

Abstract
Metabolism plays a key role in many major human diseases. Generation of high-throughput omics data has ushered in a new era of systems biology. Genome-scale metabolic network reconstructions provide a platform to interpret omics data in a biochemically meaningful manner. The release of the global human metabolic network, Recon 1, in 2007 has enabled new systems biology approaches to study human physiology, pathology, and pharmacology. There are currently over 20 publications that utilize Recon 1, including studies of cancer, diabetes, host-pathogen interactions, heritable metabolic disorders, and off-target drug binding effects. In this mini-review, we focus on the reconstruction of the global human metabolic network and four classes of its application. We show that computational simulations for numerous pathologies have yielded clinically relevant results, many corroborated by existing or newly generated experimental data.
Keywords: human metabolism, systems biology, constraints-based modeling
Introduction
Historically, biology and biochemistry have a reductionist framework where individual components are carefully enumerated and usually studied independently. The emergence of high-throughput data, or ‘omics’ data, over the past decade has led to a wave of new technologies that allow measurement of almost all cellular components. Next generation DNA sequencing can elucidate the genome sequence while transcription, proteomic, and metabolomic profiling are becoming common technologies that measure mRNA, proteins, and small metabolites respectively under a specific condition. The generation of ‘omics’ data is becoming relatively easy and cost efficient, but the interpretation and physiological assessment of the massive amounts of data has been a daunting challenge [1].
Molecular systems biology takes a holistic approach by trying to analyze and interpret high-throughput data. Genome-scale metabolic reconstructions are a common denominator in molecular systems biology by providing a computational platform to analyze generated data as well as probe molecular networks through simulation [2, 3]. Reconstructing a genome-scale network involves determining and formally organizing the known biochemical transformations in a cell or organism (Figure 1) [4]. The biochemical network can be studied as is or can be converted into a mathematical format for predictive simulations using constraint-based modeling [5] and flux balance analysis [6].
Figure 1.
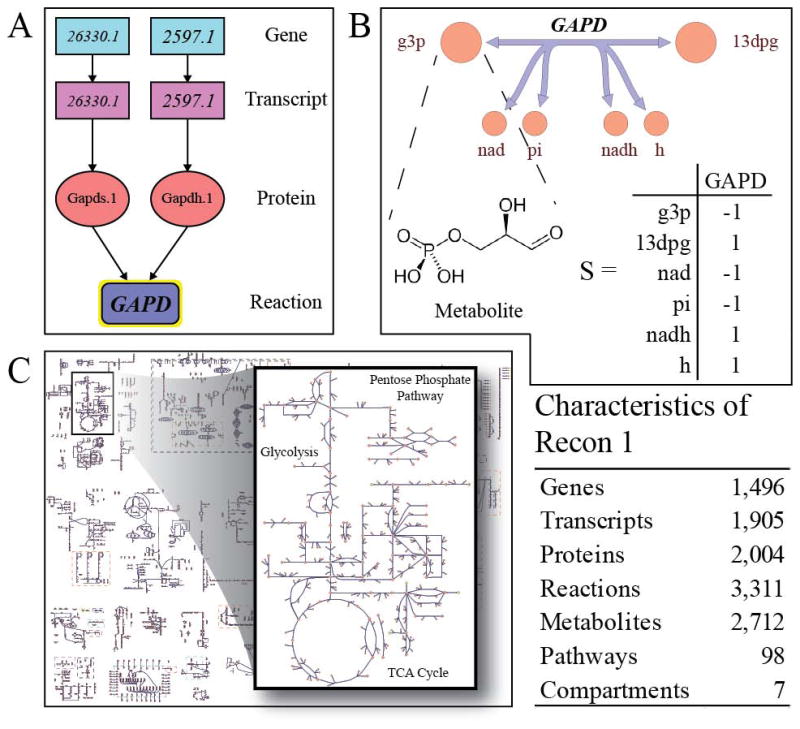
Recon 1 is a global human metabolic network reconstruction comprised of the known biochemical and physiologic data. A) Gene protein reaction associations can be represented in Boolean logic and are used to define a mechanistic genotype phenotype relationship. This is essential for determining phenotypes of genetic perturbations as well as understanding the underlying mechanisms of a particular phenotype. B) Recon 1 accounts for 3,311 metabolic reactions and their associated metabolites. The reactions in the network can be represented in a mathematical format called the stoichiometric matrix. C) Recon 1 is a thorough and very complex assessment of human metabolism accounting for over 1400 genes and 7 cellular compartments.
Since the reconstruction of H. influnzae in 1999 [7], there has been a rapid growth in both the number of reconstructions and their uses [2]. Today, there are numerous prokaryotic and eukaryotic genome-scale reconstructions of major pathogens (e.g., M. tuberculosis [8, 9], S. auerus [10–12], H. pylori [13, 14], S. typhimurium [15–17]) and industrially relevant bacteria (e.g., E. coli [18, 19], G. metallireducens [20, 21], T. maritima [22], B. subtilis [23–25], S. coelicolor [26, 27], S. cerevisiae [28, 29]). In 2007, the genome-scale reconstruction of the global human metabolic network was completed [30]. The reconstruction, named Recon 1, accounts for 1496 open reading frames and is not cell or tissue specific. Recon 1 provides a mechanistic basis for connecting the human genotype and phenotype.
