

Abstract
Previously we demonstrated by random saturation mutagenesis a set of mutations in the extracellular (EC) loops that constitutively activate the C5a receptor (C5aR) 1,2. In the current study, molecular modeling revealed possible conformations for the extracellular loops of the C5a receptors with mutations in the EC2 loop or in the EC3 loop. Comparison of low-energy conformations of the EC loops defined two distinct clusters of conformations typical either for strongly constitutively active mutants of C5aR (the CAM cluster) or for non-constitutively active mutants (the non-CAM cluster). In the CAM cluster, the EC3 loop was turned towards the transmembrane (TM) helical bundle and more closely interacted with EC2 than in the non-CAM cluster. This suggested a structural mechanism of constitutive activity where EC3 contacts EC2 leading to EC2 interactions with helix TM3, thus triggering movement of TM7 towards TM2 and TM3. The movement initiates rearrangement of the system of hydrogen bonds between TM2, TM3 and TM7 including formation of the hydrogen bond between the side chains of D822.50 in TM2 and N2967.49 in TM7, which is crucial for formation of the activated states of the C5a receptors 3. Since the relative large length of EC3 in C5aR (13 residues) is comparable with those in many other members of rhodopsin family of GPCRs (13-19 residues), our findings might reflect general mechanisms of receptor constitutive activation. The very recent X-ray structure of the agonist-induced constitutively active mutant of rhodopsin 4 is discussed in view of our modeling results.
Keywords: GPCR, molecular modeling, constitutive activation, C5a receptor, extracellular loops
Introduction
G-protein coupled receptors (GPCRs) constitute a vast protein superfamily of membrane proteins (almost 1000 members in humans). Their structures feature seven-helical transmembrane fragments (TM helices, TMs), the N- and C-terminal tails as well as the extra- and intracellular loops (EC and IC loops) connecting the TM helices. Binding of extracellular agonists triggers GPCR activation that involves conformational movements facilitating interaction of GPCRs with corresponding G-proteins inside the cell. Detailed knowledge of molecular mechanism of GPCR's transition from their ground conformational states to the activated states is of paramount pharmacological importance, since it has been estimated that almost 50% of the therapeutic compounds in use act on GPCRs 5.
Mutations in many types of GPCRs can result in constitutive activity, i.e., spontaneous GPCR activity in the absence of an agonist. Constitutively active mutants (CAMs) have been described for many GPCRs and are widely studied; recently, two volumes of Methods in Enzymology (vol. 484 and 485, 2010) extensively presented the current view on constitutive activity of GPCRs. It is commonly believed that the ground conformational states of CAMs mimic the activated conformations of GPCRs. For instance, constitutive activity-inducing mutations occurring in the TM helices may result in different packing of TM fragments within membrane; if they occur in the IC loops, they may change conformational possibilities of the IC loops directly facilitating interactions between CAMs and G-proteins. However, if CAMs feature mutations in the EC loops, their structural interpretation may not be straightforward.
Examples of CAMs with mutations in the EC loops are rare. For instance, of the almost 50 naturally occurring CAMs of the TSH receptor, only 3 possessed mutations in EC1 connecting TM2 and TM3 (all at position 486), 2 in EC2 connecting TM4 and TM5 (both in position 568), and 2 in EC3 connecting TM6 and TM7 (at positions 650 and 656) 6. In addition, mutation of residue K191 in EC2 of the GnRH receptor resulted in constitutive activity 7. In contrast, using the technique of random saturation mutagenesis of the EC2 loop in the complement factor 5a receptor (C5aR) in yeast, we found 24 constitutively active mutants (ranging from strong to weak CAMs) out of total number of 29 functionally active receptors 1, and one strong CAM with mutations in EC3 2. The goal of the present study was to suggest possible structural mechanisms for triggering constitutive activity in the CAMs of C5aR with mutations in EC2 or EC3 by modeling conformational possibilities of their EC loops.
Since the EC loops, especially the large EC2 loop (the average number of residues of EC2 in GPCRs being 20-35 residues 8), are highly flexible, direct interpretation of experimental structural data on GPCRs in terms of involvement of the EC loops in molecular mechanisms of activation is limited. For instance, the X-ray spectroscopy obtains only a single “frozen” snapshot of the set of possible conformations of the EC loops, and spectroscopic techniques in solution, such as NMR, produce loop structures “averaged” over the set of possible conformations. Besides, the available X-ray data on GPCRs (seventeen structures of six GPCRs referred in the minireview 9, and seven structures of GPCRs released very recently 4,10-15) clearly demonstrated that conformations of the EC loops are very different from one GPCR to another. This observation, in our view, precludes using the X-ray data for structural homology modeling of the EC loops.
Earlier, we have developed a de novo modeling procedure that restores with reasonable accuracy the conformational possibilities of the EC loops in GPCRs 16 (see also the very recent discussion on the issue 17-19). In the present study, we employed this procedure for modeling conformational possibilities of the EC loops in the series of C5aR CAMs with mutations in EC2 and EC3. Comparing the conformational space of the EC loops available for constitutively active and non-constitutively active mutants, we have determined geometrically similar conformations of the EC3 loop characteristic for CAMs that differ from conformations for non-CAMs. The major differences occur in spatial positions of the EC3 loop, as well as in subtle conformational changes of inter-residue interactions within the TM regions. Notably, hydrogen bonding between the side chains of D822.50 in TM2 and N2967.49 in TM7 was facilitated in CAMs when compared to non-CAMs. (Residues in TM helices are numbered both sequentially and according to the Ballesteros-Weinstein notation 20 throughout the paper.)
