

Abstract
In mammals, the Sirtuins are composed of seven Sir2 orthologues (Sirt1-7) with a conserved deacetylase core that utilizes NAD+ as a cofactor. Interestingly, the deacetylase core of Sirt1 by itself has no catalytic activity. We found within the C-terminal domain a 25 a.a. sequence that is essential for Sirt1 activity (ESA). Our results indicate that the ESA region interacts with and functions as an “on switch” for the deacetylase core. The endogenous Sirt1 inhibitor DBC1, which also binds to the deacetylase core, competes with and inhibits the ESA region from interacting with the deacetylase core. We discovered an ESA mutant peptide that can bind to the deacetylase core and inhibit Sirt1 in trans. By using this mutant peptide, we were able to inhibit Sirt1 activity and to increase the chemosensitivity of androgen-refractory prostate cancer cells. Therefore, the ESA region is a potential target for development of therapies to regulate Sirt1.
Introduction
Sirt1 is a member of the Sirtuin family, a group of protein deacetylases that utilizes NAD+ as a cofactor (Imai et al., 2000; Smith et al., 2000). It is the closest mammalian orthologue of yeast Sir2 (Guarente, 2000), the founding member of this family of protein deacetylases. Sirt1 plays important regulatory roles in diverse cellular processes, including stress resistance, mitochondrial function, suppression of inflammation and DNA repair (Bordone and Guarente, 2005). It has been reported that increased Sir2 activity increases lifespan in yeast (Lin et al., 2000), drosophila (Rogina and Helfand, 2004) and worms (Tissenbaum and Guarente, 2001) and that Sir2 is required for calorie restriction-mediated extension of lifespan in these organisms (Lin et al., 2000; Rogina and Helfand, 2004; Wang and Tissenbaum, 2006).
Sirt1 activity can be regulated by a number of different endogenous factors. Since the NAD+/NADH ratio is regulated by the metabolic status of the cell, it has been proposed that Sirt1 could couple the metabolic status of the cell with protein deacetylation as an NAD+ sensor (Imai et al., 2000; Landry et al., 2000; Smith et al., 2000). Sirt1 activity can also be regulated by direct interactions with other proteins. AROS (Active Regulator of Sirt1) (Kim et al., 2007) binds to the N-terminal domain of Sirt1 and activates its activity, whereas DBC1 (Deleted in Breast Cancer 1) (Kim et al., 2008; Zhao et al., 2008) binds to the deacetylase core of Sirt1 and inhibits its activity.
Regulation of Sirt1 activity, both positively or negatively, has therapeutic implications, particularly against diseases associated with aging. Increasing Sirt1 expression increases insulin sensitivity in transgenic mice (Banks et al., 2008; Bordone et al., 2007; Herranz et al., 2010; Pfluger et al., 2008) and also protects against the development of Alzheimer’s disease (Donmez et al., 2010). However, inhibiting Sirt1 function selectively in the liver decreases glucose production (Chen et al., 2008; Erion et al., 2009; Nie et al., 2009) and may be beneficial for treating type 2 diabetes. Available genetic evidence suggests that Sirt1 may have a tumor suppressor role (Deng, 2009). Increasing Sirt1 expression in transgenic mice decreases tumor development (Herranz et al., 2010) and decreasing Sirt1 expression increases tumor development (Firestein et al., 2008; Wang et al., 2008). However, Sirt1 also deacetylates and inactivates stress-response proteins such as p53 (Cheng et al., 2003; Langley et al., 2002; Vaziri et al., 2001), p73 (Dai et al., 2007; Yang et al., 2007) and the Foxo family of transcription factors (Brunet et al., 2004; Jung-Hynes et al., 2009; Motta et al., 2004; van der Horst et al., 2004; Yang et al., 2005) and can increase cell survival. Sirt1 is over-expressed in some cancers such as prostate cancer (PCa) (Huffman et al., 2007; Jung-Hynes et al., 2009; Kojima et al., 2008) and may play a role in the survival of tumor cells and in their resistance to chemotherapeutic agents (Jung-Hynes et al., 2009; Kojima et al., 2008). Indeed, inhibition of Sirt1 in PCa restores sensitivity to chemotherapeutic agents (Jung-Hynes et al., 2009; Kojima et al., 2008).
Despite the great interest and therapeutic need for the ability to regulate Sirt1 activity, the Sirt1 regulators that are currently available have severe limitations. Compounds such as resveratrol and SRT1720 have been reported to activate Sirt1 (Howitz et al., 2003; Milne et al., 2007). However, recent reports suggest that these compounds may not directly activate Sirt1 (Beher et al., 2009; Borra et al., 2005; Kaeberlein et al., 2005; Pacholec et al.). Small molecule inhibitors of Sirt1 such as Sirtinol, which targets the deacetylase core, have also been reported (Bedalov et al., 2001; Grozinger et al., 2001; Mai et al., 2005; Peck et al., 2010). However, the deacetylase core of the Sirtuins are highly similar, and as a result, these small molecules may inhibit other Sirtuins to varying degrees (Mai et al., 2005; Peck et al., 2010). Knock-down of Sirt1 using siRNA has also been used to reduce the level of Sirt1. However, Sirt1 has deacetylase-independent functions (Pfister et al., 2008), and therefore, knock-down approaches are not desirable for specifically inhibiting Sirt1 deacetylase activity.
Currently, the X-ray crystal structure of Sirt1 is not known. However, in order to design more effective Sirt1 regulators, we need to better understand the structural requirements for Sirt1 activity. In addition to the deacetylase core, Sirt1 has a long C-terminal domain that is required for deacetylase activity (Dvir-Ginzberg et al., 2011; Zhang et al., 2009). The C-terminal domain also appears to have a regulatory role since sumoylation (Yang et al., 2007) and phosphorylation (Guo et al., 2010; Kang et al., 2009; Nasrin et al., 2009; Sasaki et al., 2008) of amino acid residues in the C-terminal domain of Sirt1 have been shown to increase Sirt1 activity. Here, we investigate how the sequences in the C-terminal domain of Sirt1 affect its activity and use this information to develop a strategy to regulate Sirt1 activity.