Studying human physiology and pathology using the constraint-based modeling framework poses different challenges and opportunities than studying prokaryotes in the same framework. Thus the applications of Recon 1 are quite different than the traditional five applications of genome-scale reconstructions to the systems biology field [2, 3]. In this review, we begin with an overview on the reconstruction of Recon 1 and constraint-based modeling. Second, we review constraint-based modeling of human cells prior to the release of Recon 1. Third, we focus on the four current applications of the global human metabolic network: 1) Integration of ‘omics’ data for tissue and cell specific network reconstruction, 2) Mapping homologous genes for global mammalian network reconstruction, 3) Contextualization of ‘omics’ data from pathological and drug-treated states, and 4) Simulation of pathological and drug-treated states. The first two applications are related to how Recon 1 can be leveraged to build other related networks, while the second two are the actual application of Recon 1 and its model derivatives to studying human physiology and pathology. Finally, we discuss the future and challenges of constraint-based modeling of human metabolism.
Recon 1 Reconstruction and Details
Metabolism provides all molecules and energy to drive cellular functions and is thus foundational to all cellular processes. The global human metabolic network was reconstructed using a very strict “bottom-up” protocol that is now publicly available [4]. Utilizing the human genome sequence (Build 35), an initial set of 1865 human metabolic genes was defined. The gene annotations were used to reconstruct an automated draft network, consisting of the associated enzymes and the reactions they catalyze. The list of enzymes and reactions is generated from databases such as KEGG [31] and ExPASy [32]. To build the most accurate model possible, the initial draft was evaluated by looking for existing primary literature that showed direct physical evidence of the network components. In all, over 1500 literature sources (articles, reviews, and textbooks) were utilized in building Recon 1. Iteratively, simulations were completed to validate the basic functionality of the network. 288 known human metabolic functions were defined and constraint-based modeling was used to analyze draft builds of the network to ensure that the tests were successfully completed. The final network represents a compilation of over 50 years of biochemical and biological research into human metabolism (Figure 1).
Recon 1 accounts for 1,496 open reading frames, 2,004 proteins, 2,712 metabolites, and 3311 metabolic reactions (Figure 1C). The network is mass and charged balanced and fully compartmentalized, accounting for the cytoplasm, nucleus, mitochondria, lysosome, peroxisome, Golgi apparatus, and endoplasmic reticulum. To mechanistically define the genotype to phenotype relationship, gene-protein-reaction (GPR) annotations are provided. GPRs are a Boolean representation of the gene, transcript, protein, and reaction relationship accounting for spliced variants, isozymes, and protein complexes (Figure 1A).
A reconstruction can be converted into a mathematical format, represented by the stoichiometric matrix (Figure 1B) [33]. The stoichiometric matrix (S) consists of rows of metabolites and columns of reactions. Coefficients in the matrix represent elementally and charge balanced reaction coefficients with substrates having a negative value and products a positive value. The stoichiometric matrix is the basis from which all constraints-based modeling is done.
Genome-scale metabolic reconstructions for important organisms are often updated to account for additional genes, proteins, metabolites, and reactions. Even though Recon 1 accounts for about 1500 genes, the complexity of the human genome results in large gaps in the metabolic network. In recent years, there have been “reconstruction jamborees” [17, 34] that bring together experts in systems biology, bioinformatics, molecular biology, biochemistry, and metabolomics into a mini-conference environment to refine and reconcile existing metabolic reconstructions for an organism [35]. A reconstruction jamboree is critical in reconstructing a more expansive and accurate Recon 2.
Pre-Recon 1
We first begin with reviewing some of the constraints-based human metabolic modeling published before the release of Recon 1. The studies are limited in the size of the networks utilized, but the methods and results foreshadowed the current applications of Recon 1.
The first human metabolic network studied under the constraint-based approach was the erythrocyte. As the human erythrocyte is both readily available and relatively simple, the blood cell is optimal for initial studies. Various kinetic models have been built over the years for the human red blood cell taking glycolysis, the pentose phosphate pathway, and AMP metabolism into account [36–39]. More recently, a kinetic model of the human erythrocyte was converted into a constraint-based model and was used in an enzymopathy study [40]. Price et al. used sampling techniques that enumerate all candidate network flux states and found that by constraining enzymatic fluxes in silico, they were able to detect known glycolytic enzymes that cause hemolytic anemia. In addition, sampling flux states elucidated correlations between different enzymatic fluxes in both normal and pathological erythrocytes that are helpful in designing fluxomic experiments.
A 189-reaction network of the human cardiac myocyte mitochondrial metabolic network was built using proteomic data [41]. The maximal capabilities of the network’s ATP production, heme synthesis, and mixed phospholipid synthesis were determined. In addition, metabolic network flexibility was assessed by looking at variation in allowable flux levels in the steady state. Heme and phospholipid synthesis simulations had greater flexibility than ATP production. In addition, Vo et al. determined correlated reaction sets, or co-sets, that represent sets of reactions that have fixed ratios of fluxes. Co-sets help elucidate the systemic properties of the network as it can be shown how the fluxes of a group of reactions “move” together. Thiele et al. used the same human mitochondrial network to study metabolic states of normal, diabetic, ischemic, and dietetic conditions [42]. Applying external constraints for the normal and diseased phenotypes allowed prediction of intracellular fluxes, which were corroborated by experimental data. Interestingly, the metabolic reconstruction showed that some observed differences in enzyme activity could be explained solely through stoichiometry, rather than regulation. Finally, Jamshidi et al. used the human mitochondrial network to study the systemic effects of morbid, metabolic single nucleotide polymorphisms (SNPs) [43]. Known inborn errors of human metabolism were obtained from the Online Mendelian Inheritance in Man database [44] and integrated with the GPR associations of the mitochondrial network. At the same time, the co-sets of the network were determined. Jamshidi et al. postulated and then showed that SNPs of reactions in the same co-set have similar pathologies.
The methods used to study human physiology and pathology using constraint-based modeling before the introduction of Recon 1 are very similar to some of the methods that are in current use: 1) Randomized sampling is used to study defects in genes and proteins and 2) Co-sets are used to relate and study systemic network effects. However, the increased scope and detail of Recon 1 has provided new opportunities in studying human metabolism.
