

Abstract
Most of our knowledge of sex-chromosome evolution comes from male heterogametic (XX/XY) taxa. With the genome sequencing of multiple female heterogametic (ZZ/ZW) taxa, we can now ask whether there are patterns of evolution common to both sex chromosome systems. In all XX/XY systems examined to date, there is an excess of testis-biased retrogenes moving from the X chromosome to the autosomes, which is hypothesized to result from either sexually antagonistic selection or escape from meiotic sex chromosome inactivation (MSCI). We examined RNA-mediated (retrotransposed) and DNA-mediated gene movement in two independently evolved ZZ/ZW systems, birds (chicken and zebra finch) and lepidopterans (silkworm). Even with sexually antagonistic selection likely operating in both taxa and MSCI having been identified in the chicken, we find no evidence for an excess of genes moving from the Z chromosome to the autosomes in either lineage. We detected no excess for either RNA- or DNA-mediated duplicates, across a range of approaches and methods. We offer some potential explanations for this difference between XX/XY and ZZ/ZW sex chromosome systems, but further work is needed to distinguish among these hypotheses. Regardless of the root causes, we have identified an additional, potentially inherent, difference between XX/XY and ZZ/ZW systems.
Keywords: sex chromosome, duplication, retrogene
Introduction
Sex chromosomes have evolved independently multiple times in many plant and animal taxa (Bull 1983; Charlesworth and Mank 2010). Most model organisms, including mammals and Drosophila, have XX/XY karyotypes, in which the female is homogametic (XX) and males are heterogametic (XY). Conversely, in ZZ/ZW systems, the males are homogametic (ZZ), and females are heterogametic (ZW). ZZ/ZW species are found among lepidopterans, birds, and schistosomes, among others. In both types of systems, sex chromosomes are inherited differently from autosomes. In XX/XY systems, the Y chromosome is male specific, whereas the X is present twice as often in females than it is in males. In ZZ/ZW systems, the W chromosome is female specific and the Z chromosome occurs twice as often in males.
Regardless of whether males or females are heterogametic, the evolution of sex chromosomes is thought to occur in a similar fashion (Charlesworth 1991; Steinemann S and Steinemann M 2005; Bachtrog 2006; Ellegren 2011). The most common model for the evolution of sex chromosomes can be summarized as follows: first, a pair of autosomes acquires either a dominant or dosage-dependent sex-determining locus. Selection favors tight linkage between the sex-determining locus and sexually antagonistic alleles (i.e., alleles that are beneficial in one sex but detrimental in the other), which favors the suppression of recombination near the sex-determining locus. In some taxa, recombination is suppressed only in a small region near the sex-determining locus, whereas in other taxa, the nonrecombining region spreads from the area near the sex-determining locus to the majority or the entirety of the sex chromosomes. Within the nonrecombining region, the allelic combinations that selection can act upon on the Y (or W) are limited. The nonrecombining region of the Y (or W) gradually degrades as genes become inactivated through frameshift mutations and the accumulation of premature stop codons and transposable elements. As more of the chromosome stops recombining, an increasing proportion of the Y (or W) becomes hemizygous and subject to gradual degeneration. The lack of recombination may gradually spread across the full length of the chromosome, leading to fully heteromorphic sex chromosomes.
Because sex chromosomes and autosomes are inherited differently, genes on sex chromosomes will have a different selective environment than autosomes (Vicoso and Charlesworth 2006). As the nonrecombining region spreads along the length of the chromosome, selection pressures will also change. Genes that were once found equally often in males and females will become restricted to a single sex or will occur more often in one sex than the other. For genes that are necessary in both sexes, the cessation of recombination and eventual divergence between male and female gametologs may drive them to change chromosomal location. Indeed, the timing of gene movement off the mammalian Y coincides with the timing of several inversions thought to prevent recombination between the X and Y (McLysaght 2008).
In addition to gene movement off the Y chromosome, several different XX/XY animal taxa, including seven Drosophila species (Betran et al. 2002; Meisel et al. 2009; Vibranovski et al. 2009), multiple mammalian species (Emerson et al. 2004; Vinckenbosch et al. 2006; Potrzebowski et al. 2008), Anopheles gambiae (Toups and Hahn 2010), and Tribolium castaneum (Pease JB, Hahn MW, unpublished data) exhibit an excess of gene duplication events from the X chromosome to the autosomes. Furthermore, relocated genes—genes that were originally duplicated by either RNA- or DNA-based events in which ancestral copy is lost—also show excess movement off the X chromosomes in multiple XX/XY systems (Meisel et al. 2009; Vibranovski et al. 2009; Moyle et al. 2010). Although it is unclear what is causing this excess movement involving the X chromosome, several hypotheses have been proposed. Because sex chromosomes are inherited differently than autosomes, sex-specific and sexually antagonistic gene content should differ between sex chromosomes and autosomes (Rice 1984; but see Fry 2010). Dominant or partially dominant female-beneficial/male-detrimental alleles are predicted to spread more efficiently if they are X linked, whereas recessive male-beneficial/female-detrimental genes are predicted to spread more easily if X-linked because they are masked in heterozygous females (Rice 1984). Gene duplication from the X to the autosomes may therefore resolve sexual antagonism (Ellegren and Parsch 2007; Connallon and Clark 2011; Gallach and Betran 2011); however, the sexual antagonism hypothesis would also predict an excess of movement onto the X, which has been observed in mammals (Emerson et al. 2004; Potrzebowski et al. 2010), but not Drosophila (Betran et al. 2002; Meisel et al. 2009), A. gambiae (Toups and Hahn 2010), or T. castaneum (Pease JB, Hahn MW, unpublished data). Another potential cause of movement off the X is to escape from meiotic sex chromosome inactivation (MSCI). The X chromosome is precociously inactivated in the later stages of spermatogenesis in multiple XX/XY species (Lifschytz and Lindsley 1972; Hense et al. 2007; Kemkemer et al. 2011). According to this hypothesis, X-derived genes that are duplicated to the autosomes are selectively retained to perform either essential or novel functions in the male germ line (Wang 2004). Consistent with this hypothesis, in mammals and Drosophila, duplicated genes tend to be testis-biased (Betran et al. 2002; Emerson et al. 2004; Dai et al. 2006; Vinckenbosch et al. 2006; Bai et al. 2007; Potrzebowski et al. 2008; Meisel et al. 2009; Vibranovski et al. 2009).
