

Abstract
Gene duplication occurs by either DNA- or RNA-based processes; the latter duplicates single genes via retroposition of messenger RNA. The expression of a retroposed gene copy (retrocopy) is expected to be uncorrelated with its source gene because upstream promoter regions are usually not part of the retroposition process. In contrast, DNA-based duplication often encompasses both the coding and the intergenic (promoter) regions; hence, expression is often correlated, at least initially, between DNA-based duplicates. In this study, we identified 150 retrocopies in rice (Oryza sativa L. ssp japonica), most of which represent ancient retroposition events. We measured their expression from high-throughput RNA sequencing (RNAseq) data generated from seven tissues. At least 66% of the retrocopies were expressed but at lower levels than their source genes. However, the tissue specificity of retrogenes was similar to their source genes, and expression between retrocopies and source genes was correlated across tissues. The level of correlation was similar between RNA- and DNA-based duplicates, and they decreased over time at statistically indistinguishable rates. We extended these observations to previously identified retrocopies in Arabidopsis thaliana, suggesting they may be general features of the process of retention of plant retrogenes.
Keywords: retroposition, gene expression, gene duplication
Introduction
Gene duplication is a source of evolutionary novelty (Ohno 1970) and occurs through either DNA- or RNA-based processes. DNA-based processes may lead to the duplication of entire genomes (polyploidy), large chromosomal segments (Prince and Pickett 2002) or, occasionally, individual genes. In contrast, RNA-based processes typically result in the duplication of a single gene. These genes are duplicated by the reverse transcription of an RNA intermediate, followed by integration into DNA.
RNA-based duplication produces new gene copies or “retrocopies.” Retrocopies have traditionally been considered nonfunctional (“dead on arrival”) because they often lack the regulatory elements of their source (or “parental”) genes and contain functional disruptions, such as 5′ truncations, frameshift mutations, and in-frame terminations (Vanin 1985). Analyses of the human genome have revealed that RNA-based duplication is common, with over 3,590 retrocopies in total, but 84% of these retrocopies contain functional disruptions (Vinckenbosch et al. 2006). Nonetheless, a subset of retrocopies is expressed and may have evolved unique functions, particularly in the male germ line (Betran et al. 2002; Emerson et al. 2004; Potrzebowski et al. 2008). Retrocopies also contribute to novel functions within chimeric genes, such as the jingwei gene of Drosophila (Long and Langley 1993). Thus, while the majority of retroposition events result in processed pseudogenes, retroposition may play a significant role in the evolution of novelty.
Expressed retrocopies (or “retrogenes”) require regulatory elements for transcription. It is thought that retrogenes acquire regulatory functions by one of three mechanisms (Kaessmann et al. 2009). The first is the use, or even co-option, of the regulatory elements of nearby genes. For example, some retrogenes have inserted into introns and are expressed as a fusion transcript under the control of the regulatory elements of the native gene (Long and Langley 1993; Wang et al. 2002). The second mechanism is the acquisition of de novo regulatory elements. One potential source of de novo elements is CpG islands, which sometimes have the capacity to promote transcription (Yamashita et al. 2005; Okamura and Nakai 2008). Finally, retrogenes may inherit promoters and enhancer elements from their parental genes. This is most likely to occur if there is leaky transcription from a start site far upstream from the parental gene, so that retroposition includes 5′ regulatory sequences along with coding regions, resulting in a new gene with regulatory properties similar to the parental gene. Although parental genes have not typically been considered a source for regulatory elements, a recent study has suggested that the retroposition of parental regulatory sequences does occur (Okamura and Nakai 2008).
These three possible modes of promoter acquisition predict different patterns of expression divergence between a retrogene and its parental gene. If, for example, the retrogene has acquired new regulatory elements from nearby genes or from de novo sequences, then the expression patterns of the retrogene and its parental gene should be uncorrelated. Conversely, the retrogene and the parental gene are expected to have correlated patterns of gene expression if the retrogene inherits regulatory regions. Although some studies have documented tissue-specific expression of retrogenes (Marques et al. 2005; Shiao et al. 2007; Rosso et al. 2008a, 2008b), few studies have compared expression between retrogenes and their putative parental genes, particularly on a genome-wide scale.
In this study, we examine the expression of retrogenes in the genome of rice, Oryza sativa L. ssp. japonica. We have chosen to study rice for three reasons. First, a previous study has documented that retrocopies are abundant in rice (Wang et al. 2006). Second, retrocopies tend to be expressed more often in plants than in animals. Over 80% of retrocopies in rice and poplar are either expressed or inferred to be functional by structural and evolutionary characteristics (Wang et al. 2006; Zhu et al. 2009) compared with only 30% in the human genome (Vinckenbosch et al. 2006). Finally, rice has abundant genomic resources, including genomic sequences of both ssp. japonica and ssp. indica (International Rice Genome Sequencing Project 2005; Yu et al. 2005) and annotation resources (Tanaka et al. 2008).
We first identify retrocopies in the rice genome through an extensive genomic survey, using conservative approaches to maximize the probability that our designation of retrogenes and their parental genes are accurate. We then examine gene expression in seven rice tissues—leaf, root, shoot, panicle before and after flowering, seed, and callus—by generating high-throughput sequences of mRNA (mRNAseq). Unlike microarray-based approaches, mRNAseq does not rely on the predefined gene annotations, allowing us to accurately assess expression of unannotated retrocopies. Finally, we utilize mRNAseq data both to compare gene expression patterns between retrogenes and their parents and to compare patterns of expression divergence between RNA-based duplicates and DNA-based duplicates. Overall, we find that expression patterns are surprisingly well conserved between retrogenes and their parental genes, but the molecular and evolutionary mechanisms underlying this correlation are not yet clear.