Results
A small region in the C-terminal domain is essential for Sirt1 activity (ESA)
Sirt1, as well as Sir2 orthologues in fruit flies and worms, has a long C-terminal domain (Figure S1A). We made a series of truncation mutations in mouse Sirt1 starting from the C-terminal end and tested their deacetylase activities against Lys 382 acetylated-p53 (Ac-p53) (Figure 1A and Figure S1B). Henceforth, the a.a. residue number corresponds to that of mouse Sirt1. C-terminal deletions from a.a. 737 to a.a. 655 did not affect the deacetylase activity but a deletion to a.a. 640 completely abolished the deacetylase activity, indicating that the C-terminal boundary of a region that is essential for deacetylase activity is a.a. 655. We also tested Sirt1 containing internal deletions in the a.a. 611-680 region (Figures 1B and S1B). A deletion from a.a. 611 to a.a. 630 did not affect the deacetylase activity but a deletion to a.a. 640 completely abolished the deacetylase activity, indicating that the N-terminal boundary of an essential region is a.a. 631. To investigate whether a.a. 631-655 is essential for Sirt1 deacetylase activity (ESA), we deleted the 25 a.a. region from Sirt1 (ΔESA). As shown in Figures 1B and S1B, ΔESA Sirt1 did not have any deacetylase activity against either Ac-p53 or native Ac-histone H3. The complete absence of any deacetylase activity in ΔESA Sirt1 indicates that the ESA region (a.a.631-655) in the C-terminal domain, despite being far removed from the conserved Sirtuin domain (a.a. 236-490), is an essential component of the deacetylase and is not simply a modulator.
Figure 1. Identification of a region essential for Sirt1 deacetylase activity (ESA) in the C-terminal domain.
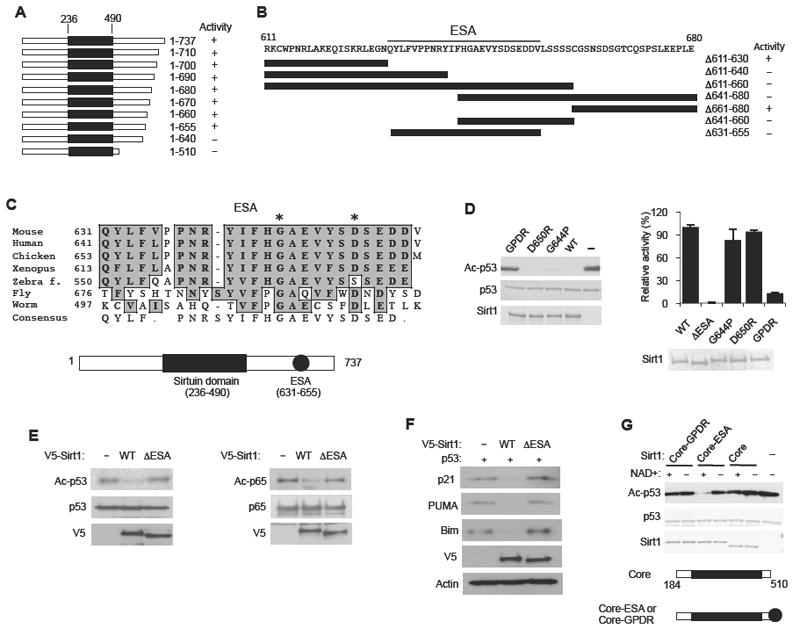
(A) A summary of the deacetylase activity of recombinant Sirt1 containing the indicated C-terminal truncations, measured by using Ac-p53 as substrate. The conserved Sirtuin domain is shown as a black rectangle. See also Figure S1A and B.
(B) A summary of the deacetylase activity of recombinant Sirt1 containing internal deletions (indicated by black bars) in the C-terminal domain. Original data for Sirt1 activity are shown in Figure S1B. The ESA region is underlined.
(C) Evolutionary conservation of the ESA region. Gly 644 and Asp 650 are indicated with asterisks (*). The locations of the Sirtuin domain and the ESA are shown below. See Figure S1C for a comparison of the C-terminal sequences of vertebrate Sirt1.
(D) Mutations of the ESA region abolish Sirt1 activity. (Left) Deacetylation reactions were performed with recombinant Sirt1 containing either a single mutation of Gly 644→Pro (G644P) or Asp 650→Arg (D650R) or a double mutation (GPDR) of these two amino acid residues. Ac-p53 is visualized by immunoblotting with an antibody specific for Ac-K382 of p53. (Right) Deacetylation reactions were performed with WT and mutant Sirt1 (including ΔESA) using 3H-Ac-H4 as the substrate. Sirt1 activity was quantified by measuring the levels of O-[3H]acetyl-ADP-ribose that was liberated from deacetylase reaction by using scintillation counting (N=4). Results are expressed as the mean ± s.e.m
(E) The ESA region is essential for Sirt1 activity in vivo. (Left) H1299 cells were transiently co-transfected with an expression vector for p53 and an expression vector for either WT Sirt1 or ΔESA Sirt1. Ac-p53 was visualized by immunoblotting with Ac-K328 (p53) antibody. (Right) Hela cells were transiently co-transfected with an expression vector for Flag-tagged NF-κB p65 (p65) and an expression vector for either V5-tagged WT Sirt1 or ΔESA Sirt1. Ac-p65 was visualized by immunoblotting with anti-Ac-Lys antibody after immunoprecipitating with Flag antibody. Transfection with an empty vector (-) was used as a negative control.
(F) Sirt1-mediated suppression of p53 activity requires the ESA region. The expression levels of p53 activated genes p21, PUMA and Bim were visualized in H1299 cells co-transfected with an expression vector for p53 and an expression vector for either WT Sirt1 or ΔESA Sirt1. Transfection with an empty vector (-) was used as a control.
(G) The ESA region is sufficient to confer activity to the decetylase core. Deacetylation reactions were performed with recombinant deacetylase core (Core, a.a. 184-510), which includes additional sequences flanking the conserved Sirtuin domain (a.a. 236-490), as well as recombinant deacetylase core fused either to the ESA region (Core-ESA) or to the ESA region containing the GPDR double mutation (Core-GPDR).
We analyzed the amino acid sequence of the Sirtuins to see if the ESA region is present in other Sirtuins. It is not present in Sirt2-7 or in yeast Sir2, which have a very short C-terminal domain. However, Sirt1 orthologues in multi-cellular organisms have conserved ESA region (Figure 1C). Although sequences that directly flank the ESA region are also evolutionarily conserved, the overall sequences of the C-terminal domain are poorly conserved (Figure S1C).