Methods
All modeling procedures used in the present study were developed earlier and were described in details elsewhere 3,16,21. Only significant differences in calculation protocols related to the present study are outlined below. Sequential steps of the general calculation procedure are described in the Results and Discussion section.
Energy calculations were performed employing the ECEPP/2 force field with rigid valence geometry 22,23 and trans-conformations of Pro residues; residues Arg, Lys, Glu and Asp were regarded as charged species. Energy calculations were routinely performed with the value of the macroscopic dielectric constant ε of 80 mimicking, to some extent, hydrophilic environment of the extracellular loops.
The possible ground states of the TM region of C5aR found previously 3 were used as templates for mounting the extracellular loops on the rigid four-residue stems of the TM helices further represented as the 20-residue fragments (see Results and Discussion for more details). The stems were kept in place by the system of the harmonic restraint potentials between Cα-atoms (U0 = 10 kcal/mol*Å2). The same type of harmonic restraint potentials was employed also for keeping together the ends of the 20-residue TM helical fragments at some steps of the calculation procedure (the first run of energy calculations for the EC1+EC2+EC3+TM constructs, see below). The highly conserved disulfide bridge between the C1093.25 in the TM3 helical stem and C188 in EC2 was represented in the templates by the standard ECEPP harmonic potentials for the disulfide bond. The initial sets of low-energy conformations of the EC2 and EC3 loops used in our calculations were the same as found earlier for C5aR 21.
Each separate run of energy calculations involved energy minimization over all dihedral angles of the loops and, at the second run for the EC1+EC2+EC3+TM constructs, also over all dihedral angles of TM helices. Repacking of spatial arrangements for each side chain in the TM region and in the EC loops was performed at each convergence step of energy minimization by an algorithm developed previously 24. Finally, all rms values throughout this paper were calculated between the heavy atoms of the loop backbones with either TM stems or TM helices overlapped.
Results and Discussion
Mutants of C5aR selected for modeling
Table I lists sequences and functional response of 26 C5a receptors with mutations in EC2 or EC3 obtained by our random saturation mutagenesis 1,2 selected for modeling. We have divided them into four groups. The first group consists of strong constitutively active receptors R25, R17, R115, R111, R127, R125 and R128 (notation according to Ref. 1). The second group is the collection of non-CAMs R12, R6, R27 and R20; the WT receptor also belongs to this group. Modeling results obtained for these two groups were used for determining structural features characteristic for CAMs and non-CAMs and for deducing the loop conformations common either for CAMs or for non-CAMs (the CAM and non-CAM clusters, see below). The third group (R18, R10, R8, R53, R102, R105, R109, R104 and R118) comprised receptors possessing constitutive activity that was weaker than for receptors in the first group. The previous three groups are mutated in EC2, whereas the fourth group consists of receptors that contain mutations in EC3, namely K13, K97, K129, K132, K17 and K127 2. Loop conformations found by modeling for the third and fourth groups of mutants were further compared to the CAM and non-CAM clusters to determine whether is it possible to predict constitutive activity for mutants in these groups by modeling results.
Table I.
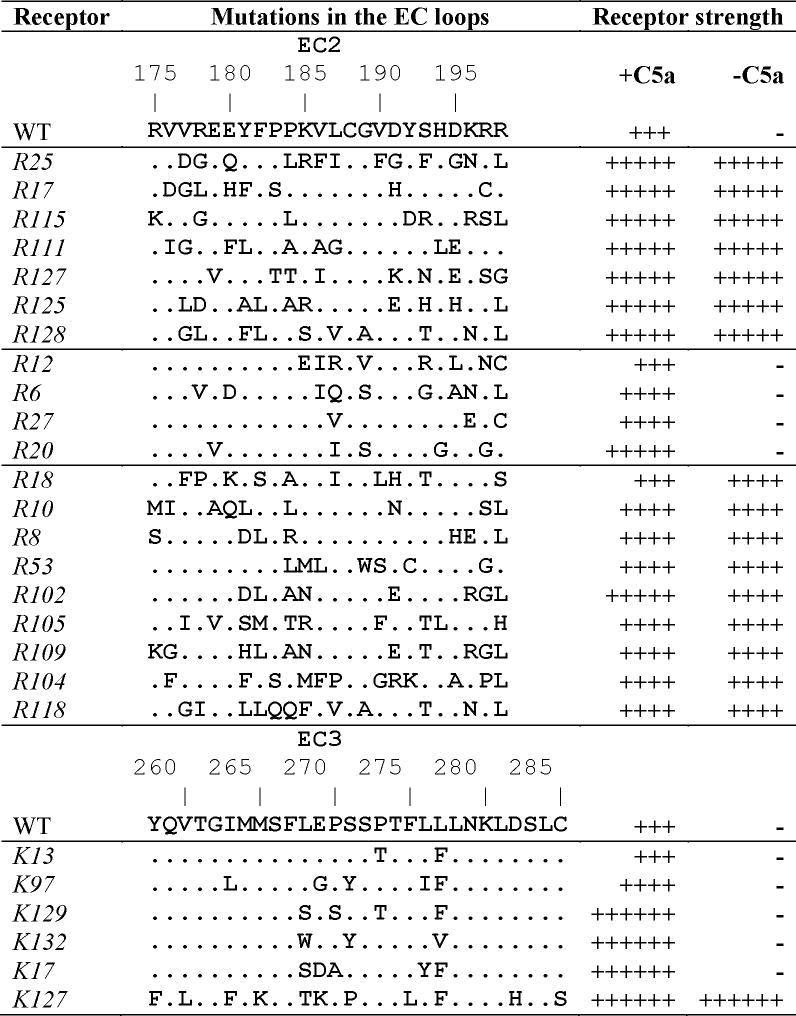
|
Receptors were co-expressed in BY1142 yeast with and without the C5a ligand. Receptor signaling strength was measured by the ability of yeast to grow in the absence of histidine and at the various concentrations of 3-aminotriazole (AT) 27: ++++++, growth on 50 mM AT; +++++, on 20 mM AT; ++++, on 10 mM AT; +++, on 5 mM AT.
