

Abstract
Epithelial cells organize into various tissue architectures that largely maintain their structure throughout the life of an organism. For decades, the morphogenesis of epithelial tissues has fascinated scientists at the interface of cell, developmental, and molecular biology. Systems biology offers ways to combine knowledge from these disciplines by building integrative models that are quantitative and predictive. Can such models be useful for gaining a deeper understanding of epithelial morphogenesis? Here, we take inventory of some recurring themes in epithelial morphogenesis that systems approaches could strive to capture. Predictive understanding of morphogenesis at the systems level would prove especially valuable for diseases such as cancer, where epithelial tissue architecture is profoundly disrupted.
Keywords: Morphogenesis, cancer biology, epithelial cells
Epithelial tissues define the boundary that separates our bodies from of our world. They protect us from microbial pathogens and secrete proteins that help to digest our food and feed our young. Epithelia absorb nutrients from what we consume and excrete waste to detoxify our bloodstream. Together, epithelial cells bear the brunt of physical-chemical insults imposed by our environment, and consequently epithelial tumors (carcinomas) comprise roughly 90% of all human cancers.
To achieve their breadth of function, epithelial tissues have evolved elaborate and diverse cellular architectures. The regulated interplay of epithelial form and function is part of a more-overarching question of tissue morphogenesis. Namely, how do precise multicellular structures arise during development or remodeling, and how do these structures become disrupted during disease? Answering this question is complicated, because many factors are involved that span molecular, cellular, and tissue scales.
Due to its multifaceted regulation and hierarchical organization, epithelial morphogenesis should be fertile ground for contributions from systems biologists. However, in the decade since “systems biology” was coined,1 morphogenesis has received little attention from a systems perspective. Perhaps this is because it is more difficult to break down the process into its fundamental units, as is commonly done with signaling pathways or gene-expression circuits.2
In this Advanced Review, we seek to define some of the core processes and modules that are evoked repeatedly during the morphogenesis of different epithelial tissues. Our focus is on mature epithelia because of their link to cancer. Thus, we do not discuss early morphogenetic programs in development, such as formation of the neural tube and ventral furrow, where transient epithelial cell states are involved. Another caveat is that the mechanisms drawn from the literature are not guaranteed to generalize across all epithelial tissues. We will therefore be careful to note the specific cell type used in each study. Nevertheless, the hope is that by organizing the biology of epithelial morphogenesis in this way, we can begin to define themes that apply to multiple epithelial-cell types.
We start with the basic morphologies that epithelial tissues can adopt, and then follow with the major inputs and outputs that drive their organization. Next, we discuss how epithelial morphologies and patterns are disrupted at the single-cell and tissue levels during tumorigenesis. We conclude with sections dedicated to experimental and computational models that have helped guide our understanding of epithelial morphogenesis.
Epithelial morphologies
Epithelial cells show characteristic organization at multiple length scales. We introduce two-dimensional epithelial cell sheets by using apico-basal and planar-cell polarity proteins as intracellular landmarks. We then discuss cellular and molecular pathways implicated in higher-order organization of epithelia into three-dimensional tubes, branches, and acini.
Sheets
Most normal epithelia exist as a thin sheet that displays asymmetry along the apical-basal axis (Figure 1(a)). The localization of ion channels and pumps to either the apical or basolateral membrane of epithelial cells generates a cellular gradient to transport solutes across the tissue (reviewed by Ref 3).3 The asymmetric distribution of membrane proteins is maintained by apical junctional complexes, which prevent the free diffusion of proteins between apical and basolateral domains.4 Junctional components include Par3, the adherens junction complex (consisting of E-cadherins, α-catenin and β-catenin), the Scribble group proteins (Scribble, Dlg, and Lgl) and ZO-1.5-8 These core interacting proteins influence assembly of the apical junctional complex and participate in apical–basal cell polarity, which is regulated during the morphogenesis of epithelial sheets. 9-11
FIGURE 1.
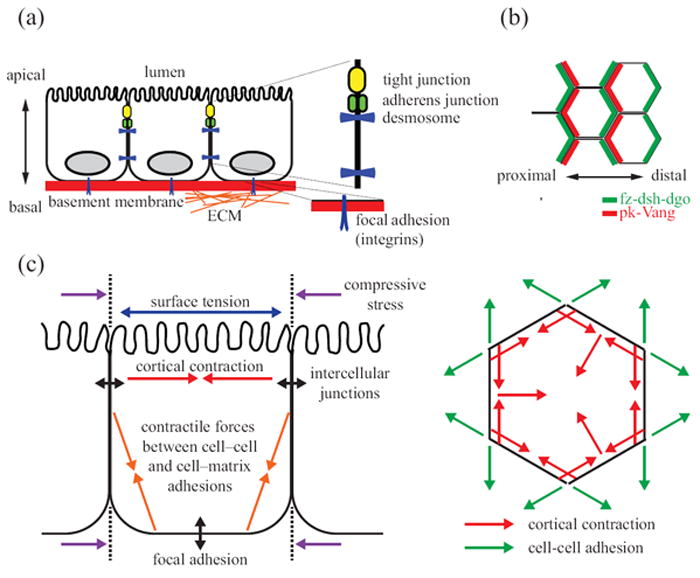
Morphology of a polarized epithelial monolayer. (a) Vertical epithelial polarization into distinct apical, basal, and lateral plasma membrane domains. Apical surfaces contain specialized microvilli for absorption and secretion. Basal surfaces bind to extracellular matrix (ECM) proteins via integrins to assemble focal adhesions. Lateral membranes contain junctional complexes (tight junctions, adherens junctions, and desmosomes) that link the adjacent cells and provide diffusion barriers. (b) Horizontal epithelial polarization of core planar cell polarity (PCP) proteins. In the Drosophila wing, fz, dsh, and dgo localize to the distal cortical domains, whereas pk and Vang localize to the proximal cortical domains. Shortly after the maximal asymmetry of PCP proteins is observed, actin-rich prehairs initiate from the distal domain of the cells. (c) Intracellular tension and intercellular forces shape epithelial morphology. Apical constriction results from the counterbalance of cortical tension and cellular adhesion. This constriction contributes line tensions at the interface between cells and serves as an energy barrier to cellular rearrangements.
Epithelial cells also can show another type of polarity, called planar cell polarity (PCP; see Table 1 for a list of abbreviations used in this Review), which is orthogonal to their apical–basal axis and in the orientation of cell-cell and cell-extracellular matrix (ECM) contact (Figure 1(b)). Asymmetric localization of mammalian PCP genes, such as Vangl2, Celsr1 and Fz6, is required for proper anterior-posterior compartmentalization of follicles in hair cells.12 The mechanisms of PCP regulation are being unraveled genetically through studies of bristle orientation in Drosophila epithelia. The signaling proteins of the core PCP group include frizzled (fz), disheveled (dsh), Van Gogh (Vang), prickle (pk), diego (dgo) and flamingo (fmi). During the development of the fly wing, the core PCP molecules are first evenly distributed near the adherens junction. Later, fz-dsh-dgo concentrate as a complex at the distal edge, whereas Vang-pk accumulate at the proximal sides. fmi co-localizes and interacts with both fz and Vang to form stable complexes.13
Table 1.
List of abbreviations used
| ADAMTS | a disintegrin and metalloproteinase with thrombospondin repeats |
| APC | adenomatous polyposis coli tumor suppressor |
| AT II | alveolar type II |
| c-FLIP | cellular caspase-8 (FLICE)-like inhibitory protein |
| CK1 | casein kinase 1 |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EMT | epithelial-to-mesenchymal transition |
| FAK | focal adhesion kinase |
| FGF | fibroblast growth factor |
| FRS2 | fibroblast growth factor receptor substrate 2 |
| Gab1 | Grb2-associated binding protein 1 |
| GAP | GTPase activating protein |
| GDNF | glial cell line-derived neurotrophic factor |
| GEF | guanine nucleotide exchange factor |
| Grb2 | growth factor receptor binding protein 2 |
| GSK3β | glycogen synthase kinase β |
| Hes | Hairy/enhancer of split |
| HGF | hepatocyte growth factor |
| HSPG | heparin sulfate proteoglycan |
| LRP | low-density lipoprotein receptor-related protein |
| MAPK | mitogen-activated protein kinase |
| MDCK | Madin-Darby canine kidney |
| MMP | matrix metalloprotease |
| MT1-MMP | membrane type 1 matrix metalloprotease |
| PCP | planar cell polarity |
| PI3K | phosphoinositide 3-kinase |
| ROCK | Rho kinase |
| RTK | receptor tyrosine kinase |
| SOS | son-of-sevenless homolog protein |
| TEB | terminal end bud |
| TGFβ | transforming growth factor β |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
Amonlirdviman and coworkers built a local-feedback model encoding behaviors of PCP proteins that was sufficient to describe the local alignment of PCP complexes.14 However, this model has difficulty reconciling PCP mutant phenotypes in other contexts.15 A more in-depth biochemical understanding of the relationships between PCP proteins may be required to build computational models that are broadly predictive.
Another important morphology of epithelial sheets is the columnar shape of individual cells (Figure 1(c)). The regulation of cell shape in epithelial sheets is crucial for many facets of morphogenesis. For example, the change in cell shape driven by apical constrictions is required for epithelial remodeling during tube formation of ventral furrow cells during Drosophila gastrulation.16 Columnar epithelial cell shape is controlled by two major mechanical forces: cortical tension and cell-cell adhesion.17 Cortical tension arises from the force generated within the cytoskeleton by actin-myosin-II interactions. Tension is exerted on the cortical F-actin network by myosin-II, which has been implicated in cell elongation during anaphase18 and in establishing the characteristic hexagonal shape of epithelial cells in proliferating intestinal epithelial cysts.19 Cortical tension is counter-balanced by adhesive forces from neighboring cells. When taken together, cells in an epithelium tend to minimize contact surface energy, resulting in a polygonal, cobblestone shape when viewed from above.20 The principle of energy minimization also applies to the shapes that epithelial sheets will adopt.21-23 Therefore, despite its simple architecture, the epithelial sheet is rich in cell and molecular organization that is important for proper function.
Tubes
Absorptive and secretory epithelia must arrange themselves macroscopically into tubes to provide conduits for directed transport within the organism. Tubes are comprised of polarized epithelial cell monolayers that surround a central lumen (Figure 2(a)). The apical epithelial surface lines the lumen, the basal surface faces the surrounding tissue and the lateral surface connects adjacent cells through intercellular junctions. Despite the often-specialized function of epithelial tubes in different tissues, tubulogenesis itself occurs in only a few ways.
FIGURE 2.
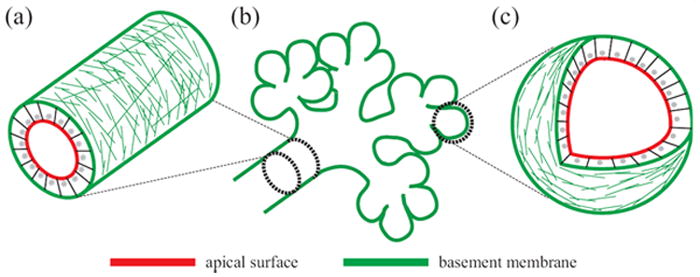
Building epithelial tissue architecture from tubes, branches, and acini. (a) Epithelial tubes are cylindrical monolayers with inner apical surfaces and outside basal surfaces that surround a central lumen. (b) Branched tubular networks result from epithelial extensions and bifurcations into the surrounding stroma. (c) Hollow spherical acini often terminate complex networks of branched tubes.
Many epithelial tubes are formed by remodeling pre-existing polarized epithelial sheets.24, 25 Two related mechanisms of formation of epithelial tubes from polarized cells are wrapping and budding.26 With wrapping, an epithelial sheet invaginates and curls, forming a final tube that is parallel to and separated from the surrounding epithelia, as during tube formation in the C. elegans digestive tract.27 With budding, cells migrate out and form a new tube. The new tube extends orthogonally from the original epithelial sheet. Budding is the principal means for tubulogenesis in the Drosophila trachea.28 Elsewhere, tubes form by polarization of epithelial precursors and exocytosis of vesicles or by removal of inner cells through apoptosis.24, 25 Unlike wrapping and budding, this mechanism requires initiation of a polarized epithelium. Polarization can be regulated both by ECM signals29 and by cell division.30, 31 Recent in vitro studies have shown that Rab-mediated membrane traffic and polarity complexes can cooperate to generate the apical surface and lumen de novo.32 Directional trafficking works both upstream and downstream of polarity proteins, suggesting a positive-feedback mechanism for generating polarity.
After cells have assembled into cylindrical cords, lumens can be created by eliminating cells in the center (cavitation) or by forming lumens de novo at apical membrane sites between cells (cord hollowing). 26 Cavitation occurs, for example, in mice during the formation of submandibular and mammary glands33, 34 and will be further discussed below. Cord hollowing occurs in rapidly polarizing Madin-Darby canine kidney (MDCK) cysts,35 during formation of the zebrafish gut,36 and during pancreatic tubulogenesis.37 In the zebrafish gut, small lumens then combine into a single lumen by a process that involves paracellular ion transport.36 Single lumen specification is genetically regulated by the expression of ion transporters, which control luminal fluid accumulation and promote the coalescence of multiple small lumens.
Branches
Tubular networks are joined at bifurcations that initiate as branch points during organ development (Figure 2(b)). Branching morphogenesis is a process whereby the epithelium elongates against the stroma or mesenchyme. Epithelial branches can be achieved by very different strategies depending on the tissue and the organism. Most commonly, branching involves cell proliferation together with cell migration, and both processes will be discussed below. However, tracheal branching in Drosophila38 and submandibular gland branching in mice39 do not require proliferation. Conversely, the early stages of lung branching in mammals occur normally even when migration is severely impaired.40 Thus, branched tissues may exploit proliferation or migration depending on the context, but neither appears universally required.
Ewald and coworkers used long-term confocal imaging and organotypic breast-epithelial culture techniques to visualize branching morphogenesis in vitro.41 In this culture model, branch morphogenesis initiates from the sprouting of highly proliferative terminal end buds (TEBs) of the mammary gland. TEBs extend from a duct and are partially depolarized. During branch elongation, junction-connected luminal epithelial cells advance collectively, whereas loosely connected myoepithelial cells appear to restrain elongating ducts. Thus, elaboration of the mammary ductal tree may result from antagonism between advancing TEBs and a belt of myoepithelia that locally constricts elongation.
Mechanical stress transmitted by the ECM has also been implicated in branch formation. For example, ECM degradation in embryonic mouse lung enables the adjacent epithelium to invade as a bud.42 Migratory epithelial cells and invasive cancer cells have been reported to generate surface-localized protease membrane type 1 matrix metalloproteinase (MT1-MMP), which causes degradation of local pre-existing ECM and reduces mechanical resistance.43, 44 Tumor-associated myoepithelial cells, which contact the basement membrane directly, deposit additional basement membrane components around the duct to help maintain the polarity of luminal epithelial cells after branch formation.45 ECM regulation thus plays an important role in epithelial branching.