Application 1: Integration of high-throughput data for model construction
As Recon 1 is a global human metabolic network, efforts have been focused on constructing cell and tissue-specific models to study their unique physiology (Figure 2.i). In order to do so, high-throughput data, such as transcriptomics and proteomics, are integrated with Recon 1 to determine the cell specific reactions needed to build a cell specific model. Algorithmic approaches have been developed to quickly build cell and tissue specific models. Other publications have utilized a more rigorous manual approach, while some have combined both automated and manual steps for building cell and tissue specific models.
Figure 2.

The four major applications of the global human metabolic network Recon 1. I) Utilizing high-throughput data, Recon 1 can be tailored to cell and tissue-specific networks. The process has been done both algorithmically and manually. II) Similarly, Recon 1 has been transformed into other mammalian reconstructions, particularly M. musculus. The high overlap of homologous genes in Recon 1 with similar mammals allows for reconstructing accurate mammalian models quickly. III) High-throughput data can be interpreted by mapping the data onto Recon 1’s metabolic network backbone. This process has been done to study pathological and drug-treated states. IV) Recon 1 can be used to simulate and predict phenotypes, providing biological clues to physiology and pathology as well as guiding experimental design.
First, there are three main methods for algorithmically tailoring Recon 1 to a cell or tissue specific model under the context of high-throughput data. Gene Inactivity Moderated by Metabolism and Expression (GIMME) is an algorithm that tailors a reconstruction to a context-specific network, based on the cellular state of the supplied expression profiling data, while maintaining a user determined functional flux [45]. GIMME takes gene expression as an input and determines active and suppressed reactions based on the GPR associations and a specific expression level threshold. GIMME removes inactive reactions and then reinserts removed reactions that are required for the predetermined functional flux. Though GIMME was initially used for building context-specific models of a particular expression state, it has been used to build cell and tissue specific models from Recon 1. Chang et al. reconstructed a renal metabolic model from transcriptomic data using the GIMME algorithm [46]. An organ specific objective function was built using scientific literature that represented the renal functions of reabsorption and secretion. A manual assessment was done with primary literature to validate the physiological renal functions for accuracy.
Similarly, another algorithm by Shlomi et al. integrates high-throughput data with Recon 1 to determine tissue specific models by matching reaction and pathway length with the metabolic network [47], rather than matching functional flux as GIMME. Thus, the algorithm does not require a pre-determined objective like the GIMME algorithm. Shlomi et al. showed that their method could be applied to building automated reconstructions of 10 human tissues and that the final models were quite different than the used expression data, showing that post-transcriptional up- and down-regulation is very important.
Very recently, a new method called the model-building algorithm (MBA) was used to reconstruct a human liver metabolic network from Recon 1 [48]. Unlike the GIMME and Shlomi et al. algorithms, MBA utilizes many forms of high-throughput data, phenotypic data, and literature knowledge derived from any state the tissue or cell is in to construct a model that is not specific to a particular expression state. The MBA distinguishes Recon 1 into three reaction sets: 1) high probability core reactions (defined through manual literature curation), 2) medium probability reactions (defined by high-throughput data), and 3) remaining reactions. The final tissue-specific network contains all the high probability core reactions, as many of the medium probability reactions, and as few of the remaining reactions to build a functional model. The final liver model was used to simulate inherited hepatic metabolic disorders and metabolite transport to predict changes in biofluids. MBA was also utilized to build a core network model of cancer metabolism for the NCI-60 cancer cell lines to predict selective drug targets [49].
Though algorithmic approaches are quick in analyzing high-throughput data and are fairly accurate in predicting cell and tissue-specificity, a manually built reconstruction is inherently more accurate. A human alveolar macrophage metabolic network was reconstructed using transcriptomic data and both the GIMME and Shlomi et al. algorithms [50]. Macrophages are known to have a varied expression and physiological states. Thus, the initial algorithm-derived reconstructions were not adequate to represent macrophage physiology and a final network was built through a rigorous manual curation process with proteomic data and primary literature. For reactions with no available literature or data, the results from the algorithm were used. The final model was used to study host-pathogen interactions with M. tuberculosis.
Several cell and tissue-specific models were manually built from the ‘omics’ data derived reactions in Recon 1. In general for these networks, transcriptomic and/or proteomic data were mapped to Recon 1 to determine an initial set of reactions for the cell or tissue-specific model. Rigorous manual curation through primary literature and iterative debugging simulations is done to arrive at an accurate final model. Vo et al. built a core fibroblast model consisting of cytosol and mitochondria reactions from transcriptomic data. The final model was used for analyzing carbon 13 flux data from normal and Leigh’s syndrome fibroblasts [51]. A second hepatic metabolic network was concurrently released with the MBA-derived model called HepatoNet 1 [52]. The network represents a compilation of 1500+ literature sources and omics data. The final model accomplishes 123 hepatic physiological metabolic functions. Finally, Lewis et al. constructed metabolic models to study brain metabolism [53]. Unlike the other described cell and tissue specific reconstructions, brain metabolism had to be partitioned into different cell types accounting for interactions between astrocytes and neurons. Three different multicellular models were built representing different neuron types and their interactions with surrounding astrocytes.
There are numerous applications of these cell and tissue-specific models. Cell and tissue-specific networks provide more accurate models of physiology and pathology than the global human metabolic network [48, 52]. Tissue-specific modeling is in its infancy. There is a need to formulate a standard operating procedure for constructing cell and tissue-specific networks with a focus on a community effort to reconstruct networks of interest. In sections three and four, we discuss applications of the mentioned networks for analysis and simulation of pathological and drug-treated states.