Selection of the template for the TM region
At initial steps of modeling (see below), the EC loops were restored on the rigid four-residue helical stems (TM2 95-98 and TM3 107-109 for EC1; TM4 169-172 and TM5 199-202 for EC2; and TM6 264-267 and TM7 281-284 for EC3). The stems were parts of the seven-helical template representing the TM region of C5aR in the ground state.
However, as we showed earlier, several configurations of TM helices (configurations A1-4) may be considered as possibilities for the ground state models of C5aR 3. The configurations are very close to each other (with the rms values from 0.54 to 1.29 Å) and differ from the recently determined X-ray structures of the TM bundles of GPCRs by the rms of ca. 3 Å 3. Spatial positions of the TM2-TM3 stems were geometrically similar (by rms cut-off of 1 Å) for all four configurations. At the same time, in regard of spatial positions of the TM4-TM5 stems, configuration A1 was similar to A3 (A1 ≈ A3) and configuration A2 was similar to A4 (A2 ≈ A4), whereas in regard of spatial positions of the TM6-TM7 stems configuration A1 was similar to A2 and A3 but not to A4 (A1 ≈ A2 ≈ A3). Accordingly, the template based on configuration A1 may be used instead of A3 for restoration of EC2, and instead of A2 and A3 for restoration of EC3. Also, the template based on configuration A4 may be used instead of A2 for restoration of EC2. Therefore, two most different templates may be used for restoring both EC2 and EC3, namely A1 (or A3) and A4. One can expect that sampling of the conformational space for the EC loops mounted onto the two most different templates (specifically, configurations A1 and A4 were selected) will cover possible conformations of the loops mounted onto the two other templates, A2 and A3, as well.
All four configurations were used as templates in calculations performed for WT and R25 yielding different numbers of low-energy conformations of the EC loops selected at the sequential steps of modeling. In both cases, however, the number of low-energy conformations modeled employing template A1 was significantly higher than that for A4 (e.g., 144 vs. 3 for WT and 80 vs. 3 for R25 upon modeling of the EC1+EC2+EC3 package; see below). Therefore, only configuration A1 was used for modeling of all other receptors as the template yielding more comprehensive sampling of the EC loop conformational space.
Sequential steps of modeling
For all receptors, the first step of modeling was to determine the low-energy conformations of the mutated EC2 loops (or the EC3 loops for the receptors in the fourth group of Table I) starting from the sets of low-energy conformations found previously for WT C5aR, namely from 339 conformations for EC2 and from 44 conformations for EC3 21. The next step involved clustering the selected EC2 or EC3 conformations by their geometrical similarity (spatial positions of fragments 172-199 or 264-284) within the rms cut-off of 4 Å (3 Å for EC3). Clusters obtained for EC2 or EC3 in mutant receptors were combined with the clusters of the low-energy conformations of the other loops in WT, namely with 3 clusters for EC1 (the energy cut-off of 10 kcal/mol and the rms cut-off of 3 Å), 27 clusters for EC2 and 21 cluster for EC3 into the EC1+EC2+EC3 “packages” consisting of the EC loops and the corresponding helical stems, including the TM1 38-40 stem. At the next step, fragments of the TM helices of the equal length of 20 residues (TM1 38-57; TM2 79-98; TM3 107-126; TM4 153-172; TM5 199-218; TM6 248-267; and TM7 281-300) were added to the low-energy structures of the EC1+EC2+EC3 packages. The resulting constructs (EC1+EC2+EC3+TM) were subjected to energy minimization by two sequential runs.
In the first run, the dihedral angles in the TM helices were frozen at the same values as in the TM template and the harmonic potentials between the ends of the TM helices were added to keep the TM bundle together. In the second run, the selected EC1+EC2+EC3+TM structures were re-minimized without any additional restraints and with all dihedral angles allowed to vary. Finally, the low-energy structures of EC1+EC2+EC3+TM constructs were selected for further geometrical comparisons.
Table II lists the numbers of conformations selected at the each step of modeling by the uniformly selected energy cut-offs, which are also shown in Table II. In some cases, different energy cut-offs were applied to prevent yielding the too large/small numbers of the resulting conformations; these cases are also noted in Table II.
Table II. Numbers of conformations selected at each calculation step by specified energy cut-offs (ΔE, kcal/mol).
| Receptor | EC2 (ΔE < 30) or EC3 (ΔE < 15), numbers of clusters | EC1+EC2+EC3 (ΔE < 30) | EC1+EC2+EC3+TM | |
|---|---|---|---|---|
| First run (ΔE < 70) | Second run (ΔE < 50) | |||
| R25 | 29 | 80 | 27 | 15 |
| R17 | 17 | 76 | 12a | 6 |
| R115 | 25 | 280 | 47 | 32 |
| R111 | 20 | 60 | 15 | 12 |
| R127 | 31 | 33 | 11a | 6b |
| R125 | 18 | 227 | 41 | 24 |
| R128 | 28 | 146 | 38 | 10 |
|
| ||||
| WT | 27 | 144 | 27 | 17 |
| R12 | 36 | 161 | 33 | 17 |
| R6 | 25 | 93 | 19 | 13 |
| R27 | 29 | 268 | 61 | 19 |
| R20 | 33 | 77 | 25 | 10 |
|
| ||||
| R18 | 30c | 111 | 31 | 24 |
| R10 | 25c | 204 | 45 | 31 |
| R8 | 29c | 259 | 80 | 19 |
| R53 | 21c | 118 | 41 | 12 |
| R102 | 49c | 187 | 57 | 36 |
| R105 | 18c | 80 | 30 | 14 |
| R109 | 33c | 308 | 71 | 22 |
| R104 | 10b | 146 | 44 | 28 |
| R118 | 17c | 67d | 13 | 6 |
|
| ||||
| K13 | 14 | 110 | 24 | 11 |
| K97 | 11 | 182 | 56 | 20 |
| K129 | 11 | 298 | 81 | 18 |
| K132 | 13 | 169 | 48 | 20 |
| K17 | 10 | 69 | 16 | 14 |
| K127 | 9 | 53 | 15c | 9 |
ΔE < 100 kcal/mol;
ΔE < 70 kcal/mol;
ΔE < 25 kcal/mol;
ΔE < 40 kcal/mol.