Materials and Methods
Retrogene Detection
We obtained 40,353 protein sequences of O. sativa ssp. japonica cv. Nipponbare as well as 9,966 ab initio predicted genes from the Rice Annotation Project Database (RAP-DB) (Tanaka et al. 2008). If there were two or more identical protein sequences in a locus, we selected one sequence with the longest transcript. We removed sequences related to transposable elements (TEs) by two methods. First, given the repeat-masked genome sequence (IRGSP build 5) (http://rapdb.dna.affrc.go.jp/), we discarded a sequence if over 40% of the sequence was masked. Second, we conducted BlastN searches (Altschul et al. 1997) with “-e 1.0e-10” option against RetrOryza sequences (Chaparro et al. 2007); we discarded genes if over 40% of the sequences were covered by any RetrOryza sequence. After this process, we were left with 46,235 nonredundant and non-TE–related protein sequences. We removed TE-related sequences from consideration because we did not want to base our inferences on TEs misannotated as genes or gene fragments captured by TEs.
These 46,235 genes were culled to make an initial set of potential parental genes, based on two criteria: 1) the CDS started with methionine, ended with a stop codon, and spanned 100 or more codons and 2) the transcribed region contained one or more introns of ≥70 bp in length with consensus GT-AG splicing motifs. If a gene had only one intron, we selected the sequence as a potential parental gene only if it had no poly-A tract within 500 bp downstream, defining the poly-A tract as a 20 bp window containing 16 or more adenines. After this culling procedure, we identified 19,235 potential parental genes.
To find retrocopies, we mapped the protein sequences of the 19,235 potential parental genes to the rice genome, using TFASTY with default parameters (Pearson et al. 1997). We retained only homologous hits that showed ≥35% identity, covered ≥70% of the parental genes, and lost one or more introns. If the parental gene had only one intron, we retained the hit only if the intron was lost and if there was a poly-A tract within 500 bp downstream. An intron was deemed to be missing if 10 bp upstream and downstream sequences of the exon/intron boundary were successfully aligned without any gaps. We also discarded hits that contained insertions of ≥70 bp relative to the CDS of a parental gene. If hits overlapped on the genome, we selected the hit with the highest identity and sequence coverage.
We applied three additional filters to our set of putative retrocopies. First, we recognized that these retrocopies could be the product of the DNA-based duplication of an intronless gene. To eliminate this possibility, we conducted all-against-all FASTA search, querying the 46,235 nonredundant protein sequences with putative retrocopies. We retained only the homologous hits that showed the highest identity and sequence coverage to the set of potential parental genes. Second, we aligned the remaining retrocopies to their putative parental genes and estimated dS and dN with the modified Nei–Gojobori method (Zhang et al. 1998). We discarded retroparent pairs with dS values (>2.0). For individual gene pairs, the deviation of the dN /dS ratio from 1.0 was measured in HYPHY (Pond et al. 2005), by estimating the likelihood of a constrained (dN /dS = 1.0) and a free model. The HYPHY analyses were based on the HKY85 model of nucleotide substitution (Hasegawa et al. 1985). Finally, we discarded retrocopies that were tandemly located with parental genes, based on the criteria of Hanada et al. (2008).
Identifying DNA-Based Duplicates
For comparison's sake, we identified pairs of duplicate genes. To identify DNA-based duplicates for comparison, we used a procedure similar to that described in Makova and Li (2003). First, all nonredundant protein sequences were subjected to BlastP with default settings. We retained pairs of sequences if 1) the alignable region between them was >80% of the longer protein and 2) the identity (I) between them was I ≥ 30% when the alignable region was longer than 150 aa and I was ≥0.01n + 4.8L0.32[1 + exp(−L/1000)] (Rost 1999) for all other protein pairs, where n = 6 and L is the alignable length between the two proteins. Based on these pairings, gene families were generated by the Markov Cluster Algorithm (http://micans.org/mcl/).
For each gene family, we calculated dS for all pairwise combinations and selected the pair with the smallest dS. We then proceeded by selecting independent pairs (with no overlap with pairs that had already been selected) with increasing dS. Any duplicate gene pairs with dS > 2 were excluded. We also discarded duplicate gene pairs with dS < 0.05 because it was difficult to estimate expression level accurately when sequence identity between two gene copies was too high. Finally, we discarded tandemly located duplicate genes (Hanada et al. 2008).
Expression Analysis
We performed mRNAseq on seven tissues of O. sativa ssp. japonica cv. Nipponbare: callus, leaf, root, panicle before flowering, panicle after flowering, seed, and shoot. We obtained seeds (accession number “JP229579”) from the GENE bank in the National Institute of Agrobiological Sciences, Japan (http://www.gene.affrc.go.jp/about_en.php). The seeds were germinated in a growth chamber at 28 °C under a 16-h light/8-h dark regime. Seven days after germination, shoots and roots were collected. In the meantime, plants were grown in paddy fields, where leaves (7 days before heading to 7 days after flowering) and panicles (7 days before and 0–7 days after heading to flowering) were collected. Callus was induced on N6D medium according to a previous protocol (Ozawa 2009). For RNA extraction from each plant tissue, at least 10 plants were collected, immediately frozen in liquid nitrogen, and mixed, to minimize the effect of transcriptome unevenness among plants. Total RNA was extracted from each tissue with the RNeasy Plant mini kit (Qiagen), and cDNA was synthesized with mRNAseq 8-sample prep kit (Illumina) (Mizuno et al. 2010). We constructed cDNA libraries for the seven tissues individually and sequenced single ends of each on the Illumina GAIIx platform for four separate runs (three at 36 cycles and one at 76 cycles). The sequence data have been submitted to the DNA Data Bank of Japan (DDBJ) Sequence Read Archive (DRA) (http://trace.ddbj.nig.ac.jp/dra/index_e.shtml) under accession numbers DRR001024–DRR001051.
After sequencing, we discarded 3′-end nucleotides of low quality reads so that every read was at least 35 bp in length and had three or more successive nucleotides with the quality score ≥ 20 at the 3′-end. We also trimmed sequencing adapters. Quality and adapter trimming were performed by customized C and Perl programs. Trimmed reads were mapped to the genome sequence with the bwa program (Li and Durbin 2009) using default settings. Only uniquely mapped reads with ≤2 mismatches were retained for further analyses. For each retrocopy, parental gene and DNA-based duplicate gene, we calculated the number of reads per kilobase (RPK) with a customized Perl program.
