

Abstract
The coupling of protein energetics and sequence changes is a critical aspect of computational protein design, as well as for the understanding of protein evolution, human disease, and drug resistance. In order to study the molecular basis for this coupling, computational tools must be sufficiently accurate and computationally inexpensive enough to handle large amounts of sequence data. We have developed a computational approach based on the linear interaction energy (LIE) approximation to predict the changes in the free energy of the native state induced by a single mutation. This approach was applied to a set of 822 mutations in 10 proteins which resulted in an average unsigned error of 0.82 kcal/mol and a correlation coefficient of 0.72 between the calculated and experimental ΔΔG values. The method is able to accurately identify destabilizing hot spot mutations however it has difficulty in distinguishing between stabilizing and destabilizing mutations due to the distribution of stability changes for the set of mutations used to parameterize the model. In addition, the model also performs quite well in initial tests on a small set of double mutations. Based on these promising results, we can begin to examine the relationship between protein stability and fitness, correlated mutations, and drug resistance.
Keywords: LIE, protein stability, ΔΔG prediction, PLOP, AGBNP, free energy
Introduction
Proteins are molecular machines whose thermodynamic stability and fitness are encoded in their amino acid sequence. Mutations can change the energetic landscape of a protein and thereby alter its structure and function. The coupling of sequence changes to protein energetics is a critical aspect of computational protein design, and is necessary for a complete understanding of protein evolution, human disease, and drug resistance. The computational protein design field employs sequence modifications to design novel protein folds, to modify thermostability and enzymatic activity, and to redesign protein-protein interfaces.1–5 On the other hand, studies of sequence evolution have focused on examining the balance between stability and fitness of proteins found in nature6, for example, the role of the “stability/activity” tradeoff mechanism for residues located in protein active sites.7–9. Furthermore, directed evolution experiments have designed proteins with novel functions through a series of functional but destabilizing replacements accompanied by a series of compensatory mutations highlighting the crucial role that stabilizing mutations play in the “evolvability” of a protein.6,10,11 This compensatory stabilizing mechanism is also quite prevalent in the evolution of drug resistance.12,13 The acquisition of drug resistance has been linked to primary mutations which cause the resistance at the cost of stability and are correlated with accessory mutations which restore activity and stability, as for example in HIV protease.14 Additional work by Ishikita and Warshel has shown how effective drug resistant mutations maintain catalytic efficiency and hence the local instability within the active site while weakening the binding affinity to a target drug.15 Therefore, it is important to understand how sequence changes can alter the thermodynamic stability and fitness of proteins to relate these effects to drug resistance and disease6,16,17.
In order to tackle these problems, computational approaches must be sufficiently accurate to capture the underlying energetics and capable of handling large amounts of sequence data. Methods such as free energy perturbation and thermodynamic integration are in principle the most accurate of these approaches but are limited to a small number of mutations.18–20 More efficient computational mutagenesis methods based on approximations to the free energy change, are used to predict protein stabilities for large databases of proteins. These methods are differentiated by their free energy function which can be categorized as knowledge-based/statistical21, empirical22,23 or physics-based24,25 potentials. These methods also vary in the extent of the conformational sampling used to model the structural changes induced by the mutation. Some approaches only model the mutated residue using a fixed backbone22,23 while other methods include flexibility either through side chain repacking and backbone relaxation26,27 or the generation of an ensemble of structures.25 Lastly, the unfolded state is treated differently by these free energy methods; unfolded state effects have been included implicitly in the coefficients of the energy function22, explicitly represented by a specific term in the energy potential23,26,28 or modeled as a short peptide of the original structure.24,25
Many of these approximate methods, however, demonstrate limited accuracy according to a recent survey by Potapov et al.29 In this report, they claim that these methods are “good on average and not in the details.” It is reported that the best method only achieved a correlation coefficient between experimental (ΔΔGexp) and calculated (ΔΔGcalc) relative free energies of folding (compared to wild type) of 0.59 on a large set of single point mutations.1 Since many of the most relevant biological problems involve multiple mutations, limitations with single point mutations may translate into large errors, especially since these methods are not typically tested with more than one mutation. Therefore, there is a need for further development of protein stability models with greater predictive accuracy.
A possible alternative to rigorous free energy methods on one hand and purely empirical approaches in the other are linear interaction energy models.30,31 LIE approaches have a foundation in linear response theory32–34 and the linear response approximation35; physically motivated interaction energy estimators which only require the knowledge of the endpoints of a particular process are used to estimate the free energy change. This approach has been mainly applied to protein-ligand binding problems.30,35–41 Typically, the LIE formulations use the following functional form to calculate the free energy of binding (ΔGb), 30,31
| (1) |
where ΔVvdw, ΔVelec and ΔA are differences between the quantities measured for the ligand complexed with the receptor and ligand free in solution. ΔVvdw and ΔVelec are the van der Waals and electrostatic interaction energies of the ligand with its environment. ΔA is the change in surface area between the receptor-ligand complex and the free ligand. Typically, these energies have been obtained from Molecular Dynamics or Monte Carlo simulations of the receptor-ligand complex and free ligand in explicit solvent. Recent studies in our lab and elsewhere have applied this formalism to studies in implicit solvent with approximate and more rigorous derivations.39–41 α, β, γ, and δ are adjustable parameters which are obtained by fitting to experimental binding data.
Linear response has also been used to study protein stability and protein-protein interactions.42–45 Previously, Warshel applied the linear response approximation to the calculation of absolute protein stabilities using an electrostatic energy function scaled with “focused” dielectric constants.42,43 In addition, the LIE method has been used to calculate the absolute and relative binding affinities of protein-protein interfaces that contained different mutations to a crucial residue for binding (treating the mutated residue as a ligand).44,45 Building on these ideas and our earlier work with LIE models, we have devised a LIE approach to calculate relative protein stabilities between wild type and single point mutations using the LIE method. For the protein stability problem, we consider the free energy change for replacing one protein residue with another in the folded and unfolded states (Figure 1). The difference between the corresponding wild-type and mutant energetic estimators can be used to construct a linear interaction energy model for the free energy of folding analogous to the LIE equation for the binding free energy.
Figure 1.
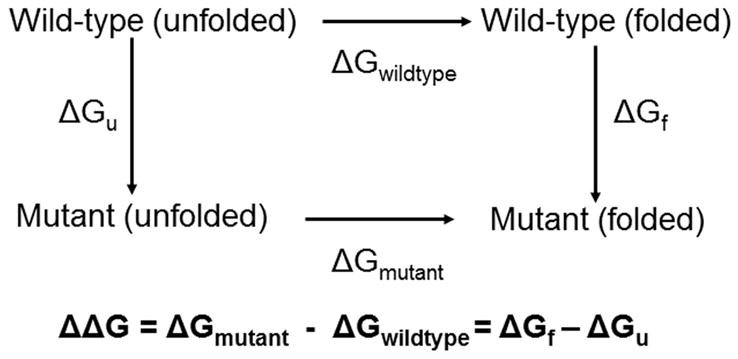
Thermodynamic cycle for calculating the relative changes in protein stability (ΔΔG). The ΔGwild-type and ΔGmutant are the free energy difference between the folded and unfolded state for the wild-type and mutant, respectively. The ΔGf and ΔGu is the free energy difference between mutant and wild-type in the folded and unfolded state, respectively. The relative free energy can be expressed as either the difference between ΔGwild-type and ΔGmutant or as the difference between ΔGf and ΔGu.
In this work, we have developed a computational approach based on the LIE method using the protein local optimization program46 (PLOP) for conformational sampling, together with the Analytical Generalized Born plus Nonpolar (AGBNP) implicit solvent model47 and OPLS force field48,49 to predict the changes in the free energy of the native state induced by single point mutations. In the following section, we derive the LIE equations used to calculate protein stability. For the initial tests of this method, we performed side chain prediction for the structural models of the wild type and mutant structures and applied the LIE protein stability formulation on 822 mutations from 10 proteins. We found that a separate model based on mutation residue types (e.g. charged vs uncharged) showed improved results compared to a model which did not distinguish between residue types. All models were validated with jack-knife prediction tests. This approach has resulted in a correlation coefficient and an average unsigned error between the ΔΔGexp and ΔΔGcalc of 0.72 and 0.82 kcal/mol respectively, which are among the best results reported to date for current protein stability prediction methods.
Materials and Methods
This section describes the methodology used to develop the LIE protein stability protocol. This involved the selection of single point mutations for our benchmark set, structure prediction for the wild-type and mutant structures, and derivation of the LIE equations for the calculation of relative protein stabilities. We also summarize in this section the analysis procedures used to evaluate the results of the LIE protein stability approach.