Loop conformations characteristic for CAMs and non-CAMs
Visual inspection of the low-energy structures of the EC1+EC2+EC3+TM constructs showed that main geometrical differences between them were in spatial positions of the EC3 loops. For each of the modeled C5aR mutants, the EC structures may be roughly divided into two broad groups, namely those with either “closed” or “opened” EC3 conformations, i.e., turned either towards or away from the TM helical bundle. Accordingly, we compared all obtained structures to each other by the rms values calculated for EC3 (fragment 267–281). For comparison, all EC1+EC2+EC3+TM structures were superimposed on the common template selected earlier for the 20-residue TM fragments (see above). The goal of comparison was to determine possible conformations of EC3 geometrically similar for all strong CAMs (or for all non-CAMs), i.e., determining whether it would be possible to find clusters of conformations of EC3 (consisting of one conformation for every CAM or non-CAM), with geometrical pair-wise differences between all conformations in the cluster not exceeding an rms cut-off of 5.0 Å.
For CAMs belonging to the first group (the strong CAMs), only one such cluster was found; the pair-wise rms values between corresponding common conformations of the seven different mutants (R25, R17, R115, R111, R127, R125 and R128) varied from 2.1 Å (between R25 and R128) to 4.6 Å (between R125 and R128). In this cluster, the EC3 loop was “closed”, i.e., turned towards the TM bundle and EC2. For non-CAMs (WT, R12, R6, R27 and R20), comparison also found only one cluster of common conformations. This non-CAM cluster featured the “opened” EC3 loop turned outwards TM bundle with the rms values between conformations in the cluster varying from 3.1 Å (between R12 and R20) to 4.9 Å (between R12 and R6).
The two clusters are depicted in Figs 1A (the CAM cluster) and 1B (the non-CAM cluster), respectively. The rms values averaged over all conformations within each cluster were 3.1 Å for the CAM cluster and 3.5 Å for the non-CAM cluster. For comparison, the average rms value calculated between the clusters was 8.4 Å, which demonstrates clear distinction between the CAM cluster and the non-CAM cluster.
Figure 1.

Cartoons of the loop structures in the CAM (A) and non-CAM (B) clusters. TM stems are included. EC1 is shown in grey, EC2 in white and EC3 in dark grey.
Structural features characteristic for CAM and non-CAM clusters
Due to the large number of amino acid substitutions in the EC2-mutated receptors, the structures within the CAM and non-CAM clusters displayed residue-residue interactions varying from receptor to receptor. Nevertheless, some specific types of interaction motifs for EC3 were characteristic either for the CAMs or for the non-CAMs.
Table III lists possible contacts between the side chains of EC3 and all other side chains in the structures of the CAM and non-CAM clusters for the first and second groups of mutants, respectively. (A contact was defined as occurrence of any inter-atomic distance between residues less than 4.5 Å.) The data in Table III suggest several differences in the systems of inter-residue interactions in the CAM and non-CAM clusters. First, the central fragment of EC3 (S272-L277) in the structures of the CAM cluster more likely interacted with the residues from the central part of EC2, fragment 178-194. Second, the systems of hydrogen bonding involving the side chains of EC3 were different in the CAM and non-CAM clusters (see contacts shown in bold in Table III; a possible hydrogen bond was defined as the O…H distance less than 3.5 Å). Specifically, within the CAM cluster, the hydrogen bonding of E269 with R2005.36 in TM5 was characteristic for R127, R25, R17 and R128, while K185 in EC2 was involved in hydrogen bonds with S272 and T274 in R17, and the N279 side chain formed hydrogen bonds with S2837.36 in TM7 in R125. The latter interaction was also characteristic for the non-CAM cluster (in WT and R12), as well as interactions of N279 with K185 and S189 (both in EC2) in R20. For illustration, see Fig. 2 depicting most essential residue-residue interactions between EC2 and EC3 (and within EC2) for R17.
Table III.