In order to determine the background level of expression, we investigated the number of mRNAseq reads mapping to intergenic regions. To do this, we first selected intergenic regions of ≥3 kb in length and extracted 1 kb from the midpoint of the regions. To ensure these were not protein coding regions, we conducted BlastX search against the nonredundant protein database (nr) of the National Center for Biotechnology Information, with a cutoff E value of 10−10 and discarded sequences with hits. For the remaining 1,885 1-kb intergenic sequences, we calculated the number of overlapping mRNAseq reads. The read counts were then normalized using the edgeR package (Robinson et al. 2010). Based on these analyses, we determined that the mean RPK value per tissue in intergenic regions was 10; this RPK value defined the background level.
Tissue Specificity
In order to evaluate the tissue specificity of each gene, we calculated Shannon entropy based on RPK values (Kadota et al. 2006). We estimated one-step Tukey biweight tbw (Mosteller and Tukey 1977; Sachs 1994) for each gene, which was defined as
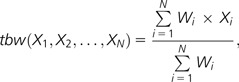 |
where N = 7 is the number of tissues and Xi is the RPK value for the ith tissue. The weight, Wi, was given by
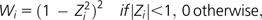 |
where Zi is given by
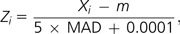 |
where m is a median of the RPK values and MAD is a median absolute deviation. We applied 5 as a multiplicative factor and added 0.0001 to avoid division by zero. Before calculating tbw, we assigned a randomly chosen value of <10−5 to the tissues with zero RPK values to avoid zero median. We identified tissue-specific expression for each gene using Ueda's Akaike's Information Criterion–based method (Kadota et al. 2003).
Arabidopsis thaliana Retrogene Analyses
We obtained 63 pairs of retrocopies and parental genes of A. thaliana from Zhu et al. (2009). Protein sequences of the retrogenes and parental genes were aligned by ClustalW2 (Chenna et al. 2003), and codon alignments were constructed by PAL2NAL (Suyama et al. 2006). We estimated dS (Zhang et al. 1998) and discarded pairs with dS > 2.0, resulting in 48 pairs. Protein and nucleotide sequences were retrieved from TAIR (http://www.arabidopsis.org/). For expression analyses, we obtained microarray expression data from 55 samples (Schmid et al. 2005; Matsuda et al. 2010). We calculated the Pearson product-moment correlation coefficients (R) across the 55 samples for the 24 (of 48) pairs with expression data for both the retrocopy and the parental gene.
Results
The Number, Age, and Structure of Retrocopies
We began with 46,235 nonredundant rice protein sequences and defined 19,235 of them as potential parental genes based on intron and coding characteristics (see Materials and Methods). Mapping this group of potential parental genes to the japonica genome, we identified 150 retrocopies that arose from 143 parental genes (supplementary table S1, Supplementary Material online). None of the retrocopies contained introns, and the vast majority (93%) lost ≥2 introns relative to their putative parental genes. Of the 150 retrocopies, 147 contained target site duplications (TSDs) of 7–60 bp repeats within 500 bp upstream or downstream; TSDs are a common by-product of the retroposition process (Vanin 1985) but may also simply be an inherent sequence characteristic. In addition, 21 retrocopies possessed poly-A tails, which is another strong indicator of retroposition.
We estimated the number of synonymous substitutions per site (dS) between each retrocopy and its parental gene. dS values ranged from 0.0 to 2.0, but few pairs had dS values < 0.4 (fig. 1). The frequency distribution of dS featured a peak at ∼0.7, corresponding to a divergence time of ∼54 Myr based on a rate estimate of 6.5 × 10−9 substitutions per site per year in the grasses (Gaut et al. 1996). These results imply that the retrogenes are considerably older than the 0.44 Myr divergence between indica and japonica (Ma and Bennetzen 2004). To verify this implication, we compared the 150 retrocopies against the indica genome sequence using BlastN; 137 of the retrocopies were shared between japonica and indica with ≥90% nucleotide identity and ≥70% sequence coverage. Another 12 retrocopies were located within insertions in japonica relative to indica, and one retrocopy was not found in indica.
FIG. 1.—

The distribution of the number of synonymous substitutions per site (dS) for the 150 retrocopies compared with their parental genes.
The age of retrocopies suggests they may be functionally conserved. This suggestion is supported by estimates of the ratio of nonsynonymous to synonysmous changes (ω) between retrogenes and parental genes. Mean for the 150 retroparent gene pairs was 0.51 (supplementary table S1, Supplementary Material online), which is far less than the neutral expectation of ω = 1.0. For individual gene pairs, 123 of 150 (82%) had an estimated ω significantly <1.0 at P value < 0.01 (supplementary table S1, Supplementary Material online).
Despite this conservation, the 150 retrocopies did encompass structural changes relative to their parental genes. For example, 90 of the 150 retrocopies contained either a premature stop codon or a frameshift replacement. These 90 have a significantly higher value (0.56) than the 60 intact retrocopies . The difference in is significant (Wilcoxon rank sum [WRS] test, P < 10−3), suggesting that intact and modified retrocopies differ in levels of constraint. In addition, eight retrocopies contributed to modified gene structures (fig. 2 and supplementary table S1, Supplementary Material online). Of these, six evolved new 5′-exons or untranslated regions (UTRs) and two added 3′-exons. Seven of the eight new structures were confirmed by full-length cDNA (flcDNA) sequences from rice, and the remaining retrocopy matched an flcDNA from maize. In addition, flcDNAs indicated potential intronization in three additional retrocopies. In these copies, a portion of the coding region of the parental gene apparently became an intron after retroposition (fig. 2 and supplementary table S1, Supplementary Material online). However, none of the three obey the canonical GT/AG rule for splice sites. For one of the three loci (fig. 2), there were flcDNA variants with and without the intron, suggesting that the newly emerged intron is unstable and not constitutively spliced.
FIG. 2.—

Examples of retrogenes with altered structures compared with their parental genes. (A) The retrocopy (bottom) has evolved a new 5′ UTR relative to its parental gene (top). (B) The retrocopy (bottom) evolved a new 3′ exon consisting of a part of protein coding region. (C) An example of an intronization event. There are two cDNA variants in the retroposed region (Os07t0693100-01 and Os07t0693100-02). Part of the Os07t0693100-01 transcript is missing in Os07t0693100-02, presumably due to an intronization event. In all figures, the blue boxes indicate annotated protein coding regions and the gray boxes indicate UTRs. Red histograms under the retrogene structures diagram the numbers of mRNAseq reads associated with the retrogenes.
