

INTRODUCTION
Writing about a biological subject based on one species can limit the scope of the discussion, but in the case of DNA methylation and Arabidopsis, the restriction is entirely appropriate. Unlike many other popular model organisms, Arabidopsis has retained and embellished a multi-layered methylation system that contributes to gene and transposon silencing, imprinting, and genome stability. Many of the findings from Arabidopsis are applicable to other eukaryotes. The field has relied in large part on forward and reverse genetic screens and the study of methylation and its consequences at one or a few loci. These sorts of experiments are still providing novel insights, especially when combined with the natural genetic variation present within Arabidopsis thaliana. Additionally, the field is increasingly moving towards genomics to understand the interplay between methylation, demethylation, chromatin state, and gene expression.
Cytosine can be methylated at the carbon five position, and in plants this can occur on any cytosine regardless of the sequence context (Figure 1). In general, 5-methylcytosine is associated with transcriptional silencing. How this is achieved is still not well understood. Methylation can block transcription factor binding and prevent transcription or it can recruit chromatin-modifying complexes that mediate silencing through changes to the underlying histones. In this chapter we provide an introduction to the mechanisms of DNA methylation and demethylation and consider the biological relevance of these activities at different types of sequences. First, we provide a brief background on how methylation status is empirically determined, then address in more detail the enzymes and systems that add and remove it, and the interaction of DNA methylation with chromatin. Finally, we consider possible regulatory roles of DNA methylation in Arabidopsis.
Figure 1.
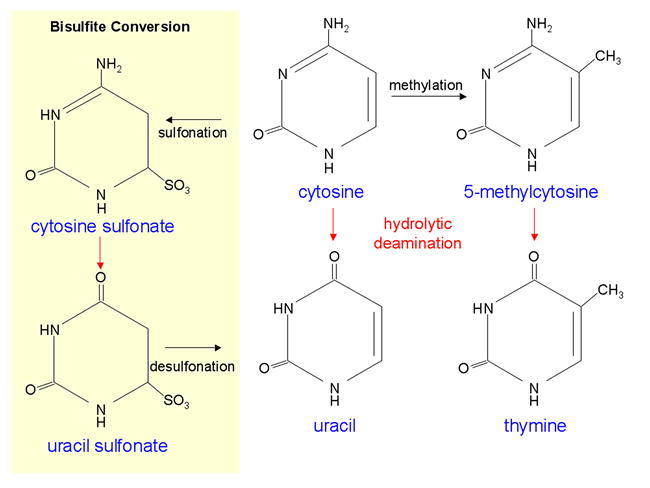
Chemical structure of cytosine and 5-methylcytosine.
Methyltransferases add a methyl group onto the 5 carbon of cytosine to generate 5-methylcytosine. Hydrolytic deamination converts cytosine to uracil and 5-methylcytosine to thymine (5-methyluracil). Treatment of single-stranded DNA with sodium bisulfite leads to sulfonation of cytosine to form cytosine sulfonate, which is then deaminated to uracil sulfonate and desulfonated by alkali treatment to uracil. The methyl group protects 5-methyl-cytosine from sulfonation.
Measuring Methylation
If you're interested in methylation at a certain sequence, how do you go about measuring it? Several methods of varying utility have been devised to determine the methylation status of specific sequences and entire genomes; most rely on chemical differences between 5-methylcytosine and cytosine or on the ability of restriction enzymes and antibodies to discriminate between the two.
Several restriction enzymes are inhibited by methylation in CG, CHG, or CHH (H = A, T, or C) sequence contexts. Genomic DNA can be digested with a methylation sensitive restriction enzyme and the sequence of interest probed by Southern blot, with the amount of methylation determined by the completeness of digestion. The major disadvantage to this method is that the methylation status of only a few nucleotides can be queried at once. Methylation is often found in clusters in Arabidopsis. Although clusters of methylation are highly heritable, differences do arise at individual nucleotides within those regions even in closely related lines (Tran et al., 2005a). Given the potential for site-to-site variability in DNA methylation, drawing any conclusions about methylation based on one or a few enzyme sites can be tenuous.
The McrBC enzyme is also of use; it imprecisely cleaves methylated DNA if there are methylated sites containing a methyl-cytosine preceded by a purine within 40 bp to 3 kb of one another (Sutherland et al., 1992). After genomic DNA is digested with McrBC, regions of interest can be amplified by PCR—the more highly methylated a sequence is the less it will be amplified (Vaughn et al., 2007). An advantage of both of the methods that employ methylation-sensitive enzymes is that all molecules can be assayed simultaneously.
Affinity purification is an attractive route for assaying methylation. A commercially available 5-methylcytosine antibody can be used to pull down the methylated fraction of the genome. Particular sequences can then be analyzed by comparing PCR amplification between input and pull-down fractions or between genotypes. The methyl-binding domain (MBD) from mammals has been used in a similar manner, but it only binds methylated CG sites.