Selection of Mutations
A benchmark set of single point mutations was created from available ΔΔGexp data in the Protherm database50 and from Guerois et al.22 Mutations were selected if the wild type residue had a larger van der Waals volume than the mutant residue, the ΔΔGexp measurement was performed between a pH of 5 and 9 and an X-ray structure existed for the wild-type protein; this was based on criteria applied by Guerois et al.22 Lastly, we required that the stability measurements were made within a relatively narrow temperature range (between 17°C and 37°C ) since we neglect effects of temperature variation in the LIE function.
From the filtered set, the 10 proteins with the largest number of mutations were selected for our current study (822 mutations) (Table 1). The proteins included in this study are: staphyloccal nuclease (PDB ID:1STN51), barnase (PDB ID:1BNI52), FK506 binding protein (PDB ID:1FKJ53), chymotrypsin inhibitor 2 (PDB ID:2CI254), protein L (PDB ID: 1HZ655), human tyrosine-protein kinase c-Src (PDB ID:1FMK56), human lysozyme (PDB ID:1REX57), bovine pancreatic inhibitor (PDB ID: 1BPI58), fibronection (PDB ID: 1TEN59) and T4-lysozyme (PDB ID:2LZM60).
Table 1.
Proteins used for LIE protein stability calculations
| Protein | %α | %β | Nmut |
|---|---|---|---|
| 1stn | 24 | 30 | 391 |
| 1bni | 22 | 23 | 91 |
| 1fkj | 14 | 37 | 31 |
| 2ci2 | 22 | 28 | 58 |
| 1hz6 | 35 | 48 | 57 |
| 1fmk | 5 | 41 | 49 |
| 1rex | 40 | 12 | 41 |
| 1bpi | 21 | 26 | 40 |
| 1ten | 0 | 51 | 39 |
| 2lzm | 7 | 66 | 25 |
%α = percentage residues in an α-helical conformation in the whole protein; % β = percentage of residues in β-sheet conformation in the whole protein; Nmut= number of single point mutations;
Structure preparation
Structural models of the single point mutants were built using the torsional angle sampling implementation in the protein local optimization program (PLOP)46. PLOP is typically used for its side chain and loop prediction capabilities in homology modeling problems46,61,62, force field/implicit solvent evaluation studies 63–66 and protein-ligand binding problems involving the modeling of receptor-induced fit effects67. We have performed a side chain prediction test on a database of approximately 2190 polar side chains found in 30 proteins using the AGBNP implicit solvent model47 with the OPLS-AA force field48,49 and measured the accuracy of the predictions by calculating the heavy atom root mean squared deviation (RMSD) of the side chain of each predicted rotamer state relative to its corresponding minimized X-ray side chain rotamer. Using a RMSD cutoff of 1.5 Å, the side chain prediction accuracy was 79% and 75% with and without the crystal environment respectively. These results provide further evidence that the PLOP program can be employed with the OPLS-AA/AGBNP force field to predict the side chain rotamer geometries of the wild-type and mutant residue with good fidelity.
1) Minimization of structures
Minimizations were performed on all of the proteins using PLOP with the OPLS-AA/AGBNP force field. The Truncated Newton algorithm68 was employed with a RMS tolerance of 0.5 kcal/mol Å using the default settings in PLOP.
2) Wild-type and mutant folded structure
The side chain conformations for both the wild-type and mutant forms were built using the side chain prediction algorithm in PLOP. Mutant structures were based on the wild-type structure and only differed at the mutated residue position. There are several steps involved in this algorithm incorporated into PLOP. The native backbone of the protein is held fixed while a conformational search is performed using a highly detailed rotamer library developed by Xiang and Honig69. Rotamers are eliminated based on an adjustable overlap factor which is a measure of the extent of the “clash” of the rotamer with the residues of the protein which are held fixed. The remaining structures are scored based on a reduced non-bonded energy and clustered in torsional space using the K-means algorithm70. The lowest energy rotamer is selected from the clustering procedure and the side chain rotamer is minimized using the TN algorithm68. This procedure can be performed with or without the crystal packing environment. Side chain prediction was performed on the wild type and mutated residue of interest without crystal packing which should result in the preferred rotamer conformation in solution.
3) Unfolded state
The unfolded state was represented using a tetrapeptide model of the local environment of the residue of interest. This peptide contained the two neighboring residues on either side and was capped with an acetyl (ACE) and n-methyl amino group (NME). The peptide was modeled with the wild-type or mutated residues fixed in the folded state conformation. If the mutated residue was close to the N-terminus or C-terminus (1–2 residues away), the charge was maintained on the corresponding terminus of the unfolded state model.
Scoring
Energies were calculated using the OPLS AA/AGBNP force field. The AGBNP model contains the analytical pairwise descreening implementation of the Generalized Born (GB) model71 and a non-polar hydration term (Gnp).47
The polar solvation energy (Gel) is estimated using the GB equation
| (2) |
where εin is the dielectric constant of the interior of the solute, εw is the dielectric constant for water, qi and qj are the charges on atom i and j and
| (3) |
where Bi and Bj are the Born radii of atoms i and j and rij is the distance between atoms i and j. The non-polar term contains two components: Gcav and Gvdw.72,73
| (4) |
Gcav accounts for the work required to make a cavity in solution and Gvdw accounts for the solute-solvent dispersive vdw forces. In Equation 5, the cavity component is a function of the surface area of atom i (Ai) and the surface tension parameter assigned to atom i (γi) while the van der Waals dispersionterm is expressed as a function of an adjustable van der Waals dispersion parameter (αi)47, the Born radii of atom i (Bi), the radius is of a water molecule (Rw) and
| (5) |
where ρw = 0.033428 Å3 is the number density of water at standard conditions and εiw and σiw are OPLSforce field Lennard Jones parameters for solute-solvent interactions with oxygen atom of TIP4P water.74
LIE formulation for Protein Stability
The concepts behind the LIE protein stability model are derived from the ideas of linear response theory and the linear response approximation.32–35 Protein stability is a measure of the free energy difference between the folded and the unfolded state (Figure 1). In this case, we are interested in determining the protein stability of the wild type (ΔGwildtype) and the mutant (ΔGmutant) in order to calculate the relative stability (ΔΔG). As illustrated in Figure 1, ΔΔG is equal to the differences of the alchemical free energies of transforming the wild-type residue into the mutated residue within the unfolded (ΔGu) and folded (ΔGf) states.
According to the LIE formalism, the free energy change at each step can be expressed as:
| (6) |
where Δ<V> is the change in the residue-environment average interaction energy in going from wild-type to mutant corresponding to the step of interest (for example, the electrostatic interaction energy for the transformation of the electrostatic interactions from those of the wild-type residue to those of the mutant).
We can write for the free energy of mutation in the folded state (ΔGf) :
| (7) |
where Δ<VLJ> is the change in the van der Waals interaction energy for intramolecular interactions within the protein, Δ<Vel> is the change in the electrostatic interaction energy, Δ<Gel> is the change in the polar solvation interaction energy, Δ<Gcav> is the change in the non-polar energetic component for cavity formation and Δ<Gvdw> is the change in the non-polar van der Waals dispersion energy between solvent and the solute. α, β, γ, δ, and ε are the corresponding LIE coefficients for the energetic estimators.
A similar equation applies to ΔGu yielding the following expression for the change in folding energy:
| (8) |
where ΔΔGcalc is the free energy difference between the stability of the mutant and wild-type protein. ΔΔ<VLJ> and ΔΔ<Vel> are the intramolecular interaction energy contributions to the stability change and ΔΔ<Gel>, ΔΔ<Gcav> and ΔΔ<Gvdw> are the solute-solvent interaction energy contributions to the stability change.
We define the interaction energy between a given residue and its environment as the difference between the total energy of the protein and the total energy of the protein without the residue of interest. This definition, which for pairwise decomposable potentials, reduces to the sum of pairwise interactions between the atoms of the residue and the other protein atoms, is a generalization of the interaction energy which is applicable for non-pairwise decomposable potentials such as the implicit solvent solvation free energy. The goal is to estimate the side chain’s contribution to the (free) energy of each (folded/unfolded) state. The energy of state-1 was first calculated for the structure with all of the solute-solute and solute-solvent interactions present. The second step was to eliminate interactions of the side chain for the second state in the calculation. The non-bonded and polar solvation energy terms were turned off by setting the partial charges and the well depth of the Lennard Jones’s equation to zero for side chain atoms (starting with Cβ atom). The non-polar hydration was turned off by settng γi and αi to zero. The difference between state-1 and state-2 represents the side chain’s interaction energy with the other residues of the protein and the solvent. Since the same structure is used for the state 1 and state 2, the covalent energy terms cancel out. This was done for both the folded and unfolded state model.
The LIE calculation for mutations involving glycine and proline is different since the partial charges of the backbone are different from the other residues. When the partial charges of the HA3 atom of the glycine or the side chain of the proline are set to zero, the backbone is left with an excess charge. For all other residues, the backbone remains neutral after the charges are turned off. For the second state corresponding to glycine and proline, we created a model where the partial charges of all the atoms in the residue were set to zero. Hence, the difference between state-1 and state-2 includes the electrostatic and reaction field interaction energy for the side chain and backbone of the residue in both the folded and unfolded state model. By taking the difference between the folded and unfolded state, the effect of turning off the charges on the backbone is eliminated.