Cell-cell and cell-ECM interactions are complemented by diffusible growth factors that act as instructive chemotactic cues for branch formation. Glial cell line-derived neurotrophic factor (GDNF), epidermal growth factor (EGF), and fibroblast growth factor (FGF) drive kidney branching,46 whereas EGF and hepatocyte growth factor (HGF) are critical branching factors in the mammary gland and lung.47, 48 By contrast, transforming growth factor β (TGFβ) is a potent inhibitor of branching in luminal epithelia.49 Nelson and colleagues used micropatterning together with computational modeling to reveal that branches form at sites of low local concentration of inhibitory autocrine morphogens, such as TGFβ.50 These results illustrate how tissue geometry controls the local microenvironment to regulate branching morphogenesis.
Acini
Branched epithelial trees terminate in secretory or absorptive epithelial acini that increase surface-to-volume ratios of the organ (Figure 2(b)). Their formation typically requires an explicit cavitation process to convert proliferating buds into mature acini (Figure 2(c)). Cavitation starts with the polarization of cells on the periphery, followed by lumen clearance of cells in the center. Cavitation is observed in 3D models of mammary acini (MCF10A cells),51, 52 in kidney cell-culture models (MDCK cells),35 and also in mouse TEBs in vivo.34 Interestingly, both cavitation and cord hollowing occur in MDCK cells, depending on cell polarization efficiency,35 suggesting that cavitation may act as a backup hollowing mechanism for epithelia that polarize slowly during morphogenesis.
Clearance of the lumen during cavitation is strongly influenced by the pro- and anti-apoptotic proteins of the Bcl-2 family. For example, overexpression of Bcl-2 or Bcl-XL inhibits caspase-3 activation and delays lumen formation in 3D breast epithelial cultures.51 Bim, a key Bcl-2 sensor of integrins and EGF receptor deprivation, is induced after cell detachment and contributes to programmed cell death in MCF10A 3D culture.53 Inhibition of Bim expression significantly delays apoptotic cell death of the central cells,54 and in vivo disruption prevents apoptosis and delays lumen clearing in mouse TEBs.34 Functional loss of another BH3-only protein, Bmf, provides protection from cell death during MCF10A anoikis and 3D morphogenesis.55 Thus much of Bcl-2-family regulation associated with apoptotic clearance is linked to stress caused by ECM detachment. However, programmed cell death is not the only mechanism for lumen clearance because cavitation eventually takes place even when apoptosis is blocked. Upregulated prodeath ligands such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) have been shown to cause autophagy in ECM-deprived cells.56 Therefore, clearance may involve a mixed cell-death program that ensures complete cavitation.
Epithelial cell outputs
Epithelial tissue morphology arises from the phenotypic responses of individual cells. These cellular “outputs” organize into major categories that control cell orientation, its microenvironment, and overall population size.
Proliferation and apoptosis
As with nearly all tissues, total cell numbers are tightly regulated during the development and maintenance of epithelia. The expansion, contraction, and homeostasis of epithelial cell populations are determined by the rates of cell proliferation and cell death. Recent work suggests that the timing and location of these events is perhaps more critical than the quantitative extent of proliferation or death during epithelial morphogenesis. For example, during in vitro mammary branching morphogenesis, epithelial proliferation is transiently increased in the vicinity where new ducts have initiated.57 If proliferation is constitutively high, then branching morphogenesis is suppressed and epithelia grow as spheroids.58
Similar results have been reported for apoptotic cell death during breast-epithelial acinar morphogenesis.52 Apoptosis is a critical event during luminal clearance of epithelial spheroids, and cell-death inhibition synergizes with hyperproliferation-inducing oncogenes to cause luminal filling.51 However, apoptosis must also be suppressed during the preceding phase of proliferation and among matrix-attached cells that eventually form the final acinar structure. Yerbes et al. recently demonstrated the tight requirement for apoptosis control by perturbing the levels of c-FLIP, an antiapoptotic protein that is transiently induced during morphogenesis. 55, 59 As expected, c-FLIP overexpression caused delayed luminal clearance in a manner similar to that reported for Bcl-2 overexpression.51 But surprisingly, the authors found that stable knockdown of c-FLIP prevented acinar formation altogether,59 suggesting that apoptosis must first be suppressed and then selectively induced to give rise to normal acinar morphology. These findings together suggest that a systems-level understanding of morphogenesis will be aided by computational models that explicitly consider the timing of cell outputs.60, 61
Extracellular matrix remodeling
Epithelia are kept separated from the adjacent tissue stroma by a special form of ECM called basement membrane. The removal and deposition of basement membrane is critical for epithelial branching morphogenesis.62 Basement membranes are highly self-assembled sheets in which glycoproteins and proteoglycans are cross-linked to laminins and collagen IV fibrils along the basal side of epithelial cells. Basement membrane degradation is catalyzed by metalloproteinases, including the matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase with thrombospondin repeats (ADAMTS) families. The local composition, turnover, and mechanics of basement membrane together govern tissue organization.
Membrane-tethered MMPs (MT-MMPs) play an especially important role in branching morphogenesis.63 MT1-MMP degrades fibrillar collagen as an initial cue for cancer cell migration.44 MT2-MMP-dependent proteolysis of collagen IV is linked with epithelial proliferation to coordinate the morphogenetic program of the submandibular gland.64 ADAMTSs act as the dominant secreted metalloproteinases cleaving large chondroitin-sulfate proteoglycans during morphogenesis.65 For example, three ADAMTS family members, Adamts5, Adamts9, and Adamts20 cooperate in limb morphogenesis to influence cell apoptosis and the regression of interdigital tissues.66 The tethering of MT-MMPs allows basement membrane degradation to be localized to individual cells, whereas ADAMTSs may require diffusion from the cell surface to gain access to their bulky substrates.
In addition to ECM degradation, myoepithelial cells in breast tissue secrete basement membrane components (including laminins, nidogen 1, perlecan and type IV collagen) between the cells and the ECM to form the secondary ECM around the new branch.67 Myoepithelial cells also produce maspin, an inhibitor of ECM-degrading proteases.68 Thus, epithelial tissues can upregulate basement membrane constituents by active secretion or inhibition of turnover.
Single-cell migration
Single-cell migration is complex and heterogeneous during morphogenesis. Epithelial cells migrate along a basement membrane, not through interstitial tissues, and often stop upon terminal differentiation. Yet, most cell migration involves the cyclic repetition of the same steps: edge protrusion, interaction with the ECM, translocation of the cell body, and retraction of the cell rear in response to ECM ligands.69 Individual cells transiently form and resolve cell-cell contacts during single-cell migration, whereas collective migration occurs with stringent cell-cell adhesions that silence migration activity of cells within the group. Cell-cell junctions are tightly controlled during migration and this control can be regulated by other factors. For example, TGFβ activation is associated with the loss of cell-cell adhesions and single-cell movement in breast cancer, while the inhibition of TGFβ signaling reverts these cells to collective migration.70
Single-cell migration in embryonic development usually involves an epithelial-to-mesenchymal transition (EMT), but dedifferentiation is rare in maturing tissue. Nevertheless, singly motile cells are often observed within epithelial tissues, such as during branching morphogenesis of ureteric buds and salivary glands, 71, 72 tissue remodeling of the small intestine, and polarization of breast epithelial acini. 57, 73, 74 The migration of single epithelial cells occurs within a larger group of nonmotile epithelia and is typically not coordinated with other cells that happen to be moving at the same time. However, “orchestrated” single-cell rearrangements have been documented in the Wolffian duct epithelia of the ureteric bud and in engineered mammary epithelial tubules. 75, 76 Currently, there is a lack of molecular tools that can target single-cell migration without affecting collective migration or other processes involving the cytoskeleton. Thus, although single-cell migration is undoubtedly an important phenotype, its precise quantitative role during morphogenesis is difficult to define.
Collective cell migration
Collective cell migration is distinct from single-cell migration in that cells in the same group remain connected with cell-cell junctions and coordinately exhibit front-rear polarity during movement.77 Among cells at the leading edge, force generation occurs by engagement of integrins with ECM at the free edge and cadherin-mediated cell-cell junctions with neighboring cells. Cell-generated contractile forces are regulated by a “tug-of-war” between cell-cell and cell-matrix adhesions during sheet migration in 2D and branching in 3D, both of which involve collective mechanisms. For example, cadherin cell-cell contacts in MCF7 breast epithelial monolayers are modulated by translocation of the focal adhesion protein vinculin from cell-matrix adhesions to cell-cell junctions.78 Conversely, increasing matrix stiffness elevates Rho kinase (ROCK)-generated contractility and focal adhesion formation in mammary epithelial cells cultured in 3D matrix, which in turn weakens adherens junctions and disrupts morphogenesis.79
Various epithelial tissues restore their sheet-like organization by collective migration during wound healing.80, 81 Cell migration at the healing front is coupled with epithelial proliferation within the sheet during wound closure. Mechanical force is a prominent factor in collective 2D migration. Leading cells initiate lamellipodia into the wound area, which transmit force to follower cells by Rho-dependent mechanisms.82, 83 When the leading cells at the front edge extend protrusions, cells away from the wound edge form basal membrane protrusions, which together generate the main traction forces for motility.84 Thus, collective 2D migration does not originate exclusively from the “pulling” of the leading cells at the wound edge or the “pushing” of proliferating cells away from the edge, but by the tensile stresses that accumulate globally in the tissue.85
In 3D, collective migration is a dominant mechanism of cellular reorganization during tube elongation and branching morphogenesis. In the zebrafish lateral line primordium and Drosophila trachea, a group of leading cells guides a strand-like collective stalk during branching.86, 87 Collective migration occurs without leading cell protrusions, such as filopodia and lamellipodia.57 Developing tissues often further require proteolytic mechanism to remove ECM barriers.88 To date, much of the work in 3D migration has focused on the importance of diffusible factors. In the trachea, branching by collective migration is initiated in cells at the tip of each branch upon stimulation with FGF.89 FGF-induced collective migration has also been implicated in branching of primary organotypic 3D cultures of mouse mammary glands. Notably FGF also promotes canonical 2D wound healing in epithelial sheets,90 suggesting that the same factor may promote collective migration differently in 2D-vs-3D contexts.
Cell polarization
In 3D in vitro cultures of MDCK epithelial cysts, apicobasal polarity of mature epithelia is established before cell division completes. A discrete apical surface develops between dividing daughter cells after spindle orientation has been established by the Rho-family GTPase, Cdc42.91 Cdc42 together with Rac1 (another small G protein) are thought to determine the early steps in epithelial polarization. Rac1 and Cdc42 are essential during the establishment of apicobasal polarity in MDCK cysts.92, 93 In response to cell-matrix interactions, β1-integrins orient the apical pole of polarized cysts via Rac1 activation.29 In 3D cultures of an intestinal epithelial cell line (Caco-2), Cdc42 regulates spindle orientation and apical surface positioning during cell division.91 Rich1, a Cdc42-specific GTPase-activating protein (GAP), associates with components of the apical-polarity network and maintains tight-junction assembly between MDCK epithelial cells through its regulation of Cdc42.94 Conversely, the Cdc42-specific guanine nucleotide exchange factors (GEFs), tuba and intersectin2, serve as key activators of Cdc42 to control spindle pole orientation during cyst formation.95, 96
The initial cell-cell adhesion and cell-ECM contacts provide the first spatial cues for cells to distinguish apical from basolateral surfaces. For instance, in MDCK cells the post-Golgi vesicles containing basolateral membrane proteins are targeted directly to the site of E-cadherin-mediated cell-cell contacts, while apical proteins are excluded.97 In addition, cell-ECM contacts, such as those between collagen I and β1-integrins, can prime basement membrane deposition by Rac1-dependent mechanisms to reinforce cell polarity.29 This cell-ECM priming is critical, because apicobasal polarity inverts when β1-integrin or Rac1 function is blocked.29, 92 Interestingly, polarity inversion in MDCK cells is not a default pathway but instead is regulated by another GTPase, RhoA, along with its effectors, ROCK1 and myosin II.98 A morphologically similar inversion of glandular epithelial polarity has been detected during tumor progression,99 suggesting cells may occasionally lack the appropriate priming ECM in vivo.
Although stable in mature tissues, apicobasal polarity is dynamic and heterogeneous during morphogenesis and tissue remodeling in vitro. For example, cells within the TEB-like region of cultured mammary glands partially lose apicobasal polarity during expansion and branching.57 Apicobasal polarity is also transiently downregulated during HGF-induced tubulogenesis in MDCK cysts.100 The dynamics of cell polarity link tightly to the proliferative and differentiation states of epithelial cells. 10 Thus, cell polarity should be considered among the earliest epithelial outputs, setting the stage for many phenotypes critical for morphogenesis.
Epithelial cell inputs
The precise signature of epithelial cell outputs during development is determined by a host of extracellular factors that act as “inputs” to shape cell fate and tissue organization. Growth factors and cytokines diffuse through tissues to exert long-range control, whereas paracellular signals are sent by an epithelial cell’s immediate neighbors. In addition, mechanical inputs are now widely recognized to influence cell fate and tissue function.101 When possible, we focus on systems models and systems experiments in the literature that have focused on the factors known to be important for epithelial morphogenesis.
Diffusible factors
Hepatocyte growth factor
Hepatocyte growth factor (HGF) is a fibroblast-derived factor that promotes motility, proliferation, and tubule formation in epithelial cells.102, 103 HGF binds to the receptor tyrosine kinase (RTK) proto-oncogene c-Met, leading to receptor dimerization and autophosphorylation.104-106 c-Met tyrosine phosphorylation recruits multi-protein docking complexes containing growth factor receptor binding protein 2 (Grb2) and son-of-sevenless homolog protein (SOS) that promote Ras–MAPK activation (Figure 3(a)).107, 108 Additionally, activated c-Met dimers bind directly to Grb2-associated binding protein 1 (Gab1), which induces downstream epithelial cell motility through its interaction with the Crk-like adaptor protein.109, 110 Gab1 also serves to recruit phosphoinositide 3-kinase (PI3K), resulting in lipid signaling that activates the anti-apoptotic AKT pathway.111, 112
FIGURE 3.