Application 2: Mapping homologous genes for model construction
Studying animal models is an integral part of clinical and pharmacological research. Thus, it is important to fully characterize the metabolism of common animal models, such as the lab mouse or rat. Some work has been done in generating metabolic reconstructions of mammals, including B. taurus [54] and M. musculus [55–58]. However, these models are relatively limited in scope compared to the gene coverage of the human metabolic reconstruction. Recently, researchers have leveraged the global human metabolic network to construct mammalian metabolic networks by homologous gene mapping (Figure 2.ii). Just as the global human metabolic network can be tailored to cell and tissue specific network derivatives through the integration of high-throughput data, Recon 1 can be transformed into global metabolic reconstructions of similar mammals. This undertaking is possible due to the high sequence homology between many mammalian genomes and the human genome. There are currently two publications on homologous model constructions.
There are a high number of orthologous genes between the human genes in Recon 1 and the mammalian genomes. Thus, Sigurdsson et al. reconstructed a genome-scale metabolic network for M. musculus and four other draft reconstructions for R. norvegicus, Canis lupus familiaris, Pan troglodytes, and Bos taurus [59]. Draft reconstructions were built using the homologous mappings and the M. musculus network was manually curated and validated to build a final reconstruction using established protocols [4]. The mouse metabolic network was used to determine essential genes for viability and to study the systemic effect of lipoprotein lipase deficiency. Thus, similar approaches for model reconstruction can be undertaken for other human related mammals quickly and comprehensively. In another work, Bekaert et al. studied copy number alterations across multiple mammalian species [60]. They found that copy number alterations were not due to genetic drift. The main difference in copy number between mammalian genomes was in transporter genes. The authors concluded that copy number alterations are due to many complex factors including: dosage-related adaptations, new functions for enzymes, gene duplication, and genetic drift.
It is very labor intensive to reconstruct mammalian metabolic networks from scratch. Determining orthologous genes of Recon 1 and using manual model curation represents a quick and accurate method to build related mammalian networks. Networks for the lab mouse and rat can be used alongside experimental models for experimental design and biological discovery.
Application 3: Contextualization of high-throughput data from pathological and drug-treated states
In the two previous sections, we have focused on the application of Recon 1 for model construction. The remainder of this review focuses on using genome-scale metabolic reconstructions, either Recon 1 or its cell and tissue-specific derivatives, to study human physiology, pathology, and pharmacology. Metabolism plays a prominent role in many disorders and diseases including cancer, diabetes, obesity, infection, neural disorders, and inherited gene and enzyme deficiencies. Current publications have used Recon 1 and its derivatives for: 1) interpreting high-throughput data and experiments for biological discovery, or 2) mathematically simulating phenotypes to guide experiments. In particular, these techniques have been used to study pathology and drug effects. In this section, we focus on interpreting high-throughput data using Recon 1 and its cell and tissue specific derivatives (Figure 2.iii).
A reconstructed network of core fibroblast metabolism was used to study the phenotype of physiologically normal and Leigh’s syndrome fibroblasts [51]. Utilizing time-course extracellular metabolomics, the flux rates of substrate uptake and secretion rates were calculated. The transport rates were integrated with the model to determine intracellular fluxes through randomized sampling, which were validated by existing biochemical studies. Vo et al. found that Leigh’s syndrome cells had slower and less flexible ATP metabolism. In addition, simulations of mutations in succinate cytochrome c reductase captured the observed changes in the time-course metabolomics data of Leigh’s syndrome.
Contextualization of transcription data on a host-pathogen network has also been done to determine differences in infection types of M. tuberculosis [50]. A host-pathogen network was built combining the Recon 1-derived human alveolar macrophage model and an existing network for M. tuberculosis [8] to model acute infection of M. tuberculosis within the host phagosome. Transcription data from three infection states (latent, pulmonary active, and meningeal active) was integrated with the network using the GIMME algorithm [45] and helped elucidate stark differences in active reactions in both the host and pathogen. Some of the differentially active pathways in M. tuberculosis are known to be potential drug targets and thus drug targeting for M. tuberculosis infections should be chosen by infection type.
Recon 1 has been used to interpret transcriptomic data related to insulin sensitivity. Capel et al. tracked obese women during a dietary intervention program and took biopsies of adipose tissue throughout the program [61]. Expression profiling was done on the adipocytes and macrophages in the samples and Recon 1 was used to determine significantly changed metabolites that were associated with significantly altered reactions. The authors found that expression response varies not only between macrophages and adipocytes from adipose tissue, but also during different phases of weight loss. Similarly in another study, transcriptomic data from skeletal muscle of type 2 diabetes patients was integrated with Recon 1 to determine significantly altered reactions and reporter metabolites [62]. The reporter metabolites range from TCA cycle, oxidative phosphorylation, and lipid metabolites. Several of the reported metabolites are candidates for novel biomarker candidates.
Disease heterogeneity is a common problem in medicine where diagnostics do not have enough resolution to distinguish individuals. Recon 1 can be used to understand the mechanistic link between a clinical phenotype and underlying processes. Deo et al. used the reactions and pathways in Recon 1 to interpret biofluid metabolomic profiles of oral glucose tolerance tests in normal individuals and pre-diabetics [63]. They found that the differences in the response by solute carriers can help distinguish pre-diabetics from individuals in a more mechanistic and tangible approach than previous diagnostics.
Recon 1 can also be used to study drug effects. As metabolism plays a key role in human disease, drug-target interactions in metabolic pathways are just as important in regulating human physiology. Many types of drugs inhibit metabolic enzymes and the systemic effect can be interpreted and simulated with Recon 1 and its cell and tissue-specific derivatives. Gene expression data from the NCI-60 cancer cell lines [64] was integrated with Recon 1 for multiple drug and cancer cell lines [65] using a variant of the Shlomi et al. algorithm [47]. The calculated networks were used to determine the functional link between metabolic reactions to predict new drug targets.