Bisulfite sequencing provides the most detailed data about methylation of particular cytosines in a sequence. Treatment of restriction digested or sheared single-stranded DNA with sodium bisulfite followed by desulfonation converts cytosine to uracil but leaves 5-methylcytosine intact (Figure 1) (Frommer et al., 1992; Clark et al., 1994). A region of interest, usually less than 700 base pairs, is then amplified in a strand-specific manner by PCR; after cloning and sequencing of the PCR product the original cytosines will be read as Ts, and 5-methylcytosines as Cs. Comparison to the reference sequence allows determination of the methylation status of each cytosine. Although bisulfite sequencing is the gold standard for methylation analysis, biases can be introduced at several points in the procedure, including PCR amplification and cloning of the PCR products (Warnecke et al., 2002). Sequencing the PCR product directly using pyrosequencing can eliminate one aspect of this bias, as long as information about individual molecules is not required (Tost and Gut, 2007). Because bisulfite treatment produces DNA strands that are no longer complementary, two separate PCR reactions must be performed if methylation information for each strand is desired. This information might be particularly relevant in plants since asymmetric cytosines are methylated and thus the number of potentially methylated sites differs on each strand. Hairpin bisulfite PCR cleverly solves this problem by joining the complementary strands of DNA using a hairpin linker, allowing them to be assessed simultaneously (Laird et al., 2004), but has so far not been applied in plant studies.
Many of the above methods can be, or have already been, extended to whole genome methylation analysis if combined with microarrays or high-throughput sequencing (Yazaki et al., 2007; Zilberman and Henikoff, 2007). As the technology has improved, the definition of what constitutes “genome-wide” and “high-resolution” methylation mapping has also changed. Initial efforts to globally map methylation in Arabidopsis relied on methylation-sensitive enzymes and microarrays of limited genomic coverage (Tompa et al., 2002; Lippman et al., 2004; Tran et al., 2005a; Tran et al., 2005b), but provided important information about methylation location at a gross level. Methylation has also been mapped by immunoprecipitating the methylated fraction of the genome with a 5-methylcytosine antibody or MBD and hybridizing the bound or unbound DNA to tiling microarrays. This has allowed detailed examination of methylation patterns across the genome and within specific regions (Zhang et al., 2006; Penterman et al., 2007c; Zilberman et al., 2007). Methylation mapping in methyltransferase, demethylase, and RNA interference mutants is giving us a broader understanding of how methylation patterns are established, maintained, and potentially changed. Recently, methylation has been mapped genome-wide at single base resolution by bisulfite-treating DNA, sequencing it using Solexa high-throughput sequencing technology, and aligning the sequences back to the genome (Cokus et al., 2008). This has provided a level of detail unprecedented for any organism. Another new method, which requires only small amounts of DNA, involves hybridizing bisulfite-treated DNA to tiling microarrays (Reinders et al., 2008). It is expected that the use of high-throughput sequencing or microarrays in combination with affinity purification or bisulfite treatment will continue to further refine the Arabidopsis methylation map in different genotypes, tissues, or conditions.
Once a methylation pattern is determined, deriving meaning from it can be challenging. Often differences are compared between wild type and mutant plants or between different conditions. Although differences might be statistically significant, biological significance is harder to assess. As will become clear in the following sections, large questions still remain as to how methylation exerts the influence that it does, or if methylation at particular sequences is relevant to biological function.
Methylation Location
The Arabidopsis genome contains methylation at 24% of CG sites, 6.7% of CHG and 1.7% of CHH (Cokus et al., 2008). How is this methylation distributed? Staining an Arabidopsis interphase nucleus with the DNA dye DAPI reveals several bright spots referred to as chromocenters. Immunostaining with a 5-methylcytosine antibody results in a strikingly similar distribution (Soppe et al., 2002; Fransz et al., 2003; Tariq et al., 2003). On each chromosome, chromocenters contain repetitive DNA sequences consisting of the 178 base pair centromere tandem repeat sequences flanked by pericentromeric heterochromatin, which is largely composed of repetitive elements, including DNA transposons and retroelements. Each chromocenter encompasses several megabases of DNA. On chromosomes 2 and 4 additional chromocenters are formed from the nucleolar organizing regions (NOR), which consist of arrays of repeated 45S rDNA genes (Pikaard, 2002). It is clear that the preferred location for DNA methylation is at repetitive DNA sequences (Figure 2). This holds true outside of chromocenters, as most euchromatic transposons are also highly methylated, and within chromocenters, where genes surrounded by methylated transposons can themselves remain free of methylation (Lippman et al., 2004; Zhang et al., 2006; Zilberman et al., 2007).
Figure 2.
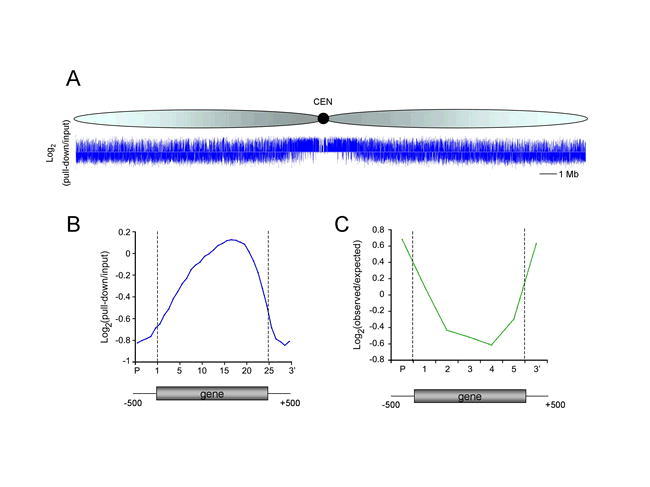
Distribution of DNA methylation in Arabidopsis.
