

Abstract
The Pd-catalyzed asymmetric allylic alkylation (AAA) is one of the most useful and versatile methods for asymmetric synthesis known in organometallic chemistry. Development of this reaction over the past 30 years has typically relied on the use of an allylic electrophile bearing an appropriate leaving group to access the reactive Pd(π-allyl) intermediate that goes on to the desired coupling product after attack by the nucleophile present in the reaction. Our group has been interested in developing alternative approaches to access the reactive Pd(π-allyl) intermediate that does not require the use of an activated electrophile, which ultimately generates a stoichiometric byproduct in the reaction that is derived from the leftover leaving group. Along these lines, we have demonstrated that allenes can be used to generate the reactive Pd(π-allyl) intermediate in the presence of an acid cocatalyst, and this system is compatible with nucleophiles to allow for formation of formal AAA products by Pd-catalyzed additions to allenes. This article describes our work regarding the use of oxindoles as carbon-based nucleophiles in a Pd-catalyzed asymmetric addition of oxindoles to allenes (Pd-catalyzed hydrocarbonation of allenes). By using the chiral standard Trost ligand (L1) and 3-aryloxindoles as nucleophiles, this hydrocarbonation reaction provides products with two vicinal stereocenters, with one being quaternary, in excellent chemo-, regio-, diastereo-, and enantioselectivities in high chemical yields.
Introduction
Catalytic asymmetric allylic alkylation is a powerful and versatile method for the construction of chiral organic molecules in nonracemic form. The development of this reaction has been examined by numerous researchers and the nucleophile scope has been developed to allow for the use of carbon, nitrogen, oxygen, sulphur, and phosphorous nucleophiles to construct C-C, N-C, O-C, S-C, and P-C bonds, respectively, in an asymmetric fashion.1 Furthermore, because of the usefulness of the AAA process, the mechanism of the Pd-catalyzed AAA reaction has been widely explored (Scheme 1).1 In the general reaction scheme, an allylic electrophile bearing an appropriate leaving group (LG) is reacted with a nucleophile (NuH) in the presence of a chiral Pd-complex as the catalyst. Ionization of the allylic leaving group by the Pd(0) salt leads to the electrophilic Pd(π-allyl) complex that undergoes nucleophilic attack by the nucleophile to regenerate the active Pd(0) catalyst and form the desired Nu-C bond. It is important to note however, that often times a stoichiometric amount of base is required to deprotonate Nu-H to generate the reactive nucleophile. This then in turn leads to formation of a stoichiometric amount of the conjugate acid, which remains at the end of the AAA reaction. Additionally, because the reactive Pd(π-allyl) intermediate in these reactions must be generated from an allylic electrophile bearing the appropriate leaving group, the AAA reaction also generates byproducts derived from the leaving group in addition to the desired Nu-C bond formation.
Scheme 1.

General Pd-Catalyzed AAA Reaction
Due to the ever increasing challenge in organic chemistry to prepare chiral molecules in enantiomerically pure form while conserving valuable natural resources, our group has been focused on the development of new atom economical2 methods for asymmetric synthesis.3 According to the philosophy of atom economy,2 the most efficient reactions are coupling reactions whereby all the atoms in the starting materials remain in the desired product, and any additives required to affect the transformation are used in only catalytic amounts. Because the general Pd-catalyzed AAA reaction produces byproducts resulting from the leaving group and because a stoichiometric amount of base is often present, the AAA reaction does not completely fit the ideal definition of atom economy. In an effort to address these issues in Pd-catalyzed AAA chemistry, our group4 and others5 have demonstrated that the reaction products of AAA can be accessed through a mechanistically distinct reaction: the Pd-catalyzed additions of nucleophiles to allenes6 (Scheme 2). In this process, an acid co-catalyst is used in conjunction with the Pd(0) catalyst to generate a Pd(II)-hydride intermediate that goes on to hydropalladate the allene and generate an analogous Pd(π-allyl) complex to that formed in the AAA reaction. This intermediate can then be attacked by Nu-H to generate the formal AAA product while regenerating the acid co-catalyst.4 Thus, this Pd-catalyzed addition of nucleophiles to allenes (Scheme 2) represents an atom-economical method to access formal AAA adducts.
Scheme 2.

General Pd-Catalyzed Addition of Nucleophiles to Allenes
In an effort to extend the scope of nucleophiles that participate in the Pd-catalyzed addition of nucleophiles to allenes, we decided to begin exploring oxindoles as nucleophiles. Importantly, 3,3-disubstituted oxindoles are prevalent in a wide array of biologically relevant natural products (Scheme 3).7 Because of the importance of this structural motif, and due to the significant challenge in preparing quaternary stereocenters in an asymmetric fashion,8 there has been a significant interest by the chemical community to develop methodologies that lead to 3,3-disubstituted oxindoles in an asymmetric fashion.9–18 Asymmetric syntheses of these oxindole units have been realized using the intramolecular Heck reaction,9 α-arylation10 or α-alkylation11 reactions, intramolecular acyl migration,12 Pd-catalyzed cyanoamidation,13 Pd- and Mo-catalyzed allylic alkylation,14 aldol15 processes, cycloaddition reactions,16 Claisen rearrangements,17 and through the use of a variety of organocatalytic methods.18 Thus, we reasoned that a catalyst controlled addition of oxindole nucleophiles to allenes would be an important addition to this list of reactions to enable preparation of 3,3-disubstituted oxindoles in nonracemic form. Because this process represents the formal addition of a C-H bond across the olefin of an allene, this reaction has also been called the hydrocarbonation of allenes.5b This article describes our work at developing an asymmetric Pd-catalyzed hydrocarbonation of allenes by oxindoles, which is unique to the other methods listed for the preparation of 3,3-disubstituted oxindoles in that two vicinal stereocenters are formed with high relative and absolute stereochemical control.
Scheme 3.

Natural Products Containing 3,3-Disubstituted Oxindole Motifs
Results and Discussion
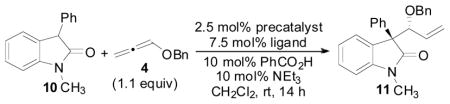
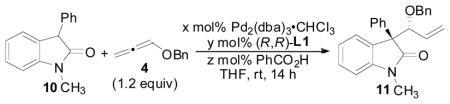
Reaction optimization
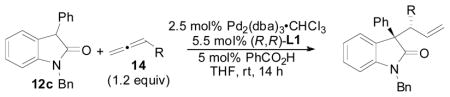
The general reaction (Figure 1) for the allene hydrocarbonation between benzyloxyallene 4 and oxindole 8 would be expected to result in the formation of 9. This process represents a significant challenge when optimizing this reaction in that appropriate conditions must be obtained to control diastereoselectivity, enantioselectivity, and regioselectivity in addition to overall chemical yield. Thus, the nature of the R- and R′-groups of 8, the Pd(0) precatalyst, the chiral ligand employed, the acid co-catalyst, the solvent, the concentration, and any additional additives are variables that can be modulated to obtain the desired reactivity and selectivity. We chose to begin our initial optimization of this reaction by examining the Pd-precatalysts (Table 1) while employing the chiral diphenylphosphino benzoic acid-based ligands (Figure 1, L1–L4) developed in our laboratory as the added chiral ligand.19
Figure 1.

General Allene Hydrocarbonation Reaction with Oxindole Nucleophiles and the Chiral Ligands Examined
Table 1.
Initial Optimization of Pd-Precatalyst and Chiral Ligand
 | |||||
|---|---|---|---|---|---|
| entry | precatalyst | ligand | % yielda | d.r.b | eec |
| 1 | none | none | NR | n.a. | n.a. |
| 2 | [Pd(allyl)Cl]2 | (R,R)-L1 | 68 (93) | 4.2:1 | 66 |
| 3 | [Pd(allyl)Cl]2 | (R,R)-L2 | 60 (82) | 1.4:1 | 65 |
| 4 | [Pd(allyl)Cl]2 | (R,R)-L3 | 65 (88) | 1.6:1 | 63 |
| 5 | [Pd(allyl)Cl]2 | (R,R)-L4 | 74 (88) | 1:2.7 | 15 |
| 6d | [Pd(allyl)Cl]2 | (R,R)-L1 | 75 (88) | 4.2:1 | 66 |
| 7d | Pd(OTFA)2 | (R,R)-L1 | NR | n.a. | n.a. |
| 8d | Pd(OAc)2 | (R,R)-L1 | 61 (95) | 4.6:1 | 67 |
| 9d | Pd2(dba)3-CHCl3 | (R,R)-L1 | 80 (94) | 6.2:1 | 78 |
| 10e | Pd2(dba)3-CHCl3 | (R,R)-L1 | 92 | 6.4:1 | 80 |
Determined by 1H NMR with respect to mesitylene as the internal standard, number in parantheses refers to yield based on unreacted 10.
Determined by 1H NMR of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
Using 1.2 equiv 4.
