongated cell morphology
Measurements of TGF-β–treated NMuMG cells were made using images of cells that were stained for F-actin and nuclei and were acquired using a 40× objective. The lengths of the major and minor cell axes were measured using Zeiss software (Zeiss LSM Image Browser). The ratios of the major axis to the minor axis of cells were used to determine the degree of elongated cell morphology. For each experiment, between 30 and 40 cells of each cell type were measured. The data were statistically analyzed using one-way ANOVA followed by Dunnett's multiple comparison post test (Prism 4).
Spinning disk confocal and time-lapse microscopy
NMuMG cells grown on glass coverslips were imaged at 37°C using a 40× Plan-fluor ELWD/0.6 air objective (phase contrast) or a 60× Plan Apochromat TIRF/1.45 oil immersion objective (fluorescence) on an inverted microscope system (Nikon Eclipse TE2000 Perfect Focus System; Nikon Instruments, Melville, NY), equipped with a spinning-disk confocal scanner unit (CSU10; Yokogawa, Newnan, GA), a 488-nm solid-state laser (LMM5; Spectral Applied Research, Richmond Hill, Canada), multipoint stage (MS-2000; Applied Scientific Instruments, Eugene, OR), a CoolSnap HQ2 cooled charge-coupled device (CCD) camera (Photometrics, Tucson, AZ), and camera-triggered electronic shutters controlled with NIS-Elements Imaging Software (Nikon). For short-term videos, cells were imaged after 48 h of TGF-β treatment in medium supplemented with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5, and images were captured every 1 min. For long-term videos, cells were imaged after 1 h (phase contrast) or 6 h (fluorescence) of TGF-β treatment in medium supplemented with 10 mM HEPES, pH 7.5, in 5% CO2, and images were captured every 10 min. Images for presentation in figures and videos were processed with a 2 × 2 Gaussian low-pass filter to reduce high-frequency pixel noise, and an unsharp mask filter (7 × 7 kernel size; scaling factor 0.5 for short-term videos, or 13 × 13 kernel size; scaling factor 0.7 for long-term videos) to enhance dim features using NIS-Elements Imaging Software.
In vitro wound-healing migration assays and time-lapse microscopy
Monolayers of NMuMG cells grown in six-well plates were wounded using a plastic pipette tip 48 h after the initiation of TGF-β treatment, washed twice with serum-free medium, and replenished with fresh medium. Cells were imaged at 37°C in 5% CO2 using a 10× Hoffmann modulation objective on a Zeiss Axiovert S-100 microscope. Images were captured every 15 min, beginning immediately after wounding and ending 20 h after wounding, using a Spot RT Slider cooled CCD camera (2.3.0; Diagnostic Instruments, Sterling Heights, MI) operated with Openlab software (Improvision, PerkinElmer). Wound-area measurements were determined using ImageJ software. The area of a single wound was calculated as the average of three different cell-free areas from the same wound. The migratory rates were determined by the total decreased wound area from 0 h to 20 h after wounding. For each condition, wounds from four independent experiments were measured. The decreased wound areas for each time point were statistically analyzed using one-way ANOVA followed by Newman–Keuls multiple comparison post test (Prism 4).
Matrigel Transwell invasion assays
NMuMG cells grown in the presence of TGF-β for 48 h were resuspended in DME medium supplemented with 0.2% FBS and were seeded in the upper chamber onto rehydrated Growth Factor Reduced Matrigel Matrix–coated inserts (BD Biosciences, San Diego, CA). The lower chamber was filled with DME medium supplemented with 10% FBS, and the invasion chambers were incubated for 21 h at 37°C in 5% CO2. Noninvading cells were removed from the upper surface of the membrane with a cotton-tipped applicator. Cells were fixed with methanol for 5 min at −20°C, and nuclei were stained with Hoechst 33342 (Invitrogen) for 5 min. Membranes were mounted onto glass slides with Fluorescence Mounting Medium (Dako, Glostrup, Denmark). Cell nuclei were imaged using a 10× Plan-Neofluar/0.3 air objective on a Zeiss Axiophot epifluorescence microscope, and images were captured using a CoolSnap HQ2 camera operated by Micro-Manager software (University of California, San Francisco). Cells were counted using ImageJ software. The number of cells invading per field of view for one membrane was calculated as the average of seven different fields of view from the same membrane. For each cell type, a total of at least five membranes were counted from three independent experiments. The data were statistically analyzed using one-way ANOVA followed by Bonferroni's multiple comparison post test (Prism 4).
Supplementary Material
Acknowledgments
We thank Amanda Marusich for technical assistance and members of the Barber and Wittmann labs for constructive discussions. We thank Rik Derynck, Jay Debnath, Hal Chapman, Roland Wedlich-Sölder, and Francisco Sanchez-Madrid for providing us with reagents and Samy Lamouille for technical assistance and helpful suggestions. This work was supported by a Canadian Cancer Society Research Institute Fellowship to J.H. and National Institutes of Health Grants GM079139 to T.W. and GM58642 to D.L.B. Work was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR16490 from the National Center for Research Resources, National Institutes of Health.