Expression Divergence between Retrogenes and Their Parental Genes
To investigate the transcriptional activity of the 150 retrocopies and their parental genes, we sampled seven tissues of rice with mRNAseq. In total, we generated 278,060,869 sequence reads, of which 60% (166,511,914) were uniquely mapped on the japonica genome (table 1). The mapped reads comprised ∼7.8 Gbp in total and ranged from 887 Mbp to 1.4 Gbp among the seven tissues. On average, our mRNAseq data resulted in ∼21-fold coverage of the rice transcriptome for each tissue (table 1).
Table 1.
Number of Short-Reads Derived by RNAseq and Summary of Mapping Results
| Tissue | Total No. of Short Reads | No. of Uniquely Mapped Reads | Total Nucleotide Length of the Mapped Reads (bp) | Fold Coverage of the Uniquely Mapped Reads against RAP-Annotated Regions |
| Callus | 36,642,482 | 23,506,559 | 1,071,781,348 | 20.2 |
| Leaf | 33,537,231 | 19,334,815 | 886,753,671 | 16.7 |
| Panicle (before flowering) | 39,438,983 | 22,845,156 | 1,051,085,731 | 19.8 |
| Panicle (after flowering) | 46,071,816 | 28,541,187 | 1,392,167,801 | 26.2 |
| Root | 33,307,824 | 20,493,666 | 927,208,012 | 17.5 |
| Seed | 48,619,562 | 27,646,800 | 1,364,148,154 | 25.7 |
| Shoot | 40,442,971 | 24,143,731 | 1,135,190,328 | 21.4 |
| Total | 278,060,869 | 166,511,914 | 7,828,335,045 | 147.4 |
We further assessed coverage by examining expression in the 22,973 loci that are both supported by rice flcDNAs (Kikuchi et al. 2003) and unrelated in sequence to TEs. By applying a background level determined from intergenic regions (see Materials and Methods), we determined that 22,279 (or 97.0%) of the loci were expressed in one or more tissues. Broadening the gene sample to include ab initio predicted genes, we detected expression in 32,164 (76.9%) of 41,847 non-TE–related loci. These results suggest that our mRNAseq data are sufficient to examine the rice transcriptome, including retrocopies.
Of our 150 retrocopies, 100 were expressed above the background level in one or more tissues; we deem these 100 transcribed retrocopies as retrogenes. Of the remaining 50 retrocopies, 32 were covered by one or more mRNAseq reads or by other transcriptional evidence, such as cDNA and expressed sequence tags (ESTs), which suggests that these retrocopies could be transcribed either at levels below the detection limits of our study or in tissues not included in our study. The remaining 18 retrocopies had no transcriptional evidence from any data source.
We sought to compare expression patterns between the 100 retrogenes and their parents, but 13 parental genes had no expression in our mRNAseq data. For the remaining 87 gene pairs, we investigated patterns and levels of expression. For expression level, we calculated the maximum number of RPK (mRPK) among the seven tissues for each gene. The mean mRPK value of the retrogenes (880.2) was significantly lower than that of parental genes (3,283.9) (WRS test, P < 0.005). Parental genes were also expressed at significantly higher levels, on average, than all other expressed, non-TE–related genes (1,5940.0; WRS test, P < 10−3). In contrast, mean mRPK values did not differ between retrogenes and all other non-TE–related genes (P > 0.1). Thus, parental genes are expressed at significantly higher levels than the genomic average.
To compare expression patterns between a retrogene and its parental gene, we calculated the Pearson product-moment correlation coefficient (R) between genes, using observed RPK values from the seven tissues. Thirty-eight of the 87 retroparent pairs exhibited slightly or strongly negative correlations, with the most negative being −0.58 (supplementary table S1, Supplementary Material online). However, 56% had R > 0.00, and some R values were strongly positive (supplementary table S1, Supplementary Material online). For example, ten retrocopies had R values > 0.90, and an additional 31 pairs had R values > 0.50. We graphed expression values for some of the highly correlated pairs (fig. 3), visually verifying that these retrogenes are expressed at lower levels than their parental genes but in a similar pattern across tissues.
FIG. 3.—

Diagram of gene expression counts for the parental gene (solid line) and the retrogene (dashed line) in seven tissues (CA = callus, LE = leaf, PBF = panicle before flowering, PAF = panicle after flowering, RO = root, SE = seed, SH = shoot). The diagrams beneath each graph represent the structure of the parental gene (top) and its retrocopy (bottom).
Given the correlation in gene expression between retrogenes and their parental genes, we next investigated the relationship between R and evolutionary time, as measured by molecular divergence (dS). To do this, we followed precedent and first transformed R by the equation Y = log((1 + R)/(1 − R)) (Gu et al. 2002; Makova and Li 2003; Li et al. 2009). Plotting Y against dS produced a clear negative correlation (fig. 4; R = −0.30, P = 0.005), implying that newer retrogenes are expressed more like their parental genes than are older retrogenes. To verify the significance of the negative correlation (fig. 4), we bootstrapped samples from the original data and calculated R for each of 10,000 resampled data sets. The resulting distribution was significantly negatively skewed, and the 95% confidence intervals (CIs) did not include zero. In fact, only 19 (0.2%) of the 10,000 resampled data sets had a correlation coefficient ≥ 0.0 (supplementary fig. S1, Supplementary Material online). To sum, the expression of retrogenes and parental genes tend to be positively correlated, and this correlation degrades as a function of evolutionary time, as measured by dS.
FIG. 4.—

The relationship between the correlation in gene expression between pairs of genes, as represented by Y [=log((1 + R)/(1 − R))], and molecular divergence (dS). (A) RNA-based duplicates (Retrogenes). (B) DNA-based duplicates.