Using 1.2 equiv of freshly prepared 4.
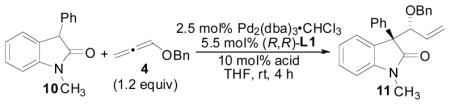
Initial optimization of the precatalyst and ligand was examined with oxindole 10 using benzoic acid as the acid cocatalyst in conjunction with an equimolar amount of NEt3 relative to the PhCO2H cocatalyst (Table 1). We have previously shown that this type of acid/base combination typically gives improved reaction rates when employing nucleophiles that are less acidic compared to Meldrum’s acid derivatives in the allene AAA reaction.4b Gratifyingly, use of the [Pd(allyl)Cl]2 dimer as the precatalyst with the standard Trost ligand (entry 2) led to formation of the desired branched product 11 in good d.r. and in moderate enantioselectivity for our initial attempts at this reaction. Not surprisingly, in the absence of a Pd-precatalyst and ligand (entry 1), no reaction occured. Use of the other commonly employed Trost ligands (entries 3–5) offered no improvements in stereocontrol, and the yield could be improved by increasing the amount of 4 present in the reaction mixture (entry 6). Ultimately, Pd2(dba)3-CHCl3 was identified as the optimal precatalyst for this reaction affording the product in 6.4:1 dr and 80% ee (entry 10).
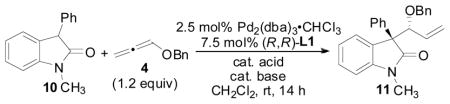
With Pd2(dba)3-CHCl3 and (R,R)-L1 identified as the optimized catalyst system, an examination of mixtures of acid and base additives were conducted (Table 2). Interestingly, the reaction proceeded without any additional acid additive, implying that the oxindole itself is a good enough acid to effect the reaction (entry 1). However, when 10 mol% PhCO2H and 10 mol% NEt3 were employed as additives, both the di-astereo- and enantioselectivity was improved (entry 1 vs 2). Varying the ratio of PhCO2H/NEt3 did not have a significant affect on stereoselectivity (entries 2–4). However, when PhCO2H was used in excess, the enantioselectivity was slightly better (entry 3), whereas when NEt3 was used in excess, the diastereomeric ratio of the product decreased (entry 4). When the base was changed to KOtBu, the reaction did not proceed to full conversion, and the stereoselectivity diminished (entry 5). Lastly, combinations of hippuric acid and KOtBu were examined because this system has previously been shown to be useful in the allene AAA employing azalactone nucleophiles,4b,e however, this led to incomplete conversion along with decreased diastereo- and enantioselectivities (entry 6–7).
Table 2.
Examination of Acid and Base additives in the Allene Hydrocarbonation Reaction
 | |||||
|---|---|---|---|---|---|
| entry | acid (mol%) | base (mol%) | % yielda | d.rb | % eec |
| 1 | none | none | 91 | 2.6:1 | 34 |
| 2 | PhCO2H (10) | NEt3 (10) | 92 | 6.4:1 | 80 |
| 3 | PhCO2H (20) | NEt3 (10) | 92 | 6.1:1 | 83 |
| 4 | PhCO2H (10) | NEt3 (20) | 96 | 5.7:1 | 80 |
| 5 | PhCO2H (10) | KOtBu (5) | 87 (>99) | 5.6:1 | 74 |
| 6 | hippuric acid (10) | KOtBu (5) | 88 (97) | 4.9:1 | 74 |
| 7 | hippuric acid (20) | KOtBu (5) | 78 (98) | 4.5:1 | 75 |
Determined by 1H NMR with respect to mesitylene as the internal standard, number in parantheses refers to yield based on unreacted 10.
Determined by 1H NMR of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
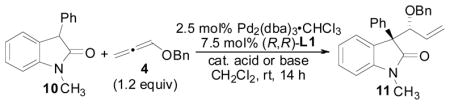
Our observations on the effects of acid and base additives on reaction efficiency described in Table 2 led us to further examine the nature of the acid/base additives employed in the reaction in an effort to improve selectivity (Table 3). Not surprisingly, the acid or base additive employed in the reaction had a significant impact on reaction selectivity; however, it is notable that regardless of the additive used, the reaction proceeds in good to excellent chemical yields.
Table 3.
Effect of Acid Strength on the Allene Hydrocarbonation Reaction
 | |||||
|---|---|---|---|---|---|
| entry | Additive (mol%) | pKaa | % yieldb | d.r.c | % eed |
| 1 | hippuric acid (10) | 3.61 | 76 | 5.9:1 | 77 |
| 2 | PhCO2H (10) | 4.22 (11.1) | 95 | 6.2:1 | 84 |
| 3 | AcOH (10) | 4.76 (12.3) | 92 | 3.4:1 | 55 |
| 4 | tBuOH (10) | 17 (29.4) | 98 | 4.5:1 | 58 |
| 5 | NEt3(10) | 10.75 (9.00)e | 99 | 2.1:1 | 22 |
| 6 | TMGf (10) | (13.6)e | 96 | 1.2:1 | −68 |
| 7 | tBuOK (5) | (29.4)e | 80 | 1.3:1 | −60 |
Value in H2O, number in parantheses refers to value in DMSO.
Determined by 1H NMR.
Determined by 1H NMR of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
pKa of the conjugate acid.
Tetramethylguanidine.
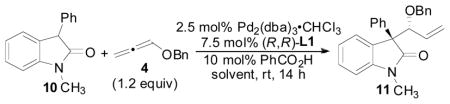
At this point in our optimization studies, the optimal conditions for the allene hydrocarbonation reaction allowed for formation of the desired product in 95% yield, 6.2:1 d.r., and 84% ee (Table 3, entry 2). We next began to examine the effect of solvent on the reaction (Table 4). Indeed, a significant solvent effect was observed. Reactions in polar solvents such as CH3CN, DMSO, and DMF gave full conversion and excellent yields, but very poor stereoselectivities (entries 1–3). In less polar solvents, stereoselectivities were better, but reactions usually did not give full conversion with THF being a notable exception to this trend (entries 4–10). Thus, by using THF as the solvent, not only was excellent yield (94%) obtained, but also excellent diastereo- and enantioselectivity was observed (13:1 d.r. and 84% ee, entry 8).
Table 4.
Solvent Effect on the Allene Hydrocarbonation Reaction
 | ||||
|---|---|---|---|---|
| entry | solvent | % yielda | d.r.b | % eec |
| 1 | MeCN | 98 | 1.1:1 | −18 |
| 2 | DMSO | 96 | 1.4:1 | −17 |
| 3 | DMF | 91 | 1.4:1 | −29 |
| 4 | CH2Cl2 | 88 (92) | 5.8:1 | 80 |
| 5 | dioxane | 74 (86) | 8.1:1 | 44 |
| 6 | Et2O | 81 (86) | 9.6:1 | 64 |
| 7 | DME | 72 (89) | 11:1 | 79 |
| 8 | THF | 94 | 13:1 | 84 |
| 9 | toluene | 74 (84) | 10:1 | 54 |
| 10 | benzene | 72 (89) | 10:1 | 39 |
Determined by 1H NMR with respect to mesitylene as the internal standard; number in parantheses refers to yield based on unreacted 10.
Determined by 1H NMR analysis of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
Effects related to reaction concentration and catalyst loading were next examined in the reaction (Table 5). Addition of catalytic amounts of TBAT or (n-hexyl) 4NBr, additives that are known to promote the rate of π-σ-π equilibration of Pd(π-allyl) intermediates,1,20 was determined to be detrimental to both diastereoselectivity and enantioselectivity (entries 2 and 3, respectively). The effect of the catalyst loading of PhCO2H was examined at 0.1 M concentration using 2.5 mol% Pd2(dba)3-CHCl3 and 7.5 mol% (R,R)-L1 (entries 1 and 4–7). Importantly, lower loadings of PhCO2H led to improvements in both yield and selectivity, while when large quantities of PhCO2H were present, poor conversions and low stereoselectivities were obtained. The Pd-loadings could be dropped to 1.0 mol% with comparable results (entry 8), and a significant concentration effect was also observed (compare entries 5 and 10–13). As the concentration of the reaction was increased, both the diastereo- and enantioselectivity decreased, but at lower concentration, for example 0.01 M (entry 13), the highest stereoselectivity (19:1 d.r. and 93% ee) was obtained. Lastly, the metal:ligand ratio was modulated (compare entries 5, 14, and 15), and a slight enhancement in selectivity was observed when using a metal to ligand ratio of 1:1.1 (entry 15). Additionally, reducing the temperature of the reaction to either 4 °C or −20 °C did not lead to any improvements (entries 16 and 17).
Table 5.