Within the ESA region, there are several amino acid residues that are more conserved than others (Figure 1C). To maximally change the properties of the ESA region with the fewest substitutions, we mutated two of the most conserved amino acid residues (*), Gly 644→Pro (G644P) and Asp 650→Arg (D650R), either individually or together (GPDR) (Figure 1D, Left). We chose G644 because Gly, being the smallest amino acid, allows flexibility to the peptide and substituting it with proline introduces a kink in the peptide structure. By changing Asp 650 to Arg, we changed a negatively charged residue to a positively charged residue. Sirt1 with a single mutation of either residue had deacetylase activity but Sirt1 with a double mutation (GPDR) had very little deacetylase activity. To more accurately quantify the deacetylase activity of these Sirt1 mutants (including ΔESA Sirt1), we used [3H]Ac-histone H4 as the substrate and measured by scintillation counting O-[3H]acetyl-ADP-ribose that was liberated from deacetylase reaction (Figure 1D, Right). We found that ΔESA Sirt1 had no detectable activity, but Sirt1 with either G633P or D650R mutation had slightly reduced, but near normal, activity. The GPDR mutation reduced Sirt1 activity by almost 10-fold. To demonstrate that the ESA region is essential for Sirt1 activity in vivo, we transiently expressed p53 with either WT or ΔESA Sirt1 in H1299 cells (Figure 1E, Left). We found that WT Sirt1, but not ΔESA Sirt1, was able to deacetylate p53. Similar results were seen with p65 (NF-κB), another Sirt1 substrate (Yeung et al., 2004) (Figure 1E, Right). In order to confirm that the ESA region is essential for Sirt1’s ability to suppress p53 activity, we visualized the expression levels of p53 target genes p21, PUMA and Bim in cells expressing p53 and either WT or ΔESA Sirt1. As shown in Figure 1F, ΔESA Sirt1 failed to suppress the expression of p53 target genes.
To determine whether the ESA region is sufficient to restore deacetylase activity, we utilized a.a. 184-510, which will henceforth be labeled as the deacetylase core to distinguish it from the Sirtuin domain (a.a. 236-490). We chose a.a. 184 as the N-terminal end of the deacetylase core because a previous report has shown that deletion of a.a. 184-236 abolished Sirt1 deacetylase activity (Malik et al., 2010). As shown in Figure 1G, attaching the ESA region to the deacetylase core (Core-ESA) restored deacetylase activity, but attaching the GPDR-mutated ESA region to the Core (Core-GPDR), did not. Therefore, the deacetylase core and the ESA region are essential and sufficient for Sirt1 deacetylase activity.
The C-terminal domain of Sirt1 is disordered in structure
For the ESA region, which is distant from the deacetylase core, to function as part of the deacetylase, one would expect the C-terminal domain to be flexible and disordered. To confirm this, we analyzed the structure of the C-terminal fragment using NMR. The 2D 1H-15N HSQC spectra of 15N-labeled a.a. 610-737 and 15N-labeled a.a. 491-737 fragments exhibited typical random coil signatures characterized by narrow dispersion of amide proton chemical shifts (Figure S2A, B). The latter exhibited a larger number of cross peaks than a.a. 610-737 fragment in the spectra due to its larger size, but the dispersion of the 1HN chemical shifts remained the same. In addition, the cross peaks arising from a.a. 610-737 did not change their chemical shifts in the spectra of a.a. 491-737, implying the absence of a long-range interaction between a.a. 610-737 region and the extended region in a.a. 491-737. When the NMR sample was prepared at pH 7.4, many cross peaks disappeared due to the exchange with water, which is commonly observed in unstructured proteins and peptides. Consistent with this, circular dichroism spectra obtained for a.a. 491-737 (both wild-type and GPDR mutant) indicated that it was mostly (> 95 %) disordered in solution (Figure S2C, D).
The ESA region interacts with the deacetylase core of Sirt1
Since the region between the Sirtuin domain and the ESA region is not essential for Sirt1 activity, and the C-terminal domain is almost entirely disordered and presumably flexible, we hypothesized that the position of the C-terminal domain relative to the deacetylase core may not be critical for the deacetylase activity. To test this hypothesis, we moved a large portion of the C-terminal domain (a.a. 610-737), including the ESA region, to the N-terminal side of the deacetylase core (C-Core, Figure 2A). We found that C-Core Sirt1 had significant deacetylase activity, suggesting that the position of the ESA region relative to that of the deacetylase core is not critical for deacetylase activity.
Figure 2. The ESA interacts with the deacetylase core.
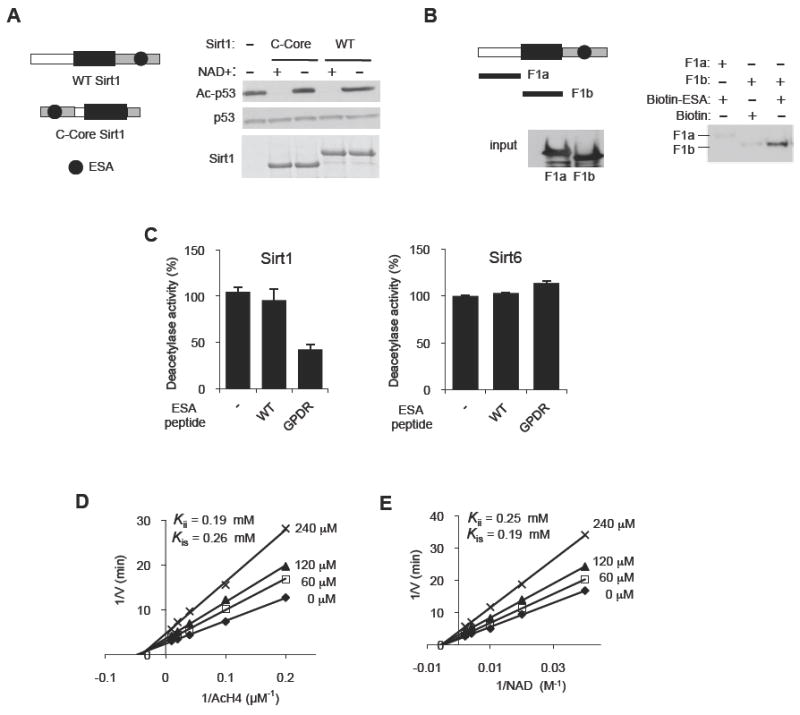
(A) The position of the C-terminal domain relative to the deacetylase core is not critical for Sirt1 deacetylase activity. Deacetylation reactions were performed with WT Sirt1 or Sirt1 in which the C-terminal domain (gray, a.a. 611-737) was placed on the N-terminal side of the deacetylase core (C-Core, a.a. 184-510). See Figure S2A-D for the disordered nature of the C-terminal domain of Sirt1.