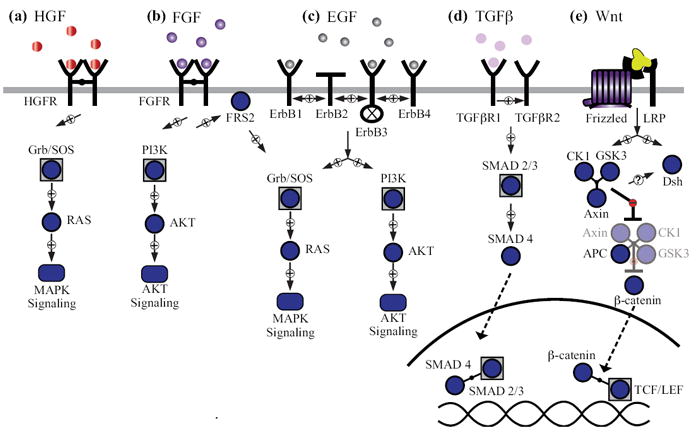
Diffusible factors involved in epithelial morphogenesis. (a) Hepatocyte growth factor (HGF) signaling. Ligand binding to the HGF receptor (HGFR) leads to tyrosine autophosphorylation and Ras–MAPK (mitogen-activated protein kinase) signaling. (b) Fibroblast growth factor (FGF) signaling. Ligand binding to the FGF receptor (FGFR) leads to tyrosine autophosphorylation and recruitment of fibroblast growth factor receptor substrate 2 (FRS2) to activate Ras–MAPK and phosphoinositide 3-kinase (PI3K)–AKT signaling. (c) Epidermal growth factor (EGF) signaling. Ligand binding to ErbB receptors leads to homo- and heterodimerization, tyrosine autophosphorylation, and signaling through the MAPK and AKT pathways. ErbB2 does not bind EGF-family ligands, and ErbB3 is catalytically inactive. (d) Transforming growth factor β (TGFβ) signaling. Ligand binding leads to heterodimerization of TGFβ receptor I (TGFβRI) and TGFβ receptor II (TGFβRII). TGFβRII transphosphorylates TGFβRI, which phosphorylates SMAD2/3 allowing them to heterodimerize with SMAD4 and translocate into the nucleus to modulate gene expression. (e) Wnt signaling. Ligand binding to the frizzled (Fz) receptor recruits the low-density lipoprotein receptor-related protein (LRP) coreceptor, which downregulates the β-catenin destruction complex. This results in β-catenin accumulation in the cytoplasm and its translocation to nucleus where it binds to T-cell factor/lymphoid enhancer factor (TCF/LEF) and initiates transcription.
The HGF-c-Met signaling pathway has been examined computationally with logic-based Boolean networks that reflect published protein-protein interaction data.113 By applying this model to microbial pathogenesis, HGF-induced pathways were fused with H. pylori signaling to examine c-Met activation during infection. Boolean approaches thus provide a convenient means for combining diverse signaling sub-networks, as has recently been shown in hepatocytes.114
Fibroblast growth factor
The fibroblast growth factor (FGF) signaling pathway contains 22 known ligands and four FGF receptors (FGFRs), leading to a large functional diversity for the signaling network.115, 116 FGF signaling plays a crucial role in epithelial morphogenesis as an inducer of branching and differentiation in lung epithelia.117 Under normal conditions, FGF is sequestered within the ECM and requires the presence of heparin sulfate proteoglycans (HSPGs) to bind to the appropriate FGFR and initiate signaling.118 HSPGs act as reservoirs for FGF ligands promoting their dimerization to induce higher-affinity FGFR binding, receptor dimerization, and RTK activation.116 Dimerized FGFRs phosphorylate and bind fibroblast growth factor receptor substrate 2 (FRS2), which recruits the Grb2–SOS complex leading to Ras–MAPK pathway activation (Figure 3(a)).119, 120 FGF ligand binding to FGFR additionally causes FGFR ubiquitylation, Src-mediated endocytic trafficking, and receptor downregulation.121-123
Like many RTKs, FGFR contains multiple tyrosine residues that become autophosphorylated during activation. The precise sequence of these autophosphorylation events is difficult to characterize but is important for quantitative modeling of the overall signaling network.124 To address this, Furudi et al. developed an approach for FGFR autophosphorylation that combines native gel electrophoresis with mass spectrometry.125 The authors used native gels to determine the stoichiometries and kinetics of tyrosine phosphorylation together with mass spectrometry to identify specific phosphotyrosine residues. By fitting their data to a kinetic model, Furudi et al. found that the first tyrosine is phosphorylated the fastest, with each subsequent phosphotyrosine accumulating at half the rate of the one before it. These empirical differences in autophosphorylation kinetics have not yet been implemented in quantitative models of FGFR signaling.126
Epidermal growth factor
Epidermal growth factor (EGF) is the founding member of a family of ligands that promote epithelial proliferation.127 EGF and EGF-like ligands directly or indirectly bind to the epidermal growth factor receptors, ErbB1-4. Canonical EGF signaling is very similar to the RTKs introduced above: ligand binding causes receptor autophosphorylation, which creates binding sites for the adaptors Shc and Grb2–SOS (activating Ras–MAPK), and the p85 subunit of PI3K (activating AKT signaling through the p110 catalytic subunit of PI3K) (Figure 3(a)). Added complexities arise from the properties of the individual receptors—ErbB2 does not bind ligands and ErbB3 lacks tyrosine-kinase activity—and from their differing abilities to homo- or heterodimerize and traffic intracellularly upon activation.128
Signaling downstream of ErbB receptors is the best characterized of all RTKs, rendering it ideal for computational modeling.129 Various models have been built to emphasize specific facets of the biology, such as MAPK signaling emanating from different ErbB adaptor proteins,130 ligand-specific ErbB activation and signaling,131 and crosstalk among ErbB-induced immediate-early genes.132 EGF-family signaling is therefore a prime candidate for future multiscale models that link molecular networks to multicellular organization.
Transforming growth factor β
Transforming growth factor β (TGFβ) signaling plays a critical role in inhibiting cell proliferation and exerts homeostatic control of cell numbers in many epithelial tissues.133-137 TGFβ ligands bind to a heterodimer complex of two receptor serine-threonine kinases, TGFβRI and TGFβRII. Upon ligand binding, the TGFβRII serine phosphorylates TGFβRI to activate the kinase,138 leading to the subsequent phosphorylation of the SMAD transcription factors, SMAD2 and SMAD3 (Figure 3(b)).139 Before TGFβR stimulation, SMAD2/3 proteins are sequestered in the cytoplasm, and upon phosphorylation, SMAD2/3 translocates to the nucleus with the co-SMAD, SMAD4, to initiate downstream transcriptional activity.140 As with the RTKs, signaling activity is terminated by the ubiquitylation and downregulation of TGFβRs through endocytic trafficking and subsequent lysosomal degradation.141 The exact mechanism of surface receptor regulation remains unclear, but it is an important aspect of TGFβR signaling, as constitutive internalization can entirely sequester TGFβRs in the cytoplasm.142
To examine the signaling properties conferred by TGFβR trafficking, Vilar and coworkers built a simple two-compartment model of the network.143 The authors predicted that regulation of constitutive-vs.-ligand-induced TGFβR degradation in endosomes could determine transient-vs.-sustained SMAD responses to TGFβ ligands. Validation of this prediction awaits quantitative measurements of TGFβR trafficking, like those available for the ErbB receptors.144
Wnt
Wnt comprises a family of lipid-modified ligands that promote proliferation and differentiation of epithelial cells during morphogenesis.145-149 There are 20 different Wnt ligands in humans that bind to Frizzled (Fz) receptors and the co-receptor, low-density lipoprotein receptor-related protein (LRP).150-152 In the absence of Wnt, the key effector protein of the pathway, β-catenin, is subject to constitutive proteolysis by a destruction complex composed of two scaffolding proteins (Axin and APC) and two kinases (GSK3β and CK1) (Figure 3(c)).153-157 Binding of Wnt to Fz-LRP translocates the destruction complex to the plasma membrane along with a third scaffolding protein, disheveled (Dsh), which leads to inhibition of the destruction complex and the consequent destabilization of β-catenin levels in the cytoplasm.158-161 Stabilized β-catenin accumulates in the nucleus and acts as a coactivator for TCF/LEF family proteins to induce gene expression.162-165 Thus, among diffusible factors important for epithelial morphogenesis, the Wnt pathway is unique in that protein localization and stability act as the major signal transducers, with phosphorylation playing a supporting role.
A quantitative differential equations-based model of the canonical Wnt pathway has been defined by biochemical reconstitution of the core signaling elements.166, 167 Using frog egg extracts to study the Wnt pathway in a test tube, the authors were able to avoid complications associated with protein localization in cells and focus on the phosphorylation-degradation reactions. The stability of β-catenin is directly controlled by phosphorylation, but it can also be indirectly modulated by the scaffold Axin, which is degraded in response to Wnt.168, 169 The model provided explanations for why scaffold destruction would be beneficial for Wnt signal transmission. More generally, it highlights classical biochemical approaches as important-but-underutilized tools for systems-biology research.170
Paracellular factors
E-cadherin
Information is directly transmitted between neighboring cells by cell-cell signaling receptors. The major homotypic receptor in epithelia cells is E-cadherin, a Ca2+-dependent receptor that is critical for cell-cell recognition and adhesion (Figure 4(a)).171 The cytoplasmic domain of E-cadherin connects to the actin cytoskeleton through α- and β-catenin to preserve cell shape and polarity. In MDCK cells, cadherin–catenin complexes influence signaling predominantly through small GTPases. Rac-induced lamellipodia activity is suppressed as E-cadherin accumulates, and Rho-dependent actomyosin contractility increases during de novo contact formation once cytoskeletal connections have formed.172
FIGURE 4.
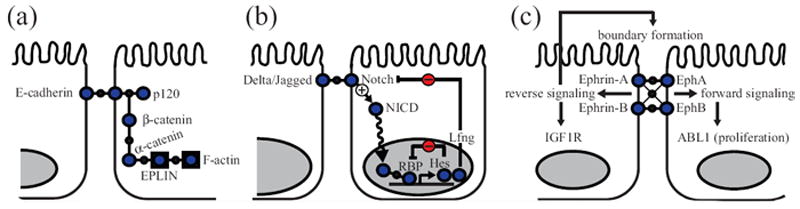
Intercellular communication between adjacent epithelial cells via cell–cell signaling receptors. (a) E-cadherin signaling. E-cadherins form extracellular homophilic complexes between neighboring cells. p120 catenin binds to the juxtamembrane domain of E-cadherin and stabilizes E-cadherin by preventing clathrin-mediated endocytosis. β-catenin binds to E-cadherin. α-catenin shuttles between β-catenin and epithelial protein lost in neoplasm (EPLIN), and connects the E-cadherin complexes to F-actin. (b) Delta-Notch signaling. Notch binds to its ligand (Delta or Jagged) on adjacent cells, proteolytically releasing the Notch intracellular domain (NICD). NICD moves to the nucleus and interacts with DNA-binding protein RBP (recombination signal-binding protein for immunoglobulin kappa J region) along with several coactivators to regulate gene expression. Multiple Notch target genes, such as Hairy/enhancer of split (Hes) and lunatic fringe (Lfng), are negative regulators of the Notch pathway. These feedbacks can give rise to oscillations in gene expression. (c) Eph–ephrin signaling. Eph–ephrin binding on neighboring cells leads to bidirectional phosphotyrosine signaling. Eph-expressing cells activate Eph RTK activity together with other tyrosine kinases, such as ABL, for each forward signaling. In ephrin-expressing cells, other RTKs such as insulin-like growth factor receptor (IGF1R) are transactivated. Ephrin-induced reverse signaling is critical for boundary formation.
Loss of E-cadherin is sufficient to promote an EMT in breast epithelial cells.173 EMT may promote metastasis of solid tumors,174 and many EMT inducers directly or indirectly suppress E-cadherin expression.174-177 The dynamics of E-cadherin–β-catenin interactions and its regulation of EMT were incorporated into a multi-scale model of cell–cell adhesion.178 Within this model, regulation of cell-cell adhesion by E-cadherin was sufficient to describe single-cell detachment from neighboring cells. Conversely, restoration of E-cadherin in invasive carcinomas has been shown to reduce the metastatic potential of transformed MCDK cells and human breast carcinoma cells.179 Therapies that can restore E-cadherin protein levels may be effective in cancers where E-cadherin transcription is suppressed.
Delta-Notch
Epithelial cells also signal to adjacent cells through heterotypic interactions involving the Notch receptor (Figure 4(b)). Notch binds to one of its ligands (Delta or Jagged) on a neighboring cell, causing proteolytic cleavage of the receptor. This cleavage releases an intracellular domain of Notch1 that then translocates into the nucleus and induces gene expression. Among Notch-dependent genes is a group of cyclic transcriptional repressors, called the Hairy/enhancer of split (Hes) family, which oscillate in expression by repressing their own transcription.180 An additional layer of negative feedback is provided by the Notch-modifying glycosyltransferase enzyme, Lunatic Fringe, which is an inhibitor of Notch signaling and also a Notch target gene.181 These negative feedback loops are thought to contribute to the different responses of cells to varying levels of Notch stimulation. For example, in breast epithelial cells, high Notch activity suppresses cell proliferation and cell-ECM adhesion, whereas low Notch activity causes hyperproliferative acini and maintains matrix adhesion.182 By virtue of these dose-dependent actions, Notch acts as a morphogen during development, whose function may be subverted during tumorigenesis.183
Computational models are investigating whether Delta-Notch negative-feedback loops are sufficient to generate the long-range patterns in signaling observed during development. Webb and Owen have compared several of the prevailing models that incorporate different non-cell autonomous properties of Delta-Notch signaling.184 In some contexts, high Delta expression in one cell suppresses Delta expression in its neighbors through Notch by a process called lateral inhibition.185 In other contexts, the reverse happens through a process called lateral induction.186 Surprisingly, the authors found that both mechanisms are capable of generating long-range patterns for certain feedback strengths. Further computational work on Delta-Notch signaling could be used to identify perturbations to signaling or feedback that would distinguish whether lateral inhibition or lateral induction is the dominant non-cell autonomous mechanism.
Eph-Ephrin
Adjacent epithelial cells communicate bidirectionally through interactions between Eph-family tyrosine kinases and their signaling ligands, the Ephrins (Figure 4(c)). Binding of Ephs to Ephrins on neighboring cells leads to receptor clustering, which increases phosphotyrosine signaling in both cells. Eph-Ephrin signaling typically acts as a repulsive cue that defines cellular boundaries between tissues and organs. A recent systems-level study by Jorgensen et al. examined the signaling activated bidirectionally by EphB2 and Ephrin-B1 engagement.187 By differentially labeling EphB2- and Ephrin-B1-expressing cells, the authors found that tyrosine kinase signaling in the two cells is asymmetric. Tyrosine kinase signaling in EphB2 cells emanated directly from EphB2 and from the nonreceptor tyrosine kinase ABL1. Conversely, in Ephrin-B1-expressing cells, the major tyrosine kinase that became activated was the insulin-like growth factor receptor, IGF1R. Eph-Ephrin-mediated segregation and epithelialization have been described as a tumor suppressor in colorectal cancers.188 The repulsive interactions between EphB-positive colorectal tumor cells and surrounding EphrinB-positive normal cells leads to segregation of the transformed cells within the tumor.189 This segregation may act as a tumor suppressive mechanism that inhibits cancer invasion.