Effects of Reaction Concentration, Catalyst, Ligand, and Acid Loadings on the Allene Hydrocarbonation Reaction
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | Conc.a | x | y | z | % yieldb | d.r.c | % eed |
| 1 | 0.1 M | 2.5 | 7.5 | 10 | 94 | 13:1 | 84 |
| 2e | 0.1 M | 2.5 | 7.5 | 10 | >99 | 4.5:1 | 52 |
| 3f | 0.1 M | 2.5 | 7.5 | 10 | 94 | 10:1 | 80 |
| 4 | 0.1 M | 2.5 | 7.5 | 2.5 | >99 | 11:1 | 82 |
| 5 | 0.1 M | 2.5 | 7.5 | 5 | 97 | 13:1 | 84 |
| 6 | 0.1 M | 2.5 | 7.5 | 50 | 61 (91) | 5.8:1 | 60 |
| 7 | 0.1 M | 2.5 | 7.5 | 100 | 55 (96) | 3.5:1 | −6 |
| 8 | 0.1 M | 0.5 | 1.5 | 1 | 94 | 11:1 | 82 |
| 9 | 0.1 M | 5 | 15 | 10 | >99 | 13:1 | 80 |
| 10 | 0.2 M | 2.5 | 7.5 | 5 | >99 | 10:1 | 74 |
| 11 | 0.5 M | 2.5 | 7.5 | 5 | 96 | 6.5:1 | 64 |
| 12 | 0.05 M | 2.5 | 7.5 | 5 | >99 | 14:1 | 89 |
| 13 | 0.01 M | 2.5 | 7.5 | 5 | 90 | 19:1 | 93 |
| 14 | 0.1 M | 2.5 | 6.0 | 5 | 95 | 14:1 | 87 |
| 15 | 0.1 M | 2.5 | 5.5 | 5 | 97 | 14:1 | 86 |
| 16g | 0.1 M | 2.5 | 5.5 | 5 | 95 | 11:1 | 80 |
| 17h | 0.1 M | 2.5 | 5.5 | 5 | >99 | 4.6:1 | 53 |
With respect to 10.
Determined by 1H NMR with respect to mesitylene as the internal standard; number in parantheses refers to yield based on unreacted 10.
Determined by 1H NMR analysis of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
20 mol% tetrabutylammonium difluorotriphenylsilicate (TBAT) employed.
20 mol% (n-hexyl)4 NBr employed.
Reaction performed at 4 °C.
Reaction performed at −20 °C.
After having optimized conditions in hand that afforded the desired allene hydrocarbonation product in excellent yield, diastereoselectivity, and enantioselectivity (up to 90% yield, 19:1 d.r. and 93% ee: Table 4, entry 13), we lastly wanted to study the effect of the benzoic acid cocatalyst by examining various substituted benzoic acid derivatives in the reaction (Table 6). A slight enhancement in stereoselectivity was observed when using either naphthoic acid or benzoic acid derivatives bearing electron withdrawing groups on the aromatic ring (entries 7–12). Overall, the best results were obtained by using 1-naphthoic acid or para-nitrobenzoic acid (entries 8 and 10). However, since using these two acids only yielded a slight improvement, we chose to keep using benzoic acid as the acid additive for convenience.
Table 6.
Effect of Substituted Benzoic Acid Cocatalysts on the Allene Hydrocarbonation Reaction
 | |||||
|---|---|---|---|---|---|
| entry | acid | pKaa | % yieldb | d.r.c | % eed |
| 1 | PhCO2H | 4.22 | 95 | 13:1 | 84 |
| 2 | p-MeO-C6H4CO2H | 4.90 | 97 | 13:1 | 86 |
| 3 | p-Me-C6H4CO2H | 4.36 | 96 | 13:1 | 84 |
| 4 | p-Ph-C6H4CO2H | -- | 99 | 13:1 | 85 |
| 5 | o-MeO-C6H4CO2H | 4.20 | 93 (97) | 11:1 | 84 |
| 6 | o-Me-C6H4CO2H | 3.91 | >99 | 13:1 | 84 |
| 7 | 2-naphthoic acid | 4.17 | 99 | 15:1 | 85 |
| 8 | 1-naphthoic acid | 3.69 | >99 | 15:1 | 87 |
| 9 | p-Cl-C6H4CO2H | 3.99 | 95 | 15:1 | 84 |
| 10 | p-NO2-C6H4CO2H | 3.44 | 98 | 16:1 | 87 |
| 11 | o-Cl-C6H4CO2H | 2.92 | 90 (95) | 15:1 | 86 |
| 12 | o-NO2-C6H4CO2H | 2.17 | 87 (98) | 15:1 | 86 |
| 13 | C6F5CO2H | -- | 70 (84) | 13:1 | 81 |
Value in H2O.
Determined by 1H NMR with respect to mesitylene as the internal standard; number in parantheses refers to yield based on unreacted 10.
Determined by 1H NMR analysis of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
Reaction scope
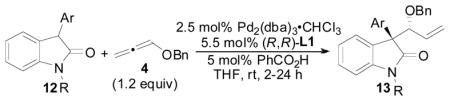
With the optimized reaction conditions in hand, we next examined the substrate scope of the Pd-catalyzed allene hydrocarbonation reaction employing 3-aryloxindoles as nucleophiles (Table 7). Previous Pd- and Mo-catalyzed AAA reactions between allylic substrates and oxindoles have shown that the N-substituent of the oxindole has a significant impact on reactivity and selectivity in the AAA reaction.14 Thus, we first examined various N-substituents on oxindole 12 (entries 1–6). N-alkyl substituted oxindoles gave the highest levels of stereocontrol, with the N-Bn and N-PMB substituted oxindoles affording enhanced stereoselection relative to the N-CH3 substituted oxindole. Additionally, when employing an N-Boc substituted oxindole, poor di-astereoselectivity and enantioselectivity was obtained (entry 5); however, it is notable that by switching to ligand L4, the diastereoselectivity could be improved, favoring the opposite diastereomer,21 albeit as a racemic mixture (entry 6).22
Table 7.
Reaction Scope Employing 3-Aryloxindoles as Nucleophile
 | |||||
|---|---|---|---|---|---|
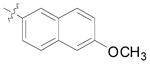
| entrya | R | Ar | % yieldb | d.r.c | % eed |
| 1 | CH3 | Ph (10) | 92 | 13:1 | 87 |
| 2 | H | Ph (12a) | 70 | 1.5:1 | n.d. |
| 3 | PMB | Ph (12b) | >99 | 14:1 | 90 |
| 4 | Bn | Ph (12c) | 95 | 16:1 | 93 |
| 5 | Boc | Ph (12d) | 99 | 1:1.2 | 0 |
| 6e | Boc | Ph (12d) | 99 (92) | 1:8.0 | 0 |
| 7 | Bn | p-Ph-C6H4-(12e) | 96 (95) | 13:1 | 93 |
| 8 | Bn | p-F-C6H4-(12f) | 96 | 13:1 | 90 |
| 9 | Bn | p-Cl-C6H4-(12g) | >99 | 11:1 | 84 |
| 10 | Bn | p-Me-C6H4-(12h) | >99 (93) | 18:1 | 86 |
| 11 | Bn | p-MeO-C6H4-(12i) | 92 (84) | 17:1 | 89 |
| 12 | Bn | p-Me2N-C6H4-(12j) | 86 | 12:1 | 85 |
| 13 | Bn | m-Me-C6H4-(12k) | 92 | 17:1 | 90 |
| 14 | Bn | o-Me-C6H4-(12l) | 40 | 1:1.2 | n.d. |
| 15 | Bn | o-MeO-C6H4-(12m) | 19 | 1:2.0 | n.d. |
| 16 | Bn |
 (12n) |
95 | 15:1 | 93 |
| 17 | Bn |
(12o) |
98 | 6.8:1 | 78 |
| 18f | Bn | (12o) | 90 (91) | 7.8:1 | 83 |
| 19 | Bn |
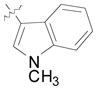 (12p) |
64 | 6.5:1 | 84 |
| 20g,h | Bn | (12p) | (94) | 7.0:1 | 88 |
| 21g,i,j | Bn |
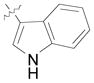 (12q) |
(71) | 7.7:1 | 91 |
| 22g,i,k | PMB |
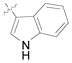 (12r) |
(80) | 7.4:1 | 91 |
0.2 mmol scale (0.1 M) relative to 12 at rt for 2 to 24 h.
Determined by 1H NMR with respect to mesitylene as the internal standard; number in parantheses refers to isolated yield.
Determined by 1H NMR analysis of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
Ligand L4 was used.
p-nitrobenzoic acid was used in place of PhCO2H.
1-naphthoic acid was used in place of PhCO2H.
1.56 mmol scale.
7.5 mol% of (R,R)-L1 was used.
1.73 mmol scale.
2.71 mmol scale.