An important application of genome-scale modeling is to interpret generated high-throughput data in a contextualized biochemical framework. The initial studies show that Recon 1 can be used for data integration of metabolite and transcription profiling data to study human physiology, pathology, and pharmacology.
Application 4: Simulation of pathological and drug-treated states
Genome-scale reconstructions allow simulation of phenotypes through constraint-based analysis and flux balance analysis [6]. In addition, GPR associations provide a mechanistic basis connecting the genotype with the phenotype (Figure 1A). In this section, we focus on the application of simulation to predict and form a mechanistic basis for pathological and drug-treated states (Figure 2.iv).
The systemic effect of inherited inborn errors of metabolism has been studied using Recon 1. The Online Mendelian Inheritance of Man (OMIM) database [44] catalogues all known hereditary morbid single nucleotide polymorphisms (SNPs). Shlomi et al. simulated SNPs by reducing the flux of the affected reactions in Recon 1 [66]. They found that there was a systemic effect on the network, particularly in changes to the variability of the substrate uptake and secretions by Recon 1. Thus the network-based predictions can be used to determine changes in biofluid metabolite levels revealing potential biomarkers. In another study, Veeramani and Bader calculated flux correlations between reactions in Recon 1, showing that flux linked reactions had similar morbid SNP phenotypes [67]. This work is very similar to that done previously on the human mitochondria network [43].
Similar to the SNP studies and the earlier enzymopathies of erythrocytes, Gille et al. used HepatoNet 1 to study hepatic enzyme deficiencies [52]. Genes, and associated reactions, were knocked out systematically to determine the effect on liver metabolic functions. The authors found that for 80 enzymes and transporters that were computationally predicted to be essential for at least one metabolic function, clinical symptoms have been reported in literature.
In addition to SNP and enzyme deficiencies, a recent study looked at the systemic effect of imprinted genes on metabolism [68]. In particular, Sigurdsson et al. focused on maternal deletion of ATP10A and showed that there was an anabolic effect, which corroborated with the clinical phenotype.
Recon 1 has also been used to study cancer metabolism and potential pharmaceuticals. Folger et al. calculated drug targets and their synergies using the generic cancer metabolic network and Recon 1 [49]. They were able to predict selective targets that preferentially affected the cancer model, taking advantage of metabolic auxotrophies. The method was further leveraged to predict selective drug targets in renal-cell cancer, which are deficient in fumarate hydratase. A pathway for heme biosynthesis and degradation was implicated as essential in renal-cell cancer, but not normal cells. The computational prediction was followed up with experimental validation in both cell lines and mouse models [69].
In addition, the Warburg effect has been computationally studied as it relates to cancerous tumors [70]. Shlomi et al. use an enzyme solvent capacity consideration [71] alongside constraint-based modeling. Increasing in silico growth rate with the new solvent capacity constraints forces an increase in glycolytic flux, providing a possible mechanism for the Warburg effect. In addition, Recon 1 recapitulated known characteristics of cancer cells including oncogenic progression and a preference for glutamine uptake.
Genome-scale metabolic reconstructions have also been used to study neural disorders. The multi-cellular models of brain metabolism were used to study the differences between neuron types in Alzheimer’s disease [52]. Key enzyme deficiencies were simulated and Lewis et al. were able to metabolically differentiate one of the cell types (GABAergic) from the other three. Their findings corroborate with the clinical phenotype as GABAergic neurons are known to be less metabolically affected by Alzheimer’s disease.
As systems biology represents a holistic approach to studying human pathology, Recon 1 has been used to study comorbidity and epistasis. Comorbidity refers to an unrelated concomitant pathology to a primary disease. Using Recon 1 and the KEGG database as knowledge-bases, Lee et al. built a metabolic disease network that accounts for flux correlations between reactions, topology, and associated diseases [72]. Simulations showed that diseases associated within correlated reaction sets showed higher comorbidity, were more prevalent in the population, and resulted in a higher chance of death. In an another work that focuses on the topology of the metabolic network, Imielinski et al. studied epistasis by calculating reaction sets that would synergistically deactivate cellular functions [73]. In particular, knockout reaction sets were calculated for deactivating fumarase, which is known to play a role in cancer. The authors found that there are many knockout reaction sets and the reactions involved within a set can be topologically distant.
Genome-scale metabolic reconstructions have also been used to simulate drug effects. In particular, the renal metabolic model reconstructed by Chang et al. was used to study off-target drug binding and the systemic consequences [46]. The renal metabolic model was integrated with structural bioinformatics techniques that allow prediction of ligand binding to proteins in the metabolic network. Thus, a drug’s systemic effect on the kidney’s physiological function can be simulated on a genome-scale level. This allows for easy off-target drug screening before potential testing in drug trials. In another drug related study, Costa et al. built a computational approach utilizing Recon 1, tissue expression profiles, and subcellular localization information to predict genes that if controlled by drugs would elicit a phenotypic response [74]. The approach can also be used to determine morbid genes.
Constraint-based modeling and Recon 1 allows simulating normal and diseased human phenotypes. GPR associations allow for linking the genotype and phenotype in simulations to form a mechanistic basis to physiology, pathology, and pharmacology. Recon 1 has been used in many studies to determine the underlying mechanisms of a particular disease phenotype as well as in predicting the phenotype of genetic perturbations.