A) Methylation profiling of Arabidopsis chromosome 1 indicates that methylation is concentrated around the centromeres (CEN) but is prolific in chromosome arms as well. Methylated DNA was isolated by immunoprecipitation with a 5-methylcytosine antibody, labeled with Cy3, and then hybridized to tiling microarrays along with Cy5-labeled input DNA. The log2 ratio of pull-down to input DNA is plotted.
B) Methylation is distributed away from gene ends. Arabidopsis genes were divided into 25 equal segments and the average log2 of methyl-IP DNA to input DNA plotted. Figure adapted from Penterman et al., (2007). Copyright © 2007 by the National Academy of Sciences.
C) Methylation accumulates in the 5′ and 3′ ends of genes in DNA glycosylase mutants. Genes hypermethylated in ros1 dml2 dml3 mutants (n=85) were divided into 5 equal segments and the log2 ratio of observed hypermethylated segments to those expected by chance was plotted. Figure adapted from Penterman et al., (2007). Copyright © 2007 by the National Academy of Sciences.
Transposable elements (TEs) invade genomes and increase in copy number, with strong potential for detriment to the host. All organisms have adopted mechanisms to keep transposable elements silent, including RNA-based chromatin silencing, histone modifications, DNA methylation, or a combination thereof (Slotkin and Martienssen, 2007). The bias of methylation toward repetitive DNA sequences suggests that one of methylation's primary functions is to silence the transcription of transposable elements (Zilberman and Henikoff, 2004; Gehring and Henikoff, 2007). In Arabidopsis, transposons are generally methylated throughout their length at cytosines in all sequence contexts, although distinct patterns do emerge at individual loci (Lippman et al., 2003; Lippman et al., 2004; Zhang et al., 2006; Zilberman et al., 2007).
Studies of individual genes and of methylation patterns genome-wide have revealed that genic sequences also contain considerable amounts of methylation, although mainly in the CG sequence context (Tran et al., 2005a; Zhang et al., 2006; Zilberman et al., 2006; Vaughn et al., 2007; Cokus et al., 2008). It is estimated that at least a third of genes contain some methylation within their coding sequences. Although coding region methylation alone can lead to gene silencing (Hohn et al., 1996; Chawla et al., 2007), most of these genes are transcribed very well and are distributed across all gene ontology categories. Clearly, methylation is not acting to silence these genes – so what is its impact there? There is an intimate relationship between methylation and transcription. Unlike in transposons, methylation shows a distinct distribution within genes (Figure 2). Methylation is depleted at both the 5′ and 3′ ends of coding sequences (Zhang et al., 2006; Zilberman et al., 2006; Vaughn et al., 2007). The distribution of methylation within genes is the inverse of the distribution of RNA polymerase II distribution (Zilberman et al., 2007). This suggests that methylation at the 5′ and 3′ ends of genes could be inhibitory to transcription, potentially interfering with initiation or termination. Indeed, genes that are methylated in their promoter and 5′ coding sequences have some of the lowest transcription rates in the genome. Moderately transcribed genes are more likely to be methylated than those with low or high expression (Zhang et al., 2006; Zilberman et al., 2007). This suggests that the process of transcription itself, if it is not too slow, or not too fast, can promote methylation. One interesting, but unexplained, observation from these studies is that genes are more likely to be methylated the closer they are to centromeres, although this does not depend on how close a gene is to a transposable element (Zilberman et al., 2007). This suggests there might be a chromosome-level organization of DNA methylation.
DNA Methyltransferases: Adding Methylation
The Arabidopsis enzymes that add a methyl group to cytosine were discovered through genetic screens for mutations that reduced methylation at centromeres, relieved silencing of endogenous genes or transgenes, or by sequence similarity to methyltransferases discovered in other organisms. These genes have been identified for a decade and several excellent reviews are available (Rangwala and Richards, 2004; Chan et al., 2005; Goll and Bestor, 2005). All known cytosine 5-methyltransferases belong to a single family with several subfamilies. There are three subfamilies of DNA methyltransferases in Arabidopsis: CG maintenance methyltransferases, chromomethylases, and the de novo methyltransferases. Multiple genes exist for each enzyme class, but only one enzyme of each type appears to be active; the other genes are either expressed at low levels or contain stop codons in various backgrounds, and none have been recovered in mutant screens. The crystal structures of bacterial cytosine methyltransferases in complex with DNA have been solved (Suck, 1994). The cytosine base is flipped out of the DNA helix into the enzyme active site, where an S-adenosyl-L-methionine (SAM) cofactor donates a methyl group to the carbon 5 position. Disruptions of enzymes that affect the SAM cycle reduce DNA methylation in Arabidopsis (Tanaka et al., 1997; Rocha et al., 2005; Mull et al., 2006; Jordan et al., 2007).