In general, a wide variety of aryl groups were tolerated in the Pd-catalyzed allene hydrocarbonation reaction affording products in excellent yields, diastereoselectivities, and enantioselectivities. However, one limitation were aryl groups bearing ortho-substitution (entries 14 and 15) whereby these oxindoles gave poor conversion in the reaction, presumably due to increased steric interactions presented by the ortho-substituent. Additionally, it should be pointed out that the opposite diastereomer was slightly preferred in these cases. Notably, heterocyclic aromatic groups were also tolerated well in the reaction (entries 17–22), and the use of p-nitrobenzoic acid or 1-naphthoic acid in place of benzoic acid in these systems was used to provide improved results (entry 17 vs 18 and entry 19 vs 20). Also, it is notable that the aryl substituent on the oxindole can be an unprotected indole (entries 21 and 22) or a thienyl group (entries 17 and 18), which is a testament to the mildness of this methodology.
Since 3-aryloxindole nucleophiles having ortho-substitution on the aromatic ring proceeded with low conversion and poor stereoselectivities using the first set of optimized reaction conditions, we initiated a short optimization study on this class of nucleophile (Table 8). Because we presumed that the nucleophilic attack was the difficult step in the catalytic cycle due to the increased steric hinderance associated with the presence of the ortho-substituent on the aromatic ring, we initially examined solvents of increased polarity since these should help to increase the rate of nucleophilic attack in the reaction. Gratifyingly, by changing the reaction solvent to DMF or MeCN, the reaction with these more hindered nucleophiles could be achieved in excellent yield, diastereoselectivity, and enantioselectivity (entries, 3, 4, and 6). Again, it is important to note that the opposite diastereomer was preferred in these reactions, and the standard Trost ligand was still the optimal chiral ligand for this class of nucleophiles (compare entry 6 vs 7–9).
Table 8.
Reaction Optimization Employing ortho-Substituted Aryl Rings
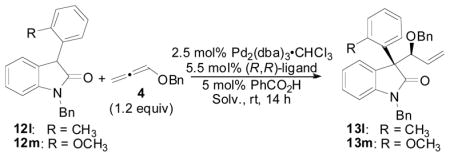 | ||||||
|---|---|---|---|---|---|---|
| Entrya | 12 | ligand | Solv. | % yieldb | d.r.c | % eed |
| 1 | 12l | L1 | THF | 40 | 1:1.2 | -- |
| 2 | 12l | L1 | CH2Cl2 | 42 | 2:1 | -- |
| 3 | 12l | L1 | DMF | 75 | 11:1 | 90 |
| 4 | 12l | L1 | MeCN | >99 (93) | 13:1 | 95 |
| 5 | 12m | L1 | THF | 19 | 1:2 | 11 |
| 6 | 12m | L1 | MeCN | 95 (84) | 10:1 | 95 |
| 7 | 12m | L2 | MeCN | 10 | 2.4:1 | -- |
| 8 | 12m | L3 | MeCN | 84 | 2.5:1 | 65 |
| 9 | 12m | L4 | MeCN | 46 | 1.7:1 | −35 |
Reactions were performed on a 0.2 mmol scale (0.1 M) relative to 12. The absolute stereochemistry of 13l and 13m was not determined
Determined by 1H NMR; number in parantheses refers to isolated yield.
Determined by 1H NMR analysis of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
We next examined the scope of the allene coupling partner in the hydrocarbonation reaction employing 3-aryl oxindole nucleophiles (Table 9). All alkoxyallenes examined (entries 1–3, and 7) participated well in the reaction giving products in excellent yields. In particular, the p-methoxybenzyloxyallene led to excellent diastereo- and enantioselectivity in a similar manner to that when 4 was used (Table 9, entry 1 vs Table 7, entry 4). Alkoxyallenes bearing simple straight chain alkyl groups were tolerated in the reaction affording the desired products in good levels of diastereoselectivity and enantioselectivity (entries 2 and 7). However, alkoxyallenes containing branching (i.e. Cy, entry 3) led to decreased stereoselectivity in the reaction. Other allenes such as allenamides (entries 4 and 5) and a 1,1-disubstituted allene (entry 6) gave no reaction.
Table 9.
Scope of the Allene Coupling Partner
 | ||||
|---|---|---|---|---|
| entrya | allene | % yieldb | d.r.c | % eed |
| 1 |
|
>99 (96) | 17:1 | 87 |
| 2 |
|
>99 | 8.0:1 | 78 |
| 3 |
|
>99 | 2.7:1 | 27 |
| 4 |
|
NR | -- | -- |
| 5 |
|
NR | -- | -- |
| 6 |
|
NR | -- | -- |
| 7e |
|
(62) | 6.5:1 | 86 |
Reactions were performed on a 0.2 mmol scale (0.1 M) relative to 12c.
Determined by 1H NMR with respect to mesitylene as the internal standard; number in parantheses refers to isolated yield.
Determined by 1H NMR analysis of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
Reaction performed using oxindole 12q using 7.5 mol% (R,R)-L1 and 5 mol% 1-naphthoic acid at rt for 63 h.
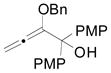
In addition to 3-aryloxindoles, we briefly began to examine the use of 3-alkyloxindoles as nucleophiles for the allene hydrocarbonation reaction (Table 10). When N-CH3 substituted oxindole 15 was subjected to the optimized conditions for the hydrocarbonation reaction employing 3-aryloxindoles, no reaction was observed (entry 1). We hypothesized that this was likely due to the fact that 3-alkyloxindoles are less acidic relative to 3-aryloxindoles, and therefore, a significant concentration of the enol tautomer may not be present to allow for useful rates in the nucleophilic addition step. Thus, to increase the acidity of the oxindole, an N-Boc substituted 3-alkyloxindole was examined (entries 2–7). Indeed, reactivity was improved, however, poor diastereoselectivities were obtained. Furthermore, it was also noted that use of MeCN as solvent gave a significant improvement in reaction yield (entries 6 and 7). Again, the increased polarity of the solvent likely facilitates the reaction by increasing the rate of nucleophilic addition by allowing for improved keto-enol tautomerization. Next, N-alkyl substituted 3-alkyloxindoles were re-examined using MeCN as the solvent (entries 8–14). By employing an N-Bn protected oxindole (entry 10), the reaction proceeded in good yield (87%) with a useful diastereoselectivity (4.2:1 d.r.) and moderate enantioselectivity (59% ee). These results could be slightly improved by increasing the catalyst loading (entry 11). Use of the other known chiral Trost ligands did not lead to any improvements in the reaction (entries 12–14). These results verify that the less acidic 3-alkyloxindoles can be applied as nucleophiles in the allene hydrocarbonation reaction albeit with only moderate levels of enantioselection at this time.
Table 10.
Use of 3-Alkyloxindoles in the Allene Hydrocarbonation
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | Solv. | ligand | % yielda | d.r.b | % eec |
| 1 | CH3 | THF | L1 | NR | -- | -- |
| 2 | Boc | THF | L1 | 96 | 1.4:1 | -- |
| 3 | Boc | THF | L2 | 41 (93) | 1:1.4 | -- |
| 4 | Boc | THF | L3 | 86 (>99) | 1.1:1 | -- |
| 5 | Boc | THF | L4 | 36 (99) | 1.0:1 | -- |
| 6 | Boc | MeCN | L1 | 95 | 1:1.5 | -- |
| 7 | Boc | MeCN | L4 | 92 | 1.5:1 | -- |
| 8 | CH3 | MeCN | L1 | 20 (95) | -- | -- |
| 9d | CH3 | MeCN | L1 | 53 (99) | 4.2:1 | -- |
| 10 | Bn | MeCN | L1 | 87 (98)e | 4.2:1 | 59 |
| 11d | Bn | MeCN | L1 | 84 (95) | 4.4:1 | 63 |
| 12 | Bn | MeCN | L2 | NR | -- | -- |
| 13 | Bn | MeCN | L3 | 10 (90) | -- | -- |
| 14 | Bn | MeCN | L4 | 38 (88) | 1:1.7 | -- |
Determined by 1H NMR using mesitylene as the internal standard; number in parantheses refers to yield based on unreacted 15.
Determined by 1H NMR analysis of the unpurified reaction mixture.
Determined by chiral HPLC analysis.
Reaction performed using 5 mol% Pd2 (dba)3-CHCl3 and 11 mol% (R,R)-L1.
Isolated yield was 80%.
Stereochemical assignment
The relative stereochemistry produced in the allene hydrocarbonation reaction employing oxindoles was assigned by X-ray crystallography. Reaction product 11 could be recrystallized from hexanes/ethyl acetate mixtures to provide crystalline material as a single enantiomer (Scheme 4). Xray analysis confirmed the anti relationship between the aryl substituent and the benzyl ether group (Figure 2).
Scheme 4.

Recrystallization of an Oxindole 11
Figure 2.