Future
High-throughput data generation has recently become an important focus in biology and medicine. Unfortunately, more omics data is being generated than can be properly analyzed [1]. Genome-scale metabolic reconstructions have become a central component of metabolic systems biology as they allow for a method to systematically and objectively interpret high-throughput data in a network context. In addition, genome-scale metabolic reconstructions provide a method to simulate and predict phenotypes. With the recent release of the global human metabolic reconstruction, it is now possible to study human physiology and pathology in a systems context.
Recon 1 represents the most comprehensive picture of human metabolism. The global human metabolic network was reconstructed from the human genome sequence (build 35) and was manually curated and physiologically validated. Recon 1 has been used to build cell and tissue specific derivatives as well as reconstructions for other mammals, particularly M. musculus, through homologous gene mapping. There are currently over 20 publications utilizing the model to predict or analyze human physiology, pathology, and drug-effects. Novel experimental design and validation of predictions have been slower to emerge, though the selective drug target of renal-cell cancer has been validated [69]. If the progress of study of human metabolism with Recon 1 is compared to the history of the uses of microbial models [2, 3, 75] one would expect that these are initial studies and a prolific field is likely to grow.
There are however many challenges ahead. First, reconstructions of key organisms have been periodically updated (E. coli and S. cerevisae) accounting for additional genes, reactions, and metabolites. The human genome sequence has been updated to build 37 and there are many dead end and gaps in Recon 1 that can be filled with newly available genomic and biochemical data. Due to the incredible complexity of the human genome, this is a difficult undertaking for “Recon 2” but must be completed. Second, as more and more human cell and tissue specific models are released, the pipeline for constructing them quickly and accurately must be elucidated. Manually built models would be ideal, especially for key cells and tissues, but algorithms and combinations of manual and algorithm approaches will play a role in completing reconstructions for all human cell types. Third, the reconstructions on brain metabolism and the host-pathogen interactions of M. tuberculosis highlight the importance of intercellular interactions in simulating human metabolism. As more tissue-specific models are released, they must account for the different cell types within a tissue as well as the interaction with other tissues. Finally, current constraint-based modeling techniques are mostly applied under certain physiological conditions, mostly cellular growth. Using computational techniques for studying human physiology must be implemented properly as well as new techniques need to be developed.
Acknowledgments
We would like to thank Roger Chang for suggestions and discussions on the manuscript. This work was supported through a Grant from the National Institutes of Health (GM068837).
Footnotes
Conflicts of interest
The authors declare that they have no conflict of interest.
References
- 1.Palsson B, Zengler K. The challenges of integrating multi-omic data sets. Nat Chem Biol. 2010(6):11–787. doi: 10.1038/nchembio.462. [DOI] [PubMed] [Google Scholar]
- 2.Oberhardt MA, Palsson BO, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feist AM, Palsson BO. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotech. 2008;26(6):659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5(1):93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, Herrgard MJ. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox. Nat Protocols. 2007;2(3):727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- 6.Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28(3):245–8. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards JS, Palsson BO. Systems properties of the Haemophilus influenzae Rd metabolic genotype. Journal of Biological Chemistry. 1999;274(25):17410–6. doi: 10.1074/jbc.274.25.17410. [DOI] [PubMed] [Google Scholar]
- 8.Jamshidi N, Palsson BO. Investigating the metabolic capabilities of Mycobacterium tuberculosis H37Rv using the in silico strain iNJ661 and proposing alternative drug targets. BMC Syst Biol. 2007;1:26. doi: 10.1186/1752-0509-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beste DJ, Hooper T, Stewart G, et al. GSMN-TB: a web-based genome-scale network model of Mycobacterium tuberculosis metabolism. Genome Biol. 2007;8(5):R89. doi: 10.1186/gb-2007-8-5-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker SA, Palsson BO. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two-dimensional annotation. BMC Microbiol. 2005;5(1):8. doi: 10.1186/1471-2180-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinemann M, Kummel A, Ruinatscha R, Panke S. In silico genome-scale reconstruction and validation of the Staphylococcus aureus metabolic network. Biotechnol Bioeng. 2005;92(7):850–64. doi: 10.1002/bit.20663. [DOI] [PubMed] [Google Scholar]
- 12.Lee DS, Burd H, Liu J, et al. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J Bacteriol. 2009;191(12):4015–24. doi: 10.1128/JB.01743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schilling CH, Covert MW, Famili I, Church GM, Edwards JS, Palsson BO. Genome-scale metabolic model of Helicobacter pylori 26695. Journal of Bacteriology. 2002;184(16):4582–4593. doi: 10.1128/JB.184.16.4582-4593.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thiele I, Vo TD, Price ND, Palsson B. An Expanded Metabolic Reconstruction of Helicobacter pylori (iIT341 GSM/GPR): An in silico genome-scale characterization of single and double deletion mutants. J Bacteriol. 