(B) The ESA binds to the Sirtuin domain. Pull-down assays were performed after incubating biotin-tagged ESA peptide immobilized on streptavidin beads with either F1a (a.a. 1-236) or F1b (a.a. 236-490) fragments. Streptavidin-immobilized biotin without the peptide is used as negative control. F1a or F1b bound to the ESA peptide is shown above, and the input levels of F1a and F1b are shown below.
(C) The GPDR mutant ESA peptide can inhibit the deacetylase activity of Sirt1 (left panel), but not of Sirt6 (right panel), in trans. The deacetylase activity of recombinant Sirt1 and Sirt6 was measured by a fluorometric assay in the presence of 150 μM of either the WT ESA peptide, or the GPDR-mutant ESA peptide (GPDR) or no peptide (-) (N=4). Results are expressed as the mean ± s.e.m. See also Figure S2E-G.
(D, E) The GPDR-mutant ESA peptide inhibits Sirt1 in a noncompetitive manner. A Lineweaver-Burk plot derived from a fluorometric measurement of WT Sirt1 activity in the absence or presence of 0 (◆), 60 (□), 120 (▲) or 240 (×) μM of the GPDR-mutant ESA peptide for the indicated concentration range of Ac-H4 (D) and NAD+ (E). Inhibition constants (Kii or Kis) for the GPDR peptide are shown for both Ac-H4 and for NAD+ substrates. For each inhibition analysis, the concentrations of the indicated substrate and the inhibitors were varied while the concentration of the other substrate was kept constant (500 μM NAD+ for (D) and 100 μM Ac H4 for (E)). Each experiment was performed in triplicates. To calculate the inhibition constants, nonlinear mixed-effects model fitting function in the programming language R, nlme, was used for data fitting against double-reciprocal form of the noncompetitive model equation as described in the Experimental Procedures section.
These findings suggest that the C-terminal domain may function by tethering the ESA region to the deacetylase core, looping out the intervening sequence. To test this hypothesis, we first examined whether the ESA region interacts with the Sirtuin domain. We incubated streptavidin-immobilized ESA peptide with two recombinant fragments from Sirt1, F1a (a.a. 1-235) and F1b (the Sirtuin domain, a.a. 236-490), and performed a pull-down assay. As shown in Figure 2B, F1b, but not F1a, bound to immobilized ESA peptide, suggesting that ESA interacts with the Sirtuin domain of Sirt1. We repeated this pull-down assay, but this time, we used the GPDR-mutated ESA (GPDR-ESA) peptide. We found that the GPDR-ESA peptide also bound to the deacetylase core (Figure S2E, top). Interestingly, the ESA-deacetylase core interaction was inhibited in the presence of competing GPDR-ESA peptide (Figure S2E, bottom), suggesting that the ESA peptide and the GPDR-peptide compete for the same or overlapping binding site in the deacetylase core. However, it is formally possible that the competing GPDR-ESA peptide inhibited the ESA-deacetylase core interaction by binding to the ESA peptide and forming a dimer rather than by binding to the deacetylase core. To rule out this possibility, we incubated GST, GST-ESA or GST-Sirtuin domain (a.a. 236-490) with streptavidin immobilized GPDR-ESA peptide and performed pull-down experiments (Figure S2F). GST-Sirtuin domain bound to the GPDR-ESA peptide but GST-ESA or GST alone did not, suggesting that the GPDR-ESA peptide did not dimerize with the ESA peptide.
Since the GPDR-ESA peptide can inhibit the ESA-deacetylase core interaction, it is possible that a molar excess of the GPDR-ESA peptide may suppress Sirt1 activity in trans. An addition of 240-fold molar excess (150 μM) of the GPDR-ESA peptide decreased Sirt1 activity by >60% against acetylated-histone H4 (Ac-H4) (Figure 2C). The GPDR-ESA peptide also inhibited Sirt1 activity against Ac-p53 (Figure S2G). To determine if the inhibitory effect of the GPDR-ESA peptide is specific to Sirt1, we added it to a Sirt6 reaction (Figure 2C). We found that the GPDR-ESA peptide had no effect on Sirt6 activity. Since the GPDR-ESA peptide was able to inhibit deacetylase activity, it was possible that the WT ESA peptide would be able to restore activity to the deacetylase core in trans. However, addition of the ESA peptide (150 μM) did not restore activity to the deacetylase core (data not shown). Taken together, these findings indicate that the GPDR-ESA peptide can specifically inhibit Sirt1 in trans.
In order to understand the mechanism by which the GPDR-mutated ESA peptide is inhibiting Sirt1 activity, we performed deacetylase reactions by using varying concentrations of GPDR-mutated ESA peptide and the two substrates, Ac-H4 and NAD+. The Lineweaver-Burk plot we generated indicated that the GPDR-ESA peptide is a noncompetitive inhibitor against both Ac-H4 (Kii = 0.19 mM, Kis = 0.26 mM) (Figure 2D) and NAD+ (Kii = 0.25 mM, Kis = 0.19 mM) (Figure 2E). These findings suggest that the GPDR-ESA peptide inhibited Sirt1 activity in trans without directly competing with either Ac-H4 or NAD+ and suggest that the ESA region alters the conformation of the deacetylase core.