Interestingly, the excess movement of duplicated genes off X chromosomes is limited primarily to retrotransposed genes (“retrogenes”). Retrotransposition occurs when the messenger RNA (mRNA) from a gene is reverse transcribed and inserted back into a random position in the genome (Hollis et al. 1982; Karin and Richards 1982; Ueda et al. 1982). DNA-mediated duplicated genes are not duplicated in excess off the X in mammals (Han and Hahn 2009) or Drosophila (Meisel et al. 2009; but see Zhang et al. 2010 for conflicting results due to different criteria for identifying retrogenes). Because of the differences in the duplication process, retrogenes are more likely to move between chromosomes than are DNA-mediated duplicates. Furthermore, newly retrotransposed genes tend to be testis-biased, regardless of their chromosomal location (Marques et al. 2005; Vinckenbosch et al. 2006; Meisel et al. 2009). This suggests that retrotransposition is essential for genes to move off the X chromosome in XX/XY species. However, the observed excess of gene relocations off the X chromosomes identified in Drosophila and mammals (Meisel et al. 2009; Vibranovski et al. 2009; Moyle et al. 2010) occurred through both DNA-based and RNA-based mechanisms.
Despite the near-universality of patterns of gene movement off X chromosomes, to date, no study has examined this phenomenon in ZZ/ZW systems. Some studies of ZZ/ZW systems have found many similarities in the evolution of sex chromosomes, such as the existence of evolutionary strata in both mammalian and avian sex-specific chromosomes (Lahn and Page 1999; Ellegren and Carmichael 2001; Handley et al. 2004; Nam and Ellegren 2008). However, other studies have revealed important differences, such as a lack of global dosage compensation in ZZ/ZW lineages (Ellegren et al. 2007; Itoh et al. 2007; Zha et al. 2009; Vicoso and Bachtrog 2011; but see Walters and Hardcastle 2011). There are several a priori reasons to expect that there should be an excess of movement off the Z. Sexual antagonism should be common regardless of sex chromosome karyotype, and while exact predictions about movement depend on the dominance of mutations, unless that dominance changes between XY and ZW systems, the expectations are the same. Furthermore, the Z chromosome is known to undergo MSCI in at least some taxa (Schoenmakers et al. 2009). However, given the differences in selection pressures on the sex chromosomes in male and female heterogametic taxa, it may also be the case that this pattern does not occur in ZZ/ZW systems.
In this study, we examine gene movement in two independently evolved ZZ/ZW systems: in birds, specifically, the chicken (Gallus gallus) and the zebra finch (Taeniopygia guttata) and in lepidopterans, specifically, the silkworm (Bombyx mori). Because patterns of gene movement seem to be universal among XX/XY systems, understanding the forces that drive this process would seem to require an examination of ZZ/ZW systems.
Materials and Methods
Retrotransposition Events in the Avian and Lepidopteran Lineages
Retrotransposed genes result when processed (intronless) mRNA intermediates are reverse transcribed and reinserted into the genome. Genes that are duplicated through retrotransposition result in one copy with introns (the original copy or “parent”) and one copy without introns (the duplicated copy or “daughter”). We identified all retrotransposition events (i.e., parent–daughter pairs) in both the chicken and silkworm genomes.
Data, including gene IDs, sequences, exon number, and chromosomal location, for the silkworm genome were downloaded from KAIKObase (v2.0; Shimomura et al. 2009). As all individuals sequenced to construct the genome were male, we expect no difference in the quality of the assembly of the Z chromosome versus the autosomes (Xia et al. 2004). We calculated local alignment scores for all pairs of peptide sequences within a species using mpiBLAST (v1.5.0; http://www.mpiblast.org/). BlastP hits with a bit score <200 were removed, and remaining genes were clustered using MCL (v10.201; van Dongen 2000). These gene clusters represent putative paralogs sets. Clusters without both an intronless retrogene and intron-containing parent gene were excluded. The remaining clusters with only two genes were tabulated as a retrogene-parent gene pairing. In addition to clusters with a single retrogene and a single parent gene, clusters with multiple possible retrogenes or parent genes were allowed as long as 1) all retrogenes appeared on the same chromosome, 2) none of the possible parent genes appeared on the same chromosome as the retrogene, and 3) the possible parental genes were either all on autosomes or all on the Z chromosome. Each multigene cluster was counted as a single event. This allowed us to count retrogenes that had been later duplicated (often tandem duplication in B. mori) as one retrotransposition event and to count events as autosome-to-Z or Z-to-autosome even when the specific autosomal parent or daughter could not be determined from a set of closely related putative parents. Clusters where the parental genes could have come from either an autosome or the Z were discarded, as were all clusters with retrogenes on multiple chromosomes to avoid biasing results toward autosome-to-autosome movement. We calculated sequence identity between paralogous pairs using the Needleman–Wunsch algorithm (Python module nwalign v0.3.1). The remaining duplicates were then filtered to have at least 50% sequence identity and an alignment overlap of at least 70%. In clusters with multiple potential parents or retrogenes, the highest sequence identity match was considered when filtering. In order to ensure that our results were not due to conservative filtering criteria or small sample size, we repeated the analyses with less stringent bit-score cutoffs of 100 and 50 and a minimum alignment overlap of 50% and minimum sequence identity of 40%. All data sets produced the same patterns of gene movement.