2005;187(16):5818–5830. doi: 10.1128/JB.187.16.5818-5830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raghunathan A, Reed J, Shin S, Palsson B, Daefler S. Constraint-based analysis of metabolic capacity of Salmonella typhimurium during host-pathogen interaction. BMC Syst Biol. 2009;3:38. doi: 10.1186/1752-0509-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.AbuOun M, Suthers PF, Jones GI, et al. Genome scale reconstruction of a Salmonella metabolic model: comparison of similarity and differences with a commensal Escherichia coli strain. J Biol Chem. 2009;284(43):29480–8. doi: 10.1074/jbc.M109.005868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thiele I, Hyduke DR, Steeb B, et al. A community effort towards a knowledge-base and mathematical model of the human pathogen Salmonella Typhimurium LT2. BMC Syst Biol. 2011;5:8. doi: 10.1186/1752-0509-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feist AM, Henry CS, Reed JL, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3(121) doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biology. 2003;4(9):R54.1–R54.12. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahadevan R, Bond DR, Butler JE, et al. Characterization of Metabolism in the Fe(III)-Reducing Organism Geobacter sulfurreducens by Constraint-Based Modeling. Appl Environ Microbiol. 2006;72(2):1558–1568. doi: 10.1128/AEM.72.2.1558-1568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun J, Sayyar B, Butler JE, et al. Genome-scale constraint-based modeling of Geobacter metallireducens. BMC Syst Biol. 2009;3:15. doi: 10.1186/1752-0509-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Thiele I, Weekes D, et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009;325(5947):1544–9. doi: 10.1126/science.1174671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh YK, Palsson BO, Park SM, Schilling CH, Mahadevan R. Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data. J Biol Chem. 2007;282(39):28791–9. doi: 10.1074/jbc.M703759200. [DOI] [PubMed] [Google Scholar]
- 24.Goelzer A, Bekkal Brikci F, Martin-Verstraete I, Noirot P, Bessieres P, Aymerich S, Fromion V. Reconstruction and analysis of the genetic and metabolic regulatory networks of the central metabolism of Bacillus subtilis. BMC Syst Biol. 2008;2:20. doi: 10.1186/1752-0509-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henry CS, Zinner JF, Cohoon MP, Stevens RL. iBsu1103: a new genome-scale metabolic model of Bacillus subtilis based on SEED annotations. Genome Biol. 2009;10(6):R69. doi: 10.1186/gb-2009-10-6-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borodina I, Krabben P, Nielsen J. Genome-scale analysis of Streptomyces coelicolor A3(2) metabolism. Genome Res. 2005;15(6):820–9. doi: 10.1101/gr.3364705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alam MT, Merlo ME, Hodgson DA, Wellington EM, Takano E, Breitling R. Metabolic modeling and analysis of the metabolic switch in Streptomyces coelicolor. BMC Genomics. 2010;11:202. doi: 10.1186/1471-2164-11-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forster J, Famili I, Fu PC, Palsson BO, Nielsen J. Genome-Scale Reconstruction of the Saccharomyces cerevisiae Metabolic Network. Genome Research. 2003;13(2):244–53. doi: 10.1101/gr.234503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mo ML, Palsson BO, Herrgard MJ. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst Biol. 2009;3:37. doi: 10.1186/1752-0509-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duarte NC, Becker SA, Jamshidi N, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci U S A. 2007;104(6):1777–82. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(Database issue):D355–60. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–8. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palsson BO. Systems biology: properties of reconstructed networks. New York: Cambridge University Press; 2006. [Google Scholar]
- 34.Herrgard MJ, Swainston N, Dobson P, et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat Biotechnol. 2008;26(10):1155–60. doi: 10.1038/nbt1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiele I, Palsson BO. Reconstruction annotation jamborees: a community approach to systems biology. Mol Syst Biol. 2010;6:361. doi: 10.1038/msb.2010.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rapoport TA, Heinrich R, Jacobasch G, Rapoport S. A linear steady-state treatment of enzymatic chains. A mathematical model of glycolysis of human erythrocytes. Eur J Biochem. 1974;42(1):107–20. doi: 10.1111/j.1432-1033.1974.tb03320.x. [DOI] [PubMed] [Google Scholar]
- 37.Brumen M, Heinrich R. A metabolic osmotic model of human erythrocytes. Biosystems. 1984;17(2):155–69. doi: 10.1016/0303-2647(84)90006-6. [DOI] [PubMed] [Google Scholar]
- 38.Joshi A, Palsson BO. Metabolic dynamics in the human red cell. Part I--A comprehensive kinetic model. Journal of Theoretical Biology. 1989;141(4):515–28. doi: 10.1016/s0022-5193(89)80233-4. [DOI] [PubMed] [Google Scholar]
- 39.Mulquiney PJ, Bubb WA, Kuchel PW. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: in vivo kinetic characterization of 2,3-bisphosphoglycerate synthase/phosphatase using 13C and 31P NMR. Biochem J. 1999;342(Pt 3):567–80. [PMC free article] [PubMed] [Google Scholar]
- 40.Price ND, Schellenberger J, Palsson BO. Uniform Sampling of Steady State Flux Spaces: Means to Design Experiments and to Interpret Enzymopathies. Biophysical Journal. 2004;87(4):2172–86. doi: 10.1529/biophysj.104.043000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vo TD, Greenberg HJ, Palsson BO. Reconstruction and functional characterization of the human mitochondrial metabolic network based on proteomic and biochemical data. J Biol Chem. 2004;279(38):39532–40. doi: 10.1074/jbc.M403782200. [DOI] [PubMed] [Google Scholar]
- 42.Thiele I, Price ND, Vo TD, Palsson BO. Candidate metabolic network states in human mitochondria: Impact of diabetes, ischemia, and diet. J Biol Chem. 2005;280(12):11683–95. doi: 10.1074/jbc.M409072200. [DOI] [PubMed] [Google Scholar]
- 43.Jamshidi N, Palsson BO. Systems biology of SNPs. Mol Syst Biol. 2006;2:38. doi: 10.1038/msb4100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM) Nucleic Acids Res. 2009;37(Database issue):D793–6. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker SA, Palsson BO. Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol. 2008;4(5):e1000082. doi: 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang RL, Xie L, Bourne PE, Palsson BO. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput Biol. 2010;6(9):e1000938. doi: 10.1371/journal.pcbi.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shlomi T, Cabili MN, Herrgard MJ, Palsson BO, Ruppin E. Network-based prediction of human tissue-specific metabolism. Nat Biotechnol. 2008;26(9):1003–10. doi: 10.1038/nbt.1487. [DOI] [PubMed] [Google Scholar]