The LIE equation requires five LIE coefficients for the energy terms. This fitting was performed using the multiple linear regression module in the statistical analysis program R. The whole set of 822 single point mutations was used for training and testing. Jack knife tests as described below were performed by iteratively training on 95 % of the data and testing on the remaining 5 % of the data set. We have developed two models for ΔΔG prediction (Table 2 and Table 3). For Model-1, five coefficients were obtained while for Model-2, five coefficients were obtained for each of the three mutation residue types (neutral, charged and glycine/proline). Coefficients were eliminated on the basis of p-values (p < 0.05 to reject the null hypothesis for a particular coefficient). If coefficients of terms with complimentary physical effects (e.g. electrostatic and reaction field interaction energies or intramolecular and solute-solvent van der Waals interaction energies) were of the same magnitude, their corresponding energy estimators were combined and refit. In this work, most of the models were fit to two coefficients except for the neutral residue model (Model-2).
Table 2.
Fitting results for Neutral, Charged, and Glycine/Proline mutations, Individually and Combined.
| Residue Type | Model # | np | α | β | γ | δ | ε | < | error | >jack | rjack |
|---|---|---|---|---|---|---|---|---|---|
| All | 1 | 2 | 0.32 | 0.07 | 0.07 | 0.00 | 0.00 | 0.95 | 0.60 |
| Neutral | 2 | 5 | 0.17 | 0.13 | 0.21 | −0.19 | −0.94 | 0.83 | 0.67 |
| Charged | 2 | 2 | 0.17 | 0.07 | 0.07 | 0.00 | 0.00 | 0.73 | 0.38 |
| Glycine/Proline | 2 | 2 | 0.53 | 0.08 | 0.08 | 0.00 | 0.53 | 0.94 | 0.75 |
np= number of LIE coefficients; α, β, γ, δ and ε are the LIE coefficients;
|error|jack= average unsigned error for jack-knife validation; rjack = pearson correlation coefficient for the jack knife validation;
The average unsigned error for the jack knife validation is in units of kcal/mol.
Table 3.
Comparison of the results for different mutation residue types using Model 1 and Model 2.
| Residue-type | # of Mutations | Model-1 | Model-2 |
|---|---|---|---|
| All | 822 | 0.61 (0.94) | 0.72 (0.82) |
| Neutral | 448 | 0.60 (0.88) | 0.69 (0.80) |
| Charged | 175 | 0.38 (0.84) | 0.45 (0.72) |
| Glycine/Proline | 199 | 0.72 (1.15) | 0.77 (0.94) |
The correlation coefficient is listed with the average absolute error in parenthesis (in units of kcal/mol).
We evaluated the predictive value of each of the models in this study (Table 2). We employed a jack knife approach where the corresponding jack knife Pearson correlation coefficient (rjack) and average absolute error (<|error|jack>) are reported. For Model-1 and Model-2, fitting resulted in rjack and <|error|jack> values that were very close to their values for the entire data set (Table 1) which suggests that these models are not biased by particular points in their training sets; this was not the case for Model-2(charged). For Model-2(charged), the corresponding rjack and <|error|jack > were slightly worse than the values reported for the entire data set which is indicative of some type of bias in our training set (Table 1). Nevertheless, Model-2 demonstrates more accuracy and precision than Model-1 even though Model-2 has more parameters (Model-1 has 2 parameters while Model-2 has 9 parameters).
Multiple Mutations
A small set of double mutations from serine protease inhibitor was taken from the Protherm database. These mutations were previously tested with the Eris program;26 this approach uses a hybrid knowledge-based/physical energy function and allows for backbone flexibility. Our structural models for the wild-type and mutant state were built using the multiple side chain prediction module in PLOP. LIE calculations were performed using LIE equations and coefficients developed for Model-2. In 16 of the 17 cases, the double mutant was composed of two mutations of the same residue type so the coefficients for that specific residue type were selected for that ΔΔG calculation. For the double mutant with different mutations types (T58AE60A), we employed the coefficients developed for the charged residue model in the LIE equation for protein stability since electrostatic interactions are likely to dominate the effect of the double mutation.
Analysis
Both Pearson correlation coefficients and average absolute errors between the calculated and experimental ΔΔG were the statistical measures used to evaluate the different models and groups of mutations in this study. Nevertheless, we note that the Pearson correlation coefficient is the gold standard used to evaluate the quality of the stability change calculations since the absolute error is highly influenced by the relatively small range of the ΔΔG data.
In Table 3, we summarize the performance of the LIE algorithm at identifying two different types of mutations: stabilizing/destabilizing and hot-spot mutations similar to Potopov et al..29 Stabilizing mutants had ΔΔGexp < 0 kcal/mol while destabilizing mutants had ΔΔGexp > 0 kcal/mol. Hot spot mutations had |ΔΔGexp| > 2 kcal/mol while non-hot spot mutations had |ΔΔGexp| < 2 kcal/mol. The stability ranges of the ΔΔGcalc and ΔΔGexp values were compared using the following measures: accuracy, sensitivity and specificity, which were functions of the number of true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN). Accuracy is calculated using the following expression:
| (9) |
which evaluates the number of correct predictions relative to the total number of predictions. Sensitivity evaluates the number of correctly identified true positives relative to the total number of positives. Sensitivity is calculated with the following expression:
| (10) |
Specificity evaluates the number of correctly identified true negatives relative to the total number negatives. Specificity is calculated with the following expression:
| (11) |
In the analysis summarized in Table 4, hot spot residues are defined as positives while non-hot spot residues are defined as negatives.
Table 4.
Prediction of stabilizing/destabilizing and hot spot mutations
| Stabilizing/Destabilizing Mutations | Hot spot Mutations | |||
|---|---|---|---|---|
| Model | Accuracy (%) | Accuracy (%) | Sensitivity (%) | Specificity (%) |
| 1 | 87 | 79 | 54 | 90 |
| 2 | 89 | 82 | 66 | 90 |
Stabilizing mutations had ΔΔGexp < 0 and destabilizing mutations had ΔΔGexp > 0.
Hot spot mutations had |ΔΔGexp|> 2 and non-hot spot mutations had |ΔΔGexp|< 2. Accuracy measures how many ΔΔGcalc have been predicted to be in the same direction of stability as the ΔΔGexp.
DSSP 75 was used to classify the secondary structure of the wild type residues of each protein. Surface areas were calculated using the Shrake-Rupley algorithm76. The fraction of the surface area exposed for each wild type residue (SAres) was calculated with this expression
| (12) |
where SAf is the surface area of the wild-type residue and the SAexp is the surface area of the completely solvent exposed residue. We approximated SAexp by calculating the surface area of the wild-type residue in a tetrapeptide (GXG where the X represents the wild-type residue) capped with an acetyl (ACE) and n-methyl amino group (NME). Buried residues were defined as having less than 10% of their side chain exposed and exposed residues were characterized as having more than 50% of their side chain exposed.
Results
Calculation of Stability Changes Using Model-1 and Model-2
In this work, we tested the LIE protein stability approach on 822 single point mutations and compared the calculated results with corresponding experimental results. This set contained large to small mutations from 10 different proteins which contained varying secondary structural content. The first LIE protein stability model (Model-1) was parameterized using the data from all of the mutations (Table 1). The correlation coefficient and the average absolute error between the calculated and experimental ΔΔG were 0.61 and 0.94 kcal/mol, respectively (Table 3 and Figure 2a). These results are comparable with the best performing programs available today as reported by Potopov et al. 29
Figure 2.

Calculated ΔΔG (ΔΔGcalc) versus experimental ΔΔG (ΔΔGexp) for Model-1 (A) and Model-2 (B). Model-1 was trained on all mutation types and Model 2 was trained on separate mutation types (Model-2) (B). The dotted black line corresponds to the x=y line and the solid black line corresponds to the least squared fit line between ΔΔGexp and ΔΔGcalc. The correlation coefficients were 0.61 and 0.72, for Model 1 and Model 2, respectively (See Table 1).
The second LIE protein stability model (Model-2) was parameterized to treat different types of mutations. We divided our mutations into three groups: neutral, charged and glycine/proline mutations. The neutral group training set contained mutations where the side chain of the wild-type and the mutant residue had a zero net charge. The charged group training set contained mutations involving ionizable residues (R,K,D,E) for the wild-type or mutant residue. The rationale behind separating the charged and neutral mutations is that the magnitude of the electrostatic and reaction field energies are much larger for charged groups and will dominate the fit of the electrostatic and polar solvation LIE coefficients. Secondly, for the Generalized Born type implicit solvent models the polarization of charged residues is underestimated within the protein compared to neutral residues resulting in the overstabilization of salt bridges. As a result, implicit solvent models have treated charged and neutral residues separately using different internal dielectric constants.43,65 The remaining residue group contains mutations where the wild type residue is being mutated to a glycine or where proline is the wild type residue being mutated to a smaller residue. These mutations typically cause the largest changes to the structure and conformational entropy of the protein backbone and therefore should be treated differently than other mutations.