Substrate stiffness and cortical tension
The tissue organization of cells is governed not only by chemical signaling networks but also by the mechanical properties of the surrounding environment. For example, breast epithelial cells undergo ductal morphogenesis when cultured in a soft low-density matrix but not when the same matrix is crosslinked and under higher tension.190 Chemically equivalent ECMs promote different cytoskeletal organization depending on the ECM rigidity.191 ECM tension is detected by a filamin-β1-integrin complex in mammary epithelial cells.190 Substrate stiffness provides the initial trigger for a cascade of mechanotransduction processes that influence cell contractility, migration, and cell-fate decisions. Adhesion of integrins leads to contraction of the cytoskeleton through myosin II, and the resulting actomyosin contractile force deforms either the adhesion complex (if the substrate is rigid) or the substrate (if the substrate is soft). Stiff ECM clusters integrins more effectively, which activates proliferation via ERK and cell contractility via Rho and ROCK to disrupt normal morphogenesis of breast epithelia.192 Increased ECM tension is particularly relevant for tumorigenesis. For example, collagen crosslinking is enhanced in mouse models of breast cancer through upregulation of lysyl oxidase, which promotes malignancy.193 A major challenge lies in understanding the interplay between ECM stiffness and ECM remodeling.
Epithelial heterogeneity in morphogenesis
The textbook cytoarchitecture of many epithelial tissues is reasonably simple. However, the grossly similar appearance of epithelial cells in a tissue belies a widespread heterogeneity that is observed at the molecular level in single cells. For example, among a large pool of proliferating cells in an expanding mammary ductal tree, only a small percentage of cells have the capacity to generate ductal outgrowths containing the luminal and myoepithelial lineages that comprise normal tissue (Figure 5(a)).194, 195
FIGURE 5.
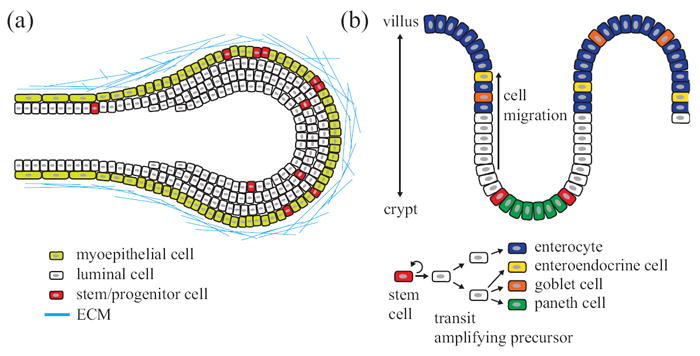
A subset of the molecular and phenotypic heterogeneities among epithelia in the breast and small intestine. (a) Breast tissue contains an outer layer of myoepithelial cells and an inner layer of luminal epithelia. Stem/progenitor cells are distributed throughout the organ and multiple stem cells can contribute to branching morphogenesis. (b) The crypt–villus unit of the small intestine. Paneth cells lie at the bottom of the crypt. Stem cells located above paneth cells produce transit-amplifying precursors that migrate and differentiate to enterocytes, enteroendocrine cells, goblet cells, or paneth cells.
Aside from multilineage progenitors, luminal-restricted and myoepithelial-restricted progenitors have also been identified in breast tissue. Luminal-restricted progenitors express a hybrid set of markers between tissue stem cells and differentiated luminal epithelia.196 For instance, luminal progenitor cells express luminal cytokeratin 8/18 but also cytokeratin 5/6, a basal cytokeratin that marks cells with in vivo repopulating capacity.197 Commitment to the luminal epithelial lineage is enforced by transcription factors, such as GATA3 and FOXA1, which drive expression of hormone receptors and luminal mucins.196, 198 Thus, a tissue made up of two phenotypically distinct cell types (luminal and myoepithelial cells) requires at least five molecular subtypes to establish and maintain it (Figure 5(a)). Many other intermediate cell types have been described, and more await to be identified.199
Molecular heterogeneity also exceeds phenotypic heterogeneity in the intestinal epithelium (Figure 5(b)). The small intestinal epithelium is organized as a cell monolayer, which bends to form crypts and villi. Rapid self-renewal takes place in crypts, whereas terminal differentiation occurs in villi. The cytoarchitecture of the intestine is more complex than the breast, in that there are four distinct lineages (enterocytes, goblet cells, enteroendocrine cells, and paneth cells). However, as in the breast, there exist many intermediate molecular states before lineage commitment.200 Cells expressing the leucine-rich repeat-containing G-protein-coupled receptor Lgr5 together with CD133 act as the long-lived, multipotent stem cells that reside at the crypt base to generate all cell types of the small intestine.201, 202 Single Lgr5-expressing cells cultured in vitro form intestinal organoids containing all four cell lineages.203 A second, more-quiescent population of intestinal stem cells resides higher in the crypt and is marked by expression of Bmi1.204 Snippert et al. and Lopez-Garcia et al., using independent labeling methods, uncovered that most intestinal stem cells divide symmetrically each day and stochastically adopt stem or transit-amplifying fates.205, 206 The rates at which stem cells divide and differentiated cells move to the villus are almost the same, thereby maintaining intestinal-crypt homeostasis.
In breast, intestine, and other tissues, the main limitation in discovering heterogeneities important for lineage progression is that cells in different molecular “states” are often not microscopically distinguishable. To address this challenge, we recently developed a method for globally identifying genes with co-regulated, heterogeneous expression in cell populations.207 In a 3D culture model of a clonal MCF10A breast-epithelial cell line, 10–15% of all genes were found to be highly non-uniform during morphogenesis. Although explicit cell differentiation is not thought to occur in this in vitro model, the profiling approach could in principle be applied to epithelial development in situations where key intermediate cell states have not been defined at the molecular level.
Disruption of epithelial morphology in carcinoma
Cellular input-output relationships are substantially perturbed during epithelial tumorigenesis. Consequently, this leads to profound morphological changes at the single-cell and tissue levels. Since the discovery of oncogenes and tumor suppressors, the field of cancer biology has largely adopted a gene-centric approach to the disease.208, 209 However, there are multiple classic examples where morphology and microenvironment trump genetics in promoting or suppressing tumorigenesis.210-212 Indeed, half of the hallmarks and enabling characteristics of cancer213, 214 are associated with clear misregulation of the core processes described in this Review (Table 2). We discuss a few salient examples below that illustrate how epithelial cancers evolve to occupy two tissue locations—the acinar lumen and the surrounding stroma—that epithelial cells do not.
Table 2.
| Cancer hallmark | Morphogenetic defect | Misregulated core process |
|---|---|---|
| Sustaining proliferative signaling213 |
|
|
| Resisting cell death213 |
|
|
| Evading growth suppressors213 |
|
|
| Activating invasion and metastasis213 |
|
|
| Genome instability and mutation214 |
|
|
Single cells
Normal epithelial cells in tissues show a cobblestone morphology and apicobasal polarity that are stably maintained by extensive cell-cell contacts and adhesion to basement membrane (Figure 1).215 However, during development, epithelial cells will convert to a mesenchymal phenotype in situations where dramatic cellular rearrangements are required.174 Mesenchymal cells are characterized by their spindle-shaped morphology, lack of cell-cell contacts, and migration through the basement membrane. Some have argued that EMT is important for epithelial tumor progression, because transformed cells must escape the constraints of the ECM and their neighbors to invade the surrounding tissue (Table 2).
Hogan et al. recently examined the fate of single epithelial cells undergoing the early transformation steps that can result in an EMT.216, 217 Using a tetracycline-inducible Ras in MDCK cells, the authors found that single Ras transformants were mostly extruded apically from a monolayer of normal epithelial cells. Apical extrusion was promoted by Cdc42 and ROCK in Ras-transformed cells and by E-cadherin in adjacent normal cells. Disrupting either of these pathways caused Ras-transformed cells to invade the basolateral side of the epithelial monolayer in a manner reminiscent of early invasive tumors. This work suggests that normal epithelial tissues may possess tumor-suppressive mechanisms that are not cell autonomous.
Epithelial tissues
Epithelial tumors disrupt the sheet-like morphology of normal epithelia (Figure 1, 2) by occupying the tissue lumen or by invading the basement membrane. Clearance of centrally located cells, which do not have matrix contact, ordinarily involves explicit cell-death mechanisms, such as anoikis.51 However, most carcinomas display anoikis resistance and are anchorage-independent for growth and survival (Table 2). Activation of ErbB2 in 3D breast epithelial acini induces anoikis resistance 53 and leads to an increase in proliferation and luminal filling.218 ErbB2 and other proliferation-inducing oncoproteins inhibit Bcl-2-family members (Bim, Bmf) that normally promote anoikis.34, 53, 54 In addition to preventing anoikis, ErbB2 can rescue matrix-detached cells from metabolic defects, which are associated with autophagy and luminal clearance.219 The intersection of metabolism and cancer has been reinvigorated over the past several years.220 By comparison, metabolic regulation during morphogenesis has been largely unexplored.
The process of invasion involves multiple steps requiring tumor cells to alter their cell-cell and cell-ECM adhesions (Table 2). Focal adhesion kinase (FAK) is recruited at an early stage to focal adhesions and acts as a pleiotropic signaling platform for cell adhesion, motility, and anchorage-independent growth. Thus, misregulated FAK activity has been implicated in tumor invasion and metastasis. FAK mRNA and protein are significantly elevated in invasive and metastatic breast tumor specimens,221 and FAK is essential for oncogenic transformation and invasion induced by ErbB2-ErbB3 receptor signaling in various breast carcinoma lines.222 One mechanism by which FAK promotes cancer invasiveness is by upregulating pericellular proteolysis of the ECM. FAK increases expression of MMP2 and MMP9 through Rac1 and c-jun N-terminal kinase activation223 and decreases endocytosis of MT1-MMP.224 FAK also cooperates with the nonreceptor tyrosine kinase Src in KM12C colon cancer cells to disrupt E-cadherin within adherens junctions.225 E-cadherin downregulation by FAK and Src is an important event for other invasion promoters, such as TGFβ.226 FAK inhibitors are actively being explored as anticancer therapeutics.227 FAK catalytic activity is required for fibroblast motility but not proliferation,228 suggesting that such inhibitors may synergize with conventional therapies if the same mechanism holds for epithelia.
Culture models of epithelial morphogenesis
Existing systems-biology approaches are most effective when cells can be easily observed and perturbed at the molecular level.170 One indication that morphogenesis is well suited for such approaches is the wealth of culture models available for exploring how epithelial tissues are shaped. As with any model, these cultures are imperfect, reflecting only how epithelial cells can organize and not necessarily how they form tissues in vivo. Nevertheless, they represent an important transition point for bridging systems biology and systems physiology.229
In vitro models
The form and function of all epithelial tissues are defined in three dimensions.230 However, standard in vitro culture creates an artificial 2D monolayer of cells. To provide a more natural epithelial context, 3D in vitro cultures were developed.
Most 3D culture protocols call for matrigel as the primary extracellular support.231 Matrigel is prepared from the stroma-rich Engelbreth-Holm-Swarm tumor grown in the mouse peritoneum. Matrigel provides cytokines (esp. TGFβ-family) and basement membrane proteins (esp. laminin, collagen IV, heparin sulfate) that are distinct from the ECM proteins that preferentially adsorb to tissue culture plastic. Matrigel thus engages a unique set of integrins and signaling pathways that promote organotypic growth. Matrigel also acts as a flexible substrate upon or within which cells tend to proliferate as isotropic spheres rather than as the sheets usually observed in 2D culture.232 For example, when cultured in 3D with matrigel, many nontransformed epithelial lines self-organize into hollow spheres comprised of a monolayer of polarized cells (cysts) that have outside basement membranes contacting matrix and inside apical surfaces facing a central lumen. Two such lines – MDCK and MCF10A – are well established and widely used in 3D to study facets of epithelial morphogenesis.
MDCK cells show properties of the kidney distal tubule and collecting duct, such as the expression of carbonic anhydrase and a characteristic pattern of ionic selectivity.233 Single MDCK cells rapidly polarize when suspended in matrigel, and polarized cells then proliferate and differentiate to form a spherical cyst. MDCK cysts develop through orderly, highly stereotyped stages, making it a great model to study the intrinsic mechanisms for epithelial polarization as directed by adhesion cues. Experimental manipulation of culture media conditions combined with genetic gain/loss-of-function experiments helped to characterize the fundamental instructive roles of ECM adhesion in cell polarity.29, 92, 98, 234 MDCK polarity also drives lumenogenesis in a cyst.35 Polarity complexes and vesicular trafficking work together to generate the apical surface and lumen de novo.235 MDCK polarity is tightly controlled but is not hard-wired. Exposing MDCK cysts to HGF transiently leads to partial loss of polarity en route to tubulogenesis.100 Cells extend as cords, repolarize and then become mature tubules with a continuous lumen. These polarizations stages are also similar in renal tubules in vivo,236 making MDCK cell an useful model for studying renal cystogenesis. Though 3D MDCK cultures offer many advantages, there are drawbacks. A substantial disadvantage for studying size control is that MDCK cysts do not arrest their proliferation in 3D.237 A second inconvenience is that because of their canine origin, all RNAi-based reagents must be designed in house.238 Nonetheless, MDCK cultures remain the preferred 3D model for polarity.
Another widely used model that complements MDCK cysts is 3D culture of the immortalized human breast epithelial cell line, MCF10A.239 Seeding single MCF10A cells atop a matrigel bed and in the presence of a dilute matrigel solution promotes the formation of growth-arrested acini in ~2 weeks. The development of MCF10A acini involves the coordination of a proliferative phase in the outer cells during days 1–8 and an apoptotic phase in central cells during days 6–12.51, 52 The control of proliferation and death-vs.-survival are processes that are well suited to this model and may reflect facets of the regulation in vivo.34 Moreover, as a nontumorigenic cell line, MCF10A cells can be used to study the effects of oncogenes and tumor suppressor genes on morphogenesis. These manipulations can draw from the wealth of resources available for human genes.240, 241 Yet, the MCF10A model cannot compete with the strengths of MDCK cysts. Compared with MDCK cells, which begin polarization and lumen formation during the first cell division,242 MCF10A acini are slow to develop. In addition, the overall polarity of MCF10A is much less conclusive when compared with MDCK cells. The Golgi apparatus orients apically and ECM is deposited basolaterally in MCF10A acini, but other important polarity complexes do not appear to form endogenously.9, 218, 243 A specific disadvantage of the MCF10A model for in vivo relevance is that by using only one cell type, it omits the dual luminal-myoepithelial layers that comprise actual breast tissue (Figure 5(a)). To track cell behavior in mixed cell types, other techniques are needed to provide insight into multilayered epithelia.