Data from the chicken and zebra finch genomes were downloaded from Ensembl version 59 (Hubbard et al. 2002). Unlike the silkworm, the individual sequenced for the chicken genome was a female (Hillier et al. 2004); therefore, the Z chromosome is potentially less well assembled than the autosomes because it was sequenced only half as deeply. The zebra finch genome, however, is from a single male, so the Z should have the same coverage and therefore be as well assembled as the autosomes. The genomic data were analyzed using two different methods. In the first analysis, the data were analyzed as described above for the silkworm. We then performed a second independent analysis. In this analysis, we analyzed only the gene families clustered by the EnsemblCompara pipeline (Vilella et al. 2009) that contained two genes (one with introns and one without) that were both assigned to chromosomes. Again, we only analyzed duplicates with at least 70% of the peptide sequences aligned and had at least 50% amino acid sequence identity. To ensure that all results are robust to inclusion criteria, we also used the less restrictive criteria of 50% alignment overlap and 40% amino acid sequence identity. These two analyses performed on the avian genomes are somewhat different and analyze different subsets of data, but the patterns identified did not differ. We present results from both sets of analyses.
Duplication Events Since the Chicken–Zebra Finch Divergence
We collected data on all functional intact duplicates for both the chicken and zebra finch genomes from Ensembl version 59. Using gene tree/species tree reconciliation (Goodman et al. 1979), we identified duplication events in the chicken genome that have occurred since the split with the zebra finch. We used ClustalW2 to align duplicates (Larkin et al. 2007). As described above, we again used two stringencies of filtering criteria: first, 70% peptide sequence alignment with 50% amino acid sequence identity and second, 50% peptide sequence alignment with 40% amino acid sequence identity. We used the same process to identify duplication events in the zebra finch genome since the divergence with the chicken.
Unlike genes that are duplicated through retrotransposition, genes that move via DNA-mediated mechanisms contain introns in both the parent copy and the daughter copy. In order to determine which copy is the parent and which is the daughter, it is necessary to examine the evolution of the paralogs in a phylogenetic context. We used the location of the orthologous single-copy gene in the zebra finch genome to assign the parent copy of the chicken duplicates. We also performed the same analysis for the DNA-mediated duplication events in the zebra finch since the split with the chicken and used the location of the single copy in the chicken genome to assign the parent copy in the zebra finch.
If both the parent copy and the daughter copy each contained only one exon, it was impossible to determine if it was duplicated by a DNA-mediated or RNA-mediated mechanism. In these cases, we polarized gene movement in the same manner as DNA-mediated duplication events described above.
Gene Relocation in the Avian Lineage
We define “relocated” genes as one-to-one orthologs that are located on nonhomologous chromosomes (Meisel et al. 2009). These represent events where the original (parent) gene was lost in one lineage, usually after duplication onto a different chromosome. We identified relocated genes by first obtaining one-to-one orthologs for the chicken and zebra finch genomes from Ensembl version 59. We considered only ortholog pairs that had moved between chromosomes, though without a further outgroup, we cannot polarize the direction of movement. We aligned duplicates using ClustalW2 and used the two filtering criteria described above. Furthermore, we divided relocation events into those most likely duplicated via DNA-mediated mechanisms, RNA-mediated mechanisms, or an undetermined mechanism, based on the presence or absence of introns.
Gene Movement Analysis
The expected number of genes duplicated among chromosomes were estimated using the model presented in Betran et al. (2002). This model uses the number of genes on the chromosome containing the parental copy, the length of the chromosome that contains the daughter copy, and whether either copy fell on the autosomes or a sex chromosome. Specifically, we used the equation:
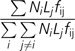 |
where i is the index of the chromosome with the parent copy, j is the index of the chromosome containing the daughter copy, Ni is the number of genes on chromosome i, Lj is the length of the chromosome j, and fij is 0.75 if j = Z and 1 if j is an autosome. From these frequencies, we will obtain the expected number of duplication events among different autosomes, from the Z to the autosomes and from the autosomes to the Z, which we can then compare with the observed gene movements. To determine if there is nonrandom gene movement, we compare observed with expected values using a χ2 goodness-of-fit test. Furthermore, because our sample sizes were often small (<20), we also conducted randomization tests similar to those presented in Emerson et al. (2004). Briefly, we computed the probability of each retrotransposition or relocation pattern under the null hypothesis that all insertions occur randomly via simulation. For each simulation, we calculated the χ2 statistic (χ2 = Σ[Di − Ei]/Ei) where i is the chromosome, Ei is the expected number of movements, and Di is the simulated number of movements. We calculated the P value as the proportion of simulated χ2 statistics that exceeded the observed χ2 statistic out of 106 iterations. Because both the parametric χ2 goodness-of-fit tests and the simulations produced qualitatively similar results for all data sets, we only show the χ2 P values below.
Expression Data
In both mammals (Marques et al. 2005; Vinckenbosch et al. 2006) and Drosophila (Meisel et al. 2009), retrogenes, regardless of chromosomal location, tend to have testis-biased expression. In ZZ/ZW systems, depending on dominance, we expect that ovary-biased genes would be selected to move onto or off of the Z. We tested for both testis-biased and ovary-biased expression among both the silkworm and chicken retrogenes. We defined testis-biased and ovary-biased genes as having a significantly higher expression in the testis or ovaries as compared with somatic tissues.
Microarray data for the silkworm were downloaded from the SilkDB Microarray Browser (Xia et al. 2007). The original data were collected using a custom oligonucleotide array (with 70-nt oligomers). Data were available for eight somatic tissues (anterior/median silk gland, the posterior silk gland, fat body, midgut, integument, hemocyte, malphigian tubule, and head) as well as the testis and ovary, from individuals on day 3 of the fifth instar. These data were normalized using a linear model normalization using four confirmed housekeeping genes. Further details on array design and processing are available in Xia et al. (2007).
We first identified all tissue-biased genes using a one-way analysis of variance, with a significance level of P < 0.001. We then used Tukey’s Honestly Significant Difference test to identify the testis-biased genes and ovary-biased at a significance level of P < 0.01. We used Fisher’s exact test to compare the proportion of the genome that is testis-biased to the proportion of retrogenes that are testis-biased as well the as the proportion of the genome that is ovary-biased to the proportion of retrogenes that are ovary-biased.