The three residue-type models exhibited correlation coefficients ranging from 0.45 to 0.77 and absolute errors ranging from 0.80 to 0.94 kcal/mol (Table 3 and Figure 3). The lowest correlation coefficient was from the prediction of mutations of or to charged groups (r = 0.45) however the absolute error of these mutants was quite low (0.72 kcal/mol). The largest absolute error was from predicting mutations to glycine (0.92 kcal/mol); it may be harder to reproduce the destabilizing effects of a glycine mutation since our sampling protocol is limited to side chain prediction in this work and the remainder of the protein is not allowed to relax. Nevertheless, these residue sub-types demonstrated an improvement in the correlation coefficient and absolute error between 7 and 18% with respect to an overall fit of LIE coefficients without respect to residue type (Table 3). With respect to predictions for the whole set of 822 mutations, the correlation coefficient and absolute error between the ΔΔGcalc and the ΔΔGexp is 0.72 and 0.82 kcal/mol respectively, which is superior to Model-1 (0.61 and 0.94 kcal/mol) but the real advantage/improvement of Model-2 over Model-1 is the prediction of experimental outliers as described in the following section.
Figure 3.

Calculated ΔΔG (ΔΔGcalc) versus experimental ΔΔG (ΔΔGexp) for Model-2. Model-2 involved separate fitting on mutations involving neutral (A), charged (B) and glycine/proline residues (C). The dotted black line corresponds to the x=y line and the solid black line corresponds to the least squared fit line between ΔΔGexp and ΔΔGcalc. The correlation coefficients for the neutral, charged and glycine/proline residue models were 0.69, 0.45 and 0.77, respectively (See Table 1).
Why does Model-2 perform better than Model-1?
The main difference between the performance of Model-1 and Model-2 is the improvement in the predictions of hot spot mutations (|ΔΔGexp| > 2 kcal/mol). The sensitivity measure quantifies how many hot spot mutations were correctly identified (true positive) compared to the total number of hot spot mutations predicted (true positive + false negative) and improved by 12% using Model-2 over Model-1 (Table 4). Furthermore, 80% of the calculated outliers in Model-1 are hot-spot mutations (an outlier is defined as having an absolute error larger than two standard deviations above the average absolute error of Model-1). Figure 4 shows the performance of Model-2 with the outliers of Model-1. By fitting individual mutation types (neutral, charged and Gly/Pro), 90% of these mutations demonstrate improved absolute errors relative to the results for Model-1. A notable example is E75V from Staph nuclease which had a predicted ΔΔGcalc of 6.33 and 2.70 kcal/mol with Model-1 and Model-2 respectively, relative to the ΔΔGexp value of 2.30 kcal/mol. Model-2 is able to decrease the range of the ΔΔGcalc and the number of outliers which increases the correlation between ΔΔGcalc and ΔΔGexp. The rest of the results section will focus on the analysis of results using Model-2.
Figure 4.
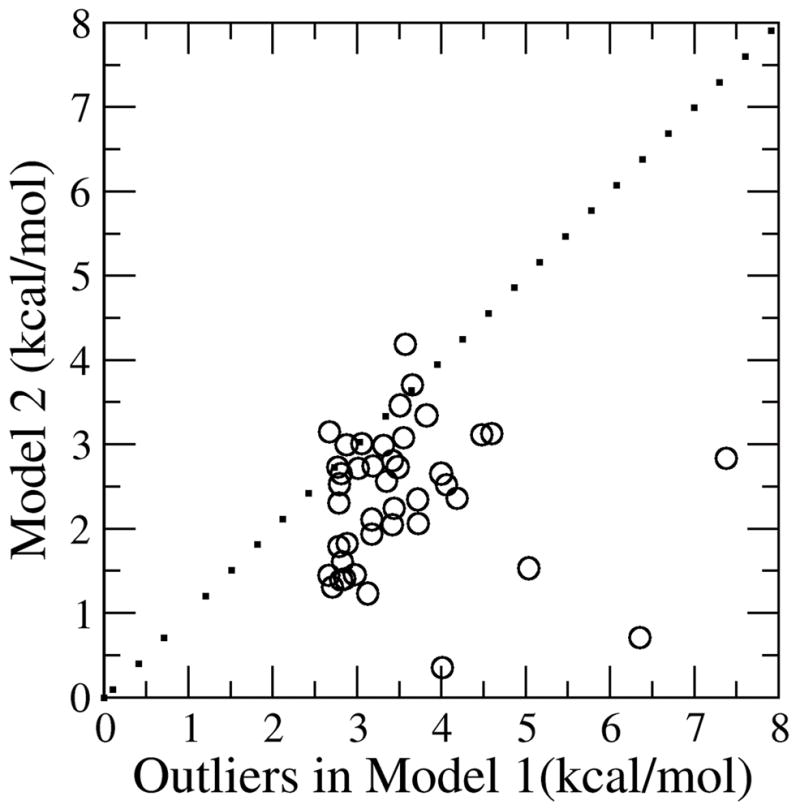
Absolute errors for Model 1 outliers using Model 2. All of the outliers had an absolute error higher than 2.5 kcal/mol using Model 1 (42 outliers). The dotted line is the x=y line. Any points below this line represent the outliers which had lower absolute errors for Model 2 compared to Model 1 (approximately 90 % in this case).
Performance of Model-2 with different types of mutations
Model-2 exhibits a very good correlation and a low average absolute error between experimental and calculated ΔΔG values (Table 3); values which are superior to those reported by Potapov et al..29 In order to further understand the predictive abilities of Model-2, we evaluated the performance of mutations categorized by the surface area exposure and secondary structure of the wild-type residue and mutation type (alanine or non-alanine mutagenesis) (Table 5). Alanine (mutagenesis) and non-alanine mutations had similar correlation coefficients but non-alanine mutations had a higher average absolute error; this group contained all of the mutations involving glycine which demonstrated the highest absolute error of all three residue types in Model-2 (Table 3). Mutations categorized by secondary structure had correlation coefficients which ranged from 0.54 to 0.75. The turn population was the smallest population of mutations which appeared to affect the value of the correlation coefficient. The greatest variation in the results was seen amongst the groups divided up based on surface area exposure. Mutations involving highly exposed residues had the lowest correlation coefficient of all the different groups (r = 0.35). The poor performance of surface exposed mutations may be due to the ΔΔG range for mutations of this type; the ΔΔG range is very small which reflects the high dielectric solvent screening of these mostly polar and charged residues. Previous studies have also reported higher correlation coefficients for mutations involving buried residues compared to exposed residues.22,25–27 In contrast, the mutations involving buried residues had the highest absolute error of all groups of mutations (average absolute error = 1.01 kcal/mol). Buried residues tend to be more sensitive to sequence changes. In this case, 57% of the buried mutations are destabilizing hot spot mutations (<|ΔΔGexp|> = 2.52 kcal/mol, Table 6) with an average absolute error of 1.12 kcal/mol.
Table 5.
The Average Unsigned Error and Correlation Coefficient between the Calculated and Experimental ΔΔG values for the Different Structural Features
| Type | NMUT | <|ΔΔGexp|> | <|ΔΔGcalc|> | < | error | > | r |
|---|---|---|---|---|---|
| Alanine mutagenesis | 385 | 1.53 | 1.46 | 0.78 | 0.72 |
| Non-Alanine | 437 | 1.70 | 1.68 | 0.85 | 0.73 |
| Beta | 310 | 2.07 | 2.05 | 0.87 | 0.73 |
| Helix | 217 | 1.60 | 1.60 | 0.79 | 0.76 |
| Turn | 90 | 1.10 | 1.03 | 0.84 | 0.54 |
| Coil | 202 | 1.20 | 1.21 | 0.76 | 0.67 |
| Exposed | 257 | 0.69 | 0.62 | 0.60 | 0.35 |
| Buried | 277 | 2.52 | 2.18 | 1.01 | 0.69 |
NMUT = number of residues;< |ΔΔGexp |>= average absolute experimental ΔΔG;< |ΔΔGcalc |>= average absolute calculated ΔΔG; <|error|>= average unsigned error (units of kcal/mol)
Table 6.
The Effect of Correct Structural Predictions on Energetic Predictions
| Structural Predictions | Energetic Predictions | Count | Conditional probability |
|---|---|---|---|
| Correct | Correct | 22 | 0.76 |
| Correct | Incorrect | 7 | 0.24 |
| Incorrect | Correct | 6 | 0.60 |
| Incorrect | Incorrect | 4 | 0.40 |
In the training set, there were 39 mutations which had corresponding structural information for the wild-type and mutant structures according to the Protherm database.