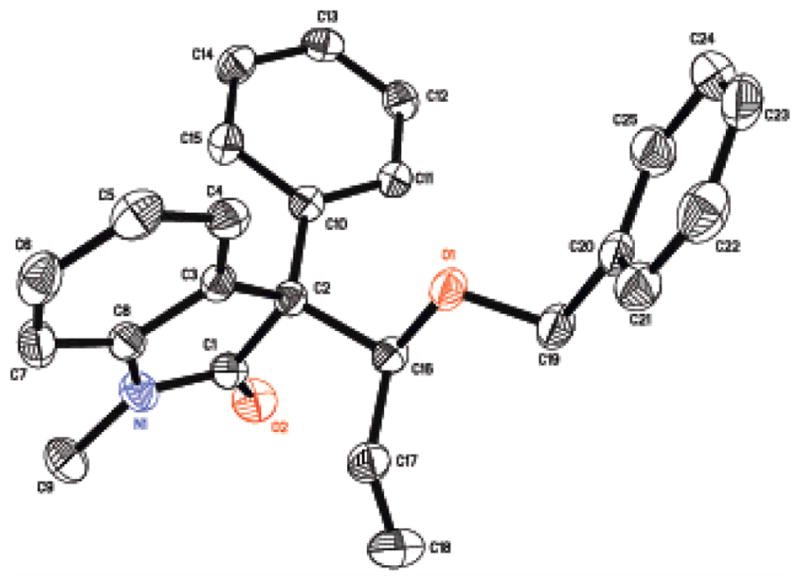
Xray Structure of Oxindole 11
Absolute stereochemistry in the reaction was verified by conversion of 17 to known material, and the optical rotation was then compared (Scheme 5). DDQ removal of the PMB-ether efficiently led to the desired allylic alcohol that was unstable to silica gel chromatography due to retro-aldol on SiO2. Therefore, the crude material was acylated directly to afford allylic acetate 18. Hydrogenolysis23 of allylic acetate 18 led to 19a and 19b as an inseparable 7:1 mixture. Comparison of the optical rotation of this mixture to that in the literature10d, 17b established the (R)-configuration at the α-quaternary stereocenter of oxindole 17. Additionally, it should be noted that partial racemization occurred during the hydrogenolysis, presumably due to Pd-catalyzed retro-allylic alkylation of 19a followed by non-selective recombination. To further confirm this assignment, the mixture of 19a:19b was ozonized and reduced17b to give known alcohol 20 in pure form. Again, comparison of the optical rotation of 20 with that in the literature9d verified the (R)-configuration at the α-quaternary stereocenter of oxindole 17. Taking this data into account along with the Xray structure of 11, ligand (R,R)-L1 furnished the hydrocarbonation product as the (R,R)-isomer.
Scheme 5.

Determination of Absolute Stereochemistry
Mechanism
Based on our observed experimental data for the optimization and substrate scope study in the allene hydrocarbonation reaction employing oxindoles, several mechanistic conclusions can be reached. While beautiful mechanistic studies1,24 have provided deeper insights to a more detailed picture of the structure of the Pd(π-allyl) intermediates with ligands L1–L4, our simple cartoon mnemonic (i.e. the “wall and flap” model)25 has proven to be very powerful and useful in predicting and understanding the sense of asymmetric induction when utilizing these ligands. A reasonable mechanistic picture for the allene hydrocarbonation reaction is given in Scheme 6. Pd-hydride 22 can be formed by Pd(0) participating as a formal base to deprotonate acid 21.26 Importantly, the conjugate base of 21 then becomes the counterion associated with Pd-complex 22. Hydropalladation of allene 4 likely occurs at the less hindered unsubstituted olefin of the allene from either the top or bottom face of the allene π-system. In general, hydropalladation would be expected to occur from the top face of the allene away from the benzyloxy group for steric reasons27 leading initially to σ-complex 23. Conversion of σ-complex 23 to the π-complexes anti-24 and anti-25 can occur through either a matched or mismatched scenario whereby in the mismatched case, the benzyloxy group is placed into the “wall” of the chiral ligand. Thermodynamically, syn π-allyl complexes are more stable; and therefore, under conditions where nucleophile trapping of anti-24 and anti-25 is slow, π-σ-π equilibration would be expected to favor the formation of complexes syn-26 and syn-27, respectively. Trapping of syn-26 and syn-27 by the nucleophile would then be expected to favor reaction with complex syn-27 because the nucleophile can approach the π-allyl electrophile from under the “flap” of the chiral ligand as apposed to approaching through the “wall” of the ligand if reacting with complex syn-26. Finally, only the branched isomer of product was observed to result from this reaction. This can be rationalized by the electronic nature of the π-allyl ligand of the Pd-intermediates whereby the carbon atom bearing the benzyloxy group can better stabilize the developing positive charge in the transition state during the nucleophilic addition step, and therefore, the incoming nucleophile is directed towards this carbon atom by electrostatics.
Scheme 6.

Reaction Mechanism and Selectivity in the Allene Hydrocarbonation Employing Oxindoles
The effect of the counterion (X−) was observed to be of extreme significance in regards to selectivity in the hydrocarbonation reaction employing oxindoles. The reaction utilizing oxindole 10 proceeded in the absence of an external acid additive implying that the oxindole nucleophile itself serves as an appropriate acid source for the reaction (Table 2, entry 1); however, in this case, only a 2.6:1 d.r. and 34% ee was obtained. The counterion associated with the Pd-intermediates in this reaction would be expected to be the conjugate base of the oxindole, a potential nucleophile for the reaction! Thus, nucleophilic trapping of the Pd(π-allyl) complex would likely be very fast, and ultimately results in poor diastereo- and enantioselectivity. When a more acidic acid is added as cocatalyst, the Pd(0) salt would be expected to react with this acid as opposed to the oxindole itself. For example, when PhCO2H was used as the acid cocatalyst, the reaction proceeded with 6.2:1 d.r. and 84% ee (Table 3, entry 2). Here, the counterion associated with the Pd-intermediates in the reaction would be benzoate, and the rate of nucleophile trapping by oxindole would be reduced since the process would no longer involve a simple collapse of a kinetically formed ion pair. Additionally, it is important to note that the presence of an aromatic ring in the counterion was important for stereoselection (Table 3 compare entries 2 and 3). When PhCO2H was replaced with AcOH, the selectivity dropped to 3.4:1 dr and 55% ee indicating that the benzoate counterion plays a significant role in organizing the transition state. We propose that a π-π stacking interaction is likely the reason for this observation. This is further supported by our results in Table 6 where the nature of the aromatic group on the carboxylic acid cocatalyst has an impact on the stereoselectivity of the reaction with naphthoic acid and electron deficient benzoic acid derivatives giving improved stereoselectivities.
Related to this observed counterion effect is the effect of solvent polarity on reaction selectivity (Table 4). Reactions in polar solvents such as MeCN, DMSO, and DMF gave ful l conversion and excellent yields, but very poor stereoselectivities (Table 4, entries 1–3), which presumably occurs because these solvents can help to separate the ion pairs and mitigate the beneficial π-π stacking effect in the transition state. In less polar solvents, stereoselectivities were better, but reactions typically did not reach full conversion with the exception of THF (Table 4, entries 4–10). Additionally, aromatic solvents, which would also be expected to affect the nature of π-π stacking, led to inferior enantioselectivities (Table 4, entries 9 and 10).
As is common in Pd-catalyzed reactions involving π-allyl intermediates,1 competitive rates of π-σ-π equilibration and attack of the Pd(π-allyl) intermediates by the nucleophile had a significant impact on selectivity in the allene hydrocarbonation reaction. For example, both diastereoselectivity and enantioselectivity were increased upon dilution of the reaction (Table 5 compare entries 5 and 10–13). Because dilution of the reaction medium reduces the rate of nucleophilic attack without affecting the rate of π-σ-π equilibration, this concentration effect is consistent with a need for the Pd(π-allyl) complexes to equilibrate prior to nucleophilic attack to obtain good stereoselectivity. Contrary to this analysis is the fact that the addition of 20 mol% TBAT or (n-hexyl)4NBr, additivies which are known to increase the rate of π-σ-π equilibration, had detrimental effects on both diastereoselectivity and enantioselectivity (Table 5, entry 2 and 3). However, it is likely that the reduction in stereoselectivity resulting from the addition of these additives is due to either counterion exchange between TBAT or (n-hexyl)4NBr and the Pd-intermediates or an increase in the rate of nucleophilic addition due to an increase in the ionic strength of the medium when these additives are present. Lastly, when the reaction was run in solvents with high polarity (MeCN, DMSO, DMF) poor stereoselectivities were obtained. This could also be the result of an increased rate of nucleophilic addition due to the increased polarity of the reaction medium.