- 48.Jerby L, Shlomi T, Ruppin E. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol Syst Biol. 2010;6:401. doi: 10.1038/msb.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Folger O, Jerby L, Frezza C, Gottlieb E, Rupin E, Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7:501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bordbar A, Lewis NE, Schellenberger J, Palsson BO, Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol. 2010;6:422. doi: 10.1038/msb.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vo TD, Paul Lee WN, Palsson BO. Systems analysis of energy metabolism elucidates the affected respiratory chain complex in Leigh’s syndrome. Mol Genet Metab. 2007;91(1):15–22. doi: 10.1016/j.ymgme.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 52.Gille C, Bolling C, Hoppe A, et al. HepatoNet1: a comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol Syst Biol. 2010;6:411. doi: 10.1038/msb.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis NE, Schramm G, Bordbar A, et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat Biotechnol. 2010;28(12):1279–85. doi: 10.1038/nbt.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seo S, Lewin HA. Reconstruction of metabolic pathways for the cattle genome. BMC Syst Biol. 2009;3:33. doi: 10.1186/1752-0509-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheikh K, Forster J, Nielsen LK. Modeling hybridoma cell metabolism using a generic genome-scale metabolic model of Mus musculus. Biotechnol Prog. 2005;21(1):112–21. doi: 10.1021/bp0498138. [DOI] [PubMed] [Google Scholar]
- 56.Evsikov AV, Dolan ME, Genrich MP, Patek E, Bult CJ. MouseCyc: a curated biochemical pathways database for the laboratory mouse. Genome Biol. 2009;10(8):R84. doi: 10.1186/gb-2009-10-8-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quek LE, Nielsen LK. On the reconstruction of the Mus musculus genome-scale metabolic network model. Genome Inform. 2008;21:89–100. [PubMed] [Google Scholar]
- 58.Selvarasu S, Karimi IA, Ghim GH, Lee DY. Genome-scale modeling and in silico analysis of mouse cell metabolic network. Mol Biosyst. 2010;6(1):152–61. doi: 10.1039/b912865d. [DOI] [PubMed] [Google Scholar]
- 59.Sigurdsson MI, Jamshidi N, Steingrimsson E, Thiele I, Palsson BO. A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst Biol. 2010;4:140. doi: 10.1186/1752-0509-4-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bekaert M, Conant GC. Copy number alterations among mammalian enzymes cluster in the metabolic network. Mol Biol Evol. 2011;28(2):1111–21. doi: 10.1093/molbev/msq296. [DOI] [PubMed] [Google Scholar]
- 61.Capel F, Klimcakova E, Viguerie N, et al. Macrophages and adipocytes in human obesity: adipose tissue gene expression and insulin sensitivity during calorie restriction and weight stabilization. Diabetes. 2009;58(7):1558–67. doi: 10.2337/db09-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zelezniak A, Pers TH, Soares S, Patti ME, Patil KR. Metabolic network topology reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput Biol. 2010;6(4):e1000729. doi: 10.1371/journal.pcbi.1000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deo RC, Hunter L, Lewis GD, et al. Interpreting metabolomic profiles using unbiased pathway models. PLoS Comput Biol. 2010;6(2):e1000692. doi: 10.1371/journal.pcbi.1000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24(3):227–35. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 65.Li L, Zhou X, Ching WK, Wang P. Predicting enzyme targets for cancer drugs by profiling human metabolic reactions in NCI-60 cell lines. BMC Bioinformatics. 2010;11:501. doi: 10.1186/1471-2105-11-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shlomi T, Cabili MN, Ruppin E. Predicting metabolic biomarkers of human inborn errors of metabolism. Mol Syst Biol. 2009;5:263. doi: 10.1038/msb.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Veeramani B, Bader JS. Metabolic flux correlations, genetic interactions, and disease. J Comput Biol. 2009;16(2):291–302. doi: 10.1089/cmb.2008.14TT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sigurdsson MI, Jamshidi N, Jonsson JJ, Palsson BO. Genome-scale network analysis of imprinted human metabolic genes. Epigenetics. 2009;4(1):43–6. doi: 10.4161/epi.4.1.7603. [DOI] [PubMed] [Google Scholar]
- 69.Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni M, Chaneton B, Adam J, Hedley A, Kalna G, Tomlinson IPM, Pollard PJ, Watson DG, Deberardinis RJ, Shlomi T, Ruppin E, Gottlieb E. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225–8. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 70.Shlomi T, Benyamini T, Gottlieb E, Sharan R, Ruppin E. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput Biol. 2011;7(3):e1002018. doi: 10.1371/journal.pcbi.1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vazquez A, Beg QK, Demenezes MA, et al. Impact of the solvent capacity constraint on E. coli metabolism. BMC Syst Biol. 2008;2:7. doi: 10.1186/1752-0509-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee DS, Park J, Kay KA, Christakis NA, Oltvai ZN, Barabasi AL. The implications of human metabolic network topology for disease comorbidity. Proc Natl Acad Sci U S A. 2008;105(29):9880–5. doi: 10.1073/pnas.0802208105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Imielinski M, Belta C. Deep epistasis in human metabolism. Chaos. 2010;20(2):026104. doi: 10.1063/1.3456056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Costa PR, Acencio ML, Lemke N. A machine learning approach for genome-wide prediction of morbid and druggable human genes based on systems-level data. BMC Genomics. 2010;11(Suppl 5):S9. doi: 10.1186/1471-2164-11-S5-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mahadevan R, Palsson BO, Lovley DR. In situ to in silico and back: elucidating the physiology and ecology of Geobacter spp. using genome-scale modelling. Nat Rev Microbiol. 2011;9(1):39–50. doi: 10.1038/nrmicro2456. [DOI] [PubMed] [Google Scholar]