METHYLTRANSFERASE1 (MET1) is the CG maintenance methyltransferase in Arabidopsis. This designation is based on sequence similarity to Dnmt1, the orthologous mammalian maintenance methyltransferase, and on the effect mutations in the gene have on DNA methylation. Like Dnmt1, MET1 contains a long N-terminal domain and a C-terminal catalytic domain. The N-terminal region contains two BAH domains (bromo-adjacent homology), which might interact with other proteins (Finnegan and Kovac, 2000; Kankel et al., 2003; Goll and Bestor, 2005). Dnmt1 methylates newly synthesized DNA strands after DNA replication, using the CG methylation pattern of the parent strand as a template (Goll and Bestor, 2005).
Methylation has been mapped globally in met1 mutants (Zhang et al., 2006; Cokus et al., 2008). More than half of the regions that are methylated in wild type are lost in met1. The effect is strongest at genes, which makes sense because they are primarily methylated in the CG context. At some repetitive sequences significant amounts of CHG and CHH methylation are also lost. Additionally, new CHG and CHH methylation (hypermethylation) appears at previously unmethylated sequences and increases as met1 plants are inbred (Jacobsen et al., 2000; Mathieu et al., 2007). Many TEs become transcriptionally active in a met1 mutant (Kato et al., 2003; Lippman et al., 2003; Zhang et al., 2006; Zilberman et al., 2006) as do sequences that are currently unannotated, which could either represent non-coding RNAs or as yet unidentified TEs (Zhang et al., 2006).
Mutations in MET1 and antisense-directed MET1 silencing cause varied and dramatic phenotypes from gametogenesis onwards. MET1 is required to maintain methylation patterns during the haploid gametophyte stage of the plant life cycle (Saze et al., 2003). In the sporophyte, met1 phenotypes include abnormal embryo patterning, narrow leaves, homeotic transformations of floral organs, altered flowering time, and reduced fertility (Finnegan et al., 1996; Kankel et al., 2003; Saze et al., 2003; Xiao et al., 2006; Mathieu et al., 2007). Some of the affected genes underlying these phenotypes have been identified. For example, met1 plants are late flowering in many backgrounds because the homeodomain gene FWA, which is normally promoter- methylated and silent during sporophyte development, becomes hypomethylated and expressed (Soppe et al., 2000; Kankel et al., 2003). Hypermethylation of the SUPERMAN and AGAMOUS genes contributes to some of the floral phenotypes (Jacobsen et al., 2000). The reasons for many of the other phenotypes remain unknown.
The other maintenance methyltransferase in Arabidopsis is CMT3 (CHROMOMETHYLASE3), a methyltransferase containing a chromodomain. This particular family of enzymes is unique to plants (Henikoff and Comai, 1998). Chromodomains bind to methylated lysines in histone tails. CMT3 also has a single BAH domain in the N-terminal region. CMT3 maintains methylation in the CHG sequence context, and genetic and molecular evidence indicates that it does so in close concert with histone H3 lysine 9 (H3K9) methyltransferases (Bartee et al., 2001; Lindroth et al., 2001; Jackson et al., 2002). In vitro, CMT3 binds the N-terminal tail of histone H3 when it is trimethylated at lysine 9 and trimethylated at lysine 27, suggesting a dependence of CHG methylation on histone methylation (Lindroth et al., 2004). However, H3 K9 trimethylation is not a common histone modification in Arabidopsis (Jackson et al., 2004; Johnson et al., 2004), so it remains to be seen if this finding is relevant in vivo. cmt3 single mutants do not have any morphological phenotypes, but when combined with a null met1 allele the outcome is severe (Xiao et al., 2006; Zhang and Jacobsen, 2006). The additional loss of CHG methylation in met1 cmt3 mutants might push the genome over a methylation threshold such that the remaining methylation, in whatever sequence context, is insufficient to accomplish its functions.
The de novo methyltransferases, DRMs (DOMAINS REARRANGED METHYLTRANSFERASES), were identified based on homology to the mammalian de novo methyltransferases Dnmt3a and Dnmt3b, although their catalytic domains are in a different order along the protein sequence (Cao and Jacobsen, 2002). The enzymes also have a N-terminal ubiquitin-associated domain. DRM2 appears to be the only functional enzyme in Arabidopsis. De novo methylation can be monitored by assaying CHH methylation, which must be maintained after DNA replication by constant targeting since it is not symmetric between complementary DNA strands. A genetic distinction can be made between establishing methylation at previously unmethylated sequences and actively maintaining asymmetric methylation. DRM2 is required for establishing methylation at all loci examined. For maintenance, DRM2 has locus-specific effects on asymmetric methylation – at some loci it is the only enzyme required, whereas at others CMT3 also acts as a de novo methyltransferase (Cao and Jacobsen, 2002). In in vitro assays, tobacco DRM preferentially methylates CHH and CHG sites, with far less activity at CG sites, and prefers unmethylated DNA over hemimethylated DNA (Wada et al., 2003). Loss of drm2 and cmt3 has little overall effect on the distribution of methylation genome-wide (Zhang et al., 2006). This is because non-CG methylation is always found in the vicinity of CG methylation (unless CG sites are depleted from the sequence) and meCG, which is the most abundant context for cytosine methylation, does not change in drm2 cmt3. drm2 cmt3 mutant plants, but not drm2 mutants, also display multiple phenotypes including small size, twisted leaves, and reduced fertility (Cao and Jacobsen, 2002). Affected genes underlying these phenotypes have not been identified.