Expression Divergence in DNA- and RNA-Based Duplicates
Previous studies have established negative correlations between Y and dS for DNA-based duplicates (Gu et al. 2002; Makova and Li 2003; Li et al. 2009). Given that our RNA-based duplicates exhibit a similar relationship, we sought to determine whether the dynamics of expression divergence differ substantially between RNA-based and DNA-based duplicates. To identify pairs of DNA-based duplicates for this analysis, we defined gene families and culled independent pairs of genes from these families (see Materials and Methods; Makova and Li 2003), resulting in 3,420 gene pairs. We then estimated dS between pairs of duplicated genes and measured Y between duplicates using our mRNAseq data.
The analysis of DNA-based duplicates yielded two notable observations. First, the mean of across 3,420 DNA-based duplicates was 0.99 compared with for the 87 RNA-based duplicates. However, this difference was not significant (WRS test, P > 0.1). To assess whether these values were significantly different from random noise, we randomly selected 87 and 3,420 pairs of genes from the set of 32,164 non-TE–related genes and calculated for these data sets. After repeating the analysis 10,000 times, we found that values of 0.81 and 0.99 were much higher than the random expectation (P < 10−3 and P < 10−4, respectively, supplementary fig. S2, Supplementary Material online). Second, Y and dS were negatively correlated for the DNA-based duplicates (R = −0.18; P < 10−15), as they were for the RNA-based duplicates (fig. 4). To assess whether the correlations in figure 4A and B were statistically different, we subsampled from the pairs of 3,420 DNA-based duplicates to produce 10,000 data sets of 87 gene pairs. We then calculated the correlation R between Y and dS for each of these data sets to estimate a CI. The interval include −0.30, indicating that the correlation between Y and dS is statistically indistinguishable (P = 0.12) and suggesting that gene expression diverges at similar rates for RNA- and DNA-based duplicates.
Promoter Expression and Divergence
What might cause the correlation in expression between a retrogene and its parent? One possible mechanism is the co-retroposition of promoter sequences with coding regions (Okamura and Nakai 2008). If promoter regions are retroposed, the retrogene and its parental gene would be expected to initially have similar expression patterns.
We assessed the probability of this mechanism in two ways. First, we investigated whether the promoter region of parental genes is transcribed. We limited the analysis to 52 parental genes that were validated by rice flcDNAs and also had 500 bp upstream sequences that did not overlap with other genes. For these 500 bp regions, the average RPK summed across tissues was 124, which is higher than the average summed RPK (70) of the 1,885 intergenic regions (see Materials and Methods and supplementary table S2, Supplementary Material online). Thus, there is evidence for leaky transcription of the promoter regions of parental genes, suggesting that retroposition of promoters is possible. However, the parental genes have no obvious tendency toward aberrantly leaky expression of regulatory regions compared with other genes because the average expression of 500 bp upstream regions was slightly higher (148 RPK) for a sample of 15,480 nonparental genes.
Second, we examined upstream sequences to search for sequence similarity between retrogenes and parental genes. There were 36 pairs in which both retrogene and parental gene were validated by rice flcDNAs and had 500 bp 5′ regions that did not overlap with adjacent genes. For these 36 pairs as well as 2,602 validated pairs of DNA duplicates, we examined divergence of the 500 bp upstream sequences with dSM (Castillo-Davis et al. 2004). The dSM metric varies from 0–1, with higher values representing greater divergence in motifs. By this metric, upstream sequences of retrogene pairs (mean dSM = 0.63) were more highly diverged than those of duplicate gene pairs (mean dSM = 0.58; WRS P < 0.05). Moreover, the dSM values for retrogene pairs did not differ significantly from pairs of rice genes chosen at random from throughout the genome (data not shown); in other words, as a group, the upstream sequence of retroparent pairs were no more closely related than randomly chosen genomic regions.
Expression and dS Analyses in A. thaliana
To begin to assess the generality of our observations, we analyzed expression correlations and molecular divergence in A. thaliana retrocopies (Zhu et al. 2009). (Retrocopies have also been characterized in poplar [Zhu et al. 2009], but only A. thaliana has sufficient genomic resources to analyze expression at this time.) We first identified the parental genes of 69 retrocopies previously identified by Zhang et al. (2005) and then estimated dS between retroparent pairs. The resulting distribution of dS revealed a peak encompassing dS ∼0.4 to ∼0.6, with few young retrocopies (fig. 5). We also compared expression patterns between retrogenes and parental genes using microarray-based expression data (Schmid et al. 2005; Matsuda et al. 2010). For the 24 retroparent pairs on the microarray, was 0.36, a value significantly larger than mean values derived from randomly generated data sets (P < 0.05). In additional, Y was negatively correlated (R = −0.13) with dS (fig. 5). The correlation was not significant (P > 0.1), but the lack of significance may be due to low sample size (n = 24).
FIG. 5.—

Analyses of retrocopies from Arabidopsis thaliana. (A) The distribution of dS between 48 retrocopies and their parental genes. (B) The relationship between Y [=log((1 + R)/(1 − R))] and molecular divergence (dS) for 24 retroparent pairs with available expression data.
Discussion
We detected 150 retrocopies in the japonica rice genome, which is far fewer than the 1,235 identified by Wang et al. (2006). The discrepancies between studies is due in part to the more stringent criteria applied in our study. First, we retained only the homologous hits between parental and retrogenes that yield dS < 2.0; 662 of 1,235 of the retrocopies from Wang et al. (2006) fell into this category. Second, we screened carefully for TE-related sequences (see Materials and Methods), to avoid possible misinferences due to misannotation or TE activity. Reanalyzing the data of Wang et al. (2006)—which was based on earlier less complete genome annotations—we found that parental cDNA sequences of 216 of the 662 retrocopies with dS < 2.0 contained masked repetitive sequences. Third, our pool of potential parental genes was more stringent. For example, we disallowed the use of genes that had only one intron and a downstream poly-A tract; more than 150 of the parental genes of the 446 non-TE–related retrocopies from Wang et al. would have been disregarded on this basis. Fourth, we did not allow retrocopies and their source genes to be tandemly duplicated because retrocopies are expected to integrate randomly in the genome rather than proximately (Zhang et al. 2002). In contrast, 126 of the Wang et al. non-TE–related retrocopies are within 100 kb of their parental gene; most of these pairs have low dS (<0.4) values, suggesting that gene conversion, which occurs more often between genes on the same chromosome (Mondragon-Palomino and Gaut 2005), may have affected evolutionary divergence. Finally, our homology criteria were stricter. We required ≥35% identity over ≥70% of the parental genes, whereas Wang et al. (2006) did not set an identity threshold.