Ex vivo models
The architecture of normal epithelium in vivo is defined by epithelial interactions with both the ECM and the stromal mesenchyme. Thus, traditional 2D and even 3D cell cultures incompletely describe epithelial morphogenesis by using a single cell type. Ex vivo cultures of tissue progenitor or entire epithelial organs seek to address this deficiency in a format that is easily observable.
Ex vivo cultures can start with entire epithelial organs, tissue fragments, or just single cells. For example, whole-corneal organ cultures using an air-interface technique have been used for evaluating ocular irritation.244-246 The major drawback of organ culture is an inability to perfuse large tissues adequately, causing a progressive decline in function during the course of an experiment.247 More recently, smaller epithelial fragments isolated from mouse mammary glands were used to study epithelial cell reorganization, collective migration, and ductal elongation.57 Fragment cultures strike a compromise by maintaining the microenvironment locally in a format that is more amenable to in vitro conditions. When multipotent progenitors can be reliably identified, ex vivo morphogenesis cultures of single primary cells may be possible. For example, single stem cells sorted from crypts of small intestine can rebuild entire crypt–villus units during long-term 3D culture.203 Single-cell cultures lack tissue stroma and must recreate an appropriate ECM, but they can be tremendously powerful for tracking differentiation in epithelial tissues.248
The major advantage of all ex vivo formats is that they are compatible with long-term time-lapse imaging techniques. Combining fragment cultures and confocal microscopy revealed a sequence of steps underlying mammary branching morphogenesis.57 Ex vivo models also enable reliable identification and tracking of single-cell behavior in mixed cell lineages.203 Last, ex vivo cultures provide control of the local cellular microenvironment in space and time.50 Advances in micropatterning and their application to morphogenesis will allow researchers to explore non-native microenvironments and their impact on tissue structure.249, 250
Computational models of epithelial morphogenesis
An indispensable component of systems-biology research is the development and use of computational models. Many recent reviews in this journal have covered modeling from the perspective of epithelial biology and cancer biology.251-253 In this section, we very briefly review the major relevant classes of models and their recent application to questions in morphogenesis and tumorigenesis.
Continuous modeling
Continuum models describe spatial tissue parameters as continuous fields that vary in time. Capturing the dual time-space dependence mathematically requires partial differential equations, which typically must be solved numerically. Nonetheless, there is a long history in applying continuous models to morphogenesis, dating back to the proposal of the morphogen theory itself.254 Within continuum models, parameters such as tissue or tumor growth rate or oxygen diffusion can be directly estimated through experiments, because they correspond to macroscopic tissue or tumor properties (Table 3). For example, a continuum model was used to examine the effects of proliferation-induced mechanical stresses and hypoxic stresses on production of proteolytic enzymes during progression of ductal carcinoma in situ (DCIS) of the breast.255 This model described how mechanical effects dominate in stimulating the protease production that alters the mechanical properties of the ductal walls and predicted the region that tumor cells are most likely to invade into the surrounding tissue.
Table 3.
Strengths and limitations of different modeling approaches
| Approach | Strengths | Limitations |
|---|---|---|
| Continuous modeling |
|
|
| Discrete modeling |
|
|
| Hybrid modeling |
|
|
Cell sheets are good tissues to study with continuum models.256 For example, an advancing enterocyte monolayer was modeled as a 1D continuum of springs to mimic physical tension between cells.257 A 2D continuum approach was adopted to model the colorectal crypt epithelium as a growing beam attached to an underlying lamina.258 Springs—buckled by the combination of cell proliferation, movement, and cellular attachment—act to model the process of crypt budding and fission. Continuum models encompass not only cell-tissue mechanics but also transport processes. Cell sheet restoration during wound repair is compatible with a reaction-diffusion model, Fisher’s equation, originally used to model population spread.259, 260 This model was further elaborated to connect receptor activation and intracellular signaling to proliferation and migration responses during 2D wound healing.261 In this way, continuum models provide a straightforward way to combine molecular and biophysical processes within a common quantitative framework.
Discrete modeling
Classic implementations of continuum models do not capture the different properties and behaviors of individual cells, which is an important facet of epithelial tissues (Table 3). One can track the autonomous behavior of single cells by discrete models based on rules describing key biological processes that depend on the microenvironment. Discrete models of morphogenesis are consistent with the historical positional-information theory of tissue patterning.262 In contrast to continuum model parameters, which can often be directly estimated from experiments, the rules of discrete models must first be defined heuristically. However, rule sets are biologically inspired and their single-cell predictions can be directly compared with experimental observations during model testing and refinement.
Several models have captured the organotypic phenotypes of in vitro cell cultures, which mimic facets of epithelial tissues. Kim et al. created an in silico study of human alveolar type II (AT II) 3D cell cultures, which revealed a set of cell operating rules that are sufficient for describing the development of cyst-like structures.263 This study was further refined to explore the underlying mechanism of AT II wound healing.264 Grant et al. also built a discrete model of 3D MDCK cysts.265 The authors revised their initial rule set based on the model’s ability to capture epithelial morphogenesis under four growth conditions that give rise to different multicellular arrangements. Interestingly, the key modification to this model was the addition of an explicit polarized cell state, whose stability depended on the local cellular and ECM environment. Thus, only a few experimental observations may be required to constrain discrete-model rule sets, provided that these constraints are stringent.
Discrete models are also easily revised to apply to different biological circumstances. For example, modeling the response of epithelial cells to contact inhibition has been used to wound healing in monolayer cell cultures.266 This study was extended to explore the effects of normal cell-cell interactions on the dynamics of population growth.267 The model predicted non-cell-autonomous effects on population dynamics and single-cell behavior when individual cells lose E-cadherin expression. Rejniak developed a single-cell-based model called IBCell (Immersed Boundary model of a Cell) to model the evolution of solid tumors.268 The model framework was subsequently adjusted to different applications, such as to represent the cross-section of normally growing epithelial acini and to study acinar development and stability. 269, 270 These studies show that local interactions between individual cells and their microenvironment guide the dynamic development of epithelial acini (Figure 2(c)). More recently, IBCell was tuned to experimental data from 3D culture of MCF10A cells.239, 271 This customized model captures the abnormal acinar morphology observed when cells express an oncogenic HER2-YVMA mutant.271, 272 In the future, it will be interesting to explore how many cancer-related morphogenetic abnormalities can be captured under a common discrete rule set.
Hybrid modeling
Discrete models thrive at single-cell simulations but struggle to handle continuous variables, such as diffusible growth factors and oxygen tension (Table 3). To address this, so-called hybrid models attempt to integrate continuum and discrete descriptions linking cell behavior with macroscopic tissue properties. Diffusible factors are described as continuum fields, with cells modeled as individual discrete elements that evolve dynamically in response to their local conditions. Hybrid models can be built from scratch or can result from the fusion of pre-existing continuum and discrete models. For example, a hybrid model of tumor growth was built from earlier continuum and discrete models to account for intratumor processes, host-tumor interactions, and cell-level heterogeneity.273
Computational resources have been developed to allow the integration of molecular, cell, and tissue scales in a hybrid framework. For example, Cickovski et al. have built a computational package, CompuCell3D, designed for hybrid modeling of morphogenesis.274 Another hybrid platform, Epitheliome, was developed to model epithelial tissue specifically and has been used to examine the effects of growth factors on epithelial development.275 Hybrid models can provide the most-realistic coupling of biophysical processes at the expense of a higher number of parameters that must be estimated at an increased computational cost.
One active research direction in the area of integrative cancer biology involves the development of hybrid models for tumor invasion. An early hybrid model was originally developed by Anderson to describe solid tumor growth based on both continuous (ECM, matrix-degrading enzymes and oxygen) and discrete (tumor cells) factors.276 This model was later extended to include cell-cell adhesion to evaluate how individual cells and cell-matrix interactions may affect tumor morphology.277 Anderson’s follow-up studies quantitatively characterized tumor invasion as a function of microenvironmental selective factors,278 linking low nutrient availability with a fingered tumor morphology.279 These studies correlate with another model, which considered individual cells interacting with surrounding nutrients and showed that tumor microenvironments cause invasive projections from solid tumors.280 Anderson and coworkers further showed by hybrid modeling that tumorigenic cells were selected under constrained growth conditions with limited nutrients.281 Aggressive phenotypes that produce tumor fingering are favored in poor, but not rich, nutrient microenvironments.282 Another hybrid model recently added a mechanical component to the finger-like tumor morphology, arising from the combined strength of cell-cell adhesion and haptotaxis.283 These models converge on a common prediction that transport limitations of diffusible factors leads to tissue instability, creating a platform for tumor evolution and invasion.
Conclusion and Future Directions
To simplify a daunting challenge, our Advanced Review has sought to break down epithelial morphogenesis into tractable elements. Many of these elements have been modeled computationally, measured quantitatively, or both, and we expect that many more will be soon. Conversely, very few of the published computational models that we discussed were explicitly focused on epithelial morphogenesis. This does not make the current models irrelevant, because it should be possible to co-opt them for morphogenesis-specific applications. For example, models of diffusible factors have been shown to make accurate predictions across different epithelial lineages and genetic backgrounds.284-286 Defining the individual elements, characterizing their biochemical properties, and modeling their emergent systems-level properties is thus an important first step.
It remains unclear whether such elements can be treated as true “modules”, which can be studied in isolation and then wired together afterwards.2 For clarity, we have avoided the topic of crosstalk between elements, but this undoubtedly occurs, and reports in the literature are too numerous to mention. One major obstacle to a more-integrative understanding is that we lack general, effective, and efficient approaches for incorporating crosstalk without getting overwhelmed by it. This hurdle will not be overcome without adopting an interdisciplinary systems approach.
Acknowledgments
We thank Barry Gumbiner and Ian Macara for critically reviewing this manuscript. Work in the Janes lab is supported by the National Institutes of Health Director’s New Innovator Award Program (1-DP2-OD006464-01), the American Cancer Society (120668-RSG-11-047-01-DMC), the Mary Kay Ash Charitable Foundation, the Pew Scholars Program in the Biomedical Sciences, and the David and Lucile Packard Foundation. Chun-Chao Wang is supported by a Breast Cancer Research Program Postdoctoral Fellowship Award from the Department of Defense (W81XWH-11-1-0037).
References
- 1.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 2.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 3.Cereijido M, Contreras RG, Shoshani L. Cell adhesion, polarity, and epithelia in the dawn of metazoans. Physiol Rev. 2004;84:1229–1262. doi: 10.1152/physrev.00001.2004. [DOI] [PubMed] [Google Scholar]
- 4.Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- 5.Tepass U, Tanentzapf G, Ward R, Fehon R. Epithelial cell polarity and cell junctions in Drosophila. Annu Rev Genet. 2001;35:747–784. doi: 10.1146/annurev.genet.35.102401.091415. [DOI] [PubMed] [Google Scholar]
- 6.Humbert PO, Dow LE, Russell SM. The Scribble and Par complexes in polarity and migration: friends or foes? Trends Cell Biol. 2006;16:622–630. doi: 10.1016/j.tcb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Itoh M, Nagafuchi A, Yonemura S, Kitani-Yasuda T, Tsukita S. The 220-kD protein colocalizing with cadherins in non-epithelial cells is identical to ZO-1, a tight junction-associated protein in epithelial cells: cDNA cloning and immunoelectron microscopy. J Cell Biol. 1993;121:491–502. doi: 10.1083/jcb.121.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.St Johnston D, Ahringer J. Cell polarity in eggs and epithelia: parallels and diversity. Cell. 2010;141:757–774. doi: 10.1016/j.cell.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, Muthuswamy SK. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135:865–878. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCaffrey LM, Macara IG. The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev. 2009;23:1450–1460. doi: 10.1101/gad.1795909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- 12.Devenport D, Fuchs E. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol. 2008;10:1257–1268. doi: 10.1038/ncb1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Mlodzik M. A quest for the mechanism regulating global planar cell polarity of tissues. Trends Cell Biol. 2009;19:295–305. doi: 10.1016/j.tcb.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amonlirdviman K, Khare NA, Tree DR, Chen WS, Axelrod JD, Tomlin CJ. Mathematical modeling of planar cell polarity to understand domineering nonautonomy. Science. 2005;307:423–426. doi: 10.1126/science.1105471. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence PA, Struhl G, Casal J. Planar cell polarity: one or two pathways? Nat Rev Genet. 2007;8:555–563. doi: 10.1038/nrg2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin AC, Kaschube M, Wieschaus EF. Pulsed contractions of an actin-myosin network drive apical constriction. Nature. 2009;457:495–499. doi: 10.1038/nature07522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lecuit T, Lenne PF. Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Nat Rev Mol Cell Biol. 2007;8:633–644. doi: 10.1038/nrm2222. [DOI] [PubMed] [Google Scholar]
