

Abstract
The title compound, C13H16N4, consists of two pyridine rings which are linked by an N,N′-dimethylmethaneamine chain. The pyridine rings adopt a twist conformation and the dihedral angle between them is 60.85 (5)°. The crystal packing is stabilized by weak C—H⋯π interactions.
Related literature
For the synthesis of the title compound, see: Kahn et al. (1945 ▶). For applications of heteroaromatic amines, see: Mehrkhodavandi & Schrock (2001 ▶); Hall et al. (1998 ▶); Lee (2003 ▶).
Experimental
Crystal data
C13H16N4
M r = 228.30
Monoclinic,
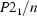
a = 11.6652 (7) Å
b = 8.3921 (5) Å
c = 12.8966 (7) Å
β = 106.634 (2)°
V = 1209.69 (12) Å3
Z = 4
Mo Kα radiation
μ = 0.08 mm−1
T = 100 K
0.12 × 0.08 × 0.08 mm
Data collection
Bruker APEXII diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.963, T max = 0.986
27844 measured reflections
2516 independent reflections
2105 reflections with I > 2σ(I)
R int = 0.050
Refinement
R[F 2 > 2σ(F 2)] = 0.038
wR(F 2) = 0.125
S = 0.96
2516 reflections
218 parameters
All H-atom parameters refined
Δρmax = 0.23 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT (Bruker, 2007 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg & Putz, 2005 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811046320/bx2378sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811046320/bx2378Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811046320/bx2378Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
Cg1 and Cg2 are the centroids of the N1/C1–C5 and N4/C9–C15 rings, respectively.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C1—H1⋯Cg2i | 0.99 (2) | 2.64 (2) | 3.484 (1) | 143 (1) |
| C6—H7⋯Cg2ii | 1.00 (2) | 2.75 (2) | 3.545 (2) | 137 (1) |
| C10—H13⋯Cg1iii | 0.95 (2) | 2.90 (1) | 3.637 (1) | 135 (1) |
Symmetry codes: (i) 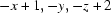 ; (ii)
; (ii) 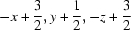 ; (iii)
; (iii) 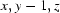 .
.
Acknowledgments
This work was supported by the "Human Resource Development (project name: Advanced track for Si-based solar cell materials and devices, project No. 201040100660)" of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korean Government Ministry of Knowledge Economy.
supplementary crystallographic information
Comment
Heteroaromatic amines based metal complexes have been extensively studied due to their numerous potential applications as catalysts, drugs, biomimetic chemistry, and so on. (Mehrkhodavandi, et al., (2001) , Hall, et al.,(1998), Lee, (2003). We are interested in the use of chelates containing pyridylamine. We report here the crystal structure of the title compound, Fig. 1, which consists of two 2-pyridyl rings which are linked together by a N,N'-dimethylmethaneamine chain .The pyridine rings adopt a twist conformation and the dihedral angle between them is 60.85 (5)°. The crystal packing is stabilized by weak C—H···π interactions, Fig. 2, Table 1.
Experimental
N,N'-dimethyl-N,N'-di(pyridin-2-yl)methanediamine was prepared by a reported method, Kahn, et al., (1945). A solution of 2-(methylamino)pyridine (5.00 g, 4.62 × 10 -2 mol) in water (50 ml) was added dropwise to 37% formaldehyde solution (1.83 ml, 2.31 × 10-2 mol) at 0°C. The reation mixture was stirred at room temperature for overnight and the white precipitate formed was filtered,washed with water and dried. It was dissolved in acetone, dried over MgSO4 and concentrated. It was recrystallized in acetonitrile. Yield: 92% (4.88 g)
Refinement
All H atoms were geometrically positioned and refined using a riding model, with C—H = 0.95 Å for the aryl, 0.99 Å for the methylene, and 0.00 Å for the methyl H atoms, respectively, and with Uiso(H) = 1.2Ueq(C) for the aryl and methylene H atoms, and 1.5Ueq(C) for the methyl H atoms.
Figures
Fig. 1.

A view of the title compound showing the labelling of the atoms. Displacement ellipsoids are shown at the 50% probability level.
Fig. 2.

Part of the crystal structure of (I) showing the formation of a C—H···π interactions.