The ESA region increases deacetylase core-substrate interaction
The deacetylase core by itself not only lacks activity but has poor interaction with substrates compared to full-length Sirt1 (Zhang et al., 2009). We examined whether the ESA region affects deacetylase core-substrate interaction by co-expressing in Hela cells Flag-p65 (NF-κB) and either V5-Sirt1 (full-length) or V5-ΔESA Sirt1. As shown in Figure 3A, full-length Sirt1 readily co-immunoprecipitated with Flag-p65 whereas ΔESA Sirt1 did so very poorly. We also examined the Sirt1-substrate interaction experiment in vitro, but this time we used GST-Ac-p53 fusion protein as substrate (Figure 3B). We found that ΔESA Sirt1 bound poorly to GST-Ac-p53 compared to full-length Sirt1. Neither full-length Sirt1 nor ΔESA Sirt1 bound to GST alone. We repeated the Sirt1-substrate interaction experiment by using Ac-H3 and Ac-H4 purified from Hela cells as substrates (Figure S3A). As with the other Sirt1 substrates, the ΔESA mutation significantly weakened the interaction with both Ac-H3 and Ac-H4. Consistent with this, fusing the ESA region to the deacetylase core increased interaction with both Ac-p53 (Figure 3C, left) and Ac-H4 (Figure 3C, right). To confirm that the ESA region itself is not binding to the substrate, we incubated streptavidin-immobilized Ac-H4 peptide with GST, GST-ESA or GST-Sirt1 fusion proteins. We found that the Ac-H4 peptide bound to full-length Sirt1, but not to either GST-ESA or GST alone (Figure 3D). We then asked whether GPDR-ESA is able to increase the deacetylase core-substrate interaction the way ESA did in Figure 3C. As shown in Figure 3E, the deacetylase core fused to GPDR ESA interacted poorly with the Ac-H4 peptide. Therefore, the GPDR mutation in the ESA region abolished both its ability to confer activity to the deacetylase core (Figure 1D, E) and also its ability to increase deacetylase core-substrate interaction.
Figure 3. The ESA increases the Sirt1-substrate interaction.

(A) Deletion of the ESA region decreases Sirt1-substrate interaction in vivo. Hela cells were transiently co-transfected with an expression vector for Flag-tagged NF-κB p65 (p65) and either an expression vector for V5-tagged full-length Sirt1 or Sirt1 with the ESA region deleted (ΔESA Sirt1). Flag-tagged NF-κB p65 was immunoprecipitated with Flag antibody and co-immunoprecipitated Sirt1 was visualized by immunoblotting with V5 antibody. The input levels of the proteins are shown on the right.
(B) Deletion of the ESA region decreases Sirt1-substrate interaction in vitro. Recombinant Flag-tagged full-length Sirt1 or ΔESA Sirt1 (a.a. 631-655 deleted) was incubated with either GST fused to Ac-p53 or GST alone. Pull-down assay was performed with anti-Flag antibody and bound GST-Ac p53 was visualized by immunoblotting with GST antibody (top). Pulled-down Flag-tagged Sirt1 (WT or ΔESA) was visualized by Coommassie staining (bottom). The light chain of the anti-Flag antibody that was used for pull-down is visible and is indicated with an arrow.
(C) The ESA region is sufficient to increase deacetylase core-substrate interaction. (Left) The ESA region increases the deacetylase core-Ac-p53 interaction. GST pull-down experiments were performed with recombinant deacetylase core fragment (Core, a.a. 184-510) and the deacetylase core fragment fused to the ESA (a.a. 631-655) (Core-ESA) and either GST-Ac p53 fusion protein or GST alone (negative control). Bound Core or Core-ESA was visualized by immunoblotting with Sirt1 antibody. Input levels of Core, Core-ESA, GST-Ac p53 and GST are also shown. (Right) The ESA region increases the deacetylase core-Ac-H4 interaction. Streptavidin-immobilized Ac-H4 peptide was incubated with either the deacetylase core (Core) or the deacetylase core fused to the ESA region (Core-ESA), and pull-down experiments were performed. Streptavidin bound to biotin without an attached peptide was used as a negative control.
(D) The ESA alone does not bind to substrate. Pull-down experiments were performed by incubating biotin-tagged acetylated-histone H4 peptide (Ac-H4) immobilized on streptavidin beads with either full-length Sirt1 (GST-FL Sirt1), ESA fused to GST (GST-ESA) or GST alone. As a negative control, we used streptavidin beads bound to free biotin. Bound GST fusion proteins were detected by immunoblotting with GST antibody. Input levels of the GST fusion proteins are shown on the right.
(E) GPDR ESA does not increase deacetylase core-substrate interaction. Pull-down experiments were performed by incubating biotin-tagged Ac-H4 that was immobilized on streptavidin beads with either the deacetylase core fragment, or the core fragment fused to the ESA (Core-ESA) or the core fragment fused to the GPDR mutant ESA (Core-GPDR). Bound core fusion proteins were visualized by immunoblotting with Sirt1 antibody. As a negative control, we used streptavidin beads bound to free biotin. Input levels of the core fusion proteins are also shown. A quantification of the levels of bound core fusion proteins is shown on the right (N=8). Results are expressed as the mean ± s.e.m.
(F) The leucine-zipper (LZ) domain of DBC1 inhibits the ESA-deacetylase core interaction. We incubated biotin-tagged ESA peptide immobilized on streptavidin beads with the deacetylase core fragment in the presence of competing 2 μg of either GST fragment or GST fused to the LZ (a.a. 243-264) domain of DBC1. The levels of bound deacetylase core and of input GST fusion proteins are shown. Streptavidin-immobilized biotin without the peptide is used as negative control. A quantification of the levels of bound deacetylase core fragment is shown on the right (N=5). Results are expressed as the mean ± s.e.m.
(G) The ESA peptide inhibits the LZ domain-deacetylase core interaction. GST pull-down experiments were performed by incubating either 2 μg GST fragment alone or GST-LZ fusion protein with the deacetylase core in the presence of either a control peptide or the ESA peptide. The levels of bound deacetylase core and that of the GST fusion proteins are shown.
(H) The interaction of the deacetylase core with the LZ domain of DBC1 is stronger than with the ESA region. Pull-down experiments were performed by incubating GST, GST-ESA or GST-LZ with the deacetylase core. The levels of bound deacetylase core and of the GST fusion proteins are shown.
The LZ domain of DBC1 and the ESA region compete for the deacetylase core
DBC1 inhibits Sirt1 activity by interacting with the deacetylase core of Sirt1 via its leucine zipper (LZ, a.a. 243-264) domain (Kim et al., 2008; Zhao et al., 2008). How DBC1 interacts with and inhibits the Sirt1 deacetylase core is poorly understood. We examined the possibility that DBC1 inhibits Sirt1 activity by preventing the ESA region from interacting with the deacetylase domain. To test this hypothesis, we first examined whether the LZ domain can inhibit Sirt1 activity. GST-LZ significantly reduced Sirt1 deacetylase activity compared to GST alone (Figure S3B). We then incubated streptavidin-immobilized ESA peptide and the deacetylase core in the presence of competing GST or GST fused to the LZ domain of DBC1 (GST-LZ). As shown in Figure 3F, GST-LZ, but not GST, prevented the ESA-deacetylase core interaction. We also performed this experiment in reverse by incubating immobilized GST-LZ and the deacetylase core in the presence of competing control peptide or ESA peptide (Figure 3G). We found that the ESA peptide, but not the control peptide, inhibited the LZ-deacetylase core interaction. The most reasonable explanation for these findings is that the ESA peptide and the LZ domain of DBC1 bind to the same or overlapping region(s) in the deacetylase core.