MET1 might also have de novo methyltransferase activity. Dnmt1, the mammalian MET1 homolog, was originally isolated based on de novo methyltransferase activity (Goll and Bestor, 2005). De novo methylation of CG sites is impaired in met1 mutants (Aufsatz et al., 2004). Additionally, drm2 cmt3 mutants retain some CHG and CHH methylation, particularly in pericentromeric heterochromatin (Cokus et al., 2008). This suggests that another enzyme(s) has de novo methylation activity.
Targeting Methylation to Specific Sequences
Now that we've introduced the enzymes that methylate DNA, we'll turn to how these enzymes are targeted to their substrates. De novo DNA methylation is guided by small interfering RNAs (siR-NAs) in a process termed RNA-directed DNA methylation (RdDM). This topic has been extensively reviewed and is the subject of much current research (Bender, 2004; Matzke and Birchler, 2005). Here we discuss only the basics.
Arabidopsis contains multiple small RNA pathways that are involved in pathogen defense, development, stress response, and the establishment of DNA methylation (Vaucheret, 2006; Chapman and Carrington, 2007). Each small RNA pathway relies on the function of an Argonaute (AGO), Dicer (DCL) and, in some instances, an RNA-dependent RNA polymerase (RdRP). The molecular functions of these enzymes have been elucidated in a number of organisms. Dicer RNA endonuclease enzymes cleave double-stranded RNAs (dsRNAs) into 21–25 nt pieces. One strand of the RNA is loaded into Argonaute complexes and then, depending on the pathway, can serve as a guide for transcript cleavage via Argonaute slicer activity or for de novo methylation. If dsRNAs do not arise naturally (e.g. from miRNA foldback precursors or overlapping sense-antisense transcripts) they can be synthesized by RNA-dependent RNA polymerases from single stranded RNA templates. Arabidopsis contains multiple genes for each of these enzymes. Many have discrete functions in particular small RNA pathways, although overlaps and substitutions between pathways do occur.
For most de novo DNA methylation at repetitive sequences, the primary players are AGO4, RDR2 (the RdRP), and DCL3. Also essential is RNA polymerase IV, a plant-specific addition to the familiar DNA-dependent RNA polymerase I, II, and III family of genes, and DRD1, a SNF2-like chromatin remodeling enzyme (Kanno et al., 2004; Herr et al., 2005; Kanno et al., 2005; Onodera et al., 2005; Pontier et al., 2005). Mutation in any of these genes prevents de novo methylation establishment at a naïve FWA transgene and causes loss of non-CG methylation at some loci where it is being actively maintained (Chan et al., 2004; Chan et al., 2006; Huettel et al., 2006). There are two large subunits for Pol IV, NRPD1a and NRPD1b, and one small subunit, NRPD2. Genetic and cell biology evidence indicates that NRPD1a and NRPD1b have distinct functions in the siRNA biogenesis and the RdDM pathway. The current model for how methylation is established is as follows: aberrant transcripts are produced from endogenous repeat loci, including transposons, in a process dependent on NRPD1a and NRPD2. RDR2 converts these transcripts into dsRNA, which DCL3 processes into 24 nt siRNAs. The siRNA is then loaded into a complex containing AGO4 and NRPD1b, which act with DRD1 and DRM2 to target the cognate DNA for methylation by DRM2 (Figure 3). NRPD1b has been shown to directly bind AGO4 via an extended C-terminal domain distinct from that found in Pol I, II, III or NRPD1a (Li et al., 2006; El-Shami et al., 2007). AGO4 can also slice transcripts generated from the target locus, producing secondary siRNAs that will continue to feed into the pathway, although this catalytic activity is not always required to maintain methylation (Qi et al., 2006).
Figure 3.
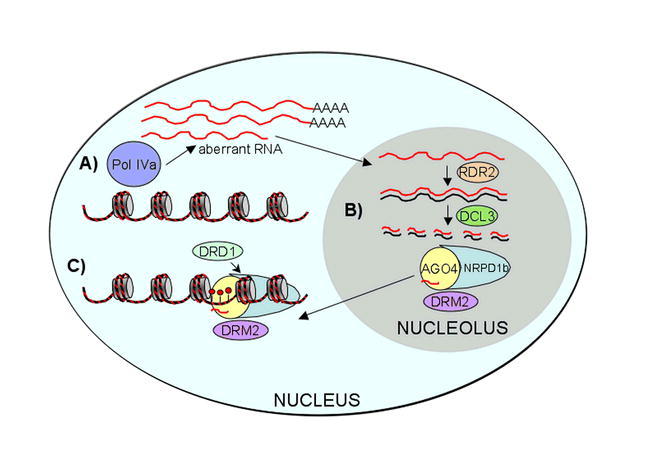
RNA directs DNA methylation.