Taking the above observations into account, the stereo-chemical outcome in the hydrocarbonation reaction can be rationalized to arise from a matched attack of the nucleophile from its Si-face onto syn-27 to furnish the major product (R,R)-13. Attack of the Si-face of the nucleophile would be expected to be preferred over attack from the Re-face to avoid a steric interaction between the oxindole and the “wall” of the chiral ligand (i.e. TS-A favored over TS-B, Scheme 6).28 Additionally, because the Pd(π-allyl) complexes need time to equilibrate prior to attack by nucleophile for high stereoselectivty (vide supra), it is unlikely that the major product would arise from reaction with anti-24 and/or anti-25 since thermodynamically syn-26 and syn-27 are significantly more stable. The switch in diastereoselectivity observed upon using oxindoles with more sterically demanding aryl groups (i.e. ortho-substituted aromatics, 12l and 12m, Table 8) can also be rationalized by comparing TS-A and TS-B. Because the aryl group must approach under the π-allyl ligand in TS-A, as the aryl group becomes more sterically encumbered, reaction through TS-B presumably is more favorable because the bulky aromatic group can avoid this destabilizing steric interaction. Also of note is that the minor diastereomers in these reactions were formed in low enantioselectivites (<30% ee). If the major reaction manifold proceeds through nucleophilic trapping of syn-27 by the Si-face of the nucleophile as we propose, then the fact that the minor diastereomer is formed in such low enantioselectivity implies that Re-face attack of the nucleophile onto syn-27 is of similar energy to the mismatched addition of the Si-face of the oxindole onto syn-26.
Application to the synthesis of the gliocladins
The gliocladins (Figure 3) are interesting indole alkaloids possessing a rare trioxopiperazine ring system in conjunction with a pyrrolidinoindoline fragment. The family of gliocladin natural products, which includes gliocladins A–C, was isolated by Usami and coworkers7c in 2004 from a strain of Gliocladium roseum OUPS-N132 that was originally obtained from the sea hare Aplysia kurodai. Gliocladins A–C showed cytotoxic activities against the P388 lymphocytic leukemia in cell culture. Among them, gliocladin C was the most potent (ED50 2.4 μg/ml).
Figure 3.

Gliocladins A (28), B (29), and C (30)
To date, there have been two total syntheses reported for (+)-30 with the first being reported by the Overman15b,29 group in 2007 and the second being reported by the Stephenson30 group in 2011. Both of these syntheses rely on a chiral pool approach to obtain chiral material in non-racemic form.31 We envisioned that the allene hydrocarbonation reaction employing a 3-indolyl substituted oxindole as the nucleophile would be well suited to prepare the all carbon quaternary stereocenter present in 30 in a catalytic asymmetric fashion, and we therefore began exploring this application of our methodology. Retrosynthetically, we envisioned that the trioxopiperazine ring system of 30 could be installed in the late stages from 31 using the elegant precedent already described by the Overman group in their synthesis of 30 (Scheme 7). The pyrrolidinoindoline fragment of 31 was envisioned to arise from a reductive cyclization of the secondary amine of 32 onto the oxindole fragment, and the primary amide of 31 could arise from the hydrolysis of the nitrile moiety of 32. The α-amino nitrile of 32 was then planned to arise from a cyanide addition to the α-benzyloxyimine formed by oxidation of the alkene of 33 followed by condensation with PMBNH2. Finally, 33 would be prepared using the allene hydrocarbonation reaction between oxindole 12q or 12r and allene 4.
Scheme 7.

Retrosynthetic Analysis of Gliocladin C
The hydrocarbonation reaction between oxindole 12q and 12r has already been described (Table 7, entries 20 and 21) and afforded the desired reaction products in 71% yield, 7.7:1 d.r., 91% ee (R = Bn) and 80% yield, 7.4:1 d.r., 91% ee (R = PMB), respectively. The diastereomeric mixture could not be separated at this point, so the mixture was carried forward. Protection of the free indole moiety could be achieved using PMBBr giving 33, which was oxidized to aldehyde 34 through dihydroxylation/periodate cleavage (Scheme 8). Condensation of 34 with PMBNH2 afforded Schiff base 35 that was then reacted with TMSCN to afford α-aminonitirile 32 in >20:1 d.r. at the newly formed stereocenter. The preferred mode of addition of the cyanide nucleophile was assigned by analogy to the model given in the literature for the addition of cyanide to α-chiral imines.32,33 In these cases, the anti-Felkin product is preferred due to a developing interaction between the SiR3 group and the forming amine, which causes a preference for the smaller hydrogen substituent to be “inside” the 4-membered ring transistion state (36).
Scheme 8.

Elaboration of 13q and 13r Towards Gliocladin C
At this point, we wanted to hydrolyze the nitrile group of 32 and subsequently install the pyrrolidinoindoline ring system using reductive cyclization. However, the hydrolysis of nitirle 32 proved nontrivial (Table 11). The use of catalytic base and 30% H2O2 initially proceeded in good yield (entry 1, 75%),34 however this result was not reproducible. Changing the amount of base, H2O2, or the solvent employed did not improve this reaction (entries 2–6). Phase transfer conditions35 were also ineffective (entry 7), and the standard base hydrolysis method using Ba(OH)2-8H2O afforded only aldimine 35q resulting from retrocyanation (entry 8). Acid hydrolysis (entry 11) and the use of Parkin’s catalyst36 (entry 12) led to poor yield and retrocyanation, respectively. Gratifyingly however, it was found that the use of urea hydrogen peroxide in place of aqueous H2O2 affected the hydrolysis reproducibly and in good yields (entries 9 and 10). It was at this point that the diastereomers resulting from the initial allene hydrocarbonation reaction could be separated by column chromatography on silica gel.
Table 11.
Hydrolysis of Nitrile 32
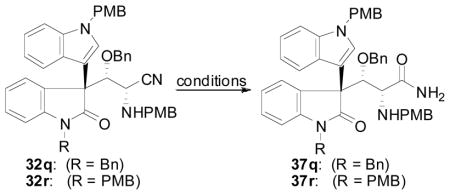 | |||
|---|---|---|---|
| entry | 32 | conditions | result |
| 1 | 32q | 26 mol% K2CO3, 10 equiv 30% H2O2, DMSO, rt | 15–75% |
| 2 | 32q | 25 mol% K2CO3, 3 equiv 30% H2O2, DMSO, rt | NR |
| 3 | 32q | 100 mol% K2CO3, 15 equiv 30% H2O2, DMSO, rt | 48% |
| 4 | 32q | 100 mol% K2CO3, 10 mol% 18-c-6, 30 equiv 30% H2O2, THF, rt | NR |
| 5 | 32q | 100 mol% K2CO3, 30 equiv 30% H2O2, TFE, rt | decomp. |
| 6 | 32q | 25 mol% K2CO3, 3 equiv 30% H2O2, MeOH, 45 °C | decomp. |
| 7 | 32q | 32 mol% Bu4NHSO4, 1 equiv 2M NaOH, 5 equiv 30% H2O2, CH2Cl2, rt | NR |
| 8 | 32q | Ba(OH)2-8H2O, DME/H2O, 65 °C | retrocyan. |
| 9 | 32q | 40 mol% K2CO3, 6 equiv urea-H2O2, DMSO, rt | 85% |
| 10 | 32r | 40 mol% K2CO3, 6 equiv urea-H2O2, DMSO, rt | 84% |
| 11 | 32q | HCl/Et2O/MeOH, rt | 45% |
| 12 | 32q | 10 mol% Parkin’s cat., EtOH/H2O (9/1), 70 °C | retrocyan. |
With access to amide 37 now realized, the next key transformation was chemoselective reduction of the lactam of 37 followed by cyclization of the pendant secondary amine moiety to install the desired pyrrolidinoindoline ring system. Unfortunately, when subjecting amide 37q to a variety of reducing agents (DIBAL, H3Al-NMe2Et, LiAlH4, LiHBEt3), no reaction was ever observed, and the starting material could be reisolated. Clearly, the primary amide will be deprotonated under these reaction conditions, and we reasoned that the amide anion may be responsible for the ineffective reduction of the lactam moiety. Therefore, we attempted to protect the amide by in situ silylation prior to reduction (Table 12). When treating 37q with 10 equiv of bis(trimethylsilyl)trifluoroacetamide (BSTFA) at 75 °C in MeCN for 1 h, followed by removal of volatile materials under reduced pressure, the silylated amide 38q was observed by 1H NMR analysis. The 1H NMR spectrum of 38q showed the presence of two trimethylsilyl moieties. This material was then subjected to a variety of reducing agents without further purification. Superhydride was effective at forming the desired reductively cyclized product (entry 1); however, the reduction did not proceed to full conversion. This may be due to competitive amide desilylation during the reduction. DIBAL reduced the oxindole moiety to the N,O-aminal, but cyclization to 39q did not occur. Therefore, the crude material was stirred with silica gel in a 4:1 mixture of MeOH:Et2O to convert the uncyclized N,O-aminal to 39q (entry 2). Again, incomplete reduction was observed when using DIBAL. Gratifyingly, using the reducing agent prepared by reacting equimolar amounts of DIBAL with n-BuLi cleanly reduced the lactam to the N,O-aminal. Subsequent acid induced cyclization of the crude material using silica gel gave the desired compound 39q in good yield (entry 3). Somewhat surprisingly, application of this procedure to the PMB-protected oxindole 39r afforded the desired product in slightly lower yield (entry 4) despite the fact that clean reduction of the oxindole to the N,O-aminal was observed by 1H NMR analysis of the crude material prior to cyclization with silica gel.