Abbreviations used:
- α-SMA
α-smooth muscle actin
- EMT
epithelial–mesenchymal transition
- ERM
ezrin/radixin/moesin
- F-actin
filamentous actin
- FAK
focal adhesion kinase
- MET
mesenchymal–epithelial transition
- p-FAK
autophosphorylated focal adhesion kinase
- p-MLC
phosphorylated myosin light chain
- ROCK
Rho-associated coiled-coil–containing protein kinase
- shRNA
short hairpin RNA
- TGF-β
transforming growth factor-β
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-02-0119) on October 26, 2011.
REFERENCES
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KA, Aakre ME, Gorska AE, Price JO, Eltom SE, Pietenpol JA, Moses HL. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 2004;6:R215–R231. doi: 10.1186/bcr778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley ST, Medina C, Kasper M, Ehrhardt C. Interplay between RAGE, CD44, and focal adhesion molecules in epithelial-mesenchymal transition of alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L548–L559. doi: 10.1152/ajplung.00230.2010. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Yoo J. Rho activation is required for transforming growth factor-beta-induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol Int. 2007;31:1225–1230. doi: 10.1016/j.cellbi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Doi Y, Itoh M, Yonemura S, Ishihara S, Takano H, Noda T, Tsukita S. Normal development of mice and unimpaired cell adhesion/cell motility/actin-based cytoskeleton without compensatory up-regulation of ezrin or radixin in moesin gene knockout. J Biol Chem. 1999;274:2315–2321. doi: 10.1074/jbc.274.4.2315. [DOI] [PubMed] [Google Scholar]
- Edlund S, Landstrom M, Heldin CH, Aspenstrom P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002;13:902–914. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecha A, Sanchez-Martin L, Puig-Kroger A, Bartolome RA, Teixido J, Samaniego R, Sanchez-Mateos P. Moesin orchestrates cortical polarity of melanoma tumour cells to initiate 3D invasion. J Cell Sci. 2009;122:3492–3501. doi: 10.1242/jcs.053157. [DOI] [PubMed] [Google Scholar]
- Faure S, Salazar-Fontana LI, Semichon M, Tybulewicz VL, Bismuth G, Trautmann A, Germain RN, Delon J. ERM proteins regulate cytoskeleton relaxation promoting T cell-APC conjugation. Nat Immunol. 2004;5:272–279. doi: 10.1038/ni1039. [DOI] [PubMed] [Google Scholar]
- Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fievet B, Louvard D, Arpin M. ERM proteins in epithelial cell organization and functions. Biochim Biophys Acta. 2007;1773:653–660. doi: 10.1016/j.bbamcr.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Gautreau A, Louvard D, Arpin M. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J Cell Biol. 2000;150:193–203. doi: 10.1083/jcb.150.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godar S, et al. Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia. 2005;7:509–521. doi: 10.1593/neo.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison N, Hendry BM, Sharpe CC. Rho isoforms have distinct and specific functions in the process of epithelial to mesenchymal transition in renal proximal tubular cells. Cell Signal. 2009;21:1522–1531. doi: 10.1016/j.cellsig.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Ilani T, Khanna C, Zhou M, Veenstra TD, Bretscher A. Immune synapse formation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J Cell Biol. 2007;179:733–746. doi: 10.1083/jcb.200707199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshamouni VG, et al. Differential protein expression profiling by iTRAQ-2DLC-MS/MS of lung cancer cells undergoing epithelial-mesenchymal transition reveals a migratory/invasive phenotype. J Proteome Res. 2006;5:1143–1154. doi: 10.1021/pr050455t. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, et al. Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nat Genet. 2002;31:320–325. doi: 10.1038/ng905. [DOI] [PubMed] [Google Scholar]
- Kitajiri S, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S, Tsukita S. Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol. 2004;166:559–570. doi: 10.1083/jcb.200402007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178:437–451. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Katakai T, Hara T, Gonda H, Sugai M, Shimizu A. Roles of p-ERM and Rho-ROCK signaling in lymphocyte polarity and uropod formation. J Cell Biol. 2004;167:327–337. doi: 10.1083/jcb.200403091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenferink AE, Cantin C, Nantel A, Wang E, Durocher Y, Banville M, Paul-Roc B, Marcil A, Wilson MR, O'Connor-McCourt MD. Transcriptome profiling of a TGF-beta-induced epithelial-to-mesenchymal transition reveals extracellular clusterin as a target for therapeutic antibodies. Oncogene. 2010;29:831–844. doi: 10.1038/onc.2009.399. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Esch F, Furthmayr H, Hall A. Rho- and rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: an essential role for ezrin/radixin/moesin proteins. J Cell Biol. 1997;138:927–938. doi: 10.1083/jcb.138.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005;118:873–887. doi: 10.1242/jcs.01634. [DOI] [PubMed] [Google Scholar]
- Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Villar E, Megias D, Castel S, Yurrita MM, Vilaro S, Quintanilla M. Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J Cell Sci. 2006;119:4541–4553. doi: 10.1242/jcs.03218. [DOI] [PubMed] [Google Scholar]
- Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–F924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, et al. Zyxin mediates actin fiber reorganization in epithelial-mesenchymal transition and contributes to endocardial morphogenesis. Mol Biol Cell. 2009;20:3115–3124. doi: 10.1091/mbc.E09-01-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–1520. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Yano H, Schaefer E, Sabe H. Different modes and qualities of tyrosine phosphorylation of Fak and Pyk2 during epithelial-mesenchymal transdifferentiation and cell migration: analysis of specific phosphorylation events using site-directed antibodies. Oncogene. 2001;20:2626–2635. doi: 10.1038/sj.onc.1204359. [DOI] [PubMed] [Google Scholar]
- Okayama T, et al. Attenuated response to liver injury in moesin-deficient mice: impaired stellate cell migration and decreased fibrosis. Biochim Biophys Acta. 2008;1782:542–548. doi: 10.1016/j.bbadis.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Oshiro N, Fukata Y, Kaibuchi K. Phosphorylation of moesin by rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J Biol Chem. 1998;273:34663–34666. doi: 10.1074/jbc.273.52.34663. [DOI] [PubMed] [Google Scholar]
- Ostapkowicz A, Inai K, Smith L, Kreda S, Spychala J. Lipid rafts remodeling in estrogen receptor-negative breast cancer is reversed by histone deacetylase inhibitor. Mol Cancer Ther. 2006;5:238–245. doi: 10.1158/1535-7163.MCT-05-0226. [DOI] [PubMed] [Google Scholar]
- Riedl J, et al. LifeAct: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RG, et al. Mesenchymal cells reactivate Snail1 expression to drive three-dimensional invasion programs. J Cell Biol. 2009;184:399–408. doi: 10.1083/jcb.200810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safina AF, Varga AE, Bianchi A, Zheng Q, Kunnev D, Liang P, Bakin AV. Ras alters epithelial-mesenchymal transition in response to TGFbeta by reducing actin fibers and cell-matrix adhesion. Cell Cycle. 2009;8:284–298. doi: 10.4161/cc.8.2.7590. [DOI] [PubMed] [Google Scholar]
- Saotome I, Curto M, McClatchey AI. Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev Cell. 2004;6:855–864. doi: 10.1016/j.devcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Henry M, Solomon F, Jacks T. RhoA-dependent phosphorylation and relocalization of ERM proteins into apical membrane/actin protrusions in fibroblasts. Mol Biol Cell. 1998;9:403–419. doi: 10.1091/mbc.9.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-Wen X, et al. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell. 2004;15:2707–2719. doi: 10.1091/mbc.E03-12-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck O, Hughes SC, Noren NK, Kulikauskas RM, Fehon RG. Moesin functions antagonistically to the Rho pathway to maintain epithelial integrity. Nature. 2003;421:83–87. doi: 10.1038/nature01295. [DOI] [PubMed] [Google Scholar]
- Takahashi E, et al. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J Biol Chem. 2010;285:4060–4073. doi: 10.1074/jbc.M109.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Sasaki T, Mammoto A, Takaishi K, Kameyama T, Tsukita S, Takai Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J Biol Chem. 1997;272:23371–23375. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- Tamura A, Kikuchi S, Hata M, Katsuno T, Matsui T, Hayashi H, Suzuki Y, Noda T, Tsukita S, Tsukita S. Achlorhydria by ezrin knockdown: defects in the formation/expansion of apical canaliculi in gastric parietal cells. J Cell Biol. 2005;169:21–28. doi: 10.1083/jcb.200410083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares AL, Mercado-Pimentel ME, Runyan RB, Kitten GT. TGF beta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev Dyn. 2006;235:1589–1598. doi: 10.1002/dvdy.20771. [DOI] [PubMed] [Google Scholar]
- Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16:1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargha R, Endemann M, Kratochwill K, Riesenhuber A, Wick N, Krachler AM, Malaga-Dieguez L, Aufricht C. Ex vivo reversal of in vivo transdifferentiation in mesothelial cells grown from peritoneal dialysate effluents. Nephrol Dial Transplant. 2006;21:2943–2947. doi: 10.1093/ndt/gfl355. [DOI] [PubMed] [Google Scholar]
- Xie L, Law BK, Aakre ME, Edgerton M, Shyr Y, Bhowmick NA, Moses HL. Transforming growth factor beta-regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res. 2003;5:R187–198. doi: 10.1186/bcr640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.