Contacts between the side chains of EC3 and other residues of the EC1+EC2+EC3+TM constructs in the structures of the CAM (top half) and non-CAM clusters (bottom half). Occurrences of hydrogen bonding are shown in bold.
| Receptor | Residues of EC3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L268 | E269 | P270 | S271 | S272 | P273 | T274 | F275 | L276 | L277 | L278 | N279 | K280 | |
| R25 | R200 | M265 | Y181 | Y258 | F190 | ||||||||
| R200 | Y192 | M265 | |||||||||||
| F193 | |||||||||||||
| R17 | F181 | R200 | K185 | K185 | K185 | M264 | L187 | ||||||
| R200 | M265 | L187 | S283 | ||||||||||
| V190 | |||||||||||||
| Y192 | |||||||||||||
| R115 | R200 | M264 | D191 | S283 | |||||||||
| F267 | M265 | ||||||||||||
| L281 | |||||||||||||
| R111 | F267 | E195 | L194 | R178 | Y192 | L194 | M264 | V190 | |||||
| E195 | P183 | R200 | M265 | S283 | |||||||||
| L194 | M265 | L281 | |||||||||||
| R127 | R200 | R197 | K191 | Y192 | T261 | S283 | |||||||
| R200 | R200 | M265 | |||||||||||
| L281 | |||||||||||||
| R125 | F267 | M265 | M265 | H194 | E191 | L187 | V190 | ||||||
| R200 | E191 | D282 | |||||||||||
| M265 | S283 | ||||||||||||
| R128 | T193 | D191 | A189 | M264 | V190 | ||||||||
| R200 | T193 | H194 | L281 | D191 | |||||||||
| R200 | T261 | ||||||||||||
| M265 | M265 | ||||||||||||
|
| |||||||||||||
| WT | M264 | F267 | K185 | L281 | T261 | L187 | |||||||
| F267 | L187 | M265 | S283 | ||||||||||
| R12 | Y192 | M264 | F267 | M264 | V189 | M264 | V189 | ||||||
| F267 | L281 | D191 | M265 | S283 | |||||||||
| L281 | |||||||||||||
| R6 | R200 | M264 | M264 | K185 | M264 | M264 | Q187 | ||||||
| F267 | S283 | ||||||||||||
| R27 | H194 | F267 | M264 | M265 | M264 | V186 | V186 | ||||||
| L281 | V187 | ||||||||||||
| S283 | |||||||||||||
| R20 | M264 | F267 | M264 | T261 | K185 | S283 | |||||||
| F267 | L281 | M264 | L187 | ||||||||||
| M265 | S189 | ||||||||||||
| L281 | |||||||||||||
Figure 2.

Essential side chain – side chain interactions within EC2 and between EC2 and EC3 in the R17 mutant. All hydrogens are omitted. Hydrogen bonds are sketched as thick dashed lines. Labels for the EC3 residues are underlined.
Our modeling demonstrated more close interactions between the residues of EC3 and EC2 in the structures of the CAM clusters rather than in structures of the non-CAM cluster (compare top and bottom halves of Table III). These interactions are of special significance, since the spatial position of EC2 within the transmembrane segments makes it likely to be involved in a conserved mechanism of receptor activation. The segment of EC2 (from 182 to 192) containing the highly conserved C188, which is involved in the disulfide bond C1093.25-C188, interacted with fragment 110-117 in TM3 both in the CAM and non-CAM clusters (the data not shown). In the CAM cluster, interactions between EC2 and EC3 led to re-orientation of TM helices; namely, the slight kink in TM2 at P902.58 was straightened and TM7 slightly rotated and moved closer to TM2 and TM3 compared to structures of the non-CAM cluster (as illustrated by Fig. 3). For instance, the average distances between Cα atoms of N2967.49 (TM7) and D822.50 (TM2) were 7.6 Å for the CAM cluster and 9.5 Å for the non-CAM cluster, and the distances between Cα atoms of N2967.49 (TM7) and N1193.35 (TM3) were 11.2 Å and 13.6 Å for the CAM cluster and for the non-CAM cluster, respectively. At the same time, the average Cα – Cα distances between D822.50 and N1193.35 for both clusters were very close, 6.7 Å and 6.6 Å, respectively.
Figure 3.

The system of hydrogen bonding between selected side chains in structures characteristic for non-CAMs (WT, left) and CAMs (R127, right). TM2, TM3 and TM7 are shown as cylindrical cartoons. All hydrogens are omitted. Hydrogen bonds are sketched as thick dashed lines.
Movement of TM7 suggested by modeling also triggered significant changes in the system of hydrogen bonds within the TM region. Possible hydrogen bonds between the side chains in the structures of both clusters are listed in Table IV. Notably, most conformations in the CAM cluster displayed hydrogen bonding between the side chains of D822.50 in TM2 and N2967.49 in TM7 (those belonging to R25, R115, R111, R127, R125 and R128), while none of the conformations in the non-CAM cluster featured this interaction. We believe this finding is noteworthy, because the D822.50-N2967.49 interaction was shown to be crucial for stabilization of the activated states of C5aR: the mutation N2967.49 A resulted in loss of constitutive activity of the strong CAM, I1243.40N/L1273.43Q 25.
Table IV. Hydrogen bonds between selected side chains in the structures of the CAM and non-CAM clusters.
| H-bonds | CAM cluster | Non-CAM cluster | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R25 | R17 | R115 | R111 | R127 | R125 | R128 | WT | R12 | R6 | R27 | R20 | |
| N48-N296 | * | |||||||||||
| D82-C86 | * | * | ||||||||||
| D82-N119 | * | * | * | * | ||||||||
| D82-S123 | * | |||||||||||
| D82-N292 | * | * | ||||||||||
| D82-C293 | * | * | ||||||||||
| D82-N296 | * | * | * | * | * | * | ||||||
| S85-N119 | * | * | * | * | ||||||||
| N119-S123 | * | * | * | * | * | |||||||
| N119-Y258 | * | |||||||||||
| N119-C293 | * | * | * | |||||||||
| S123-N292 | * | |||||||||||
| S250-N292 | * | * | ||||||||||
| S250-N296 | * | |||||||||||
The possible structural mechanism leading to formation of the D822.50-N2967.49 hydrogen bond in the CAM cluster is shown in Fig. 3, exemplified by WT (non-CAM) and R127 (CAM). In R127, TM7 moves by ca. 2 Å closer towards TM2 and TM3. As a result, the side chain of S1233.39 in R127 re-orients towards N2927.45 thus decreasing the hydrogen bonding with the side chain of N1193.35 in WT. In turn, movement of the N2927.45 side chain in R127 allows re-orientation of the N2967.49 side chain from the hydrogen bonding with the backbone carbonyl of N2927.45 (not shown in Fig. 3) towards the D822.50 – N2967.49 hydrogen bonding.