A) Transcription of a locus gives rise to Pol IVa-dependent aberrant RNA, which is shuttled to the nucleolus and is
B) converted into double-stranded RNA by the RNA-dependent RNA polymerase RDR2. DICER-LIKE 3 (DCL3) processes the dsRNA into 24 nt small interfering RNAs (siRNAs), which are loaded into AGO4 complexes that also contain NRPD1b.
C) The siRNA complex guides the de novo DNA methyltransferase DRM2. The chromatin remodeller DRD1 is also required. (Red lollipops = 5-methyl-cytosine, short squiggly lines = siRNAs).
Most, but not all, of the heterochromatic small RNAs generated from repeats are of the 24 nt size class and are dependent on RDR2 and NRPD1a (Xie et al., 2004; Lu et al., 2006; Zhang et al., 2007). For a transgene that produces a hairpin transcript, neither NRPD1a nor RDR2 is required (Kanno et al., 2005). Deep sequencing of small RNA populations in nrpd1a/1b and rdr2 mutants demonstrated that a class of siRNAs corresponding to hairpin RNAs persist, presumably because the hairpin generates a dsRNA substrate that can be directly diced (Zhang et al., 2007). While DCL3 is responsible for processing many of the methylation-directing siRNAs, DCL2 and DCL4, which generate slightly smaller siRNAs, serve functions that are redundant with DCL3 for targeting methylation (Henderson et al., 2006). For example, dcl2 dcl3 dcl4 triple mutants establish de novo methylation at naïve sequences more poorly than dcl3 mutants alone and, at some loci, are required to maintain a full level of non-CG methylation (Henderson et al., 2006). Additionally, AGO6 seems to act redundantly with AGO4 at transgene and endogenous loci (Zheng et al., 2007). Ultimately, the mechanics of how the process works seems to depend on the type of locus being examined. All repeats are not created equal: inverted repeats, tandem repeats, and dispersed repeats can have different genetic requirements for establishing and maintaining methylation.
Recently, progress has been in made in understanding where these processes take place within the cell (Figure 3). Unlike small RNAs that direct post-transcriptional gene silencing, which are found in the cytoplasm, heterochromatic 24 nt siRNAs reside in the nucleus. Many concentrate in the nucleolus (Pontes et al., 2006), the nuclear organelle responsible for the production of rRNA and ribosome assembly. Also found in the nucleolus are RDR2, DCL3, AGO4, NRPD1b, and DRM2, suggesting that siRNA processing and complex loading takes place there (Li et al., 2006; Pontes et al., 2006; Li et al., 2008a). Disruption of NRPD1a causes mislocalization of RDR2, DCL3, and NRPD1b, supporting its upstream role in the RdDM pathway (Pontes et al., 2006). AGO4 colocalizes with Cajal bodies, which are distinct nucleolar bodies involved in ribonucleoprotein (RNP) complex processing (Li et al., 2006), and with smaller AB bodies, which lie adjacent to the NORs (Li et al., 2008a). NRPD1b and DRM2 localize with AB bodies, but not Cajal bodies (Li et al., 2008a). Unlike AGO4, localization of NRPD1b to the nucleolus is actually only observed in a small percentage of nuclei (6%) (Li et al., 2006; Li et al., 2008a). It would be interesting to determine under what conditions this occurs or if these nuclei arise from a particular cell type. AGO4, NRPD1b, and DRM2 are also found more diffusely throughout the nucleus, presumably at target DNA sequences (Li et al., 2006; Pontes et al., 2006; Li et al., 2008a).
It has been proposed that Pol IVa transcribes heterochromatic methylated DNA into RNA, which is then made into dsRNA by RDR2. However, polymerase activity has not yet been demonstrated for RNA Pol IV. No DNA-dependent RNA polymerase activity is associated with the smaller subunit NRPD2 (Onodera et al, 2005). An RNA template has also been suggested as a substrate for Pol IV (Vaughn and Martienssen, 2005; Pontes et al., 2006). A conventional DNA-dependent RNA polymerase might transcribe methylated DNA into RNA. Any aberrant transcripts arising from this transcription could then become a template for Pol IVa and the resulting RNA a template for RDR2 (Pontes et al., 2006). Transcripts from methylated DNA might be more likely to be aberrant because methylated DNA can impede transcript elongation (Lorincz et al., 2004), thus reinforcing methylation at sequences that are already methylated. This begs the question of what makes an RNA aberrant. Another RdRP (RDR6) makes dsRNA from transgene RNA templates that are truncated and without poly (A) tails (Luo and Chen, 2007) or uncapped (Gazzani et al., 2004). The template for RDR2 is unknown.
An open question remains as to how siRNAs actually guide methylation. Does the siRNA bind to complementary DNA, or does it bind to a nascent RNA transcript? Data from an instance of microRNA (miRNA)-directed DNA methylation suggests that the latter is the more likely possibility (Bao et al., 2004). PHABULOSA (PHB) expression is down-regulated by a miRNA that is complementary to the spliced PHB transcript. This is correlated with methylation of the PHB gene downstream of the miRNA binding site, which only occurs when the miRNA binding site remains intact. This suggests that the miRNA base-pairs to the spliced nascent PHB transcript and directs DNA methylation, through unknown means, to the template chromosome (Bao et al., 2004).