Microarray data for the chicken were downloaded from the Gene Expression Omnibus database, accession number GSE12974. The original data were collected using a custom oligonucleotide array (with 60-nt oligomers) designed by Chan et al. (2009). Data were available for 17 somatic tissues (bursa of fabricius, cerebellum, cerebral cortex, eye, femur, gallbladder, gizzard, heart, intestine, kidney, liver, lung, muscle, skin, spleen, stomach, and thymus) as well as three reproductive tissues (ovary, oviduct, and testes). Relative expression levels for each tissue were determined using methods in Chan et al. (2009) and Zhang et al. (2004). Briefly, variance-stabilizing normalization was used to normalize all channels to each other, and subsequent measurements were arcsinh transformed. The median value across all arrays was then subtracted, resulting in relative expression ratios for each gene in each tissue compared with all other tissues. Further details for array design and processing are available in Chan et al. (2009) and Zhang et al. (2004).
The expression data for the chicken did not consist of multiple measurements per tissue and therefore are not suited to the same analysis as we have done with the silkworm. Instead, we identified testis-biased and ovary-biased genes in the chicken genome using the relative expression ratios. Genes were considered tissue biased if the relative expression ratio exceeded 1.0, which is equivalent to a linear ratio of 2.7 (Zhang et al. 2004). We again used a Fisher’s exact test to compare the proportion of the genome that is testis-biased to the proportion of retrogenes that are testis-biased as well the as the proportion of the genome that is ovary-biased to the proportion of retrogenes that are ovary biased.
Results
Retrotransposition in the Avian and Lepidopteran Lineages
When gene families were identified using MCL, we identified 22 retrotransposition events in the silkworm genome, including two movements from the Z to the autosomes and one movement from the autosomes to the Z. No excess of movement involving the Z was identified (table 1A: χ2 = 0.936, degrees of freedom [df] = 2, P = 0.626; supplementary table 5, Supplementary Material online). Varying the clustering or filtering parameters of the analysis produced the same qualitative results (supplementary table 1, Supplementary Material online). Using the same methodology, we also identified 21 retrotransposition events in the chicken genome. Of these, there was not a single movement involving the Z chromosome; as would be expected, there was no excess of movements involving the Z chromosome (table 1B: χ2 = 2.46, df = 2, P = 0.293; supplementary table 5, Supplementary Material online). Similarly, varying the clustering or filtering parameters produced the same qualitative results (supplementary table 1, Supplementary Material online). Furthermore, when we used the EnsemblCompara pipeline to identify retrotransposition events in the chicken lineage, we obtained qualitatively similar results: When we considered retrotransposition events with 50% of the peptide sequence aligned and 40% amino acid sequence identity, we identified 12 gene movements. Of these gene movements, none were off the Z chromosome and only one moved onto the Z, indicating no excess of gene movement involving the Z (supplementary table 2, Supplementary Material online: χ2 = 0.74, df = 2, P = 0.691). We identified only two retrotransposition events that had 70% of the peptide sequence aligned and 50% amino acid sequence identity, neither of which involved the Z chromosome. Similar analyses for the zebra finch genome produced qualitatively similar results (Supplemental table 3, Supplementary Material online).
Table 1.
Retrotransposition in (A) lepidopteran and (B) avian lineages
| Movement | Observed | Expected |
| (A) | ||
| Z → A | 2 | 1.1 |
| A → Z | 1 | 0.7 |
| A → A | 19 | 20.2, P = 0.626 |
| (B) | ||
| Z → A | 0 | 1 |
| A → Z | 0 | 1.2 |
| A → A | 21 | 18.8, P = 0.293 |
Table 2.
DNA-Mediated Duplications
| Movement | Observed | Expected |
| (A) | ||
| A ↔ Z | 3 | 1.5 |
| A ↔ A | 11 | 12.5, P = 0.194 |
| (B) | ||
| Z → A | 1 | 0.4 |
| A → Z | 1 | 0.4 |
| A → A | 6 | 7.2, P = 0.423 |
| (C) | ||
| A ↔ Z | 1 | 0.6 |
| A ↔ A | 5 | 5.4, P = 0.636 |
| (D) | ||
| A ←→ Z | 1 | 0.5 |
| A ←→ A | 4 | 4.5, P = 0.501 |
NOTE.—(A) All events that could be identified (no filtering criteria). (B) Pooling all unfiltered events that could be polarized from both taxa. (C) All duplication events with at least 50% peptide sequences aligned and 40% amino acid sequence identity. (D) All duplications events with at least 70% peptide sequences aligned and 50% amino acid sequence identity.
Although we detected relatively few retrotransposition events in these genomes, our numbers are only slightly smaller than those in the study of Betran et al. (2002), in which 24 retrotransposition events were identified in Drosophila melanogaster. We determined the number of movements that would be necessary to detect an excess off the Z chromosome in order to determine the statistical power of our data set. For our analysis of the 21 retrogenes in the chicken genome, 4 movements off the Z would be required to detect an excess, whereas 1 was expected (none were observed). Similarly, for the same analysis in the silkworm, in which there are 22 movements (1 onto the Z, 2 off the Z, and 19 between autosomes), 4 movements off the Z would be required to get a significant result at P < 0.05. For comparison, previous analyses of retrogene movement off the A. gambiae X chromosome found a >400% excess of movement to autosomes (Toups and Hahn 2010); therefore, it is reasonable to expect that a significant excess could have been detected here, for either data set.
Finally, in order to ensure that our analysis did not miss potential Z → A or A → Z movements, we performed an additional analysis that examined cases in which there were multiple potential daughter genes on different chromosomes and families in which the parent gene may be on the Z or the autosomes. In cases where there was a possible parent gene on the Z or the autosomes, we counted it as a Z → A movement; likewise, if daughter genes were on the Z or the autosomes, we treated it as an A → Z movement, even if these were not the most likely movements. In these cases, we are counting all ambiguous events as Z → A or A → Z. Even by analyzing a data set with the maximum potential Z-related movements, we were still unable to detect any excess gene movement involving the Z chromosome (Supplementary table 4, Supplementary Material online).