- 18.Hickson GR, Echard A, O’Farrell PH. Rho-kinase controls cell shape changes during cytokinesis. Curr Biol. 2006;16:359–370. doi: 10.1016/j.cub.2005.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanov AI, Hopkins AM, Brown GT, Gerner-Smidt K, Babbin BA, Parkos CA, Nusrat A. Myosin II regulates the shape of three-dimensional intestinal epithelial cysts. J Cell Sci. 2008;121:1803–1814. doi: 10.1242/jcs.015842. [DOI] [PubMed] [Google Scholar]
- 20.Gibson MC, Patel AB, Nagpal R, Perrimon N. The emergence of geometric order in proliferating metazoan epithelia. Nature. 2006;442:1038–1041. doi: 10.1038/nature05014. [DOI] [PubMed] [Google Scholar]
- 21.Farhadifar R, Roper JC, Aigouy B, Eaton S, Julicher F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Curr Biol. 2007;17:2095–2104. doi: 10.1016/j.cub.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 22.Rauzi M, Verant P, Lecuit T, Lenne PF. Nature and anisotropy of cortical forces orienting Drosophila tissue morphogenesis. Nat Cell Biol. 2008;10:1401–1410. doi: 10.1038/ncb1798. [DOI] [PubMed] [Google Scholar]
- 23.Sawyer JM, Harrell JR, Shemer G, Sullivan-Brown J, Roh-Johnson M, Goldstein B. Apical constriction: a cell shape change that can drive morphogenesis. Dev Biol. 2010;341:5–19. doi: 10.1016/j.ydbio.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryant DM, Mostov KE. From cells to organs: building polarized tissue. Nature Reviews Molecular Cell Biology. 2008;9:887–901. doi: 10.1038/nrm2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andrew DJ, Ewald AJ. Morphogenesis of epithelial tubes: Insights into tube formation, elongation, and elaboration. Developmental Biology. 2010;341:34–55. doi: 10.1016/j.ydbio.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lubarsky B, Krasnow MA. Tube morphogenesis: Making and shaping biological tubes. Cell. 2003;112:19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- 27.Rasmussen JP, English K, Tenlen JR, Priess JR. Notch signaling and morphogenesis of single-cell tubes in the C. elegans digestive tract. Dev Cell. 2008;14:559–569. doi: 10.1016/j.devcel.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghabrial AS, Krasnow MA. Social interactions among epithelial cells during tracheal branching morphogenesis. Nature. 2006;441:746–749. doi: 10.1038/nature04829. [DOI] [PubMed] [Google Scholar]
- 29.Yu W, Datta A, Leroy P, O’Brien LE, Mak G, Jou TS, Matlin KS, Mostov KE, Zegers MM. Beta1-integrin orients epithelial polarity via Rac1 and laminin. Mol Biol Cell. 2005;16:433–445. doi: 10.1091/mbc.E04-05-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tawk M, Araya C, Lyons DA, Reugels AM, Girdler GC, Bayley PR, Hyde DR, Tada M, Clarke JDW. A mirror-symmetric cell division that orchestrates neuroepithelial morphogenesis. Nature. 2007;446:797–800. doi: 10.1038/nature05722. [DOI] [PubMed] [Google Scholar]
- 31.Jaffe AB, Kaji N, Durgan J, Hall A. Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. Journal of Cell Biology. 2008;183:625–633. doi: 10.1083/jcb.200807121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nature Cell Biology. 2010;12:1035–U1024. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melnick M, Jaskoll T. Mouse submandibular gland morphogenesis: a paradigm for embryonic signal processing. Crit Rev Oral Biol Med. 2000;11:199–215. doi: 10.1177/10454411000110020401. [DOI] [PubMed] [Google Scholar]
- 34.Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell. 2007;12:221–234. doi: 10.1016/j.devcel.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin-Belmonte F, Yu W, Rodriguez-Fraticelli AE, Ewald AJ, Werb Z, Alonso MA, Mostov K. Cell-polarity dynamics controls the mechanism of lumen formation in epithelial morphogenesis. Curr Biol. 2008;18:507–513. doi: 10.1016/j.cub.2008.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagnat M, Cheung ID, Mostov KE, Stainier DYR. Genetic control of single lumen formation in the zebrafish gut. Nature Cell Biology. 2007;9:954–U119. doi: 10.1038/ncb1621. [DOI] [PubMed] [Google Scholar]
- 37.Kesavan G, Sand FW, Greiner TU, Johansson JK, Kobberup S, Wu X, Brakebusch C, Semb H. Cdc42-mediated tubulogenesis controls cell specification. Cell. 2009;139:791–801. doi: 10.1016/j.cell.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 38.Samakovlis C, Manning G, Steneberg P, Hacohen N, Cantera R, Krasnow MA. Genetic control of epithelial tube fusion during Drosophila tracheal development. Development. 1996;122:3531–3536. doi: 10.1242/dev.122.11.3531. [DOI] [PubMed] [Google Scholar]
- 39.Nakanishi Y, Morita T, Nogawa H. Cell proliferation is not required for the initiation of early cleft formation in mouse embryonic submandibular epithelium in vitro. Development. 1987;99:429–437. doi: 10.1242/dev.99.3.429. [DOI] [PubMed] [Google Scholar]
- 40.Oblander SA, Zhou Z, Galvez BG, Starcher B, Shannon JM, Durbeej M, Arroyo AG, Tryggvason K, Apte SS. Distinctive functions of membrane type 1 matrix-metalloprotease (MT1-MMP or MMP-14) in lung and submandibular gland development are independent of its role in pro-MMP-2 activation. Dev Biol. 2005;277:255–269. doi: 10.1016/j.ydbio.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 41.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Developmental Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore KA, Polte T, Huang S, Shi B, Alsberg E, Sunday ME, Ingber DE. Control of basement membrane remodeling and epithelial branching morphogenesis in embryonic lung by Rho and cytoskeletal tension. Developmental Dynamics. 2005;232:268–281. doi: 10.1002/dvdy.20237. [DOI] [PubMed] [Google Scholar]
- 43.Gilles C, Polette M, Coraux C, Tournier JM, Meneguzzi G, Munaut C, Volders L, Rousselle P, Birembaut P, Foidart JM. Contribution of MT1-MMP and of human laminin-5 gamma2 chain degradation to mammary epithelial cell migration. J Cell Sci. 2001;114:2967–2976. doi: 10.1242/jcs.114.16.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008;68:7247–7249. doi: 10.1158/0008-5472.CAN-08-0784. [DOI] [PubMed] [Google Scholar]
- 45.Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. Journal of Cell Science. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davies JA. Do different branching epithelia use a conserved developmental mechanism? Bioessays. 2002;24:937–948. doi: 10.1002/bies.10161. [DOI] [PubMed] [Google Scholar]
- 47.Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132:3923–3933. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu PF, Werb Z. Patterning Mechanisms of Branched Organs. Science. 2008;322:1506–1509. doi: 10.1126/science.1162783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ewan KB, Shyamala G, Ravani SA, Tang Y, Akhurst R, Wakefield L, Barcellos-Hoff MH. Latent transforming growth factor-beta activation in mammary gland - Regulation by ovarian hormones affects ductal and alveolar proliferation. American Journal of Pathology. 2002;160:2081–2093. doi: 10.1016/s0002-9440(10)61158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson CM, VanDuijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 52.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 53.Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5:733–740. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- 54.Reginato MJ, Mills KR, Becker EB, Lynch DK, Bonni A, Muthuswamy SK, Brugge JS. Bim regulation of lumen formation in cultured mammary epithelial acini is targeted by oncogenes. Mol Cell Biol. 2005;25:4591–4601. doi: 10.1128/MCB.25.11.4591-4601.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmelzle T, Mailleux AA, Overholtzer M, Carroll JS, Solimini NL, Lightcap ES, Veiby OP, Brugge JS. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci U S A. 2007;104:3787–3792. doi: 10.1073/pnas.0700115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci U S A. 2004;101:3438–3443. doi: 10.1073/pnas.0400443101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fata JE, Mori H, Ewald AJ, Zhang H, Yao E, Werb Z, Bissell MJ. The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev Biol. 2007;306:193–207. doi: 10.1016/j.ydbio.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yerbes R, Palacios C, Reginato MJ, Lopez-Rivas A. Cellular FLIP(L) plays a survival role and regulates morphogenesis in breast epithelial cells. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamcr.2010.1010.1003. [DOI] [PubMed] [Google Scholar]
- 60.Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, Sorger PK. Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol. 2008;6:2831–2852. doi: 10.1371/journal.pbio.0060299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rowe RG, Weiss SJ. Navigating ECM barriers at the invasive front: the cancer cell-stroma interface. Annu Rev Cell Dev Biol. 2009;25:567–595. doi: 10.1146/annurev.cellbio.24.110707.175315. [DOI] [PubMed] [Google Scholar]
- 64.Rebustini IT, Myers C, Lassiter KS, Surmak A, Szabova L, Holmbeck K, Pedchenko V, Hudson BG, Hoffman MP. MT2-MMP-dependent release of collagen IV NC1 domains regulates submandibular gland branching morphogenesis. Dev Cell. 2009;17:482–493. doi: 10.1016/j.devcel.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284:31493–31497. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCulloch DR, Nelson CM, Dixon LJ, Silver DL, Wylie JD, Lindner V, Sasaki T, Cooley MA, Argraves WS, Apte SS. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev Cell. 2009;17:687–698. doi: 10.1016/j.devcel.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Streuli CH. Maspin is a tumour suppressor that inhibits breast cancer tumour metastasis in vivo. Breast Cancer Res. 2002;4:137–140. doi: 10.1186/bcr437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 70.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11:1287–1296. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shakya R, Watanabe T, Costantini F. The role of GDNF/Ret signaling in ureteric bud cell fate and branching morphogenesis. Dev Cell. 2005;8:65–74. doi: 10.1016/j.devcel.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 72.Larsen M, Wei C, Yamada KM. Cell and fibronectin dynamics during branching morphogenesis. J Cell Sci. 2006;119:3376–3384. doi: 10.1242/jcs.03079. [DOI] [PubMed] [Google Scholar]
- 73.Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering M, Pawson T, et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111:251–263. doi: 10.1016/s0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 74.Pearson GW, Hunter T. Real-time imaging reveals that noninvasive mammary epithelial acini can contain motile cells. J Cell Biol. 2007;179:1555–1567. doi: 10.1083/jcb.200706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chi X, Michos O, Shakya R, Riccio P, Enomoto H, Licht JD, Asai N, Takahashi M, Ohgami N, Kato M, et al. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev Cell. 2009;17:199–209. doi: 10.1016/j.devcel.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mori H, Gjorevski N, Inman JL, Bissell MJ, Nelson CM. Self-organization of engineered epithelial tubules by differential cellular motility. Proc Natl Acad Sci U S A. 2009;106:14890–14895. doi: 10.1073/pnas.0901269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rorth P. Collective cell migration. Annu Rev Cell Dev Biol. 2009;25:407–429. doi: 10.1146/annurev.cellbio.042308.113231. [DOI] [PubMed] [Google Scholar]
- 78.Maddugoda MP, Crampton MS, Shewan AM, Yap AS. Myosin VI and vinculin cooperate during the morphogenesis of cadherin cell-cell contacts in mammalian epithelial cells. Journal of Cell Biology. 2007;178:529–540. doi: 10.1083/jcb.200612042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 80.Grose R, Hutter C, Bloch W, Thorey I, Watt FM, Fassler R, Brakebusch C, Werner S. A crucial role of beta 1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development. 2002;129:2303–2315. doi: 10.1242/dev.129.9.2303. [DOI] [PubMed] [Google Scholar]
- 81.Zelenka PS, Arpitha P. Coordinating cell proliferation and migration in the lens and cornea. Semin Cell Dev Biol. 2008;19:113–124. doi: 10.1016/j.semcdb.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 82.Omelchenko T, Vasiliev JM, Gelfand IM, Feder HH, Bonder EM. Rho-dependent formation of epithelial “leader” cells during wound healing. Proc Natl Acad Sci U S A. 2003;100:10788–10793. doi: 10.1073/pnas.1834401100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Poujade M, Grasland-Mongrain E, Hertzog A, Jouanneau J, Chavrier P, Ladoux B, Buguin A, Silberzan P. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci U S A. 2007;104:15988–15993. doi: 10.1073/pnas.0705062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Trepat X, Wasserman MR, Angelini TE, Millet E, Weitz DA, Butler JP, Fredberg JJ. Physical forces during collective cell migration. Nature Physics. 2009;5:426–430. [Google Scholar]
- 85.Ladoux B. BIOPHYSICS Cells guided on their journey. Nature Physics. 2009;5:377–378. [Google Scholar]
- 86.Haas P, Gilmour D. Chemokine signaling mediates self-organizing tissue migration in the zebrafish lateral line. Dev Cell. 2006;10:673–680. doi: 10.1016/j.devcel.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 87.Caussinus E, Colombelli J, Affolter M. Tip-cell migration controls stalk-cell intercalation during Drosophila tracheal tube elongation. Curr Biol. 2008;18:1727–1734. doi: 10.1016/j.cub.2008.10.062. [DOI] [PubMed] [Google Scholar]
- 88.Mori H, Gjorevski N, Inman JL, Bissell MJ, Nelson CM. Self-organization of engineered epithelial tubules by differential cellular motility. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14890–14895. doi: 10.1073/pnas.0901269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ikeya T, Hayashi S. Interplay of Notch and FGF signaling restricts cell fate and MAPK activation in the Drosophila trachea. Development. 1999;126:4455–4463. doi: 10.1242/dev.126.20.4455. [DOI] [PubMed] [Google Scholar]
- 90.Greenhalgh DG, Sprugel KH, Murray MJ, Ross R. PDGF and FGF stimulate wound healing in the genetically diabetic mouse. Am J Pathol. 1990;136:1235–1246. [PMC free article] [PubMed] [Google Scholar]
- 91.Jaffe AB, Kaji N, Durgan J, Hall A. Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. J Cell Biol. 2008;183:625–633. doi: 10.1083/jcb.200807121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Brien LE, Jou TS, Pollack AL, Zhang Q, Hansen SH, Yurchenco P, Mostov KE. Rac1 orientates epithelial apical polarity through effects on basolateral laminin assembly. Nat Cell Biol. 2001;3:831–838. doi: 10.1038/ncb0901-831. [DOI] [PubMed] [Google Scholar]
- 93.Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, Kulkarni S, Gish G, Virag C, Lim C, et al. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535–548. doi: 10.1016/j.cell.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 95.Qin Y, Meisen WH, Hao Y, Macara IG. Tuba, a Cdc42 GEF, is required for polarized spindle orientation during epithelial cyst formation. J Cell Biol. 2010;189:661–669. doi: 10.1083/jcb.201002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rodriguez-Fraticelli AE, Vergarajauregui S, Eastburn DJ, Datta A, Alonso MA, Mostov K, Martin-Belmonte F. The Cdc42 GEF Intersectin 2 controls mitotic spindle orientation to form the lumen during epithelial morphogenesis. J Cell Biol. 2010;189:725–738. doi: 10.1083/jcb.201002047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nejsum LN, Nelson WJ. A molecular mechanism directly linking E-cadherin adhesion to initiation of epithelial cell surface polarity. J Cell Biol. 2007;178:323–335. doi: 10.1083/jcb.200705094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu W, Shewan AM, Brakeman P, Eastburn DJ, Datta A, Bryant DM, Fan QW, Weiss WA, Zegers MM, Mostov KE. Involvement of RhoA, ROCK I and myosin II in inverted orientation of epithelial polarity. EMBO Rep. 2008;9:923–929. doi: 10.1038/embor.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Adams SA, Smith ME, Cowley GP, Carr LA. Reversal of glandular polarity in the lymphovascular compartment of breast cancer. J Clin Pathol. 2004;57:1114–1117. doi: 10.1136/jcp.2004.016980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zegers MM, O’Brien LE, Yu W, Datta A, Mostov KE. Epithelial polarity and tubulogenesis in vitro. Trends Cell Biol. 2003;13:169–176. doi: 10.1016/s0962-8924(03)00036-9. [DOI] [PubMed] [Google Scholar]