One of the apparent shortcomings of the LIE protein stability approach is distinguishing between stabilizing and destabilizing mutations. According to Table 4, Model-2 is quite successful at predicting whether a mutation is stabilizing or destabilizing (89% accuracy). Nevertheless, the sensitivity was quite different for the prediction of destabilizing and stabilizing mutations. Mutations with ΔΔGexp values greater than 0 kcal/mol were predicted with a sensitivity of 95%, in contrast, mutations with ΔΔGexp values less than 0 kcal/mol were predicted with a sensitivity of 23%. Hence, Model-2 performs poorly at correctly identifying stabilizing mutations. This is likely an effect of the distribution of ΔΔGexp values in our benchmark set used to parameterize the LIE model. It is composed of mainly destabilizing mutations (91 %); and the range of ΔΔG values for stabilizing mutations which is very small.
What effect does structure prediction have on energy prediction?
We turn next to examine how the accuracy of the structure predictions affect stability change predictions. We evaluated the accuracy of the structural predictions using the criterion for a correct structure prediction that all χ dihedrals deviated from the X-ray rotamer geometry by less than +/− 30° and an energy criterion that the absolute error between the ΔΔGcalc and ΔΔGexp was less than 1.5 kcal/mol (Table 6). Complete structural data, including both wild-type and mutant PDB structures, was available for 39 of the mutations. The distribution of mutation residue-types is different in this small set of mutations (67% neutral/18% charged/15% glycine/proline) with structural data compared to the larger benchmark set of 822 mutations (55% neutral/21% charged/24% glycine/proline). Correspondingly, the accuracy of the stability change calculations varies between these two sets; 72% of the stability changes were correctly predicted in the set where the structure of wild-type and mutant was known, in contrast, 86% of the stability changes were predicted correctly for the benchmark set of 822 mutations. Therefore, the small set includes a large percentage of mutations from the benchmark set that were challenging stability change predictions. For this set, the percentage of correct stability change predictions is reduced from 76% to 60% when the predicted side chain deviates by more than +/− 30° from the X-ray rotamer geometry (Table 6). In the cases of correct structure and incorrect stability change predictions (24%), all the mutations are from large residues (K,F, L, or Y) to alanine or glycine. Modeling additional structural reorganization and relaxation may be needed to capture the real effect of the mutation on the surrounding residues. Nevertheless, our results indicate that accurate structural models of wild-type and mutant enhance the ability to predict stability changes.
The accuracy of the stability change predictions is also affected by the range of possible energies that are sampled by a residue; this is highly dependent on the amino acid type, location of the residue and the energy function. For 60% of the incorrect structure predictions, ΔΔGcalc was predicted within +/− 1.5 kcal/mol. An explanation for this effect is that for these cases the X-ray and the incorrectly predicted rotamer state are approximately isoenergetic which results in very similar ΔΔGcalc values. For example, this effect is observed for the stability calculation of the S44A mutant in T4-Lysozyme (2LZM) where the wild-type rotamer was predicted incorrectly (Figure 5). In the X-ray structure, the hydroxyl group of the S44 side chain is solvent exposed and relatively close to a crystal water, in contrast, the S44 side chain is forming a hydrogen bond with backbone carbonyl group of N55 for the predicted rotamer (Figure 5). For the S44A mutant, the ΔΔGcalc values are 0.46 and −0.07 kcal/mol (relative to ΔΔGexp = −0.34 kcal/mol) using the minimized wild type and the predicted rotamer as the wild-type model. In both cases, this method is robust enough to predict the correct stability changes despite the different side chain geometries. Nevertheless, stability change predictions are more sensitive to the side chain conformations of charged residues which are susceptible to forming salt bridges. For the stability calculation of the K116A mutant in staphylococcal nuclease (1STN), the wild type side chain geometry was predicted incorrectly and the absolute error of the stability change was greater than 1.5 kcal/mol. In the X-ray structure, the K116 side chain is solvent exposed and distant from most residues of the protein except from a crystal water (Figure 6). In contrast, the predicted rotamer state of K116 side chain forms an ion pair with the D122 side chain. If the correct wild type rotamer state is used, the ΔΔGcalc changes from 1.02 kcal/mol to 0.24 kcal/mol which is in better agreement with experimental measurements (ΔΔGexp = −0.70 kcal/mol). The error in the ΔΔGcalc prediction of the K116G mutation appears to be a result of the erroneous salt bridge conformation formed by K116.
Figure 5.
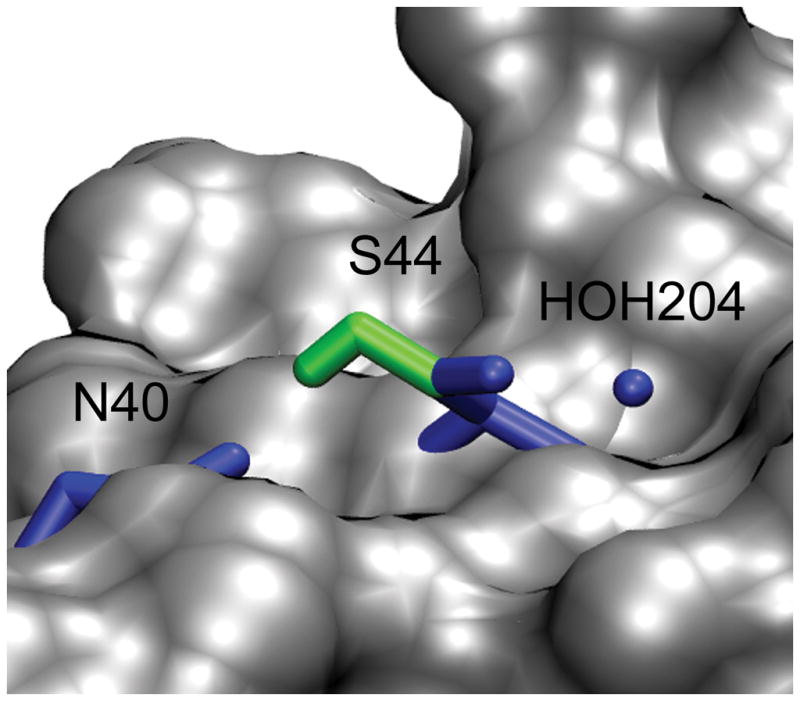
Incorrect structural predictions with accurate energy predictions. In each case, the wild-type rotamer geometry deviated from the crystal structure reference state by more than 30° for the ΔΔGcalc of the S44A mutant in T4-Lysozyme (2LZM). The X-ray model is shown in blue and the predicted models is shown in green. All of the atoms in licorice are heavy atoms except for the hydrogen of the hydroxyl group of S43.
Figure 6.
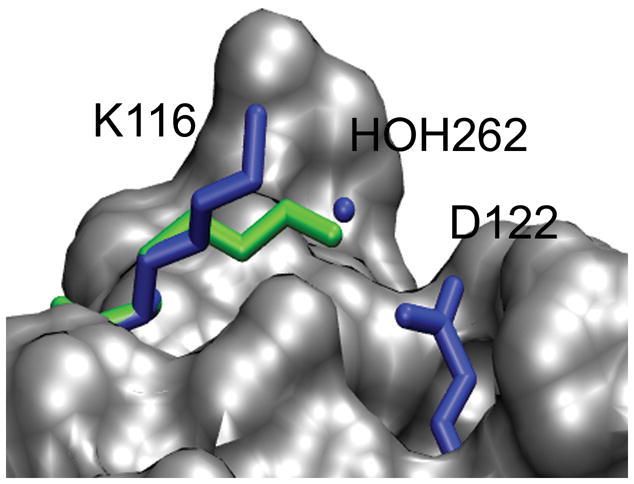
Incorrect structural predictions with incorrect energy predictions. In this case, the wild-type rotamer geometry deviated from the crystal structure reference state by more than 30° for the ΔΔGcalc of the K116A mutant (A) in staphylococcal nuclease (1STN). The X-ray model is shown in blue and the predicted model is shown in green.
Discussion
Physical interpretation of the LIE models
The LIE model approximates the free energy change with an empirical scoring function motivated by linear response theory which employs physically motivated energy estimators. In the protein stability LIE model, these estimators capture the differences between the energetic interactions of the mutant and the wild-type protein in the folded and unfolded state. For our calculations of the unfolded state, we use a local tetrapeptide model of the residue of interest with its two N- and C-termini neighboring residues. The role of this model is to capture possible residual structure in the unfolded state compared to a random coil model. By using the folded and unfolded state in our calculations, we see a significant increase in the accuracy of the stability calculations relative to only using the folded state (data not shown). Therefore, the unfolded state model does play a role in the stability calculation by screening the short range folded state interactions.