Gene Movement Since Chicken–Zebra Finch Divergence
We identified very few DNA-mediated duplicates between chromosomes for either the chicken or zebra finch genome. Therefore, we present the combined data for the chicken and zebra finch. With no minimum criteria for the proportion of duplicates that aligned or amino acid percent identity, we identified ten DNA-mediated duplicates on the chicken lineage since the split with zebra finch and four on the zebra finch lineage since the split with chicken. Of these, three movements involve the Z chromosome, whereas 11 movements are between autosomes. We found no significant excess of movement involving the Z chromosome (table 2A: χ2 = 1.69, df = 1, P = 0.194; supplementary table 6, Supplementary Material online). If we consider only the movements that can be polarized, there is also no excess onto or off of the Z chromosomes (table 2B: χ2 = 1.72, df = 2, P = 0.423; supplementary table 6, Supplementary Material online).
When we restrict our analysis by using more stringent criteria for identifying duplicates, we do not have a lot of statistical power to reject the null hypothesis. Nevertheless, we examined each data set in turn. When we consider duplicates that have a minimum of 50% of the peptide sequences aligned and 40% amino acid percent identity, we identify three movements in the chicken and three movements in the zebra finch. Of these, only one involved the Z chromosome, indicating there is no excess of genes movement involving the Z (table 2C: χ2 = 0.224, df = 1, P = 0.636; supplementary table 6, Supplementary Material online). Only three of these events can be polarized, none involving the Z chromosome. Similarly, when we consider duplicates with a minimum 70% of the peptide sequences aligned and 50% amino acid identity, one autosome-to-autosome movement is removed in the zebra finch, leaving two movements in the zebra finch, and three in the chicken. Of these, only one involved the Z, and we found no excess of genes moving involving the Z (table 2D: χ2 = 0.453, df = 1, P = 0.501; supplementary table 6, Supplementary Material online).
We were unable to identify any gene movements since the divergence of the two bird species that were clearly the result of retrotransposition in either genome. We did, however, identify two duplication events in the chicken and one duplication event in the zebra finch where both copies had a single exon. Of these three events, one involved the Z chromosome.
Gene Relocation in the Avian Lineage
We considered relocations in the avian genome that resulted from both DNA-mediated and RNA-mediated movements. (No such analysis could be done for B. mori because no closely related species has been sequenced). Our analyses of gene relocation had larger sample sizes than our analysis of gene duplicates. For movements that were likely DNA mediated, using the 50% peptide sequence alignment/40% amino acid sequence identity criteria, we identified 37 relocated pairs of orthologs. Of these, six movements involved the Z chromosome, and there was no excess of movement involving the Z (table 3A: χ2 = 1.18, df = 1, P = 0.277; supplementary table 7, Supplementary Material online). When we consider the more stringent criteria of 70% peptide sequence alignment/50% amino acid percent identity, we identify 26 relocated pairs of orthologs. Of these, four movements involved the Z chromosome; again, there was no excess of movement involving the Z (table 3B: χ2 = 0.60, d.f. = 1, P = 0.439; supplementary table 7, Supplementary Material online).
Table 3.
Gene Relocations
| Movement | Observed | Expected |
| (A) | ||
| A ↔ Z | 6 | 4.0 |
| A ↔ A | 31 | 33.0, P = 0.277 |
| (B) | ||
| A ↔ Z | 4 | 2.8 |
| A ↔ A | 22 | 23.2, P = 0.439 |
| (C) | ||
| A ↔ Z | 1 | 0.7 |
| A ↔ A | 6 | 6.3, P = 0.755 |
| (D) | ||
| A ↔ Z | 5 | 3.5 |
| A ↔ A | 28 | 29.56, P = 0.39 |
NOTE.—(A) DNA-mediated relocation events with at least 50% peptide sequence aligned and 40% amino acid sequence identity. (B) DNA-mediated relocation events with at least 70% peptide sequence aligned and 50% amino acid sequence identity. (C) RNA-mediated relocation events with at least 50% peptide sequence aligned and 40% amino acid sequence identity. (D) Combined analysis of DNA-mediated and RNA-mediated relocation events with at least 50% peptide sequence aligned and 40% amino acid sequence identity.
For movements that were likely RNA-mediated duplicates at the 50% peptide sequence alignment/40% amino acid sequence identity criteria, we identified seven movements. Of these, only one involved the Z chromosome, indicating there was no excess of movements involving the Z (table 3C: χ2 = 0.10, df = 1, P = 0.755; supplementary table 7, Supplementary Material online). There were only two movements that fit the 70% peptide sequence alignment/50% amino acid percent identity, one of which involved the Z. When we combine our results for DNA-mediated and RNA-mediated duplicates with 50% peptide sequence alignments/40% amino acid percent identity, we again detect no excess involving the Z chromosome (table 3D: χ2 = 0.71, df = 1, P = 0.396; supplementary table 7, Supplementary Material online).
Gene Expression of Retrotransposed Genes
In the silkworm, we determined that ∼16% (3,563 of 22,987) of transcripts were testis-biased. Of the retrogenes with expression data, ∼21% (4 of 19) were testis-biased. In contrast to previous studies in XX/XY taxa, this indicates that retrogenes were not significantly more likely to be testis-biased than the rest of the genome (P = 0.608, Fisher’s exact test). Similarly, in chicken, ∼17% (2,866 of 16,901) of genes were testis-biased. Of the retrogenes with expression data, ∼4% (1 of 24) were testis-biased, which is not significantly different than the rest of the chicken genome (P = 0.107, Fisher’s exact test). Additionally, we did not find evidence for an excess of ovary-biased expression for retrogenes in either the silkworm or chicken genomes. We determined that ∼2% (453 of 22,987) of the silkworm transcripts are ovary biased, and none of the retrogenes were ovary-biased indicating no ovary-biased expression (P = 1, Fisher’s exact test). We determined that ∼5% (877 of 16,901) of the chicken transcripts are ovary-biased whereas 12% (3 of 25) of the retrogenes are ovary-biased However, this difference is not significant (P = 0.128, Fisher’s exact test).