- 101.Orr AW, Helmke BP, Blackman BR, Schwartz MA. Mechanisms of mechanotransduction. Dev Cell. 2006;10:11–20. doi: 10.1016/j.devcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 102.Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a Fibroblast-Derived Epithelial Morphogen as Hepatocyte Growth-Factor. Cell. 1991;67:901–908. doi: 10.1016/0092-8674(91)90363-4. [DOI] [PubMed] [Google Scholar]
- 103.Bussolino F, Direnzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. Hepatocyte Growth-Factor Is a Potent Angiogenic Factor Which Stimulates Endothelial-Cell Motility and Growth. Journal of Cell Biology. 1992;119:629–641. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Park M, Dean M, Cooper CS, Schmidt M, O’Brien SJ, Blair DG, Vande Woude GF. Mechanism of met oncogene activation. Cell. 1986;45:895–904. doi: 10.1016/0092-8674(86)90564-7. [DOI] [PubMed] [Google Scholar]
- 105.Bottaro DP, Rubin JS, Faletto DL, Chan AML, Kmiecik TE, Vandewoude GF, Aaronson SA. Identification of the Hepatocyte Growth-Factor Receptor as the C-Met Protooncogene Product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 106.Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, Narsimhan RP, Hartmann G, Zarnegar R, Michalopoulos GK, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991;10:2867–2878. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, Graziani A, Panayotou G, Comoglio PM. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77:261–271. doi: 10.1016/0092-8674(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 108.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature Reviews Molecular Cell Biology. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 109.Weidner KM, Di Cesare S, Sachs M, Brinkmann V, Behrens J, Birchmeier W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. 1996;384:173–176. doi: 10.1038/384173a0. [DOI] [PubMed] [Google Scholar]
- 110.Sakkab D, Lewitzky M, Posern G, Schaeper U, Sachs M, Birchmeier W, Feller SM. Signaling of hepatocyte growth factor/scatter factor (HGF) to the small GTPase Rap1 via the large docking protein Gab1 and the adapter protein CRKL. J Biol Chem. 2000;275:10772–10778. doi: 10.1074/jbc.275.15.10772. [DOI] [PubMed] [Google Scholar]
- 111.Xiao GH, Jeffers M, Bellacosa A, Mitsuuchi Y, Vande Woude GF, Testa JR. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc Natl Acad Sci U S A. 2001;98:247–252. doi: 10.1073/pnas.011532898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 113.Franke R, Muller M, Wundrack N, Gilles ED, Klamt S, Kahne T, Naumann M. Host-pathogen systems biology: logical modelling of hepatocyte growth factor and Helicobacter pylori induced c-Met signal transduction. BMC Syst Biol. 2008;2:4. doi: 10.1186/1752-0509-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saez-Rodriguez J, Alexopoulos LG, Epperlein J, Samaga R, Lauffenburger DA, Klamt S, Sorger PK. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol Syst Biol. 2009;5:331. doi: 10.1038/msb.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 116.Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506–1507. doi: 10.1126/science.1105396. [DOI] [PubMed] [Google Scholar]
- 117.Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L. Targeted Expression of a Dominant-Negative Fgf Receptor Blocks Branching Morphogenesis and Epithelial Differentiation of the Mouse Lung. Embo Journal. 1994;13:3296–3301. doi: 10.1002/j.1460-2075.1994.tb06631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biology. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D, Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70:431–442. doi: 10.1016/0092-8674(92)90167-b. [DOI] [PubMed] [Google Scholar]
- 120.Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 121.Prudovsky I, Savion N, Zhan X, Friesel R, Xu J, Hou J, McKeehan WL, Maciag T. Intact and functional fibroblast growth factor (FGF) receptor-1 trafficks near the nucleus in response to FGF-1. J Biol Chem. 1994;269:31720–31724. [PubMed] [Google Scholar]
- 122.Zhan X, Plourde C, Hu X, Friesel R, Maciag T. Association of fibroblast growth factor receptor-1 with c-Src correlates with association between c-Src and cortactin. J Biol Chem. 1994;269:20221–20224. [PubMed] [Google Scholar]
- 123.Sandilands E, Akbarzadeh S, Vecchione A, McEwan DG, Frame MC, Heath JK. Src kinase modulates the activation, transport and signalling dynamics of fibroblast growth factor receptors. EMBO Rep. 2007;8:1162–1169. doi: 10.1038/sj.embor.7401097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Faeder JR, Blinov ML, Goldstein B, Hlavacek WS. Combinatorial complexity and dynamical restriction of network flows in signal transduction. Syst Biol (Stevenage) 2005;2:5–15. doi: 10.1049/sb:20045031. [DOI] [PubMed] [Google Scholar]
- 125.Furdui CM, Lew ED, Schlessinger J, Anderson KS. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol Cell. 2006;21:711–717. doi: 10.1016/j.molcel.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 126.Gaffney EA, Heath JK, Kwiatkowska MZ. A mass action model of a Fibroblast Growth Factor signaling pathway and its simplification. Bull Math Biol. 2008;70:2229–2263. doi: 10.1007/s11538-008-9342-1. [DOI] [PubMed] [Google Scholar]
- 127.Cohen S. The stimulation of epidermal proliferation by a specific protein (EGF) Dev Biol. 1965;12:394–407. doi: 10.1016/0012-1606(65)90005-9. [DOI] [PubMed] [Google Scholar]
- 128.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 129.Wiley HS, Shvartsman SY, Lauffenburger DA. Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol. 2003;13:43–50. doi: 10.1016/s0962-8924(02)00009-0. [DOI] [PubMed] [Google Scholar]
- 130.Schoeberl B, Eichler-Jonsson C, Gilles ED, Muller G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat Biotechnol. 2002;20:370–375. doi: 10.1038/nbt0402-370. [DOI] [PubMed] [Google Scholar]
- 131.Chen WW, Schoeberl B, Jasper PJ, Niepel M, Nielsen UB, Lauffenburger DA, Sorger PK. Input-output behavior of ErbB signaling pathways as revealed by a mass action model trained against dynamic data. Mol Syst Biol. 2009;5:239. doi: 10.1038/msb.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nakakuki T, Birtwistle MR, Saeki Y, Yumoto N, Ide K, Nagashima T, Brusch L, Ogunnaike BA, Okada-Hatakeyama M, Kholodenko BN. Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell. 2010;141:884–896. doi: 10.1016/j.cell.2010.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Koff A, Ohtsuki M, Polyak K, Roberts JM, Massague J. Negative Regulation of G1 in Mammalian-Cells - Inhibition of Cyclin-E-Dependent Kinase by Tgf-Beta. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- 134.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 135.Moses HL, Yang EY, Pietenpol JA. TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63:245–247. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- 136.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 137.He W, Li AG, Wang D, Han S, Zheng B, Goumans MJ, Ten Dijke P, Wang XJ. Overexpression of Smad7 results in severe pathological alterations in multiple epithelial tissues. EMBO J. 2002;21:2580–2590. doi: 10.1093/emboj/21.11.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen F, Weinberg RA. Biochemical evidence for the autophosphorylation and transphosphorylation of transforming growth factor beta receptor kinases. Proc Natl Acad Sci U S A. 1995;92:1565–1569. doi: 10.1073/pnas.92.5.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Furuhashi M, Yagi K, Yamamoto H, Furukawa Y, Shimada S, Nakamura Y, Kikuchi A, Miyazono K, Kato M. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol Cell Biol. 2001;21:5132–5141. doi: 10.1128/MCB.21.15.5132-5141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heldin CH, Miyazono K, tenDijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 141.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 142.Koli KM, Arteaga CL. Predominant cytosolic localization of type II transforming growth factor beta receptors in human breast carcinoma cells. Cancer Res. 1997;57:970–977. [PubMed] [Google Scholar]
- 143.Vilar JMG, Jansen R, Sander C. Signal processing in the TGF-beta superfamily ligand-receptor network. Plos Computational Biology. 2006;2:36–45. doi: 10.1371/journal.pcbi.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: distribution of homo- and heterodimers depends on relative HER2 levels. J Biol Chem. 2003;278:23343–23351. doi: 10.1074/jbc.M300477200. [DOI] [PubMed] [Google Scholar]
- 145.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 146.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 147.Jamora C, DasGupta R, Kocieniewski P, Fuchs E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003;422:317–322. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372:679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 149.Brisken C, Heineman A, Chavarria T, Elenbaas B, Tan J, Dey SK, McMahon JA, McMahon AP, Weinberg RA. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000;14:650–654. [PMC free article] [PubMed] [Google Scholar]
- 150.Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 151.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 152.Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407:530–535. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- 153.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 155.Papkoff J, Rubinfeld B, Schryver B, Polakis P. Wnt-1 regulates free pools of catenins and stabilizes APC-catenin complexes. Mol Cell Biol. 1996;16:2128–2134. doi: 10.1128/mcb.16.5.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xing Y, Clements WK, Le Trong I, Hinds TR, Stenkamp R, Kimelman D, Xu W. Crystal structure of a beta-catenin/APC complex reveals a critical role for APC phosphorylation in APC function. Mol Cell. 2004;15:523–533. doi: 10.1016/j.molcel.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 157.Peters JM, McKay RM, McKay JP, Graff JM. Casein kinase I transduces Wnt signals. Nature. 1999;401:345–350. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- 158.Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kimelman D, Xu W. beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- 160.Yang-Snyder J, Miller JR, Brown JD, Lai CJ, Moon RT. A frizzled homolog functions in a vertebrate Wnt signaling pathway. Curr Biol. 1996;6:1302–1306. doi: 10.1016/s0960-9822(02)70716-1. [DOI] [PubMed] [Google Scholar]
- 161.Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, Almeida K, Wang J, Doble B, Woodgett J, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008;135:367–375. doi: 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Giese K, Kingsley C, Kirshner JR, Grosschedl R. Assembly and function of a TCR alpha enhancer complex is dependent on LEF-1-induced DNA bending and multiple protein-protein interactions. Genes Dev. 1995;9:995–1008. doi: 10.1101/gad.9.8.995. [DOI] [PubMed] [Google Scholar]
- 163.Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59:3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- 164.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 165.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 166.Salic A, Lee E, Mayer L, Kirschner MW. Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell. 2000;5:523–532. doi: 10.1016/s1097-2765(00)80446-3. [DOI] [PubMed] [Google Scholar]
- 167.Lee E, Salic A, Kruger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tolwinski NS, Wehrli M, Rives A, Erdeniz N, DiNardo S, Wieschaus E. Wg/Wnt signal can be transmitted through arrow/LRP5,6 and Axin independently of Zw3/Gsk3beta activity. Dev Cell. 2003;4:407–418. doi: 10.1016/s1534-5807(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 169.Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase-3beta regulates its stability. J Biol Chem. 1999;274:10681–10684. doi: 10.1074/jbc.274.16.10681. [DOI] [PubMed] [Google Scholar]
- 170.Kirschner MW. The meaning of systems biology. Cell. 2005;121:503–504. doi: 10.1016/j.cell.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 171.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 172.Yamada S, Nelson WJ. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J Cell Biol. 2007;178:517–527. doi: 10.1083/jcb.200701058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 174.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 175.Peinado H, Marin F, Cubillo E, Stark HJ, Fusenig N, Nieto MA, Cano A. Snail and E47 repressors of E-cadherin induce distinct invasive and angiogenic properties in vivo. J Cell Sci. 2004;117:2827–2839. doi: 10.1242/jcs.01145. [DOI] [PubMed] [Google Scholar]
- 176.Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, Zhao J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007;67:7184–7193. doi: 10.1158/0008-5472.CAN-06-4729. [DOI] [PubMed] [Google Scholar]
- 177.Sobrado VR, Moreno-Bueno G, Cubillo E, Holt LJ, Nieto MA, Portillo F, Cano A. The class I bHLH factors E2-2A and E2-2B regulate EMT. J Cell Sci. 2009;122:1014–1024. doi: 10.1242/jcs.028241. [DOI] [PubMed] [Google Scholar]
- 178.Ramis-Conde I, Drasdo D, Anderson AR, Chaplain MA. Modeling the influence of the E-cadherin-beta-catenin pathway in cancer cell invasion: a multiscale approach. Biophys J. 2008;95:155–165. doi: 10.1529/biophysj.107.114678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Lochner D, Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bessho Y, Kageyama R. Oscillations, clocks and segmentation. Curr Opin Genet Dev. 2003;13:379–384. doi: 10.1016/s0959-437x(03)00083-2. [DOI] [PubMed] [Google Scholar]
- 181.Dale JK, Maroto M, Dequeant ML, Malapert P, McGrew M, Pourquie O. Periodic notch inhibition by lunatic fringe underlies the chick segmentation clock. Nature. 2003;421:275–278. doi: 10.1038/nature01244. [DOI] [PubMed] [Google Scholar]
- 182.Mazzone M, Selfors LM, Albeck J, Overholtzer M, Sale S, Carroll DL, Pandya D, Lu Y, Mills GB, Aster JC, et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci U S A. 2010;107:5012–5017. doi: 10.1073/pnas.1000896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–1525. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- 184.Webb SD, Owen MR. Oscillations and patterns in spatially discrete models for developmental intercellular signalling. J Math Biol. 2004;48:444–476. doi: 10.1007/s00285-003-0247-1. [DOI] [PubMed] [Google Scholar]
- 185.Kimble J, Simpson P. The LIN-12/Notch signaling pathway and its regulation. Annu Rev Cell Dev Biol. 1997;13:333–361. doi: 10.1146/annurev.cellbio.13.1.333. [DOI] [PubMed] [Google Scholar]
- 186.de Celis JF, Bray S. Feed-back mechanisms affecting Notch activation at the dorsoventral boundary in the Drosophila wing. Development. 1997;124:3241–3251. doi: 10.1242/dev.124.17.3241. [DOI] [PubMed] [Google Scholar]
- 187.Jorgensen C, Sherman A, Chen GI, Pasculescu A, Poliakov A, Hsiung M, Larsen B, Wilkinson DG, Linding R, Pawson T. Cell-specific information processing in segregating populations of Eph receptor ephrin-expressing cells. Science. 2009;326:1502–1509. doi: 10.1126/science.1176615. [DOI] [PubMed] [Google Scholar]
- 188.Batlle E, Bacani J, Begthel H, Jonkheer S, Gregorieff A, van de Born M, Malats N, Sancho E, Boon E, Pawson T, et al. EphB receptor activity suppresses colorectal cancer progression. Nature. 2005;435:1126–1130. doi: 10.1038/nature03626. [DOI] [PubMed] [Google Scholar]
- 189.Cortina C, Palomo-Ponce S, Iglesias M, Fernandez-Masip JL, Vivancos A, Whissell G, Huma M, Peiro N, Gallego L, Jonkheer S, et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat Genet. 2007;39:1376–1383. doi: 10.1038/ng.2007.11. [DOI] [PubMed] [Google Scholar]