Given that careful searches of the A. thaliana genome identified only 69–83 retrocopies (Zhang et al. 2005; Zhu et al. 2009), our total of 150 retrocopies seems reasonable for the ∼3-fold larger rice genome. Although our total estimated number of retrocopies in the japonica genome could be conservative, the estimate of 1,235 from Wang et al. (2006) may have been, in retrospect, too liberal.
Rice Retrocopies Tend to be Old
The dS distribution for our retrocopies produces a peak of divergence in the range from ∼0.5 to 0.9 (fig. 1). Assuming that synonymous substitutions are neutral and evolve at a rate similar to other genes (Gaut et al. 1996), this dS distribution corresponds to insertion times of ∼40 to 70 Myr. This age distribution is surprising, for two reasons. First, our conservative approach should be biased toward identifying fairly new retrocopies that have had little time to diverge from their parental genes. Second, if retrocopies are generated and eliminated in a constant manner, then younger retrocopies should be more frequently observed than older retrocopies (Lynch and Conery 2000). Taken together, the dS distribution suggests that the frequency of successful retroposition events was higher ∼40 to ∼70 Ma than in the more recent past.
The salient question is “Why?” We can think of two possibilities, neither of which can be proven at this time. The first is that the retroelements responsible for retroposition have decreased in activity over the last ∼50 Myr. Clearly some retrotransposons have been active recently. For example, many long terminal repeat (LTR) retrotransposon insertions are <∼3 Myr old (Wicker and Keller 2007; Baucom et al. 2009). However, it is difficult to conclude from this information whether LTR activity has decreased, increased, or remained the same for ∼50 Myr because older insertions are expected to be deleted rapidly from the rice genome (Ma and Bennetzen 2004). In mammals, gene retroposition is often attributed to long interspersed nuclear elements (LINEs) (Esnault et al. 2000). The extant rice genome has very few LINEs, and LINE transcriptional activity is relatively weak compared with other TEs in the rice genome (Jiao and Deng 2007). If LINEs were more active in the past, it may explain the peak of old retroposition events.
The second possibility is that successful rice retroposition events are associated with a period of genomic flux following a whole genome duplication (WGD) event ∼50 to 70 Ma (Paterson et al. 2004; Schlueter et al. 2004; Fawcett et al. 2009). The A. thaliana data provides (weak) support for this interpretation because the dS peak from ∼0.4 to 0.6 (fig. 5) corresponds to a date of 20–30 Myr (Koch et al. 2000), and this range encompasses the earliest dates estimated for the most recent polyploidy event in the A. thaliana lineage (Beilstein et al. 2010). Importantly, the two possibilities—retrotransposon activity and WGD events—may be related because polyploidy has long been recognized as a genomic stress that could promote TE activity (reviewed in Tenaillon et al. 2010).
Patterns of Retrogene Expression
Two-thirds of the 150 rice retrocopies are expressed in our mRNAseq data. This is probably an underestimate of the true proportion of expressed retrocopies because the number of tissues (7) and experimental conditions (1) were limited. Indeed, another 32 of the 150 retrocopies have some mRNAseq reads and/or EST evidence suggesting they may be expressed. The total proportion of expressed retrocopies in rice (66–88%) contrasts starkly with human retrocopies, of which only 30% are expressed (Vinckenbosch et al. 2006). One has to be careful with comparisons across studies because different studies use different methods to identify retrocopies and to assess expression. Nonetheless, the contrast between rice and humans contributes to the overarching impression that the proportion of expressed rice retrocopies is relatively high.
Studies in human and fruitflies have shown that retrogenes are expressed in more tissue-specific manners than their parental genes, and several retrocopies have evolved testis-specific expression (Marques et al. 2005; Vinckenbosch et al. 2006; Bai et al. 2007). Our rice data paint a less clear picture. For example, our retrogenes are expressed in significantly fewer tissues than their parental genes (4.8 vs. 5.6 tissues; WRS P < 0.05), but the retrogenes as a group are neither more tissue-specific (table 2) nor exhibit lower Shannon entropies (a measure of the breadth of expression) than their parental genes (data not shown).
Table 2.
Comparison of the Number of Tissue Specific Genes in Each Tissue between Retrogenes and Parental Genes
| Retrogene | Parental Gene | ||||
| Tissue | No. of Tissue-Specific Genesa | No. of Nonspecific Genes | No. of Tissue-Specific Genesa | No. Nonspecific Genes | P Value |
| Callus | 15 | 72 | 17 | 66 | 0.60 |
| Leaf | 13 | 74 | 20 | 63 | 0.13 |
| Panicle 1b | 24 | 63 | 27 | 56 | 0.48 |
| Panicle 2b | 26 | 61 | 32 | 51 | 0.24 |
| Root | 34 | 53 | 26 | 57 | 0.29 |
| Seed | 13 | 74 | 12 | 71 | 0.93 |
| Shoot | 22 | 65 | 14 | 69 | 0.18 |
Tissue-specific genes are defined based on Ueda's Akaike's Information Criterion–based method.
Panicle 1, panicle before flowering; panicle 2, panicle after flowering.
However, both expression patterns and some structural characteristics hint that retrogenes could be filling new functions. For example, 29 of our 87 retrogenes are expressed in a new tissue—that is, a tissue in which their source gene is not expressed. Similarly, 13 parental genes were not expressed in our data, when their retrocopy was expressed. With regard to structure, full-length cDNA suggest that some retrocopies contain new exons and introns (fig. 2). The intronization of exons has been identified previously in retroposed genes of mammals (Szczesniak et al. 2011), but the functional implications of intronization are not yet known.