Discussion
Gene Movement in ZZ/ZW Systems
We did not identify a statistically significant excess of RNA- or DNA-mediated duplication events off the Z chromosome in either the avian or lepidopteran genomes. As expected from previous genome analyses (Hillier et al. 2004), we identified relatively few retrotransposition events in the avian lineage in comparison with the mammalian lineage. Previous analyses have found an average of 109 functional retrotransposed genes among therian mammals since the divergence with monotremes ∼200 Ma (Potrzebowski et al. 2008), whereas we were only able to identify 21 for an equivalent time period along the avian lineage. Our analyses also demonstrate that we had sufficient statistical power to find an excess of movement if one existed, and the same methods used here were able to find significant patterns in multiple XY systems (Pease JB, Hahn MW, unpublished data). Interestingly, when each avian lineage was examined individually since their divergence, there were no retrogenes moving between chromosomes identified in the chicken, and only two identified in the zebra finch. It is hypothesized that retrogenes in mammals are formed via the reverse transcriptase provided by the LINE1 (L1) transposable element (Esnault et al. 2000). Interestingly, birds have their own LINE-like retroelements, chicken repeat 1 (CR1; Burch et al. 1993). However, the reverse transcriptase of CR1 likely cannot copy polyadenylated mRNAs (Haas et al. 2001), meaning that it cannot produce retrotransposed copies of native protein-coding genes. This lack of CR1 function is likely responsible for the lack of recent retrogenes in avian genomes.
We also did not find any excess of relocated gene pairs involving the Z chromosome between the chicken and zebra finch genomes, even though we identified a large number of relocated orthologs. Relocated genes—originally duplicated by either RNA- or DNA-based events—show excess movement off sex chromosomes in multiple XX/XY systems (Meisel et al. 2009; Vibranovski et al. 2009; Moyle et al. 2010). Unfortunately, no similar analyses of relocated genes can be done in the Lepidoptera because there is currently only one whole-genome sequence in this clade. Further sequencing—coupled with the relatively more difficult step of assigning scaffolds to chromosomes—will be necessary for a full analysis of gene movement among the butterflies and moths.
Gene Expression of Retrotransposed Genes
We were unable to identify an excess of retrotransposed genes that had testis-biased expression in the silkworm or chicken genome. Previous studies have identified testis bias as a general feature of newly retrotransposed genes, regardless of their chromosomal location (Vinckenbosch et al. 2006; Meisel et al. 2009). It is possible that the difference in our results may be related to the age of the daughter copy, as previous analyses have also found that testis-biased expression is lost over time (Vinckenbosch et al. 2006). Our gene tree reconciliation analysis in the chicken genome demonstrates that there are no newly retrotransposed genes since the divergence with the zebra finch approximately 105 Ma (van Tuinen and Hedges 2001). Although we do not have the same information on the age of duplicates for the silkworm, it is possible that the lack of testis-biased genes may be the result of a lack of recent retrotransposition events. Alternatively, the lack of testis-biased expression of retrogenes in these two taxa may be related to the ZZ/ZW system, though it is not immediately obvious why this would occur.
Hypotheses for the Lack of Movement in ZZ/ZW Systems
Many of the features of XX/XY systems that are invoked by the common hypotheses for the excess of gene movement off X chromosomes also appear to be present in ZZ/ZW taxa. We expect that sexual antagonism is acting in birds and lepidopterans; and at least for chickens, MSCI has been confirmed (Schoenmakers et al. 2009). In addition, other genomic patterns thought to be associated with gene movement—that is, the nonrandom distribution of sex-biased genes—also occur in both chicken and silkworm (Arunkumar et al. 2009; Mank and Ellegren 2009). However, we find no excess of genes moving from the Z. We propose several potential contributing factors to this pattern.
In XX/XY systems, consistent with both sexually antagonistic selection and escape from MSCI, testis-biased male associated genes more often flee the X. In these systems, newly retrotransposed genes have generally been found to be testis-biased and/or testis-expressed, regardless of the chromosomal location of daughter or parent gene (Vinckenbosch et al. 2006; Kaessmann et al. 2009; Meisel et al. 2009). Thus, in XX/XY systems, retrotransposition and selection reinforce each other to produce an excess of testis-expressed genes moving to the autosomes. In contrast, ZZ/ZW systems exhibit no excess of retrotransposed genes off the Z. Because ZZ males are homogametic, it is likely that testis-expressed genes are not selected to move off the Z (as they would be on the X). Instead, genes that would be selected to move off the Z are ovary-biased or, more precisely, female advantageous/male deleterious. As there is no known mechanism that produces an excess of ovary-biased daughter genes, no duplicated gene products can be acted upon by selection. Interestingly, in these systems, we find that retrogenes are neither testis-biased nor ovary-biased. Thus, the differing relationships between the gene duplication machinery and selective forces in XX/XY and ZZ/ZW systems could be the source of the differences we observe. However, in order to determine if this is indeed occurring, further examination of the expression patterns of retrogenes is necessary in both XX/XY and ZZ/ZW systems.
Alternatively, the mechanism (or mechanisms) that produces an excess of gene movement off the X may not operate in ZZ/ZW systems. Examination of ZZ/ZW taxa has already demonstrated important differences between XX/XY and ZZ/ZW systems, such as a lack of global dosage compensation in ZZ/ZW systems (Ellegren et al. 2007; Itoh et al. 2007) and a difference in the relative rates of molecular evolution of sex chromosomes (Ellegren and Fridolfsson 1997). Therefore, lack of gene movement in ZZ/ZW may be related to dosage compensation. However, this explanation is undermined by the observation that there is an excess of gene movement off the X chromosome in T. castaneum (Pease JB, Hahn MW, unpublished data), which has no dosage compensation (Prince et al. 2010), though the upregulation of both Xs may be an intermediate step to full dosage compensation according to some models (Vicoso and Bachtrog 2009).
An additional mechanism that may influence the movement off the Z chromosome is MSCI. In the only ZW system in which MSCI has been identified, the chicken, the process is ephemeral and lasts only from early pachytene to early diplotene phases during oogenesis (Schoenmakers et al. 2009). Assuming that MSCI occurs in ZZ/ZW taxa, and the chicken is representative of the process, it is possible that the duration of MSCI is not a strong enough selective force to produce an excess of movement off the Z. Interestingly, the link between MSCI and movement off the X chromosome in Drosophila has been questioned, as recent experimental evidence casts doubt on whether MSCI occurs in Drosophila (Meiklejohn et al. 2011; Mikhaylova and Nurminsky 2011).