Predicting constitutive activation in C5a receptors with mutations in EC loops
Our modeling found varieties of the “closed” and “opened” conformations of the EC3 loops in the C5aR mutants. Among them was the structure type characteristic for the strong CAMs (conformations in the CAM cluster), and another type of structure that was characteristic for non-CAMs (conformations in the non-CAM cluster). Comparing these two types of structures with low-energy conformations found by modeling for any other C5aR receptor with mutations in the EC loops might be, therefore, used as a tool for predicting the constitutive activation of a given mutant.
However, low-energy conformations similar to the structures of the non-CAM cluster were found even for some receptors from the first modeled group of the highly strong CAMs (R25, R17, R115, R111, R127, R125 and R128). Specifically, geometrical comparison determined at least one conformation with the structure of EC3 that was similar to all structures of the non-CAM cluster by the rms cut-off of 4.9 Å (the maximal value for the pair-wise rms between the structures in the non-CAM cluster) in R25, R115 and R125. Also, at least one conformation similar to all structures of the CAM cluster was found for most of non-CAMs from the second modeled group (WT, R12, R6 and R27). In other words, if presence or absence of structures similar to the CAM or non-CAM clusters in the sets of low-energy conformations of the C5aR mutants with mutations in EC2 is used for prediction of whether the mutants from the first and second groups may display constitutive activity, four of seven mutants would be correctly predicted as CAMs (R17, R111, R127 and R128) and one out of five receptors would be correctly predicted as a non-CAM (R20). All other mutants did not fit into a clear classification of CAMs (or non-CAMs), since their sets of low-energy conformations contained structures similar to both CAM and non-CAM clusters.
By definition of the CAM and non-CAM clusters, each of the strong CAMs from the first group contained one conformation belonging to the CAM cluster, and, vice versa, each of the non-CAMs from the second group contained one conformation belonging to the non-CAM cluster. Therefore, the above comparison was inherently biased and could not produce a wrong prediction, i.e., prediction of CAM as non-CAM and vice versa. However, when low-energy conformations found by modeling C5a receptors from two independent groups (receptors R18, R10, R8, R53, R102, R105, R109, R104 and R118 possessing weaker constitutive activity, the third group in Table I, and receptors K13, K97, K129, K132, K17 and K127 with mutations in EC3, the fourth group in Table I) were compared with the CAM and non-CAM clusters, the predictions of CAMs were still more accurate than predictions of non-CAMs. Four receptors, R18, R53, R104 and K127, were correctly predicted as CAMs, whereas only one CAM, R109, was incorrectly predicted to be a non-CAM. All other CAMs (R10, R8, R102, R105, R109 and R118) were not identified as CAMs or non-CAMs. At the same time, non-CAMs K13, K97, K129 and K132 were not discerned as either CAMs or non-CAMs, and the non-CAM K17 was incorrectly predicted as CAM.
Functional role of EC2 and EC3 in displaying constitutive activity
Our modeling suggested that the EC3 loop adopts the “closed” spatial position for triggering constitutive activity in the C5a receptors with mutations in EC2 or EC3. This stabilizes spatial position of the EC2 fragment 188-199 (“downstream” of the highly conserved residue C188). The downstream fragment of EC2 is located between the extracellular ends of TM3, TM4, TM5 and TM6, which form the pocket for the second binding site of the natural agonist of C5aR, anaphylotoxin C5a 26-28. The site consists of several residues of C5aR that interact with C5a. Based on previous docking studies, E1995.35, R2005.36 and R2065.42 in TM5 interact with Lys68, Asp69 and the C-terminal carboxyl of Arg74 in C5a, respectively 21. Our results showed that the side chains of residues E1995.35 and R2005.36 were involved in various contacts with the side chains of EC2 (mostly with fragment 190-194) in all structures from the CAM and non-CAM clusters, whereas contacts of EC2 with the side chain of R2065.42 were rare. The side chain of E1995.35 was more likely involved in hydrogen bonding with the EC2 side chains in the structures of the CAM cluster (with Y192 in R127, K185 and S193 in R111, R193 in R115, H191 in R17 (see Fig. 2), R197 in R128, and H193 in R125), than in the structures of the non-CAM cluster (with S193 in R20, and R193 and H194 in R12). On the other hand, the side chain of R2005.36 was involved in hydrogen bonding with E269 in EC3 in the structures from the CAM cluster (see Fig. 2), but not the non-CAM cluster (see Table III).
Previously, we concluded that EC2 might serve as a negative regulator of C5aR function, most likely by forming interactions that stabilize the ground state of the receptor 1. Structural features found by our modeling now allow further refinement of this concept. Mutations in EC2 initiate subtle conformational shifts in the ground state of C5aR, which facilitate the activated states of C5aR that are stabilized by interactions such as the hydrogen bond between the side chains of D822.50 and N2967.49. These shifts are triggered by interactions between EC3 and EC2 as well as between EC2 and residues in the TM region. Importantly, in the structures characteristic for constitutively active mutants, the EC3 loop in the “closed’ conformation not only stabilizes EC2, but is directly involved in interactions with the TM region including tight interactions with R2005.36 that are important for binding the natural ligand of C5aR, C5a.