Crystal data
| C13H16N4 | F(000) = 488 |
| Mr = 228.30 | Dx = 1.254 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P2yn | Cell parameters from 2516 reflections |
| a = 11.6652 (7) Å | θ = 2.1–26.5° |
| b = 8.3921 (5) Å | µ = 0.08 mm−1 |
| c = 12.8966 (7) Å | T = 100 K |
| β = 106.634 (2)° | Rod, colourless |
| V = 1209.69 (12) Å3 | 0.12 × 0.08 × 0.08 mm |
| Z = 4 |
Data collection
| Bruker APEXII diffractometer | 2105 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.050 |
| graphite | θmax = 26.5°, θmin = 2.1° |
| θ/2φ scans | h = −14→14 |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | k = −10→10 |
| Tmin = 0.963, Tmax = 0.986 | l = −16→14 |
| 27844 measured reflections | 4 standard reflections every 30 min |
| 2516 independent reflections | intensity decay: 0.0% |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.038 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.125 | All H-atom parameters refined |
| S = 0.96 | w = 1/[σ2(Fo2) + (0.1P)2] where P = (Fo2 + 2Fc2)/3 |
| 2516 reflections | (Δ/σ)max < 0.001 |
| 218 parameters | Δρmax = 0.23 e Å−3 |
| 0 restraints | Δρmin = −0.21 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.43474 (8) | 0.22148 (11) | 0.82931 (7) | 0.0194 (3) | |
| N2 | 0.61745 (8) | 0.11262 (11) | 0.82658 (8) | 0.0203 (3) | |
| N3 | 0.59838 (8) | −0.15394 (11) | 0.89105 (8) | 0.0189 (3) | |
| N4 | 0.79220 (8) | −0.22454 (12) | 0.98754 (7) | 0.0182 (3) | |
| C1 | 0.34526 (10) | 0.32394 (15) | 0.78856 (9) | 0.0211 (3) | |
| C2 | 0.33876 (11) | 0.42500 (14) | 0.70242 (9) | 0.0214 (3) | |
| C3 | 0.43186 (10) | 0.41792 (14) | 0.65514 (9) | 0.0200 (3) | |
| C4 | 0.52555 (10) | 0.31431 (14) | 0.69472 (9) | 0.0173 (3) | |
| C5 | 0.52473 (10) | 0.21664 (13) | 0.78366 (9) | 0.0160 (3) | |
| C6 | 0.71691 (12) | 0.09942 (17) | 0.78056 (12) | 0.0272 (3) | |
| C7 | 0.62117 (10) | 0.01339 (13) | 0.91939 (9) | 0.0173 (3) | |
| C8 | 0.47475 (11) | −0.19594 (16) | 0.83643 (11) | 0.0240 (3) | |
| C9 | 0.67862 (10) | −0.27049 (13) | 0.94043 (8) | 0.0163 (3) | |
| C10 | 0.64315 (11) | −0.43154 (14) | 0.93928 (9) | 0.0205 (3) | |
| C11 | 0.72811 (12) | −0.54307 (15) | 0.98755 (9) | 0.0248 (3) | |
| C12 | 0.84533 (12) | −0.49686 (15) | 1.03581 (9) | 0.0255 (3) | |
| C13 | 0.87199 (11) | −0.33721 (14) | 1.03368 (9) | 0.0221 (3) | |
| H1 | 0.2816 (13) | 0.3259 (16) | 0.8256 (12) | 0.031 (4)* | |
| H2 | 0.2701 (12) | 0.4955 (17) | 0.6790 (10) | 0.029 (4)* | |
| H3 | 0.4313 (12) | 0.4841 (17) | 0.5946 (11) | 0.028 (4)* | |
| H4 | 0.5883 (13) | 0.3065 (16) | 0.6629 (11) | 0.025 (3)* | |
| H5 | 0.7601 (15) | 0.201 (2) | 0.7856 (14) | 0.048 (5)* | |
| H6 | 0.7696 (15) | 0.022 (2) | 0.8232 (13) | 0.047 (4)* | |
| H7 | 0.6898 (15) | 0.059 (2) | 0.7046 (14) | 0.045 (4)* | |
| H8 | 0.5576 (11) | 0.0536 (14) | 0.9520 (10) | 0.018 (3)* | |