The mechanisms of methylation targeting described so far apply to CG and non-CG methylation at repeated sequences. What about the methylation in genes? Genes can contain repeats, which can also be methylated efficiently by these systems (Chan et al., 2004). However, in general genes are depleted in siRNAs (Kasschau et al., 2007) and even genes with methylation in their bodies are not enriched in siRNAs (Zhang et al., 2006). As a result, most of the methylation found within gene bodies is clusters of CG methylation with little non-CG methylation. CG methylation without non-CG methylation indicates that the sequence is not being actively targeted by siRNAs and de novo methyltransferases, but that methylation is simply being maintained by MET1. The genic meCG-bias is consistent with analysis of di- and trinucleotide content within the Arabidopsis genome (Tran et al., 2005a). 5-methylcytosine can be deaminated to thymine, and, if left unrepaired, a CG to TA transition occurs (Figure 1). CG dinucleotides are depleted in introns compared to intergenic regions, as expected if the methylation within genes is of the CG type (selection for amino acids in exons confounds analysis there). Additionally, C(A/T)G trinucleotides have the highest intronic/intergenic ratio, and C(C/G)G the lowest, indicating that the methylation found at genes for long enough periods of evolutionary time to result in sequence biases is of the CG type only. Originally, this methylation probably existed as actively maintained CG and non-CG methylation that was targeted by RdDM due to the production of RNAs that could be templates for RdRPs. As the active signal is lost, only CG methylation remains. Active signals could be lost in particular tissues, or from entire plants. A comparison of methylation between the Ler and Col accessions made by hybridizing McrBC-digested DNA to 1 kb tiling arrays found that half the genes with methylation in one accession were not methylated in the other (Vaughn et al., 2007). This speaks to the stochastic nature of the processes producing methylation in the bodies of genes. A brief period of aberrant RNA production and subsequent siRNA biogenesis could lead to CG methylation that is maintained for thousands of years.
Methylation in the Context of Chromatin
All of the processes we have discussed take place not on naked DNA, but on DNA wrapped around nucleosomes and compacted into chromatin. Changes in DNA methylation often parallel changes in histone modifications. In organisms that lack DNA methylation, like S. pombe and Drosophila, complexes containing siRNAs direct histone modifications and repressive chromatin structure (Matzke and Birchler, 2005; Slotkin and Martienssen, 2007). What impact does DNA methylation have on chromatin structure and vice versa?
In mammals, methyl-binding domain (MBD) proteins bind to methylated CpG sites and can recruit other chromatin modifying enzymes, such as histone deacetylases or histone methyltransferases (Wade, 2001). The Arabidopsis genome contains 12 MBD proteins, but so far they have been assigned little role in mediating methylation interactions (Grafi et al., 2007). Only a few can bind methylated CpG in vitro, and although some are required for normal plant development, it is not clear that this has anything to do with DNA methylation per se (Zemach and Grafi, 2003). AtMBD7 seems to be the most likely to have any role in CG methylation; it binds methylated CpG in vitro and is found nuclear-localized to chromocenters (Zemach et al., 2005). We await further elucidation of the functions of these genes.
The RdDM methylation pathway is not required to maintain methylation at centromere repeats. A screen for naturally hypomethylated centromere repeats among Arabidopsis accessions led to the discovery of a novel methylcytosine binding protein, VIM1 (Woo et al, 2007). The Bor-4 strain of Arabidopsis contains centromere repeats that are hypomethylated and decondensed compared to the standard lab strain Columbia (Col). This phenotype is caused by a deletion in the VIM1 gene, which codes for a protein with a PHD domain, RING finger domains, and a SRA domain. In Col-0, VIM1 is found throughout the nucleus but is concentrated at chromocenters. Through its SRA domain and some additional amino acids, VIM1 binds double-stranded CG or CHG methylated oligonucleotides, but not those that are CHH methylated. In in vitro pull down assays, VIM1 interacts with the centromeric H3 variant (CenH3) and with all of the core histones, except for H2A. This led the authors to propose a model whereby VIM1 binds 5-methylcytosine and then alters chromatin structure by modifying histones, although the nature of this modification remains speculative. The mammalian homologue, Np95, has a N-terminal ubiquitin ligase domain and can ubiquitinate histones in vitro (Citterio et al., 2004).
Another mechanism for VIM1's effect on methylation has been suggested by studies of Np95 in mouse ES cells. It was recently shown that Np95, which also binds histones and double stranded oligonucleotides with hemimethylated CG sites (Citterio et al., 2004; Unoki et al., 2004), recruits the Dnmt1 methyltransferase to replication foci and is required to maintain DNA methylation patterns throughout the genome (Bostick et al., 2007; Sharif et al., 2007). It remains to be seen whether VIM1 recruits MET1 or other methyltransferases to replicating methylated DNA, and, if so, whether this is general or is specific to centromeric sequences. In contrast to the global methylation defect observed in np95 mutant mouse ES cells, Arabidopsis Col-0 vim1 mutants lose methylation at centromeres but not at FWA or pericentromeric repeats (Woo et al., 2007).