In addition, recently we identified a previously unappreciated role for S272 in EC3 of C5aR and its interaction with the N terminus of C5aR 29. These studies demonstrated a new microswitch region within the C5aR for receptor activation. It is unclear how the N terminus might affect the conformations of the EC3 in the mutant receptors in the current study since the highly flexible N-termini were excluded from the modeling to avoid combinatorial explosion of sampling the possible conformations of the “N-termini + loops” packages. However, if the N-terminus helps stabilize the ground state of the receptor, we would predict that this interaction could stabilize the “open” conformation of EC3.
Now we may conclude that mutations in EC2 that destabilize the ground state of C5aR (i.e., shift equilibrium of receptor conformations to the activated state) do so in part by interacting with EC3. This conclusion is in line with the important functional role of the EC3 loop in various GPCRs (see, e.g., Ref. 30). The most frequent lengths of the EC3 loops in GPCRs belonging to the rhodopsin family were estimated as being in the regions of either 5-13 or 13-19 residues 8. The size of EC3 adopted in the C5aR modeling was 13 residues; as we showed, this length allows both the “opened” and the “closed” conformations of EC3. Longer EC3 loops would also be predicted to be able to adopt these two conformations. The average lengths of the upstream and downstream fragments of EC2 in C5aR (14 and 10 residues, respectively) are also conserved in many GPCRs 31. Therefore, the typical “closed” conformations of the EC3 loop that caps the downstream fragment of EC2 found by our modeling could be readily available in many GPCRs. Also, D822.50 and N2967.49 are highly evolutionary conservative residues (94% and 75% of conservation in the rhodopsin family 8). Taken together, these observations may indicate a possible general structural mechanism involving EC2 and EC3 in displaying constitutive activity within GPCRs belonging to the rhodopsin family.
Concluding Remarks
In the present study, de novo molecular modeling procedures developed earlier 3,16 were applied to extensive sampling of possible conformational space available for the EC loops of the C5a receptors with mutations in EC2 or EC3. The obtained low-energy conformations of the EC loops defined two distinct clusters of conformations typical either for the strong constitutively active mutants (the CAM cluster) or for non-constitutively active mutants (the non-CAM cluster). The CAM cluster featured only the “closed” structure of the EC3 loop turned towards the TM bundle, while the non-CAM cluster, on the contrary, featured only the “opened” EC3 structure turned outwards the TM bundle (see Fig. 1). This modeling predicts a role for the EC3 loop in which its contacts with EC2 and with TM3 are necessary for constitutive activity. These interactions trigger movement of TM7 towards TM2 and TM3, which may be the first step in destabilizing the ground state of the receptor. This movement initiates rearrangement of the system of hydrogen bonds between TM2, TM3 and TM7 including formation of the hydrogen bond between the side chains of D822.50 in TM2 and N2967.49 in TM7. As shown elsewhere 3, this hydrogen bond was crucial for formation of the activated states of the C5a receptors, thus providing strong support for the mechanism of constitutive activity suggested by the current de novo modeling. Given the relative large length of EC3 in C5aR (13 residues) comparable with those in many other rhodopsin family members of GPCRs (13-19 residues, which allows the “closed” conformations of EC3), the findings of this modeling might reflect general mechanisms of receptor constitutive activation. However, flexibility of EC3 may be limited by interactions with the N-terminus that was not present in our calculations.
Interestingly, the EC3 loop in the very recently released X-ray structure of the agonist-induced constitutively active mutant of rhodopsin, E1133.28N, did not show the “closed” conformation, though some EC3 residues contacted residues of EC2 (e.g., Q279 - P194) 4. A major consideration when interpreting this result is that an engineered disulfide bond C2-C282 between the N-terminus and EC3 likely limited the conformational flexibility of EC3. Also, the EC3 loop of rhodopsin is only 8 residues long, which might preclude its ability of assume a “closed” conformation. Of note, the constitutive activity of the disulfide bonded rhodopsin mutant N2C/E1133.28N/D282C was not reported in functional assays. Nevertheless, the X-ray structure featured the hydrogen bonding D2.50 – N7.49 discussed above, which occurred through an intermediate water molecule. Importantly, the mutant was co-crystallized with the C-terminal peptide of the α-transducin, which stabilized the active state of rhodopsin (see also Ref. 32, where the same conformational shift was demonstrated for opsin bound to C-terminal peptide of α-transducin).
Acknowledgments
Grant sponsor: NIH; Grant numbers GM 71634 and GM63720.