Based on Equation 6, the relative free energy change (ΔΔG) can be expressed as the difference between the residue-environment interaction energy in the folded (Δ <Vf>) and unfolded state (Δ <Vu>) in going from the wild-type to the mutant where i is one of the components of the energy.
| (13) |
The sign of ΔΔ<Vi> determines whether a mutation is stabilizing or destabilizing relative to the wild-type protein. If the ΔΔ< Vi > is positive, the wild-type is contributing more favorably to the folding free energy than the mutant (ΔΔGi > 0), in contrast, if the ΔΔ<Vi> is negative, the mutant is contributing more favorably to the folding free energy than the wild-type. The magnitude of ΔΔ<Vi> indicates how important a particular energy estimator is to determining the relative free energy change of the mutant compared to the wild-type. In order to maintain the physical interpretation of the sign in Equation 13, the LIE coefficients should be positive. This is the case for all of the models except the neutral residue model (Model-2) (Supplemental Figure 1A). Here, the ΔΔVLJ energies are shifted to more positive values since a large number of mutations in this group are buried hydrophobic residues that are mutated to smaller residues which results in an unfavorable loss of vdw contacts. ΔΔVel and ΔΔGel values are smaller but have more outliers because of the mutations involving surface exposed polar residues. All of the energies have positive coefficients except for the coefficients for the ΔΔGvdw and ΔΔGcav terms. The negative coefficient for the ΔΔGvdw term is a result of fitting coefficients for the ΔΔVLJ and ΔΔGvdw energy estimators separately; these estimators are typically fit with the same coefficient which is positive.41 Separate fitting of each energy estimator improves the results relative to a model where the ΔΔVLJ and ΔΔGvdw energy estimators are combined however the physical meaning behind each of the coefficients is lost since both estimators are highly correlated. We hypothesize that the negative value for the coefficient of the ΔΔGcav energy estimator reflects conformational reorganization effects which are not treated explicitly in our model. The corresponding coefficient absorbs the reorganization free energy difference between the wild-type and the single point mutant; this is always inversely correlated to the change in the side chain size during the mutation, as absorbed by its change in exposed surface area. In a previous protein-ligand binding study based on LIE models, negative coefficients were also observed for the surface area cavity free energy estimator41 and it was suggested that this energy term was statistically correlated with reorganization effects as well.
The model for mutations involving charged residues was originally fit using all five energetic descriptors but only required two coefficients after statistical refitting (Supplemental Figure 2). The driving force behind this model appears to be the electrostatic (ΔVele) and polar solvation (ΔGel ) energies that span the broadest ranges of all the five energies. As a result, coefficients for these energetic descriptors are smaller than the corresponding values for the neutral residue model. The resulting energy distribution range decreases dramatically when these energies are combined. This effect is well known; it results from the fact that ΔGel is a reaction field, which largely cancels the direct electrostatic energy term, ΔVele. The ΔΔGcav and ΔΔGvdw terms are less significant for fitting the charged residues since most of the charged residues are located on the surface (89%). These terms may become more significant as we add more mutation types to the model (small to large mutations).
The model for glycine/proline mutations was also originally fit with five coefficients but only required two coefficients during refitting (Supplemental Figure 3). ΔΔVLJ makes the largest contribution to this model since the mutation involves the removal of the whole side chain and the loss of the most contacts. The coefficients for the ΔΔVLJ and ΔΔGvdw energy estimators are similar for this model; this is consistent with the previous LIE derivation for protein-ligand interactions in implicit solvent41 as well as previous derivations in explicit solvent where the vdw estimator includes solute-solute and solute-solvent vdw forces scaled by one coefficient.30 Since the training set included charged residue to glycine mutations, the distributions and coefficients of the ΔΔVel and ΔΔGel energy estimators were similar to those observed for mutations involving charged residues.
Performance of Mutations involving Charged Residues
The mutations involving charged residues demonstrated the worst fit of the mutation residue-type models. Fitting was affected by the ΔΔGexp range. The ΔΔGexp range was the smallest for the mutations involving charged residues and largest for the mutations involving glycine/proline; this was similar to the trend in the corresponding correlation coefficients (Table 3 and Figure 3). The ΔΔG range (both experiment and calculation) is the largest for mutations involving glycine and proline since they typically involve large structural changes to the native state of the protein, in contrast, the smallest range is observed experimentally for mutations involving charged residues. Mutations involving charged residues are usually located in solvent exposed positions and because of solvent screening have a smaller effect on stability. Furthermore, the charged residue model training set constituted 40% of the stabilizing mutations in our data set which are the most difficult type of mutations to predict. ΔΔG predictions of stabilizing mutations were challenging because the LIE model was trained on a benchmark set containing mostly destabilizing mutations. Unfortunately, this bias is difficult to avoid since there are many more destabilizing than stabilizing mutations.77,78
Comparison with previous methods
We compared the results for single point mutations from the LIE protein stability model (Model-2) to previous results from Potapov et al.29 For the six different approaches, correlation coefficients ranged from 0.26 to 0.59 and the average absolute errors ranged from 1.00 to 1.69 kcal/mol. Based on these results, the LIE protein stability model is superior to these other approaches (r=0.72; <|error|> =0.82). Nevertheless, the original studies report significantly higher correlation coefficients for these methods, ranging from 0.62 to 0.75.22,24,25,27,79 The results from the protein survey were worse because the testing sets were larger and more diverse than in the original studies and unbiased through the removal of mutations used in the original training sets for each method. In addition, these programs may be optimized by the labs that developed them, and the comparison studies may not be using the programs in the optimum way.27
Effects of Sampling for the Mutant Structural Model
The LIE protein stability approach appears to be among the best programs available to calculate stability changes despite the lack of structural relaxation in the current version of the LIE models. In our calculations, the protein-protein interactions are on average underestimated for the folded mutant because surrounding side chains do not repack to optimize the interactions with the smaller mutant residue. Nevertheless, the reorganizational cost for repacking the side chains around the mutated residue is also not accounted for and these two effects tend to cancel. Secondly, structural relaxation may only be beneficial for certain types of mutations. Yin et al.26 and Kellogg et al27 noted that using a flexible backbone sampling improved the correlation between the calculation and experiment for mutations where the wild-type residue was smaller than the mutant residue. Lastly, structural relaxation may also cause errors in the stability change calculation if the sampling approach introduces erroneous structural changes into the mutant structures. Kellogg et al. noted that using backbone relaxation actually degraded the results for the entire set of mutations.27
Treatment of Conformational and Solvation Entropy
We note that the LIE protein stability equations are missing explicit terms for the treatment of entropy. Free energy changes produced by linear response models, such as the one employed here, implicitly include conformational entropy effects through the linear response expressions which relate potential energy differences to free energy differences. Moreover, the target of our calculations is relative stabilities resulting from the net contributions of the differences between the unfolded and folded states of the wild-type and mutant protein. The LIE models capture these relative entropic effects only in an average way. Solvent entropic effects are implicitly included in the solvation free energies modeled in this work by AGBNP effective potential, although important structural waters may not be correctly modeled under the continuum approximation on which the model is based.
Multiple mutations
Our broader interest is to apply this method to proteins with multiple mutations. We applied the LIE protein stability approach to a small group of double mutations from serine protease inhibitor using Model-2. This subset of 17 mutations is included in the Protherm database and has also been tested with the Eris approach.26 Figure 7 shows a scatter plot of calculated vs experimental ΔΔG values for this subset of 17 mutations. Surprisingly, the correlation coefficient and average absolute error (r = 0.80 and <|error|> = 0.69 kcal/mol) improve compared to the single point mutation set. Using a fixed backbone approach, a correlation coefficient between experimental and calculated ΔΔG values of 0.69 (compared to 0.64 with single point mutations on a larger set) was reported for the Eris method, which is similar to the trend in our results. The improvement of double mutations over single point mutations is probably due to a cancellation of errors. In this test set, the positive cooperativity of the ΔΔGexp for the double mutations compensates for the errors from the original model of single point mutations which underestimate ΔΔGcalc relative to ΔΔGexp. Based on these results, it appears that the LIE protein stability approach is a potentially useful tool for calculating protein stability changes caused by multiple mutations that will be improved by incorporating structural relaxation in order to accurately capture the effects of cooperativity between mutated residues.
Figure 7.