Table 12.
Reductive Cyclization of 37
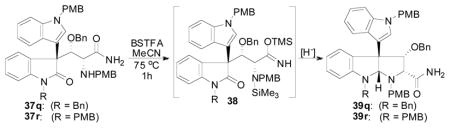 | ||
|---|---|---|
| entry | [H−] conditions | % yielda |
| 1 | 37q, 10 equiv LiHBEt3, THF | 50 (81) |
| 2 | 37q, 5 equiv DIBAL, CH2Cl2; MeOH/Et2O, SiO2, rt | 43 (52) |
| 3 | 37q, 6 equiv DIBAL/6 equiv n-BuLi, THF; MeOH/THF, SiO2, rt | 85 |
| 4 | 37r, 6 equiv DIBAL/6 equiv n-BuLi, THF; MeOH/THF, SiO2, rt | 71 |
Isolated yield. Number in parantheses refers to yield based on recovered 37.
With the successful reductive cyclization in hand, the pyrrolidinoinoline core of the gliocladins could be accessed from the allene hydrocarbonation reaction in an efficient and highly enantioselective manner. This methodology therefore represents a convenient protocol to prepare this class of functional unit and may be applied to other alkaloids possessing this structural motif. Additionally, access to gliocladin C from advanced intermediate 39 can be envisioned to arise from global N-deprotection and installation of the final trioxopiperazine ring with concomitant dehydration according to the reaction sequence described by Overman and coworkers.15b
Conclusion
In conclusion, we have developed a useful atom economical Pd-catalyzed allene hydrocarbonation reaction employing oxindoles as nucleophiles that allows for the formation of formal AAA reaction products without the use of the typical allyl equivalents bearing activated leaving groups required in AAA chemistry. This methodology proceeds with high branched regioselectivity to allow for the formation of valuable chiral oxindoles bearing two vicinal stereocenters, with one being quaternary, in excellent yields, diastereoselectivities, and enantioselectivities. The reaction conditions are extremely mild, which allows for the use of oxindole nucleophiles bearing sensitive functional groups (i.e. thienyl-groups or unprotected indoles), and sterically demanding oxindoles (i.e. ortho-substituted 3-aryloxindoles) can also be used. The choice of acid cocatalyst in the reaction has a significant impact on reaction selectivity, which we believe is the result of a Pd(II)+/conjugate base ion pair intermediate, whereby the conjugate base is involved in organizing the transition state. Additionally, the “wall-and-flap”24a,25 model for the Trost-family of chiral ligands can be used to rationalize the observed stereochemical outcome in the reactions. We have shown that this method can be extended to the less acidic 3-alkyloxindoles as well by employing reaction conditions that favor keto-enol tautomerization. Lastly, we have demonstrated the utility of this methodology by conversion of the 3,3-disubstituted oxindole products from the hydrocarbonation reaction to the pyrrolidinoindoline core of the gliocladin natural products in a convenient and efficient manner, and this may have application to other alkaloid natural products bearing this structural motif. Overall, this methodology adheres to the criteria of chemoselectivity, regioselectivity, diastereoselectivity, enantioselectivity, and atom economy.
Supplementary Material
Acknowledgments
We thank (GM-033049) National Science Foundation and National Institutes of Health for their generous support of our programs. Mass Spectra were provided by the Mass Spectrometry Regional Center of the University of California-San Francisco, supported by the NIH Division of Research Resources. We thank Johnson Matthey for their generous gift of palladium salts. J.D.S. thanks the American Cancer Society for a postdoctoral fellowship.
Footnotes
Supporting Information. Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews: Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804.Lu Z, Ma S. Angew Chem Int Ed. 2008;47:258. doi: 10.1002/anie.200605113.Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w.Trost BM, Zhang T, Sieber JD. Chem Sci. 2010;1:427.
- 2.Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 3.For reviews on atom-economic reactions, see: Trost BM, Toste FD, Pinkerton AB. Chem Rev. 2001;101:2067. doi: 10.1021/cr000666b.Trost BM. Acc Chem Res. 2002;35:695. doi: 10.1021/ar010068z.Trost BM, Frederiksen MR, Rudd MT. Angew Chem, Int Ed. 2005;44:6630. doi: 10.1002/anie.200500136.Li C. Acc Chem Res. 2010;43:581. doi: 10.1021/ar9002587.Basavaiah D, Reddy BS, Badsara SS. Chem Rev. 2010;110:5447. doi: 10.1021/cr900291g.Aubert C, Fensterbank L, Garcia P, Malacria M, Simmonneau A. Chem Rev. 2011;111:1954. doi: 10.1021/cr100376w.Corma A, Leyva-Prez A, Sabater MJ. Chem Rev. 2011;111:1657. doi: 10.1021/cr100414u.Yu DG, Li BJ, Shi ZT. Acc Chem Res. 2010;43:1486. doi: 10.1021/ar100082d.You SL, Cai Q, Zeng M. Chem Soc Rev. 2009;38:2190. doi: 10.1039/b817310a.Abu S, Shariar M, Liu RS. Chem Soc Rev. 2009;38:2269. doi: 10.1039/b807499m.McGlacken GP, Bateman LM. Chem Soc Rev. 2009;38:2447. doi: 10.1039/b805701j.Ganem B. Acc Chem Res. 2009;42:463. doi: 10.1021/ar800214s.Kumagai N, Shibasaki M. Angew Chem Int Ed. 2011;50:4760. doi: 10.1002/anie.201100918.Yadav JS, Antony A, Rao TS, Reddy BVS. J Organomet Chem. 2011;696:16.Alonso F, Foubelo F, González-Gómez TC, Martínez R, Ramón DJ, Riente P, Yus M. Mol Divers. 2010;14:411. doi: 10.1007/s11030-009-9195-z.Fustero S, Sanchez-Rosello M, del Pozo C. Pure Appl Chem. 2010;82:669.Li CJ, Trost BM. Proc Natl Acad Sci. 2008;105:13197. doi: 10.1073/pnas.0804348105.Kondo T. Synlett. 2008;5:692.Trost BM. Angew Chem Int Ed. 1995;34:259.
- 4.(a) Trost BM, Gerusz VJ. J Am Chem Soc. 1995;117:5156. [Google Scholar]; (b) Trost BM, Simas ABC, Plietker B, Jäkel C, Xie J. Chem Eur –J. 2005;11:7075. doi: 10.1002/chem.200500826. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Xie J. J Am Chem Soc. 2006;128:6044. doi: 10.1021/ja0602501. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Trost BM, Xie J. J Am Chem Soc. 2008;130:6231. doi: 10.1021/ja7111299. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Jäkel C, Plietker B. J Am Chem Soc. 2003;125:4438. doi: 10.1021/ja029190z. [DOI] [PubMed] [Google Scholar]
- 5.For non-enantioselective variants, see: Yamamato Y, Al-Masum M, Asao N. J Am Chem Soc. 1994;116:6019.Yamamoto Y. Tetrahedron Lett. 1995;36:2811.
- 6.Review: Zimmer R, Dinesh CU, Nandanan E, Kahn FA. Chem Rev. 2000;100:3067. doi: 10.1021/cr9902796.
- 7.Diazonamide A: Lindquist N, Fenical W, Van Duyne GD, Clardy J. J Am Chem Soc. 1991;113:2303.Structural revision: Li J, Burgett AWG, Esser L, Amezcua C, Harran PG. Angew Chem Int Ed. 2001;40:4770. doi: 10.1002/1521-3773(20011217)40:24<4770::aid-anie4770>3.0.co;2-t.Gliocladin: Usami Y, Yamaguchi J, Numata A. Heterocycles. 2004;63:1123.Leptosin D: Takahashi C, Numata A, Ito Y, Matsumura E, Araki H, Iwaki H, Kushida K. J Chem Soc, Perkin Trans 1. 1994:1859.Spirotryprostatin: Cui CB, Kakeya H, Okada G, Onose R, Osada H. J Antibiot. 1996;49:527. doi: 10.7164/antibiotics.49.527.Javaniside: Ma J, Hecht SM. Chem Commun. 2004:1190. doi: 10.1039/b402925a.Gelsemine: Conroy H, Chakrabarti JK. Tetrahedron Lett. 1959;(4):6.Communesin: Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M, Ito T, Hasegawa T. Tetrahedron Lett. 1993;34:2355.Physostigmine: Takano S, Ogasawara K. Alkaloids. 1989;36:225.Perophoramidine: Verbitski SM, Mayne CL, Davis RA, Concepcion GP, Ireland CM. J Org Chem. 2002;67:7124. doi: 10.1021/jo026012f.
- 8.For a review on catalytic asymmetric methods for the synthesis of quaternary stereocenters, see: Douglas CJ, Overman LE. Proc Natl Acad Sci. 2004;101:5363. doi: 10.1073/pnas.0307113101.