References
- 1.Klco JM, Wiegand CB, Narzinski K, Baranski TJ. Essential role for the second extracellular loop in C5a receptor activation. Nature Struct Mol Biol. 2005;12:320–326. doi: 10.1038/nsmb913. [DOI] [PubMed] [Google Scholar]
- 2.Klco JM, Nikiforovich GV, Baranski TJ. Genetic Analysis of the First and Third Extracellular Loops of the C5a Receptor Reveals an Essential WXFG Motif in the First Loop. J Biol Chem. 2006;281:12010–12019. doi: 10.1074/jbc.M600548200. [DOI] [PubMed] [Google Scholar]
- 3.Nikiforovich GV, Marshall GR, Baranski TJ. Simplified modeling approach suggests structural mechanisms for constitutive activation of the C5a receptor. Proteins: Structure, Function, and Genetics. 2011;79:787–802. doi: 10.1002/prot.22918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Standfuss J, Edwards PC, D'Antona A, Fransen M, Xie G, Oprian DD, Schertler GFX. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drews J. Drug discovery: A historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 6.Lado-Abeal J, Quisenberry LR, Castro-Piedras I. Identification and Evaluation of Constitutively Active Thyroid Stimulating Hormone Receptor Mutations. Methods in Enzymology. 2010;484:375–395. doi: 10.1016/B978-0-12-381298-8.00019-8. [DOI] [PubMed] [Google Scholar]
- 7.Janovick JA, Pogozheva ID, Mosberg HI, Conn PM. Salt bridges overlapping the GnRHR agonist binding site reveal a coincidence detector for GPCR activation. J Pharmacol Exp Ther. 2011 doi: 10.1124/jpet.111.180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence Analyses of G-Protein Coupled Receptors: Similarities to Rhodopsin. Biochemistry. 2003;42:2769–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mustafi D, Palczewski K. Topology of class A G protein-coupled receptors: insights gained from crystal structures of rhodopsins, adrenergic and adenosine receptors. Molecular Pharmacology. 2009;75(1):1–12. doi: 10.1124/mol.108.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chien EYT, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the Human Dopamin D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science Express. 2010 doi: 10.1126/science.science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauß N, Hofmann KP, Scheerer P, Ernst OP. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 13.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an Agonist-Bound Human A2A Adenosine Receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwara S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Galinski D, Mathiesen JM, Shah STA, Lyons JA, Gaffrey M, Gellman SH, Steyaert J, Skiniotis G, Weiss WI, Sunahara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011 doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nikiforovich GV, Taylor CM, Marshall GR, Baranski TJ. Modeling the possible conformations of the extracellular loops in G-protein-coupled receptors. Proteins: Structure, Function, and Genetics. 2010;78:271–285. doi: 10.1002/prot.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldfeld DA, Zhu K, Beuming T, Friesner RA. Successful prediction of the intra- and extracellular loops of four G-protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8275–8280. doi: 10.1073/pnas.1016951108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikiforovich GV, Taylor CM, Marshall GR, Baranski TJ. Difference between restoring and predicting 3D structures of the loops in G-protein-coupled receptors by molecular modeling. Proceedings of the National Academy of Sciences of the United States of America. 2011 doi: 10.1073/pnas.1107702108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldfeld DA, Zhu K, Beuming T, Friesner RA. Reply to Nikiforovich et al.: Restoration of the loop regions of G-protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America. 2011 doi: 10.1073/pnas.1108089108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60(1):1–19. [PubMed] [Google Scholar]
- 21.Nikiforovich GV, Marshall GR, Baranski TJ. Modeling Molecular Mechanisms of Binding of the Anaphylotoxin C5a to the C5a Receptor. Biochemistry. 2008;47:3117–3130. doi: 10.1021/bi702321a. [DOI] [PubMed] [Google Scholar]
- 22.Dunfield LG, Burgess AW, Scheraga HA. Energy Parameters in Polypeptides. 8. Empirical Potential Energy Algorithm for the Conformational Analysis of Large Molecules. J Phys Chem. 1978;82:2609–2616. [Google Scholar]
- 23.Nemethy G, Pottle MS, Scheraga HA. Energy Parameters in Polypeptides. 9. Updating of Geometrical Parameters, Nonbonded Interactions, and Hydrogen Bond Interactions for the Naturally Occuring Amino Acids. J Phys Chem. 1983;87:1883–1887. [Google Scholar]
- 24.Nikiforovich GV, Hruby VJ, Prakash O, Gehrig CA. Topographical Requirements for Delta-Selective Opioid Peptides. Biopolymers. 1991;31(8):941–955. doi: 10.1002/bip.360310804. [DOI] [PubMed] [Google Scholar]
- 25.Whistler JL, Gerber BO, Meng E, Baranski TJ, von Zastrow M, Bourne HR. Constitutive Activation and Endocytosis of the Complement Factor 5a Receptor: Evidence for Multiple Activated Conformations of a G Protein-Coupled Receptor. Traffic. 2002;3:866–877. doi: 10.1034/j.1600-0854.2002.31203.x. [DOI] [PubMed] [Google Scholar]
- 26.Mollison KW, Mandecki W, Zuiderweg ER, Fayer L, Fey TA, Krause RA, Conway RG, Miller L, Edalji RP, Shallcross MA. Identification of receptor-binding residues in the inflammatory complement protein C5a by site-directed mutagenesis. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(1):292–296. doi: 10.1073/pnas.86.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siciliano SJ, Rollins TE, DeMartino J, Konteatis Z, Malkowitz L, Van Riper G, Bondy S, Rosen H, Springer MS. Two-site binding of C5a by its receptor: an alternative binding paradigm for G protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(4):1214–1218. doi: 10.1073/pnas.91.4.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeMartino JA, Van Riper G, Siciliano SJ, Molineaux CJ, Konteatis ZD, Rosen H, Springer MS. The amino terminus of the human C5a receptor is required for high affinity C5a binding and for receptor activation by C5a but not C5a analogs. Journal of Biological Chemistry. 1994;269(20):14446–14450. [PubMed] [Google Scholar]
- 29.Rana S, Baranski TJ. Third extracellular loop (EC3)-N terminus interaction is important for seven-transmembrane domain receptor function: implications for an activation microswitch region. Journal of Biological Chemistry. 2010;285(41):31472–31483. doi: 10.1074/jbc.M110.129213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson Z, Wheatley M. The third extracellular loop of G-protein-coupled receptors: more than just a linker between two important transmembrane helices. Biochemical Society Transactions. 2004;32(Pt 6):1048–1050. doi: 10.1042/BST0321048. [DOI] [PubMed] [Google Scholar]
- 31.de Graaf C, Foata N, Engkvist O, Rognan D. Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins. 2008;71(2):599–620. doi: 10.1002/prot.21724. [DOI] [PubMed] [Google Scholar]
- 32.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455(7212):497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]