- 190.Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, Eliceiri KW, Weaver VM, Calderwood DA, Keely PJ. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20:3224–3238. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Alexander NR, Branch KM, Parekh A, Clark ES, Iwueke IC, Guelcher SA, Weaver AM. Extracellular matrix rigidity promotes invadopodia activity. Curr Biol. 2008;18:1295–1299. doi: 10.1016/j.cub.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 193.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gudjonsson T, Villadsen R, Nielsen HL, Ronnov-Jessen L, Bissell MJ, Petersen OW. Isolation, immortalization, and characterization of a human breast epithelial cell line with stem cell properties. Genes Dev. 2002;16:693–706. doi: 10.1101/gad.952602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Villadsen R, Fridriksdottir AJ, Ronnov-Jessen L, Gudjonsson T, Rank F, LaBarge MA, Bissell MJ, Petersen OW. Evidence for a stem cell hierarchy in the adult human breast. J Cell Biol. 2007;177:87–101. doi: 10.1083/jcb.200611114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 2007;9:201–209. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- 197.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 198.Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127:1041–1055. doi: 10.1016/j.cell.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009;23:2563–2577. doi: 10.1101/gad.1849509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 201.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 202.Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 204.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 206.Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–825. doi: 10.1126/science.1196236. [DOI] [PubMed] [Google Scholar]
- 207.Janes KA, Wang CC, Holmberg KJ, Cabral K, Brugge JS. Identifying single-cell molecular programs by stochastic profiling. Nat Methods. 2010;7:311–317. doi: 10.1038/nmeth.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 209.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wang F, Hansen RK, Radisky D, Yoneda T, Barcellos-Hoff MH, Petersen OW, Turley EA, Bissell MJ. Phenotypic reversion or death of cancer cells by altering signaling pathways in three-dimensional contexts. J Natl Cancer Inst. 2002;94:1494–1503. doi: 10.1093/jnci/94.19.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60:1254–1260. [PubMed] [Google Scholar]
- 212.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 214.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 215.Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 216.Oft M, Akhurst RJ, Balmain A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol. 2002;4:487–494. doi: 10.1038/ncb807. [DOI] [PubMed] [Google Scholar]
- 217.Hogan C, Dupre-Crochet S, Norman M, Kajita M, Zimmermann C, Pelling AE, Piddini E, Baena-Lopez LA, Vincent JP, Itoh Y, et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 2009;11:460–467. doi: 10.1038/ncb1853. [DOI] [PubMed] [Google Scholar]
- 218.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg L, Liu ET, Cance WG. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55:2752–2755. [PubMed] [Google Scholar]
- 222.Benlimame N, He Q, Jie S, Xiao D, Xu YJ, Loignon M, Schlaepfer DD, Alaoui-Jamali MA. FAK signaling is critical for ErbB-2/ErbB-3 receptor cooperation for oncogenic transformation and invasion. J Cell Biol. 2005;171:505–516. doi: 10.1083/jcb.200504124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, et al. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–767. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wu X, Gan B, Yoo Y, Guan JL. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev Cell. 2005;9:185–196. doi: 10.1016/j.devcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 225.Avizienyte E, Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol. 2005;17:542–547. doi: 10.1016/j.ceb.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 226.Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A, Fantoni A, Amicone L, Tripodi M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314:143–152. doi: 10.1016/j.yexcr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 227.van Nimwegen MJ, van de Water B. Focal adhesion kinase: a potential target in cancer therapy. Biochem Pharmacol. 2007;73:597–609. doi: 10.1016/j.bcp.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 228.Lim ST, Chen XL, Tomar A, Miller NL, Yoo J, Schlaepfer DD. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J Biol Chem. 2010;285:21526–21536. doi: 10.1074/jbc.M110.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Brent R. A partnership between biology and engineering. Nat Biotechnol. 2004;22:1211–1214. doi: 10.1038/nbt1004-1211. [DOI] [PubMed] [Google Scholar]
- 230.Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–610. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 231.Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15:378–386. doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 232.Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol. 2007;8:839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- 233.Montesano R, Schaller G, Orci L. Induction of epithelial tubular morphogenesis in vitro by fibroblast-derived soluble factors. Cell. 1991;66:697–711. doi: 10.1016/0092-8674(91)90115-f. [DOI] [PubMed] [Google Scholar]
- 234.Wang AZ, Ojakian GK, Nelson WJ. Steps in the morphogenesis of a polarized epithelium. II. Disassembly and assembly of plasma membrane domains during reversal of epithelial cell polarity in multicellular epithelial (MDCK) cysts. J Cell Sci. 1990;95(Pt 1):153–165. doi: 10.1242/jcs.95.1.153. [DOI] [PubMed] [Google Scholar]
- 235.Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol. 2010;12:1035–1045. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Saxen L, Sariola H. Early organogenesis of the kidney. Pediatr Nephrol. 1987;1:385–392. doi: 10.1007/BF00849241. [DOI] [PubMed] [Google Scholar]
- 237.McAteer JA, Evan AP, Gardner KD. Morphogenetic clonal growth of kidney epithelial cell line MDCK. Anat Rec. 1987;217:229–239. doi: 10.1002/ar.1092170303. [DOI] [PubMed] [Google Scholar]
- 238.Chen X, Macara IG. RNA interference techniques to study epithelial cell adhesion and polarity. Methods Enzymol. 2006;406:362–374. doi: 10.1016/S0076-6879(06)06026-5. [DOI] [PubMed] [Google Scholar]
- 239.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 240.Hariharan IK, Haber DA. Yeast, flies, worms, and fish in the study of human disease. N Engl J Med. 2003;348:2457–2463. doi: 10.1056/NEJMon023158. [DOI] [PubMed] [Google Scholar]
- 241.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 242.Schluter MA, Pfarr CS, Pieczynski J, Whiteman EL, Hurd TW, Fan SL, Liu CJ, Margolis B. Trafficking of Crumbs3 during Cytokinesis Is Crucial for Lumen Formation. Molecular Biology of the Cell. 2009;20:4652–4663. doi: 10.1091/mbc.E09-02-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Aranda V, Haire T, Nolan ME, Calarco JP, Rosenberg AZ, Fawcett JP, Pawson T, Muthuswamy SK. Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat Cell Biol. 2006;8:1235–1245. doi: 10.1038/ncb1485. [DOI] [PubMed] [Google Scholar]
- 244.Richard NR, Anderson JA, Weiss JL, Binder PS. Air/liquid corneal organ culture: a light microscopic study. Curr Eye Res. 1991;10:739–749. doi: 10.3109/02713689109013868. [DOI] [PubMed] [Google Scholar]
- 245.Foreman DM, Pancholi S, Jarvis-Evans J, McLeod D, Boulton ME. A simple organ culture model for assessing the effects of growth factors on corneal re-epithelialization. Exp Eye Res. 1996;62:555–564. doi: 10.1006/exer.1996.0065. [DOI] [PubMed] [Google Scholar]
- 246.Xu KP, Li XF, Yu FS. Corneal organ culture model for assessing epithelial responses to surfactants. Toxicol Sci. 2000;58:306–314. doi: 10.1093/toxsci/58.2.306. [DOI] [PubMed] [Google Scholar]
- 247.Lu Z, Hasse S, Bodo E, Rose C, Funk W, Paus R. Towards the development of a simplified long-term organ culture method for human scalp skin and its appendages under serum-free conditions. Exp Dermatol. 2007;16:37–44. doi: 10.1111/j.1600-0625.2006.00510.x. [DOI] [PubMed] [Google Scholar]
- 248.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2010 doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Nelson CM, Inman JL, Bissell MJ. Three-dimensional lithographically defined organotypic tissue arrays for quantitative analysis of morphogenesis and neoplastic progression. Nat Protoc. 2008;3:674–678. doi: 10.1038/nprot.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Elowitz M, Lim WA. Build life to understand it. Nature. 2010;468:889–890. doi: 10.1038/468889a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Rejniak KA, Anderson AR. Hybrid models of tumor growth. Wiley Interdiscip Rev Syst Biol Med. 2011;3:115–125. doi: 10.1002/wsbm.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Edelman LB, Eddy JA, Price ND. In silico models of cancer. Wiley Interdiscip Rev Syst Biol Med. 2010;2:438–459. doi: 10.1002/wsbm.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Smallwood R. Computational modeling of epithelial tissues. Wiley Interdiscip Rev Syst Biol Med. 2009;1:191–201. doi: 10.1002/wsbm.18. [DOI] [PubMed] [Google Scholar]
- 254.Turing AM. The chemical basis of morphogenesis. Proc R Soc Lond B Biol Sci. 1952;237:37–72. [Google Scholar]
- 255.Franks SJ, Byrne HM, Underwood JC, Lewis CE. Biological inferences from a mathematical model of comedo ductal carcinoma in situ of the breast. J Theor Biol. 2005;232:523–543. doi: 10.1016/j.jtbi.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 256.Sherratt JA, Dallon JC. Theoretical models of wound healing: past successes and future challenges. C R Biol. 2002;325:557–564. doi: 10.1016/s1631-0691(02)01464-6. [DOI] [PubMed] [Google Scholar]
- 257.Mi Q, Swigon D, Riviere B, Cetin S, Vodovotz Y, Hackam DJ. One-dimensional elastic continuum model of enterocyte layer migration. Biophys J. 2007;93:3745–3752. doi: 10.1529/biophysj.107.112326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Edwards CM, Chapman SJ. Biomechanical modelling of colorectal crypt budding and fission. Bull Math Biol. 2007;69:1927–1942. doi: 10.1007/s11538-007-9199-8. [DOI] [PubMed] [Google Scholar]
- 259.Fisher RA. The wave of advance of advantageous genes. Ann Eugenics. 1937;7:353–369. [Google Scholar]
- 260.Savla U, Olson LE, Waters CM. Mathematical modeling of airway epithelial wound closure during cyclic mechanical strain. J Appl Physiol. 2004;96:566–574. doi: 10.1152/japplphysiol.00510.2003. [DOI] [PubMed] [Google Scholar]
- 261.Haugh JM. Deterministic model of dermal wound invasion incorporating receptor-mediated signal transduction and spatial gradient sensing. Biophys J. 2006;90:2297–2308. doi: 10.1529/biophysj.105.077610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wolpert L. Positional information and the spatial pattern of cellular differentiation. J Theor Biol. 1969;25:1–47. doi: 10.1016/s0022-5193(69)80016-0. [DOI] [PubMed] [Google Scholar]
- 263.Kim SH, Yu W, Mostov K, Matthay MA, Hunt CA. A computational approach to understand in vitro alveolar morphogenesis. PLoS One. 2009;4:e4819. doi: 10.1371/journal.pone.0004819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kim SH, Matthay MA, Mostov K, Hunt CA. Simulation of lung alveolar epithelial wound healing in vitro. J R Soc Interface. 2010;7:1157–1170. doi: 10.1098/rsif.2010.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Grant MR, Mostov KE, Tlsty TD, Hunt CA. Simulating properties of in vitro epithelial cell morphogenesis. PLoS Comput Biol. 2006;2:e129. doi: 10.1371/journal.pcbi.0020129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Walker DC, Hill G, Wood SM, Smallwood RH, Southgate J. Agent-based computational modeling of wounded epithelial cell monolayers. IEEE Trans Nanobioscience. 2004;3:153–163. doi: 10.1109/tnb.2004.833680. [DOI] [PubMed] [Google Scholar]
- 267.Walker DC, Georgopoulos NT, Southgate J. Anti-social cells: predicting the influence of E-cadherin loss on the growth of epithelial cell populations. J Theor Biol. 2010;262:425–440. doi: 10.1016/j.jtbi.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 268.Rejniak KA. A single-cell approach in modeling the dynamics of tumor microregions. Math Biosci Eng. 2005;2:643–655. doi: 10.3934/mbe.2005.2.643. [DOI] [PubMed] [Google Scholar]
- 269.Rejniak KA, Anderson AR. A computational study of the development of epithelial acini: I. Sufficient conditions for the formation of a hollow structure. Bull Math Biol. 2008;70:677–712. doi: 10.1007/s11538-007-9274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Rejniak KA, Anderson AR. A computational study of the development of epithelial acini: II. Necessary conditions for structure and lumen stability. Bull Math Biol. 2008;70:1450–1479. doi: 10.1007/s11538-008-9308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Rejniak KA, Wang SE, Bryce NS, Chang H, Parvin B, Jourquin J, Estrada L, Gray JW, Arteaga CL, Weaver AM, et al. Linking changes in epithelial morphogenesis to cancer mutations using computational modeling. PLoS Comput Biol. 2010:6. doi: 10.1371/journal.pcbi.1000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, Arteaga CL. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 273.Gevertz J, Torquato S. Growing heterogeneous tumors in silico. Phys Rev E Stat Nonlin Soft Matter Phys. 2009;80:051910. doi: 10.1103/PhysRevE.80.051910. [DOI] [PubMed] [Google Scholar]
- 274.Cickovski T, Aras K, Alber MS, Izaguirre JA, Swat M, Glazier JA, Merks RM, Glimm T, Hentschel HG, Newman SA. From Genes to Organisms Via the Cell A Problem-Solving Environment for Multicellular Development. Comput Sci Eng. 2007;9:50–60. doi: 10.1109/MCSE.2007.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Walker D, Wood S, Southgate J, Holcombe M, Smallwood R. An integrated agent-mathematical model of the effect of intercellular signalling via the epidermal growth factor receptor on cell proliferation. J Theor Biol. 2006;242:774–789. doi: 10.1016/j.jtbi.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 276.Anderson AR, C M, Newman EL, Steele RJ, Thompson AM. Mathematical Modelling of Tumour Invasion and Metastasis. J Theoret Med. 2000;2:129–154. [Google Scholar]
- 277.Anderson AR. A hybrid mathematical model of solid tumour invasion: the importance of cell adhesion. Math Med Biol. 2005;22:163–186. doi: 10.1093/imammb/dqi005. [DOI] [PubMed] [Google Scholar]
- 278.Anderson AR, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 279.Gerlee P, Anderson AR. An evolutionary hybrid cellular automaton model of solid tumour growth. J Theor Biol. 2007;246:583–603. doi: 10.1016/j.jtbi.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Rejniak KA. An immersed boundary framework for modelling the growth of individual cells: an application to the early tumour development. J Theor Biol. 2007;247:186–204. doi: 10.1016/j.jtbi.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 281.Anderson AR, Hassanein M, Branch KM, Lu J, Lobdell NA, Maier J, Basanta D, Weidow B, Narasanna A, Arteaga CL, et al. Microenvironmental independence associated with tumor progression. Cancer Res. 2009;69:8797–8806. doi: 10.1158/0008-5472.CAN-09-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Anderson AR, Rejniak KA, Gerlee P, Quaranta V. Microenvironment driven invasion: a multiscale multimodel investigation. J Math Biol. 2009;58:579–624. doi: 10.1007/s00285-008-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Jeon J, Quaranta V, Cummings PT. An off-lattice hybrid discrete-continuum model of tumor growth and invasion. Biophys J. 2010;98:37–47. doi: 10.1016/j.bpj.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Schoeberl B, Pace E, Howard S, Garantcharova V, Kudla A, Sorger PK, Nielsen UB. A data-driven computational model of the ErbB receptor signaling network. Conf Proc IEEE Eng Med Biol Soc. 2006;1:53–54. doi: 10.1109/IEMBS.2006.259754. [DOI] [PubMed] [Google Scholar]
- 285.Janes KA, Reinhardt HC, Yaffe MB. Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell. 2008;135:343–354. doi: 10.1016/j.cell.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Miller-Jensen K, Janes KA, Brugge JS, Lauffenburger DA. Common effector processing mediates cell-specific responses to stimuli. Nature. 2007;448:604–608. doi: 10.1038/nature06001. [DOI] [PubMed] [Google Scholar]