Finally, there may be an unknown process that differs between the two types of sex chromosome systems. Further analyses of the mechanistic differences between X and Z chromosomes—as well as additional ZZ/ZW taxa—will be needed to distinguish among these hypotheses.
Conclusions
Although an excess of gene movement off the X chromosome appears to be the rule in XX/XY sex chromosome systems, we find that there is no such bias off the Z chromosome in either birds or lepidopterans. Escape from MSCI and sexually antagonistic selection have both been proposed to explain this phenomenon in XX/XY systems; however, although these processes are also occurring in ZZ/ZW systems, there is no corresponding gene movement. We have proposed several potential contributing factors for this difference, and further investigation into genomic processes that differ between ZZ/ZW and XX/XY will likely provide insight into these alternative explanations.
Supplementary Material
Supplementary tables 1–7 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank J. Siriwardane for assistance and D. Schrider for advice on analyses. This work was supported by National Science Foundation grant DBI-0845494 to M.W.H.
References
- Arunkumar KP, Mita K, Nagaraju J. The silkworm Z chromosome is enriched in testis-specific genes. Genetics. 2009;182:493–501. doi: 10.1534/genetics.108.099994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. A dynamic view of sex chromosome evolution. Curr Opin Genet Dev. 2006;16:578–585. doi: 10.1016/j.gde.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Bai YS, Casola C, Feschotte C, Betran E. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 2007;8:R11. doi: 10.1186/gb-2007-8-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betran E, Thornton K, Long M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002;12:1854–1859. doi: 10.1101/gr.604902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ. Evolution of sex determining mechanisms. Menlo Park (CA): Benjamin Cummings; 1983. [Google Scholar]
- Burch JBE, Davis DL, Haas NB. Chicken repeat-1 elements contain a pol-like open reading frame and belong to the non-long terminal repeat class of retrotransposons. Proc Natl Acad Sci U S A. 1993;90:8199–8203. doi: 10.1073/pnas.90.17.8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan ET, et al. Conservation of core gene expression in vertebrate tissues. J Biol. 2009;8:33. doi: 10.1186/jbiol130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of sex-chromosomes. Science. 1991;251:1030–1033. doi: 10.1126/science.1998119. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Mank JE. The birds and the bees and the flowers and the trees: lessons from genetic mapping of sex determination in plants and animals. Genetics. 2010;186:9–31. doi: 10.1534/genetics.110.117697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connallon T, Clark AG. The resolution of sexual antagonism by gene duplication. Genetics. 2011;187:919–937. doi: 10.1534/genetics.110.123729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Yoshimatsu TF, Long M. Retrogene movement within- and between-chromosome in the evolution of Drosophila genomes. Gene. 2006;385:97–102. doi: 10.1016/j.gene.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Sex-chromosome evolution: recent progress and the influence of male and female heterogamety. Nat Rev Genet. 2011;12:157–166. doi: 10.1038/nrg2948. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Carmichael A. Multiple and independent cessation of recombination between avian sex chromosomes. Genetics. 2001;158:325–331. doi: 10.1093/genetics/158.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Fridolfsson AK. Male-driven evolution of DNA sequences in birds. Nat Genet. 1997;17:182–184. doi: 10.1038/ng1097-182. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- Ellegren H, et al. Faced with inequality: chicken does not have a general dosage compensation of sex-linked genes. BMC Biol. 2007;5:40. doi: 10.1186/1741-7007-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, Kaessmann H, Betran E, Long MY. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24:363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- Fry JD. The genomic location of sexually antagonistic variation: some cautionary comments. Evolution. 2010;64:1510–1516. doi: 10.1111/j.1558-5646.2009.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallach M, Betran E. Intralocus sexual conflict resolved through gene duplication. Trends Ecol Evol. 2011;26:222–228. doi: 10.1016/j.tree.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M, et al. Fitting the gene lineage into its species lineage, a parsimony strategy illustrated by cladograms constructed from globin sequences. Syst Zool. 1979;28:132–163. [Google Scholar]
- Haas NB, et al. Subfamilies of CR1 non-LTR retrotransposons have different 5' UTR sequences but are otherwise conserved. Gene. 2001;265:175–183. doi: 10.1016/s0378-1119(01)00344-4. [DOI] [PubMed] [Google Scholar]
- Han MV, Hahn MW. Identifying parent-daughter relationships among duplicated genes. Pac Symp Biocomput. 2009;14:114–125. [PubMed] [Google Scholar]
- Handley LL, Ceplitis H, Ellegren H. Evolutionary strata on the chicken Z chromosome: implications for sex chromosome evolution. Genetics. 2004;167:367–376. doi: 10.1534/genetics.167.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hense W, Baines JF, Parsch J. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol. 2007;5:2288–2295. doi: 10.1371/journal.pbio.0050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Hollis GF, Hieter PA, McBride OW, Swan D, Leder P. Processed genes: a dispersed human immunoglobulin gene bearing evidence of RNA-type processing. Nature. 1982;296:321–325. doi: 10.1038/296321a0. [DOI] [PubMed] [Google Scholar]
- Hubbard T, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, et al. Dosage compensation is less effective in birds than in mammals. J Biol. 2007;6:2. doi: 10.1186/jbiol53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Vinckenbosch N, Long MY. RNA-based gene duplication: mechanistic and evolutionary insights. Nat Rev Genet. 2009;10:19–31. doi: 10.1038/nrg2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Richards RI. Human metallothionein genes: primary structure of the metallothionein-II gene and a related processed gene. Nature. 1982;299:797–802. doi: 10.1038/299797a0. [DOI] [PubMed] [Google Scholar]