The DBC1-deacetylase core interaction is intermolecular, whereas the ESA-deacetylase core interaction is intramolecular. Therefore, all things being equal, in order for DBC1 to be able to effectively compete with the ESA region for the binding site in the deacetylase core, it should have higher affinity for the deacetylase core than the ESA peptide. To compare the affinity of the LZ domain of DBC1 and the ESA region for the deacetylase core, we incubated the deacetylase core with either GST-LZ or GST-ESA and performed a GST pull-down assay. As shown in Figure 3H, the LZ-deacetylase core interaction was stronger than the ESA-deacetylase core interaction.
The GPDR-ESA peptide can inhibit Sirt1 in vivo
Our observation that the GPDR-ESA peptide can inhibit Sirt1 activity in trans suggested to us that the GPDR-ESA peptide may inhibit Sirt1 activity in vivo. To test this hypothesis, we transiently transfected an expression vector for Sirt1 and a 10-fold excess of an expression vector for 3 copies (3X) of tandemly fused GPDR-ESA peptide with or without the nuclear localization signal (NLS) into H1299 cells (Figure 4A). We found that the GPDR-ESA peptide with or without the NLS inhibited Sirt1 activity. Overexpression of DBC1, as expected, also inhibited Sirt1 activity.
Figure 4. The GPDR mutated ESA peptide can inhibit Sirt1 activity in vivo.
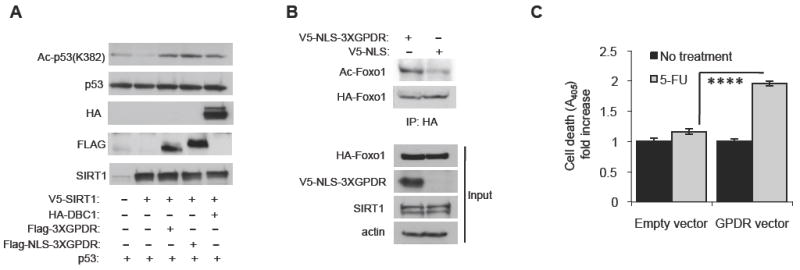
(A) The GPDR mutated ESA peptide inhibits deacetylation of p53 by Sirt1 in H1299 cells. Expression vectors for p53 and V5-tagged Sirt1 were co-transfected with a 10-fold molar excess of an expression vector for HA-tagged DBC1, Flag-tagged 3XGPDR (Flag-3XGPDR) or Flag-tagged 3XGPDR with a nuclear localization signal (Flag-NLS-3XGPDR). Levels of acetylated p53 were visualized by antibody specific for acetylated K382 in p53.
(B) The GPDR mutated ESA peptide inhibits deacetylation of Foxo1 by Sirt1 in the prostate cancer cell line DU145. An expression vector for HA-tagged Foxo1 was co-tranfected with an expression vector for either V5-tagged-NLS-GPDR (3X) peptide or V5-tagged NLS without GPDR. Acetylation of Foxo1 was visualized by immunoblotting with an antibody specific for acetylated lysine after HA-Foxo1 was immunoprecipitated with anti-HA antibody.
(C) The GPDR mutated ESA peptide restores sensitivity to the chemotherapeutic agent 5-FU. DU145 cells were co-transfected with an expression vector for V5-tagged-NLS-GPDR or control and treated with 20 μM 5-FU for two days before apoptosis levels were measured. Results are expressed as the mean ± s.e.m. ****, p<0.0001 between empty and GPDR vectors.
Androgen-refractory PCa, unlike androgen-sensitive PCa, is very resistant to treatment. To investigate whether the GPDR-ESA peptide can inhibit Sirt1 in the androgen-refractory PCa cell line DU145, we expressed V5-NLS-3XGPDR-ESA peptide and visualized deacetylation of the Sirt1 substrate Foxo1 (Figure 4B). We found that the V5-NLS-3XGPDR-ESA peptide increased Foxo1 acetylation, suggesting that it inhibited Sirt1. This result prompted us to investigate whether the GPDR-ESA peptide can increase the sensitivity of DU145 to the chemotherapeutic agent 5-flurouracil (5-FU). As shown in Figure 4C, the V5-NLS-3XGPDR-ESA peptide increased the level of apoptosis in 5-FU-treated DU145 cells by ~80%. We also measured the effect of the V5-NLS-3XGPDR-ESA peptide on the sensitivity to 5-FU in the presence of siRNA specific for either a scrambled sequence or Sirt1. As shown in Figure S4, the V5-NLS-3XGPDR-ESA peptide significantly increased (by >60%) the level of apoptosis in 5-FU-treated DU145 cells. Sirt1 siRNA alone increased 5-FU sensitivity and combining it with V5-NLS-3XGPDR-ESA further increased apoptosis by only ~10%. Taken together, these findings indicate that the V5-NLS-3XGPDR-ESA peptide can function as a Sirt1 inhibitor in vivo.
Discussion
In summary, our findings show that the ESA region functions as an “on switch” for the deacetylase core of Sirt1. DBC1 appears to inhibit Sirt1 by competing with the ESA region for the binding site in the deacetylase core. Although it was proposed that DBC1 prevents the deacetylase core-substrate interaction by steric hinderance (Kim et al., 2008; Zhao et al., 2008), our work suggests that it may also do so indirectly by preventing the ESA region from increasing the deacetylase core-substrate interaction. We discovered that an ESA peptide containing a mutation of two residues in the ESA region, G644P and D650R (GPDR), was able to bind to the deacetylase core but was unable to confer activity to it. This property of the GPDR-ESA peptide allows it to inhibit Sirt1 activity when present in molar excess and to function as an “off switch” (Figure 5).
Figure 5.