Because retrogenes are typically thought to acquire new regulatory regions, our a priori expectation was that expression patterns would be completely uncorrelated between retrogenes and parental genes. In contrast to our expectation, expression for most retroparent pairs is positively correlated (fig. 4 and supplementary table S1, Supplementary Material online) and sometimes strongly so (fig. 3). Moreover, expression diverges as a function of molecular divergence (dS) at a rate statistically indistinguishable from that of DNA-based duplicates (fig. 4; Li et al. 2009). Our power to distinguish differences may be limited due to small sample sizes of both retrocopies and tissues, but the similarity in expression divergence between RNA- and DNA-based duplicates is surprising nonetheless.
One possible mechanism underlying expression correlation between a retrogene and its source is the co-transcription and co-retroposition of 5′ promoter sequences along with coding regions (Okamura and Nakai 2008). Consistent with this mechanism, we found that upstream sequences of parental genes are, indeed, transcribed above background levels. However, parental genes have lower levels of upstream expression than the genomic average. In addition, our analysis of sequence similarity in upstream regions yielded little evidence for homology; by the measure dSM, retrogene and parental gene are no more closely related in sequence than pairs of genes taken at random from throughout the genome. Our results are similar to those from Drosophila, for which there was no clear sequence homology in promoter regions between retrogenes and their source (Bai et al. 2008). Based on these observations, it seems unlikely that the most common cause of correlated expression is retroposition of 5′ regulatory regions from the parental gene.
This leaves three possibilities for the genesis of retrogene regulation: 1) that retrogenes acquire de novo regulatory elements, 2) that retrogenes use nearby genes to drive expression, or 3) that regulatory elements are embedded within retroposed regions, such as exons and UTRs. Each of these may be true to some extent. For example, previous studies have shown that retrogenes tend to be located near other genes (Vinckenbosch et al. 2006), suggesting that nearby genes drive retrogene regulation. Similarly, the 100 rice retrogenes expressed in mRNAseq data are significantly closer (mean = 3.7 kb) to annotated genes than the 50 nonexpressed retrocopies (mean = 7.7 kb) (WRS test, P < 0.01). Yet, the acquirement of nearby promoters is unlikely to produce correlated expression patterns. Of these three possibilities, only one—embedded regulatory elements within the transcribed region—provides a mechanism to explain the correlated patterns of expression between retrogenes and parental genes.
The Characteristics of Successful Retroposition Events in Plant Genomes
It seems likely that the retrocopies identified in rice and A. thaliana by computational approaches represent only a subset of retroposition events because most events are probably lost quickly to evolutionary history. Our studies provide information about the general characteristics that may make them successful. First, in rice, these events are most likely to arise from genes that are highly expressed. Second, they are biased for events that lead to similar expression patterns, at least initially, between the retrogene and its source. Retrocopies with similar expression may be favored because altered expression patterns usually confer deleterious effects. (As an extreme example, ectopic expression of homeobox genes results in abnormal leaf development [Matsuoka et al. 1993; Lincoln et al. 1994; Muehlbauer et al. 1999]). The bias to retain events with correlated expression patterns may favor the retroposition of genes that have embedded regulatory elements or leaky transcription from upstream regions, although we believe the latter to be uncommon. Third, successful events tend to occur at certain times in genomic history. It is not hard to imagine that RNA-based gene duplication events may be more successful at a time when DNA-based duplications are also rampant—for example, during the aftermath of a WGD event.
Supplementary Material
Supplementary figures S1 and S2 and Supplementary tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors are grateful to S. Takuno, L. Yang, O. Tenaillon, and M. Tenaillon for comments and suggestions. This work was supported by Grant-in-Aid for Young Scientists (B) (20710149) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, Genomics for Agricultural Innovation (RTR0001) from the Ministry of Agriculture, Forestry and Fisheries (MAFF) of Japan, and National Science Foundation grant DEB-0723860 to B.S.G.
References
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, et al. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 2007;8:R11. doi: 10.1186/gb-2007-8-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, et al. Evolutionary origin of regulatory regions of retrogenes in Drosophila. BMC Genomics. 2008;9:241. doi: 10.1186/1471-2164-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucom RS, et al. Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res. 2009;19:243–254. doi: 10.1101/gr.083360.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilstein MA, et al. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2010;107:18724–18728. doi: 10.1073/pnas.0909766107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betran E, et al. Retroposed new genes out of the X in Drosophila. Genome Res. 2002;12:1854–1859. doi: 10.1101/gr.604902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Davis CI, et al. cis-Regulatory and protein evolution in orthologous and duplicate genes. Genome Res. 2004;14:1530–1536. doi: 10.1101/gr.2662504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaparro C, et al. RetrOryza: a database of the rice LTR-retrotransposons. Nucleic Acids Res. 2007;35:D66–D70. doi: 10.1093/nar/gkl780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, et al. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- Esnault C, et al. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24:363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- Fawcett JA, et al. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc Natl Acad Sci U S A. 2009;106:5737–5742. doi: 10.1073/pnas.0900906106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS, et al. Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci U S A. 1996;93:10274–10279. doi: 10.1073/pnas.93.19.10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, et al. Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet. 2002;18:609–613. doi: 10.1016/s0168-9525(02)02837-8. [DOI] [PubMed] [Google Scholar]
- Hanada K, et al. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 2008;148:993–1003. doi: 10.1104/pp.108.122457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, et al. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Deng XW. A genome-wide transcriptional activity survey of rice transposable element-related genes. Genome Biol. 2007;8:R28. doi: 10.1186/gb-2007-8-2-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota K, et al. Detection of genes with tissue-specific expression patterns using Akaike's information criterion procedure. Physiol Genomics. 2003;12:251–259. doi: 10.1152/physiolgenomics.00153.2002. [DOI] [PubMed] [Google Scholar]