| H9 | 0.6997 (13) | 0.0194 (15) | 0.9712 (11) | 0.023 (3)* | |
| H10 | 0.4350 (13) | −0.1019 (18) | 0.8023 (12) | 0.032 (4)* | |
| H11 | 0.4723 (12) | −0.2806 (19) | 0.7818 (13) | 0.035 (4)* | |
| H12 | 0.4316 (13) | −0.2327 (17) | 0.8900 (12) | 0.033 (4)* | |
| H13 | 0.5616 (13) | −0.4628 (16) | 0.9100 (11) | 0.028 (4)* | |
| H14 | 0.7042 (13) | −0.6532 (18) | 0.9862 (12) | 0.032 (4)* | |
| H15 | 0.9091 (13) | −0.5706 (18) | 1.0696 (11) | 0.033 (4)* | |
| H16 | 0.9557 (12) | −0.3009 (15) | 1.0693 (11) | 0.021 (3)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0190 (5) | 0.0203 (5) | 0.0189 (5) | 0.0021 (4) | 0.0054 (4) | 0.0009 (4) |
| N2 | 0.0208 (5) | 0.0187 (5) | 0.0239 (5) | 0.0052 (4) | 0.0102 (4) | 0.0059 (4) |
| N3 | 0.0177 (5) | 0.0145 (5) | 0.0211 (5) | −0.0003 (4) | 0.0003 (4) | 0.0002 (4) |
| N4 | 0.0185 (5) | 0.0185 (5) | 0.0166 (5) | 0.0017 (4) | 0.0035 (4) | 0.0008 (4) |
| C1 | 0.0178 (6) | 0.0235 (6) | 0.0218 (6) | 0.0025 (5) | 0.0051 (5) | −0.0018 (5) |
| C2 | 0.0200 (6) | 0.0186 (6) | 0.0218 (6) | 0.0038 (5) | −0.0002 (5) | −0.0010 (5) |
| C3 | 0.0260 (6) | 0.0146 (6) | 0.0170 (6) | −0.0035 (5) | 0.0021 (5) | 0.0005 (4) |
| C4 | 0.0186 (6) | 0.0159 (6) | 0.0177 (6) | −0.0029 (4) | 0.0055 (5) | −0.0019 (4) |
| C5 | 0.0178 (6) | 0.0133 (6) | 0.0162 (5) | −0.0015 (4) | 0.0037 (4) | −0.0029 (4) |
| C6 | 0.0236 (7) | 0.0254 (7) | 0.0363 (8) | 0.0077 (6) | 0.0145 (6) | 0.0075 (6) |
| C7 | 0.0197 (6) | 0.0149 (6) | 0.0159 (6) | 0.0004 (5) | 0.0030 (5) | 0.0000 (4) |
| C8 | 0.0196 (6) | 0.0204 (7) | 0.0274 (7) | −0.0006 (5) | −0.0007 (5) | −0.0002 (5) |
| C9 | 0.0208 (6) | 0.0172 (6) | 0.0120 (5) | 0.0008 (5) | 0.0063 (4) | 0.0005 (4) |
| C10 | 0.0254 (7) | 0.0185 (6) | 0.0178 (6) | −0.0024 (5) | 0.0067 (5) | −0.0015 (4) |
| C11 | 0.0400 (8) | 0.0151 (6) | 0.0208 (6) | 0.0022 (5) | 0.0111 (6) | 0.0001 (5) |
| C12 | 0.0342 (7) | 0.0219 (6) | 0.0202 (6) | 0.0119 (5) | 0.0072 (5) | 0.0039 (5) |
| C13 | 0.0224 (6) | 0.0258 (7) | 0.0173 (6) | 0.0063 (5) | 0.0044 (5) | 0.0011 (5) |
Geometric parameters (Å, °)
| N1—C1 | 1.3382 (15) | C4—H4 | 0.938 (15) |
| N1—C5 | 1.3431 (15) | C6—H5 | 0.983 (17) |
| N2—C5 | 1.3769 (15) | C6—H6 | 0.951 (18) |
| N2—C7 | 1.4485 (14) | C6—H7 | 0.998 (17) |
| N2—C6 | 1.4508 (15) | C7—H8 | 1.010 (13) |
| N3—C9 | 1.3773 (15) | C7—H9 | 0.968 (14) |
| N3—C8 | 1.4556 (15) | C8—H10 | 0.957 (16) |
| N3—C7 | 1.4563 (15) | C8—H11 | 0.996 (16) |
| N4—C13 | 1.3406 (15) | C8—H12 | 1.012 (16) |
| N4—C9 | 1.3463 (15) | C9—C10 | 1.4123 (16) |
| C1—C2 | 1.3823 (17) | C10—C11 | 1.3756 (17) |
| C1—H1 | 0.992 (15) | C10—H13 | 0.954 (14) |
| C2—C3 | 1.3907 (17) | C11—C12 | 1.3858 (19) |
| C2—H2 | 0.972 (14) | C11—H14 | 0.964 (15) |
| C3—C4 | 1.3753 (17) | C12—C13 | 1.3775 (18) |
| C3—H3 | 0.957 (15) | C12—H15 | 0.969 (15) |