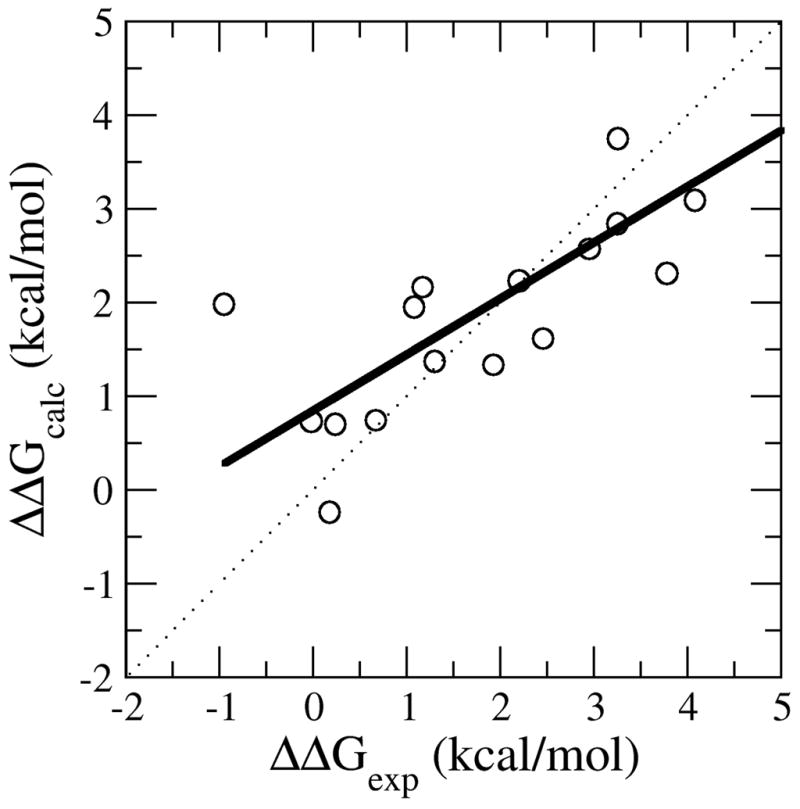
Calculated ΔΔG (ΔΔGcalc) versus experimental ΔΔG (ΔΔGexp) for a set of serine protease inhibitor (2CI2) double mutants. The dotted black line corresponds to the x=y line and the solid black line corresponds to the least squared fit line between ΔΔGexp and ΔΔGcalc. The correlation coefficient is 0.80 using Model 2.
Conclusions
In this work, we have presented an approach to calculate relative protein stabilities based on the Linear Interaction Energy model to estimate free energies and the protein local optimization program (PLOP) to sample side chain rotamer states. On a large set of single point mutations, this method leads to results that are comparable to or better than results reported for existing methods even without including structural relaxation in the calculations. Future work will focus on using a better sampling approach to allow for more extensive relaxation of the protein structure following mutations and a revised version of the AGBNP implicit solvent model80 which includes a first shell solute-solvent term and an improved model for protein salt bridge formation. With further development of this approach to model multiple mutations, we can begin to integrate sequence data and energetic information to examine the relationship between protein stability, fitness and drug resistance.
Supplementary Material
Acknowledgments
This work has been supported by a NIH postdoctoral fellowship to LW (Grant NIH 5 T90 DK070135) and by NIH GM30580. LW gratefully acknowledges Anthony Felts, Mauro Lapelosa and Omar Haq for providing helpful discussions and scripts for the analysis of the protein stability results.
Footnotes
The validity of this study was recently questioned by Kellogg et al,27 who noted that an improper sampling technique lead to the poor results for the Rosetta program (r = 0.26 for the data set). Using an optimized protocol, the results were comparable with the best programs (r = 0.62) in the original survey of protein stability methods.
References
- 1.Kortemme T, Baker D. Computational design of protein-protein interactions. Curr Opin Chem Biol. 2004;8(1):91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Lippow SM, Tidor B. Progress in computational protein design. Curr Opin Biotechnol. 2007;18(4):305–311. doi: 10.1016/j.copbio.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butterfoss GL, Kuhlman B. Computer-based design of novel protein structures. Annu Rev Biophys Biomol Struct. 2006;35:49–65. doi: 10.1146/annurev.biophys.35.040405.102046. [DOI] [PubMed] [Google Scholar]
- 4.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302(5649):1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 5.Frushicheva MP, Cao J, Chu ZT, Warshel A. Exploring challenges in rational enzyme design by simulating the catalysis in artificial kemp eliminase. Proc Natl Acad Sci U S A. 2010;107(39):16869–16874. doi: 10.1073/pnas.1010381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat Rev Genet. 2005;6(9):678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 7.Warshel A. Energetics of enzyme catalysis. Proc Natl Acad Sci U S A. 1978;75(11):5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106(8):3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 9.Nagatani RA, Gonzalez A, Shoichet BK, Brinen LS, Babbitt PC. Stability for function tradeoffs in the enolase superfamily “catalytic module”. Biochemistry. 2007;46(23):6688–6695. doi: 10.1021/bi700507d. [DOI] [PubMed] [Google Scholar]
- 10.Bloom JD, Arnold FH. In the light of directed evolution: pathways of adaptive protein evolution. Proc Natl Acad Sci U S A. 2009;106 (Suppl 1):9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fasan R, Meharenna YT, Snow CD, Poulos TL, Arnold FH. Evolutionary history of a specialized p450 propane monooxygenase. J Mol Biol. 2008;383(5):1069–1080. doi: 10.1016/j.jmb.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320(1):85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 13.Thomas VL, McReynolds AC, Shoichet BK. Structural bases for stability-function tradeoffs in antibiotic resistance. J Mol Biol. 2010;396(1):47–59. doi: 10.1016/j.jmb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haq O, Levy RM, Morozov AV, Andrec M. Pairwise and higher-order correlations among drug-resistance mutations in HIV-1 subtype B protease. BMC Bioinformatics. 2009;10 (Suppl 8):S10. doi: 10.1186/1471-2105-10-S8-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikita H, Warshel A. Predicting drug-resistant mutations of HIV protease. Angew Chem Int Ed Engl. 2008;47(4):697–700. doi: 10.1002/anie.200704178. [DOI] [PubMed] [Google Scholar]
- 16.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 17.Steward RE, MacArthur MW, Laskowski RA, Thornton JM. Molecular basis of inherited diseases: a structural perspective. Trends Genet. 2003;19(9):505–513. doi: 10.1016/S0168-9525(03)00195-1. [DOI] [PubMed] [Google Scholar]
- 18.Tidor B, Karplus M. Simulation analysis of the stability mutant R96H of T4 lysozyme. Biochemistry. 1991;30(13):3217–3228. doi: 10.1021/bi00227a009. [DOI] [PubMed] [Google Scholar]
- 19.Sun YC, Veenstra DL, Kollman PA. Free energy calculations of the mutation of Ile96-->Ala in barnase: contributions to the difference in stability. Protein Eng. 1996;9(3):273–281. doi: 10.1093/protein/9.3.273. [DOI] [PubMed] [Google Scholar]
- 20.Seeliger D, de Groot BL. Protein thermostability calculations using alchemical free energy simulations. Biophys J. 2010;98(10):2309–2316. doi: 10.1016/j.bpj.2010.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen M, Potapov V, Schreiber G. Four distances between pairs of amino acids provide a precise description of their interaction. PLoS Comput Biol. 2009;5(8):e1000470. doi: 10.1371/journal.pcbi.1000470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320(2):369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 23.Bordner AJ, Abagyan RA. Large-scale prediction of protein geometry and stability changes for arbitrary single point mutations. Proteins. 2004;57(2):400–413. doi: 10.1002/prot.20185. [DOI] [PubMed] [Google Scholar]
- 24.Pokala N, Handel TM. Energy functions for protein design: adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J Mol Biol. 2005;347(1):203–227. doi: 10.1016/j.jmb.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 25.Benedix A, Becker CM, de Groot BL, Caflisch A, Bockmann RA. Predicting free energy changes using structural ensembles. Nat Methods. 2009;6(1):3–4. doi: 10.1038/nmeth0109-3. [DOI] [PubMed] [Google Scholar]
- 26.Yin S, Ding F, Dokholyan NV. Modeling backbone flexibility improves protein stability estimation. Structure. 2007;15(12):1567–1576. doi: 10.1016/j.str.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 27.Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2010;79(3):830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97(19):10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel. 2009;22(9):553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 30.Aqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7(3):385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 31.Carlson HA, Jorgensen WL. An extended linear-response method for determining free-energies of hydration. J Phys Chem. 1995;99(26):10667–10673. [Google Scholar]
- 32.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811(3):265–322. [Google Scholar]
- 33.Levy RM, Belhadj M, Kitchen DB. Gaussian fluctuation formula for electrostatic free-energy changes in solution. J Chem Phys. 1991;95(5):3627–3633. [Google Scholar]
- 34.Figueirido F, Delbuono GS, Levy RM. Molecular mechanics and electrostatic effects. Biophys Chem. 1994;51(2–3):235–241. doi: 10.1016/0301-4622(94)00044-1. [DOI] [PubMed] [Google Scholar]
- 35.Lee FS, Chu ZT, Bolger MB, Warshel A. Calculations of antibody antigen Interactions -Microscopic and semimicroscopic evaluation of the free-energies of binding of phosphorylcholine analogs to Mcpc603. Protein Eng. 1992;5(3):215–228. doi: 10.1093/protein/5.3.215. [DOI] [PubMed] [Google Scholar]
- 36.Hansson T, Aqvist J. Estimation of binding free energies for HIV proteinase inhibitors by molecular dynamics simulations. Protein Eng. 1995;8(11):1137–1144. doi: 10.1093/protein/8.11.1137. [DOI] [PubMed] [Google Scholar]
- 37.Sham YY, Chu ZT, Tao H, Warshel A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins. 2000;39(4):393–407. [PubMed] [Google Scholar]