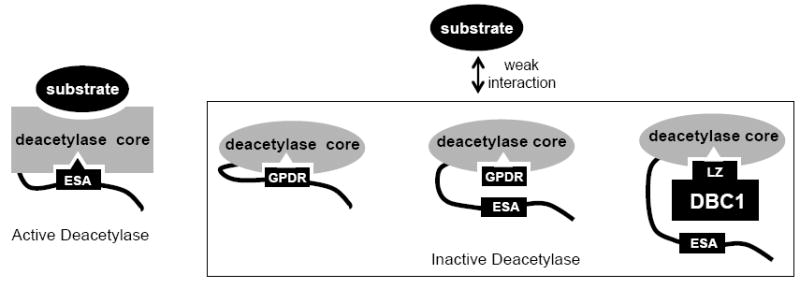
A schematic diagram of the ESA peptide switch. The ESA-deacetylase core interaction turns “on” the deacetylase core and increases its affinity for substrates. The GPDR mutated ESA peptide or the LZ domain of DBC1 bind to the deacetylase core in a manner that is different from that of the ESA region and, therefore, cannot activate it or increase its affinity for substrates. DBC1 could also sterically hinder the deacetylase core-substrate interaction (not shown).
It is curious that Sirt2-7 and yeast Sir2 do not require the ESA region for activity. The presence of the ESA region in Sirt1 and in Sir2 ortholog in worms and flies, but not in yeast Sir2, may be a reflection of a need for a more complex mode of regulation in metazoans via DBC1 or CK2, which phosphorylates residues within the ESA region and increases Sirt1 activity (Kang et al., 2009). It is also interesting that in Sirt2-7, the last exon codes for the C-terminal end of the deacetylase domain. However, in Sirt1, an additional exon (exon 8), which begins precisely where the ESA region begins, is added 3’ to what is homologous to the last exon in Sirt2-7. Thus, in mammals, evolution has not only increased the number of Sir2 orthologues (Sirt1-7) but also added a “switch exon” in the case of Sirt1.
A number of questions still remain unanswered. For example, if the LZ domain and the GPDR-ESA can bind to the deacetylase core, why do they not activate it? Also, if the GPDR-ESA peptide inhibits Sirt1 activity in trans, why does the ESA peptide not activate the deacetylase core in trans? We speculate that the ESA region has to bind to the deacetylase core with proper alignment in order to activate it, and it binds with proper alignment only if it is tethered to the deacetylase core. Another related possibility is that much higher local concentration of the ESA region is required to activate the deacetylase core in trans than to inhibit it. To fully answer these questions, X-ray crystal structures of these complexes will need to be solved. In conclusion, our work provides evidence that the deacetylase activity of Sirt1 requires a separate “switch” for activity and a proof of principle that targeting the ESA switch may constitute a strategy for the development of Sirt1-specific regulators.
Experimental Procedures
Sirt1 deacetylase activity measurement
For in vitro deacetylase assays, recombinant His-tagged WT or mutant Sirt1 (2 μg) was incubated with acetylated GST-p53 (0.2 μg) (Kang et al., 2009) and 0.5 mM NAD+ in the deacetylase buffer (50 mM HEPES at pH 7.0, 1 mM DTT, 10 mM MgCl2, 200 mM NaCl, protease inhibitor cocktail, and phosphatase inhibitor cocktail (Roche)). The reaction mixtures were incubated at 37 °C for the indicated durations and stopped by the addition of SDS sample loading buffer. The loaded amounts of Sirt1 and GST-p53 were visualized with Coomassie staining, Ponceau S staining or Western blotting. Deacetylation of Ac-p53 (K382) by Sirt1 was detected by immunoblotting with antibody specific for acetylated-p53 (Cell Signaling).
The effect of the GPDR-ESA peptide on Sirt1
For the GPDR-ESA peptide competition of WT Sirt1, enzyme activity was determined by using a Fluorometric HDAC assay kit (Millipore) according to the manufacturer’s instructions. The peptide used in this assay was comprised of di-peptide containing acetylated K16 of histone H4. The fluorescence values obtained without NAD+ during the reactions were used for both negative control and subtraction value for Sirt1 dependent fluorometric reaction values. The ESA or GPDR-ESA peptides at the concentrations indicated in the figure legends were pre-incubated with 2 μg of His-tagged Sirt1 in the deacetylase buffer described above for 5 minutes at room temperature before commencing the reaction by the addition of 100 μM fluorometric substrate and 0.5 mM NAD+ in a 40 μl final volume. After 25 minutes of incubation at 37 °C, the reaction was terminated by the addition of 20 μl of activator solution containing 8 mM nicotinamide and the mixture was further incubated at room temperature for 15 minutes. Fluorescence was read in a Victor 2 1420 multi-label counter (PerkinElmer) with an excitation wavelength of 355 nm and an emission wavelength of 460 nm.
The effect of the GPDR-ESA peptide on Sirt6
To analyze the effects of WT and GPDR ESA peptides on other Sirtuin proteins, the activity of His-tagged Sirt6 protein was measured in the presence of 150 μM of either the ESA or the GPDR-ESA peptides by using the CyLex SIRT6 Deacetylase Fluorometric Assay kit (CycLex Co.) according to manufacturer’s protocol, except using the same buffer as for the Sirt1 reaction described above.
Interaction between the ESA region and Sirt1 fragments
To measure the interaction between biotinylated peptides and the specific region of Sirt1or other protein designated in the figure legends, biotinylated peptides were bound to Streptavidin agarose resins before starting the experiments. Briefly, 100 μl biotinylated peptide (300 nmole/ml) was mixed with 1 ml Streptavidin agarose beads and incubated at room temperature for 1 hr on a rotary platform. After incubation, unbound peptide was extensively washed out using 100 volumes of PBS and peptide-bound Streptavidin resins were re-suspended with 2 ml of PBS containing 1 % Triton X-100, and protease inhibitor cocktail (Roche). For the peptide binding assay, 30 μl Streptavidin-bound peptide was mixed with 2 μg GST-fused or His-tagged proteins as indicated in the figure legends and was incubated for 2 hrs at room temperature. GST-LZ was generated by fusing the LZ domain of DBC1 (a.a. 243-264) (a gift from Zhenkun Lou) to GST. For the GST pull-down assay, 2 μg His-tagged Sirt1 fragment was incubated with 2 μg target GST-fusion protein as described in the figure legends. Unbound proteins were removed from the complex on micro-spin columns by extensively washing using PBS containing 1 % Triton X-100 and 200 mM NaCl. Bound proteins were eluted by the addition of SDS sample buffer containing β-ME, heated at 95 °C for 10 min, and collected by micro-centrifugation. Eluted samples were subjected to 4 – 12 % gradient SDS-PAGE and Western blotting against the indicated antibodies.