| C4—C5 | 1.4121 (16) | C13—H16 | 1.001 (14) |
| C1—N1—C5 | 117.83 (10) | H6—C6—H7 | 108.0 (13) |
| C5—N2—C7 | 122.07 (9) | N2—C7—N3 | 112.76 (9) |
| C5—N2—C6 | 120.81 (10) | N2—C7—H8 | 107.5 (7) |
| C7—N2—C6 | 117.12 (9) | N3—C7—H8 | 108.9 (7) |
| C9—N3—C8 | 119.99 (10) | N2—C7—H9 | 109.8 (8) |
| C9—N3—C7 | 121.18 (9) | N3—C7—H9 | 107.0 (8) |
| C8—N3—C7 | 116.06 (10) | H8—C7—H9 | 111.0 (10) |
| C13—N4—C9 | 117.83 (10) | N3—C8—H10 | 107.8 (9) |
| N1—C1—C2 | 124.54 (11) | N3—C8—H11 | 109.9 (8) |
| N1—C1—H1 | 115.4 (8) | H10—C8—H11 | 110.5 (12) |
| C2—C1—H1 | 120.0 (8) | N3—C8—H12 | 111.2 (8) |
| C1—C2—C3 | 117.13 (11) | H10—C8—H12 | 107.2 (12) |
| C1—C2—H2 | 118.3 (8) | H11—C8—H12 | 110.2 (13) |
| C3—C2—H2 | 124.6 (8) | N4—C9—N3 | 117.13 (10) |
| C4—C3—C2 | 120.11 (11) | N4—C9—C10 | 121.76 (10) |
| C4—C3—H3 | 119.2 (9) | N3—C9—C10 | 121.10 (10) |
| C2—C3—H3 | 120.7 (9) | C11—C10—C9 | 118.39 (11) |
| C3—C4—C5 | 118.62 (11) | C11—C10—H13 | 119.9 (9) |
| C3—C4—H4 | 121.2 (9) | C9—C10—H13 | 121.6 (8) |
| C5—C4—H4 | 120.1 (9) | C10—C11—C12 | 120.22 (12) |
| N1—C5—N2 | 117.75 (10) | C10—C11—H14 | 118.5 (9) |
| N1—C5—C4 | 121.76 (10) | C12—C11—H14 | 121.3 (9) |
| N2—C5—C4 | 120.49 (10) | C13—C12—C11 | 117.55 (11) |
| N2—C6—H5 | 111.2 (10) | C13—C12—H15 | 118.8 (9) |
| N2—C6—H6 | 106.0 (10) | C11—C12—H15 | 123.7 (9) |
| H5—C6—H6 | 108.4 (14) | N4—C13—C12 | 124.25 (12) |
| N2—C6—H7 | 111.2 (10) | N4—C13—H16 | 116.8 (7) |
| H5—C6—H7 | 111.7 (14) | C12—C13—H16 | 118.9 (7) |
Hydrogen-bond geometry (Å, °)
| Cg1 and Cg2 are the centroids of the N1/C1–C5 and N4/C9–C15 rings, respectively. |
| D—H···A | D—H | H···A | D···A | D—H···A |
| C1—H1···Cg2i | 0.99 (2) | 2.64 (2) | 3.484 (1) | 143 (1) |
| C6—H7···Cg2ii | 1.00 (2) | 2.75 (2) | 3.545 (2) | 137 (1) |
| C10—H13···Cg1iii | 0.95 (2) | 2.90 (1) | 3.637 (1) | 135 (1) |
Symmetry codes: (i) −x+1, −y, −z+2; (ii) −x+3/2, y+1/2, −z+3/2; (iii) x, y−1, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BX2378).
References
- Brandenburg, K. & Putz, H. (2005). DIAMOND Crystal Impact, Bonn, Germany.
- Bruker (2007). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Hall, J., Haner, R., Aime, S., Botta, M., Faulkner, S., Parker, D. & de Sousa, S. S. (1998). New J. Chem. pp. 627–631.
- Kahn, H. J., Petrow, V. A., Wien, R. & Harrison, J. (1945). J. Chem. Soc. pp. 858–861 [DOI] [PubMed]
- Lee, D.-H. (2003). J. Kor. Chem. Soc. 47, 427–431.
- Mehrkhodavandi, P. & Schrock, R. R. (2001). J. Am. Chem. Soc 123, 10746–10747. [DOI] [PubMed]
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811046320/bx2378sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811046320/bx2378Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811046320/bx2378Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report