- 38.Chen X, Tropsha A. Calculation of the relative binding affinity of enzyme inhibitors using the generalized linear response method. J Chem Theory Comput. 2006;2(5):1435–1443. doi: 10.1021/ct600071z. [DOI] [PubMed] [Google Scholar]
- 39.Zhou RH, Friesner RA, Ghosh A, Rizzo RC, Jorgensen WL, Levy RM. New linear interaction method for binding affinity calculations using a continuum solvent model. J Phys Chem B. 2001;105(42):10388–10397. [Google Scholar]
- 40.Carlsson J, Ander M, Nervall M, Aqvist J. Continuum solvation models in the linear interaction energy method. J Phys Chem B. 2006;110(24):12034–12041. doi: 10.1021/jp056929t. [DOI] [PubMed] [Google Scholar]
- 41.Su Y, Gallicchio E, Das K, Arnold E, Levy RM. Linear interaction energy (LIE) models for ligand binding in implicit solvent: Theory and application to the binding of NNRTIs to HIV-1 reverse transcriptase. J Chem Theory Comput. 2007;3(1):256–277. doi: 10.1021/ct600258e. [DOI] [PubMed] [Google Scholar]
- 42.Roca M, Messer B, Warshel A. Electrostatic contributions to protein stability and folding energy. FEBS letters. 2007;581(10):2065–2071. doi: 10.1016/j.febslet.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 43.Vicatos S, Roca M, Warshel A. Effective approach for calculations of absolute stability of proteins using focused dielectric constants. Proteins. 2009;77(3):670–684. doi: 10.1002/prot.22481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brandsdal BO, Aqvist J, Smalas AO. Computational analysis of binding of P1 variants to trypsin. Protein Sci. 2001;10(8):1584–1595. doi: 10.1110/ps.940101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almlof M, Aqvist J, Smalas AO, Brandsdal BO. Probing the effect of point mutations at protein-protein interfaces with free energy calculations. Biophys J. 2006;90(2):433–442. doi: 10.1529/biophysj.105.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55(2):351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 47.Gallicchio E, Levy RM. AGBNP: an analytic implicit solvent model suitable for molecular dynamics simulations and high-resolution modeling. J Comput Chem. 2004;25(4):479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- 48.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. [Google Scholar]
- 49.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105(28):6474–6487. [Google Scholar]
- 50.Kumar MD, Bava KA, Gromiha MM, Prabakaran P, Kitajima K, Uedaira H, Sarai A. ProTherm and ProNIT: thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Res. 2006;34(Database issue):D204–206. doi: 10.1093/nar/gkj103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hynes TR, Fox RO. The crystal structure of staphylococcal nuclease refined at 1.7 A resolution. Proteins. 1991;10(2):92–105. doi: 10.1002/prot.340100203. [DOI] [PubMed] [Google Scholar]
- 52.Buckle AM, Henrick K, Fersht AR. Crystal structural analysis of mutations in the hydrophobic cores of barnase. J Mol Biol. 1993;234(3):847–860. doi: 10.1006/jmbi.1993.1630. [DOI] [PubMed] [Google Scholar]
- 53.Wilson KP, Yamashita MM, Sintchak MD, Rotstein SH, Murcko MA, Boger J, Thomson JA, Fitzgibbon MJ, Black JR, Navia MA. Comparative X-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Crystallogr D Biol Crystallogr. 1995;51(Pt 4):511–521. doi: 10.1107/S0907444994014514. [DOI] [PubMed] [Google Scholar]
- 54.McPhalen CA, James MN. Crystal and molecular structure of the serine proteinase inhibitor CI-2 from barley seeds. Biochemistry. 1987;26(1):261–269. doi: 10.1021/bi00375a036. [DOI] [PubMed] [Google Scholar]
- 55.O’Neill JW, Kim DE, Baker D, Zhang KY. Structures of the B1 domain of protein L from Peptostreptococcus magnus with a tyrosine to tryptophan substitution. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 4):480–487. doi: 10.1107/s0907444901000373. [DOI] [PubMed] [Google Scholar]
- 56.Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385(6617):595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 57.Kohno T, Kobayashi K, Maeda T, Sato K, Takashima A. Three-dimensional structures of the amyloid beta peptide (25–35) in membrane-mimicking environment. Biochemistry. 1996;35(50):16094–16104. doi: 10.1021/bi961598j. [DOI] [PubMed] [Google Scholar]
- 58.Parkin S, Rupp B, Hope H. Structure of bovine pancreatic trypsin inhibitor at 125 K definition of carboxyl-terminal residues Gly57 and Ala58. Acta Crystallogr D Biol Crystallogr. 1996;52(Pt 1):18–29. doi: 10.1107/S0907444995008675. [DOI] [PubMed] [Google Scholar]
- 59.Weis WI, Kahn R, Fourme R, Drickamer K, Hendrickson WA. Structure of the calcium-dependent lectin domain from a rat mannose-binding protein determined by MAD phasing. Science. 1991;254(5038):1608–1615. doi: 10.1126/science.1721241. [DOI] [PubMed] [Google Scholar]
- 60.Weaver LH, Matthews BW. Structure of bacteriophage T4 lysozyme refined at 1.7 A resolution. J Mol Biol. 1987;193(1):189–199. doi: 10.1016/0022-2836(87)90636-x. [DOI] [PubMed] [Google Scholar]
- 61.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320(3):597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 62.Sellers BD, Zhu K, Zhao S, Friesner RA, Jacobson MP. Toward better refinement of comparative models: predicting loops in inexact environments. Proteins. 2008;72(3):959–971. doi: 10.1002/prot.21990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jacobson MP, Kaminski GA, Friesner RA, Rapp CS. Force field validation using protein side chain prediction. J Phys Chem B. 2002;106(44):11673–11680. [Google Scholar]
- 64.Zhu K, Pincus DL, Zhao S, Friesner RA. Long loop prediction using the protein local optimization program. Proteins. 2006;65(2):438–452. doi: 10.1002/prot.21040. [DOI] [PubMed] [Google Scholar]
- 65.Zhu K, Shirts MR, Friesner RA. Improved methods for side chain and loop predictions via the protein local optimization program:Variable dielectric model for implicitly improving the treatment of polarization effects. J Chem Theory Comput. 2007;3(6):2108–2119. doi: 10.1021/ct700166f. [DOI] [PubMed] [Google Scholar]
- 66.Felts AK, Gallicchio E, Chekmarev D, Paris KA, Friesner RA, Levy RM. Prediction of protein loop conformations using the AGBNP implicit solvent model and torsion angle sampling. J Chem Theory Comput. 2008;4(5):855–868. doi: 10.1021/ct800051k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49(2):534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 68.Zhu K, Shirts MR, Friesner RA, Jacobson MP. Multiscale optimization of a truncated Newton minimization algorithm and application to proteins and protein-ligand complexes. J Chem Theory Comput. 2007;3(2):640–648. doi: 10.1021/ct600129f. [DOI] [PubMed] [Google Scholar]
- 69.Xiang Z, Honig B. Extending the accuracy limits of prediction for side-chain conformations. J Mol Biol. 2001;311(2):421–430. doi: 10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- 70.Hartigan JA, Wong MA. Algorithm AS 136: A k-means clustering algorithm. J Roy Statist Soc Ser C. 1979;28(1):100–108. [Google Scholar]
- 71.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112(16):6127–6129. [Google Scholar]
- 72.Levy RM, Zhang LY, Gallicchio E, Felts AK. On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. J Am Chem Soc. 2003;125(31):9523–9530. doi: 10.1021/ja029833a. [DOI] [PubMed] [Google Scholar]
- 73.Su Y, Gallicchio E. The non-polar solvent potential of mean force for the dimerization of alanine dipeptide: the role of solute-solvent van der Waals interactions. Biophys Chem. 2004;109(2):251–260. doi: 10.1016/j.bpc.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 74.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 75.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 76.Shrake A, Rupley JA. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J Mol Biol. 1973;79(2):351–371. doi: 10.1016/0022-2836(73)90011-9. [DOI] [PubMed] [Google Scholar]
- 77.Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr Opin Struct Biol. 2009;19(5):596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 78.Zeldovich KB, Chen P, Shakhnovich EI. Protein stability imposes limits on organism complexity and speed of molecular evolution. Proc Natl Acad Sci U S A. 2007;104(41):16152–16157. doi: 10.1073/pnas.0705366104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33(Web Server issue):W306–310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gallicchio E, Paris K, Levy RM. The AGBNP2 implicit solvation model. J Chem Theory Comput. 2009;5(9):2544–2564. doi: 10.1021/ct900234u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.