Sirt1 kinetics calculation
The noncompetitive model defined by Cleland (Cleland, 1977) was used for data fitting. The data were plotted as reciprocal initial velocity, 1/Vo, versus reciprocal substrate concentration, 1/[S]. Nonlinear mixed-effects model fitting function in the programming language R (Lindstrom and Bates, 1990) (http://www.r-project.org) was used for data fitting against the double-reciprocal form of the noncompetitive model equation (eq 2), where Vo the is initial velocity, Vmax is the maximal velocity of a reaction, Km is the Michaelis constant, Kis = [E][I]/[EI], Kii = [ES][I]/[ESI], [S] is the substrate concentration, [I] is inhibitor concentration, [E] is the enzyme concentration, [EI] is the enzyme and inhibitor complex concentration, [ESI] is the enzyme and substrate and inhibitor complex concentration. Under our reaction conditions, the Km for Ac-H4 peptide and NAD+ was 26.8 μM and 203 μM, respectively. The catalytic constant, kcat was derived by fitting to eq 3.
| (eq 1) |
| (eq 2) |
| (eq 3) |
Using the GPDR-ESA peptide to inhibit Sirt1 activity against p53 or Foxo1 in vivo
To assess the effect of the GPDR region on the activity of Sirt1 in vivo, we transiently transfected H1299 cells with expression vectors for p53, and DU145 cells for HA-Foxo1 with or without the 3xGPDR expression vector. Forty-eight hours later, H1299 cells were exposed to 20 μM etoposide for 1 hr and DU145 cells to 0.5 mM H2O2 for 1 hr prior to harvest.
Apoptosis assay
To measure the effect of GPDR on 5-FU induced cell death, DU145 cells were transfected with scrambled (Scr) or sirt1 siRNAs by lipofectamine 2000 reagent (Invitrogen) and 16 hours later, these cells were transfected again with pcDNA 6.0 V5-NLS (control) or V5-NLS-3×GPDR expression vector by polyfectamine (Qiagen). After selection with blasticidin (2 μg/ml) for forty-eight hours, cells were treated with 20 μM 5-fluorouracil (5-FU) for 48 hours. Cell death was analyzed by measuring the amount of histone-coupled DNA fragments by using the cell death detection ELISAPLUS (Roche) kit according to the vendor’s manual.
Supplementary Material
Highlights.
The ESA region in the C-terminal domain is essential for Sirt1 activity.
The ESA region confers activity by interacting with the Sirtuin domain.
The ESA region increases Sirt1-substrate interaction.
Mutant ESA peptide inhibits Sirt1 and increases chemosensitivity in tumor cells.
Acknowledgments
This work was supported by the Intramural Research Program, National Heart Lung and Blood Institute, National Institutes of Health. We thank Alexandra Brown for help with manuscript preparation.
Footnotes
CONFLICT OF INTEREST We have no conflict of interest to declare.
References
- Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci U S A. 2001;98:15113–15118. doi: 10.1073/pnas.261574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L, Wang M. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des. 2009;74:619–624. doi: 10.1111/j.1747-0285.2009.00901.x. [DOI] [PubMed] [Google Scholar]
- Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland WW. Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv Enzymol Relat Areas Mol Biol. 1977;45:273–387. doi: 10.1002/9780470122907.ch4. [DOI] [PubMed] [Google Scholar]
- Dai JM, Wang ZY, Sun DC, Lin RX, Wang SQ. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J Cell Physiol. 2007;210:161–166. doi: 10.1002/jcp.20831. [DOI] [PubMed] [Google Scholar]
- Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5:147–152. doi: 10.7150/ijbs.5.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dvir-Ginzberg M, Gagarina V, Lee EJ, Booth R, Gabay O, Hall DJ. TNFalpha-mediated cleavage and inactivation of SirT1 in human osteoarthritic chondrocytes. Arthritis Rheum. 2011 doi: 10.1002/art.30279. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erion DM, Yonemitsu S, Nie Y, Nagai Y, Gillum MP, Hsiao JJ, Iwasaki T, Stark R, Weismann D, Yu XX, et al. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc Natl Acad Sci U S A. 2009;106:11288–11293. doi: 10.1073/pnas.0812931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021–1026. [PubMed] [Google Scholar]
- Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285:13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:1–8. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jung-Hynes B, Nihal M, Zhong W, Ahmad N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J Biol Chem. 2009;284:3823–3832. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, et al. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Kang H, Jung JW, Kim MK, Chung JH. CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS One. 2009;4:e6611. doi: 10.1371/journal.pone.0006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Kojima K, Ohhashi R, Fujita Y, Hamada N, Akao Y, Nozawa Y, Deguchi T, Ito M. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun. 2008;373:423–428. doi: 10.1016/j.bbrc.2008.06.045. [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Lindstrom ML, Bates DM. Nonlinear mixed effects models for repeated measures data. Biometrics. 1990;46:673–687. [PubMed] [Google Scholar]
- Mai A, Massa S, Lavu S, Pezzi R, Simeoni S, Ragno R, Mariotti FR, Chiani F, Camilloni G, Sinclair DA. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J Med Chem. 2005;48:7789–7795. doi: 10.1021/jm050100l. [DOI] [PubMed] [Google Scholar]
- Malik R, Kashyap A, Bansal K, Sharma P, Rayasam GV, Davis JA, Bora RS, Ray A, Saini KS. Comparative deacetylase activity of wild type and mutants of SIRT1. Biochem Biophys Res Commun. 2010;391:739–743. doi: 10.1016/j.bbrc.2009.11.130. [DOI] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R, Bordone L. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One. 2009;4:e8414. doi: 10.1371/journal.pone.0008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 2010;9:844–855. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- Pfister JA, Ma C, Morrison BE, D’Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, Minor W, Scrable H. Phosphorylation regulates SIRT1 function. PLoS ONE. 2008;3:e4020. doi: 10.1371/journal.pone.0004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci U S A. 2000;97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tissenbaum HA. Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech Ageing Dev. 2006;127:48–56. doi: 10.1016/j.mad.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV, Bhalla K, Bai W. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol. 2007;9:1253–1262. doi: 10.1038/ncb1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. Embo J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, Divekar R, McBurney MW, Braley-Mullen H, Zaghouani H, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest. 2009;119:3048–3058. doi: 10.1172/JCI38902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.