- 9.Ashimori A, Matsuura T, Overman LE, Poon DJ. J Org Chem. 1993;58:6949.Matsuura T, Overman LE, Poon DJ. J Am Chem Soc. 1998;120:6500.Lebsack AD, Link JT, Overman LE, Stearns BA. J Am Chem Soc. 2002;124:9008. doi: 10.1021/ja0267425.Dounay A, Hatanaka K, Kodanko J, Oestreich M, Overman LE, Pfeifer L, Weiss M. J Am Chem Soc. 2003;125:6261. doi: 10.1021/ja034525d.For a review on the of the asymmetric Heck reaction in the total synthesis of natural products, see: Dounay A, Overman LE. Chem Rev. 2003;103:2945. doi: 10.1021/cr020039h.Busacca CA, Grossbach D, So RC, O’Brien EM, Spinelli EM. Org Lett. 2003;5:595. doi: 10.1021/ol0340179.
- 10.(a) Lee S, Hartwig JF. J Org Chem. 2001;66:3402. doi: 10.1021/jo005761z. [DOI] [PubMed] [Google Scholar]; (b) Kundig EP, Seidel TM, Jia YX, Bernardinelli G. Angew Chem Int Ed. 2007;46:8484. doi: 10.1002/anie.200703408. [DOI] [PubMed] [Google Scholar]; (c) Luan XJ, Mariz R, Robert C, Gatti M, Blumentritt S, Linden A, Dorta R. Org Lett. 2008;10:5569. doi: 10.1021/ol8021808. [DOI] [PubMed] [Google Scholar]; (d) Luan X, Wu L, Drinkel E, Mariz R, Gatti M, Dorta R. Org Lett. 2010;12:1212. doi: 10.1021/ol1003093. [DOI] [PubMed] [Google Scholar]
- 11.(a) Lee TBK, Wong GSK. J Org Chem. 1991;56:872. [Google Scholar]; (b) Huang A, Kodanko JJ, Overman LE. J Am Chem Soc. 2004;126:14043. doi: 10.1021/ja046690e. [DOI] [PubMed] [Google Scholar]; (c) Ma S, Han X, Krishnan S, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2009;48:8037. doi: 10.1002/anie.200902943. [DOI] [PubMed] [Google Scholar]
- 12.(a) Shaw S, Aleman P, Vedejs E. J Am Chem Soc. 2003;125:13368. doi: 10.1021/ja037223k. [DOI] [PubMed] [Google Scholar]; (b) Shaw S, Aleman P, Christy J, Kampf J, Va P, Vedejs E. J Am Chem Soc. 2006;128:925. doi: 10.1021/ja056150x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hills ID, Fu GC. Angew Chem Int Ed. 2003;42:3921. doi: 10.1002/anie.200351666. [DOI] [PubMed] [Google Scholar]
- 13.Yasui Y, Kamisaki H, Takemoto Y. Org Lett. 2008;10:3303. doi: 10.1021/ol801168j. [DOI] [PubMed] [Google Scholar]
- 14.(a) Trost BM, Frederiksen MU. Angew Chem Int Ed. 2005;44:308. doi: 10.1002/anie.200460335. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Zhang Y. J Am Chem Soc. 2006;128:4590. doi: 10.1021/ja060560j. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Zhang Y. J Am Chem Soc. 2007;129:14548. doi: 10.1021/ja0755717. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Trost BM, Brennan MK. Org Lett. 2006;8:2027. doi: 10.1021/ol060298j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Adhikari S, Caille S, Hanbauer M, Ngo VX, Overman LE. Org Lett. 2005;7:2795. doi: 10.1021/ol051172+. [DOI] [PubMed] [Google Scholar]; (b) Overman LE, Shin Y. Org Lett. 2007;9:339. doi: 10.1021/ol062801y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ogawa S, Shibata N, Inagaki J, Nakamura S, Toru T, Shiro M. Angew Chem Int Ed. 2007;46:8669. doi: 10.1002/anie.200703317. [DOI] [PubMed] [Google Scholar]
- 16.Trost BM, Cramer N, Silverman SM. J Am Chem Soc. 2007;129:12396. doi: 10.1021/ja075335w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Linton EC, Kozlowski MC. J Am Chem Soc. 2008;130:16162. doi: 10.1021/ja807026z. [DOI] [PubMed] [Google Scholar]; (b) Duguet N, Slawin AMZ, Smith AD. Org Lett. 2009;11:3858. doi: 10.1021/ol901441t. [DOI] [PubMed] [Google Scholar]
- 18.(a) Tian X, Jiang K, Peng J, Du W, Chen YC. Org Lett. 2008;10:3583. doi: 10.1021/ol801351j. [DOI] [PubMed] [Google Scholar]; (b) He R, Ding C, Maruoka K. Angew Chem Int Ed. 2009;48:4559. doi: 10.1002/anie.200901277. [DOI] [PubMed] [Google Scholar]; (c) Jiang K, Peng J, Cui HL, Chen YC. Chem Commun. 2009:3955. doi: 10.1039/b905177e. [DOI] [PubMed] [Google Scholar]
- 19.Trost BM, Van Vranken DL. Angew Chem Int Ed. 1992;31:228. [Google Scholar]
- 20.(a) Crociani B, Di Biana F, Giovenco A, Boschi T. Inorg Chim Acta. 1987;127:169. [Google Scholar]; (b) Trost BM, Toste FD. J Am Chem Soc. 1999;121:4545. [Google Scholar]
- 21.Determined by Xray crystallography of 13d.
- 22.The enhanced diastereoselectivity provided by L4 is presumably due to the chirality of the ligand and not a property of ligand L4’s unusually large bite angle (110.5). When the reaction was run using xantphos as the achiral ligand (bite angle = 111.7), the product was formed in only 1.6:1 d.r. For reviews on the effects of ligand bite angles, see: Dierkes P, van Leeuwen PWNM. J Chem Soc, Dalton Trans. 1999:1519.van Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P. Chem Rev. 2000;100:2741. doi: 10.1021/cr9902704.
- 23.(a) Tsuji J, Minami I, Shimizu I. Synthesis. 1986:623. [Google Scholar]; (b) Hughes G, Lautens M, Wen C. Org Lett. 2000;2:107. doi: 10.1021/ol991170n. [DOI] [PubMed] [Google Scholar]; (c) Qi J, Porco JA., Jr J Am Chem Soc. 2007;129:12682. doi: 10.1021/ja0762339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Trost BM, Toste FD. J Am Chem Soc. 1999;121:4545. [Google Scholar]; (b) Butts CP, Filali E, Lloyd-Jones GC, Norrby P–O, Sale DA, Schramm Y. J Am Chem Soc. 2009;131:9945. doi: 10.1021/ja8099757. [DOI] [PubMed] [Google Scholar]
- 25.Review: Trost BM, Machacek MR, Aponick A. Acc Chem Res. 2006;39:747. doi: 10.1021/ar040063c.
- 26.(a) Trost BM. Chem Eur J. 1998;4:2405. [Google Scholar]; (b) Amatore C, Jutland A, Meyer G, Carelli I, Chiarotto I. Eur J Inorg Chem. 2000:1855. [Google Scholar]
- 27.(a) Hiroi K, Kato F, Yamagata A. Chem Lett. 1998:397. [Google Scholar]; (b) Gamez P, Ariente C, Gore J, Cazes B. Tetrahedron. 1998;54:14835. [Google Scholar]
- 28.A similar rational has been proposed in the asymmetric Pd-catalyzed decarboxylative AAA reaction developed in these laboratories, see: Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343. doi: 10.1021/ja9053948.
- 29.DeLorbe JE, Jabri SY, Mennen S, Overman LE, Zhang FL. J Am Chem Soc. 2011;133:6549. doi: 10.1021/ja201789v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furst L, Narayanam JMR, Stephenson CRJ. Angew Chem Int Ed. 2011;50:1. doi: 10.1002/anie.201103145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Recently, Overman and coworkers have reported a second generation synthesis of gliocladin C (reference 29) that utilizes Fu’s (reference 11c) catalytic asymmetric acyl-migration chemistry to access enantiomerically enriched material.
- 32.(a) Cativiela C, de Villegas MD, Galvez J. Tetrahedron. 1996;52:9563. [Google Scholar]; (b) Cainelli G, Giacomini D, Trere A, Galletti P. Tetrahedron Asymm. 1995;6:1593. [Google Scholar]
- 33.A similar model has been proposed in addition reactions to nitrones, see: Merino P, Lanaspa A, Merchan FL, Tejero T. J Org Chem. 1996;61:9028. doi: 10.1021/jo961293a.
- 34.Katritzky AR, Pilarski B, Urogdi L. Synthesis. 1989:949. [Google Scholar]
- 35.Cacchi S, Misiti D, Torre FL. Synthesis. 1980:243. [Google Scholar]
- 36.Ghaffar T, Parkins AW. J Mol Cat A. 2000;160:249. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.