- Kadota K, et al. ROKU: a novel method for identification of tissue-specific genes. BMC Bioinformatics. 2006;7:294. doi: 10.1186/1471-2105-7-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, et al. RNA-based gene duplication: mechanistic and evolutionary insights. Nat Rev Genet. 2009;10:19–31. doi: 10.1038/nrg2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, et al. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science. 2003;301:376–379. doi: 10.1126/science.1081288. [DOI] [PubMed] [Google Scholar]
- Koch MA, et al. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae) Mol Biol Evol. 2000;17:1483–1498. doi: 10.1093/oxfordjournals.molbev.a026248. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, et al. Expression pattern divergence of duplicated genes in rice. BMC Bioinformatics. 2009;10(Suppl 6):S8. doi: 10.1186/1471-2105-10-S6-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln C, et al. A knotted1-like homeobox gene in Arabidopsis is expressed in the vegetative meristem and dramatically alters leaf morphology when overexpressed in transgenic plants. Plant Cell. 1994;6:1859–1876. doi: 10.1105/tpc.6.12.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Langley CH. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science. 1993;260:91–95. doi: 10.1126/science.7682012. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101:12404–12410. doi: 10.1073/pnas.0403715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova KD, Li WH. Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res. 2003;13:1638–1645. doi: 10.1101/gr.1133803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques AC, et al. Emergence of young human genes after a burst of retroposition in primates. PLoS Biol. 2005;3:e357. doi: 10.1371/journal.pbio.0030357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda F, et al. AtMetExpress development: a phytochemical atlas of Arabidopsis development. Plant Physiol. 2010;152:566–578. doi: 10.1104/pp.109.148031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, et al. Expression of a rice homeobox gene causes altered morphology of transgenic plants. Plant Cell. 1993;5:1039–1048. doi: 10.1105/tpc.5.9.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, et al. Massive parallel sequencing of mRNA in identification of unannotated salinity stress-inducible transcripts in rice (Oryza sativa L.) BMC Genomics. 2010;11:683. doi: 10.1186/1471-2164-11-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon-Palomino M, Gaut BS. Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol Biol Evol. 2005;22:2444–2456. doi: 10.1093/molbev/msi241. [DOI] [PubMed] [Google Scholar]
- Mosteller F, Tukey J. Exploratory data analysis and regression. Reading (MA): Addison-Wesley; 1977. [Google Scholar]
- Muehlbauer GJ, et al. Ectopic expression of the maize homeobox gene liguleless3 alters cell fates in the leaf. Plant Physiol. 1999;119:651–662. doi: 10.1104/pp.119.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. New York: Springer; 1970. [Google Scholar]
- Okamura K, Nakai K. Retrotransposition as a source of new promoters. Mol Biol Evol. 2008;25:1231–1238. doi: 10.1093/molbev/msn071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K. Establishment of a high efficiency Agrobacterium-mediated transformation system of rice (Oryza sativa L.) Plant Sci. 2009;176:522–527. doi: 10.1016/j.plantsci.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Paterson AH, et al. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc Natl Acad Sci U S A. 2004;101:9903–9908. doi: 10.1073/pnas.0307901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, et al. Comparison of DNA sequences with protein sequences. Genomics. 1997;46:24–36. doi: 10.1006/geno.1997.4995. [DOI] [PubMed] [Google Scholar]
- Pond SL, et al. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Potrzebowski L, et al. Chromosomal gene movements reflect the recent origin and biology of therian sex chromosomes. PLoS Biol. 2008;6:e80. doi: 10.1371/journal.pbio.0060080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince VE, Pickett FB. Splitting pairs: the diverging fates of duplicated genes. Nat Rev Genet. 2002;3:827–837. doi: 10.1038/nrg928. [DOI] [PubMed] [Google Scholar]
- Robinson MD, et al. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso L, et al. Birth and rapid subcellular adaptation of a hominoid-specific CDC14 protein. PLoS Biol. 2008a;6:e140. doi: 10.1371/journal.pbio.0060140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso L, et al. Mitochondrial targeting adaptation of the hominoid-specific glutamate dehydrogenase driven by positive Darwinian selection. PLoS Genet. 2008b;4:e1000150. doi: 10.1371/journal.pgen.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- Sachs J. Robust dual scaling with Tukey's biweight. Appl Psychol Meas. 1994;18:301–309. [Google Scholar]
- Schlueter JA, et al. Mining EST databases to resolve evolutionary events in major crop species. Genome. 2004;47:868–876. doi: 10.1139/g04-047. [DOI] [PubMed] [Google Scholar]
- Schmid M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37:501–506. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- Shiao MS, et al. Origins of new male germ-line functions from X-derived autosomal retrogenes in the mouse. Mol Biol Evol. 2007;24:2242–2253. doi: 10.1093/molbev/msm153. [DOI] [PubMed] [Google Scholar]
- Suyama M, et al. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesniak MW, et al. Primate and rodent specific intron gains and the origin of retrogenes with splice variants. Mol Biol Evol. 2011;28:33–37. doi: 10.1093/molbev/msq260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, et al. The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008;36:D1028–1033. doi: 10.1093/nar/gkm978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, et al. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 2010;15:471–478. doi: 10.1016/j.tplants.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Vanin EF. Processed pseudogenes: characteristics and evolution. Annu Rev Genet. 1985;19:253–272. doi: 10.1146/annurev.ge.19.120185.001345. [DOI] [PubMed] [Google Scholar]
- Vinckenbosch N, et al. Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci U S A. 2006;103:3220–3225. doi: 10.1073/pnas.0511307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, et al. Origin of sphinx, a young chimeric RNA gene in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2002;99:4448–4453. doi: 10.1073/pnas.072066399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, et al. High rate of chimeric gene origination by retroposition in plant genomes. Plant Cell. 2006;18:1791–1802. doi: 10.1105/tpc.106.041905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Keller B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007;17:1072–1081. doi: 10.1101/gr.6214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R, et al. Genome-wide analysis reveals strong correlation between CpG islands with nearby transcription start sites of genes and their tissue specificity. Gene. 2005;350:129–136. doi: 10.1016/j.gene.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. The Genomes of Oryza sativa: a history of duplications. PLoS Biol. 2005;3:e38. doi: 10.1371/journal.pbio.0030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci U S A. 1998;95:3708–3713. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Computational identification of 69 retroposons in Arabidopsis. Plant Physiol. 2005;138:935–948. doi: 10.1104/pp.105.060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, et al. Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res. 2002;12:1466–1482. doi: 10.1101/gr.331902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, et al. Extensive structural renovation of retrogenes in the evolution of the Populus genome. Plant Physiol. 2009;151:1943–1951. doi: 10.1104/pp.109.142984. [DOI] [PMC free article] [PubMed] [Google Scholar]