- Kemkemer C, Hense W, Parsch J. Fine-scale analysis of X chromosome inactivation in the male germ line of Drosophila melanogaster. Mol Biol Evol. 2011;28:1561–1563. doi: 10.1093/molbev/msq355. [DOI] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Larkin MA, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lifschytz E, Lindsley DL. Role of X-chromosome inactivation during spermatogenesis. Proc Natl Acad Sci U S A. 1972;69:182–186. doi: 10.1073/pnas.69.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Ellegren H. Sex-linkage of sexually antagonistic genes is predicted by female, but not male, effects in birds. Evolution. 2009;63:1464–1472. doi: 10.1111/j.1558-5646.2009.00618.x. [DOI] [PubMed] [Google Scholar]
- Marques AC, et al. Emergence of young human genes after a burst of retroposition in primates. PLoS Biol. 2005;3:1970–1979. doi: 10.1371/journal.pbio.0030357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLysaght A. Evolutionary steps of sex chromosomes are reflected in retrogenes. Trends Genet. 2008;24:478–481. doi: 10.1016/j.tig.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol. 2011;9:e1001126. doi: 10.1371/journal.pbio.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Han MV, Hahn MW. A complex suite of forces drives gene traffic from Drosophila X chromosomes. Genome Biol Evol. 2009;1:176–188. doi: 10.1093/gbe/evp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova LM, Nurminsky DI. Lack of global meiotic sex chromosome inactivation, and paucity of tissue-specific gene expression on the Drosophila X chromosome. BMC Biol. 2011;9:29. doi: 10.1186/1741-7007-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle LC, Muir CD, Han MV, Hahn MW. The contribution of gene movement to the “two rules of speciation”. Evolution. 2010;64:1541–1557. doi: 10.1111/j.1558-5646.2010.00990.x. [DOI] [PubMed] [Google Scholar]
- Nam K, Ellegren H. The chicken (Gallus gallus) Z chromosome contains at least three nonlinear evolutionary strata. Genetics. 2008;180:1131–1136. doi: 10.1534/genetics.108.090324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potrzebowski L, Vinckenbosch N, Kaessmann H. The emergence of new genes on the young therian X. Trends Genet. 2010;26:1–4. doi: 10.1016/j.tig.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Potrzebowski L, et al. Chromosomal gene movements reflect the recent origin and biology of therian sex chromosomes. PLoS Biol. 2008;6:709–716. doi: 10.1371/journal.pbio.0060080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince EG, Kirkland D, Demuth JP. Hyperexpression of the X chromosome in both sexes results in extensive female bias of X-linked genes in the flour beetle. Genome Biol Evol. 2010;2:336–346. doi: 10.1093/gbe/evq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR. Sex-chromosomes and the evolution of sexual dimorphism. Evolution. 1984;38:735–742. doi: 10.1111/j.1558-5646.1984.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Schoenmakers S, et al. Female meiotic sex chromosome inactivation in chicken. PLoS Genet. 2009;5:e1000466. doi: 10.1371/journal.pgen.1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura M, et al. KAIKObase: an integrated silkworm genome database and data mining tool. BMC Genomics. 2009;10:486. doi: 10.1186/1471-2164-10-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinemann S, Steinemann M. Y chromosomes: born to be destroyed. Bioessays. 2005;27:1076–1083. doi: 10.1002/bies.20288. [DOI] [PubMed] [Google Scholar]
- Toups MA, Hahn MW. Retrogenes reveal the direction of sex-chromosome evolution in mosquitoes. Genetics. 2010;186:763–766. doi: 10.1534/genetics.110.118794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda S, Nakai S, Nishida Y, Hisajima H, Honjo T. Long terminal repeat-like elements flank a human immunoglobulin epsilon pseudogene that lacks introns. EMBO. 1982;1:1539–1544. doi: 10.1002/j.1460-2075.1982.tb01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S. Graph clustering by flow simulation. Utrecht (The Netherlands): Centers for mathematics and computer science (CWI), University of Utrecht; 2000. [Google Scholar]
- van Tuinen M, Hedges SB. Calibration of avian molecular clocks. Mol Biol Evol. 2001;18:206–213. doi: 10.1093/oxfordjournals.molbev.a003794. [DOI] [PubMed] [Google Scholar]
- Vibranovski MD, Zhang Y, Long MY. General gene movement off the X chromosome in the Drosophila genus. Genome Res. 2009;19:897–903. doi: 10.1101/gr.088609.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D. Progress and prospects toward our understanding of the evolution of dosage compensation. Chromosome Res. 2009;17:585–602. doi: 10.1007/s10577-009-9053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D. Lack of global dosage compensation in Schistosoma mansoni, a female-heterogametic parasite. Genome Biol Evol. 2011;3:230–235. doi: 10.1093/gbe/evr010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet. 2006;7:645–653. doi: 10.1038/nrg1914. [DOI] [PubMed] [Google Scholar]
- Vilella AJ, et al. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinckenbosch N, Dupanloup I, Kaessmann H. Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci U S A. 2006;103:3220–3225. doi: 10.1073/pnas.0511307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters JR, Hardcastle TJ. Getting a Full Dose? Reconsidering sex chromosome dosage compensation in the silkworm, Bombyx mori. Genome Biol. Evol. 2011;3:491–504. doi: 10.1093/gbe/evr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PJ. X chromosomes, retrogenes and their role in male reproduction. Trends Endocrinol Metab. 2004;15:79–83. doi: 10.1016/j.tem.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Xia Q, et al. Microarray-based gene expression profiles in multiple tissues of the domesticated silkworm, Bombyx mori. Genome Biol. 2007;8:R162. doi: 10.1186/gb-2007-8-8-r162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia QY, et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori) Science. 2004;306:1937–1940. doi: 10.1126/science.1102210. [DOI] [PubMed] [Google Scholar]
- Zha XF, et al. Dosage analysis of Z chromosome genes using microarray in silkworm, Bombyx mori. Insect Biochem Mol Biol. 2009;39:315–321. doi: 10.1016/j.ibmb.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Zhang W, et al. The functional landscape of mouse gene expression. J Biol. 2004;3:21. doi: 10.1186/jbiol16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE, Vibranovski MD, Krinsky BH, Long M. Age-dependent chromosomal distribution of male-biased genes in Drosophila. Genome Res. 2010;20:1526–1533. doi: 10.1101/gr.107334.110. [DOI] [PMC free article] [PubMed] [Google Scholar]