There are 12 additional SRA domain genes in Arabidopsis, three of which are part of the VIM1 family. Nine SUVH histone methyltransferase genes also contain the domain (Johnson et al., 2007). A connection between histone methylation and DNA methylation has been apparent for many years. In Neurospora, DNA methylation is lost if the histone methyltransferase DIM5 is mutated (Tamaru and Selker, 2001). In repetitive regions of the Arabidopsis genome, the distribution of H3 lysine 9 dimethylation (H3K9me2) closely parallels that of DNA methylation. Mutations in the SWI/SNF2 chromatin remodeling enzyme DDM1 cause loss of DNA methylation and H3 K9 methylation in heterochromatic sequences (Jeddeloh et al, 1999; Lippmann et al., 2004). met1 mutants also lose H3K9me2 at repetitive sequences (Soppe et al., 2002; Tariq et al., 2003; Mathieu et al., 2007). This has been interpreted to mean that CG methylation directs H3K9me2. Another possibility is that the change in histone methylation status is a byproduct of the increased transcription of repetitive elements observed in met1. For example, transcription might disrupt chromatin and evict nucleosomes, which could result in the replacement of H3 methylated at K9 with unmethylated H3. Or, transcription might prevent histone methyltransferases from gaining access to their substrates.
Three histone lysine methyltransferases KYP/SUVH4, SUVH5, and SUVH6 are required for maintenance of non-CG methylation at CMT3 target loci. The impact mutations in each of these genes has on DNA methylation differs depending on the locus (Ebbs and Bender, 2006). The SRA domain of KYP/SUVH4 and SUVH6 binds to methylated double-stranded oligonucleotides with a preference for methylated CHG and CHH sites (Johnson et al., 2007). This suggests that histone methyltransferases and CMT3 act in a positive feedback loop to maintain non-CG DNA methylation and histone H3K9 dimethylation (Johnson et al., 2007).
Most of the attention on histone modifications and DNA methylation has focused on lysine methylation of histone H3. A screen for suppressors of transgene silencing associated with DNA hypermethylation and siRNA accumulation has suggested that histone H2B monoubiquitination is negatively correlated with DNA methylation (Sridhar et al., 2007). The suppressor encodes a protein, UBP26, similar to ubiquitination-specific proteases, and is required for maintaining non-CG methylation and H3K9me2 at the target locus.
DNA methylation, although an excellent tool for keeping transposons transcriptionally silent, can be harmful if it invades gene space, particularly in promoter regions. As will be discussed in the next section, one mechanism to prevent this is genic demethylation by 5-methylcytosine DNA glycosylases. Analysis of the bonsai epimutant has suggested that histone demethylases might also function in this process. bonsai originally arose in a ddm1 mutant background due to silencing of the APC13 gene (Saze and Kakutani, 2007). Gene silencing is associated with accumulation of siR-NAs and hypermethylation of the 3′ transcribed sequence, which extends to the entire gene as ddm1 is successively inbred. APC13 sits about 1 kb upstream of a heavily methylated LINE retrotransposon element that seems to be responsible for the epigenetic effects. Hypermethylation is not induced in accessions in which the LINE element is not inserted downstream of the gene. A screen for other mutants that would cause hypermethylation at bonsai within one generation (unlike ddm1, which takes several generations) identified the IBM1 gene (Saze et al., 2008). Methylation accumulated at the 3′ end of the gene, particularly at non-CG sites, and was associated with increased H3 K9 methylation. IBM1 encodes a Jumonji-C domain-containing protein (Saze et al., 2008). Jumonji domains are found in H3K9 demethylases from diverse organisms (Klose et al., 2006). Although as of yet evidence that IBM1 is a histone demethylase is not complete, this work suggests a mechanism for “protecting” genes from nearby TEs.
DNA Demethylases: Removing Methylation
Another mechanism to protect genes from methylation targeted to repeats is to directly remove 5-methylcytosine from the DNA. Arabidopsis is the first organism in which DNA demethylases have been positively identified. Therefore a given methylation pattern can be the outcome of both methyltransferase and demethylase activities.
The plant DNA demethylases are HhH-GPD (helix-hairpin-helix —Gly/Pro/Asp) DNA glycosylases. DNA glycosylases are base excision repair proteins that typically recognize and remove damaged or mispaired bases from DNA (Fromme and Verdine, 2004). They cleave the glycosidic bond between the base and sugar-phosphate backbone and leave an abasic site in the DNA. Other enzymes then complete DNA repair; an AP endonuclease nicks the DNA backbone, DNA polymerase inserts the correct base, and DNA ligase seals the nick. DNA glycosylases are found in all organisms and repair many different types of damage, including oxidation, alkylation, and deamination. The crystal structure of several classes of glycosylases in complex with DNA has been solved. Like methyltransferases, these enzymes work by flipping the lesion base into the active site (Scharer and Jiricny, 2001; Fromme et al., 2004).
There are four members of the DNA glycosylase demethylase family in Arabidopsis: DEMETER (DME), REPRESSOR OF SI-LENCING1 (ROS1), DEMETER-LIKE2 (DML2), and DEMETER-LIKE3 (DML3) (Choi et al., 2002; Gong et al., 2002). Each of these proteins contains a HhH-GPD DNA glycosylase domain embedded within a much larger protein. The proteins share two other conserved domains that are of unknown function and unique to this class of proteins (Morales-Ruiz et al., 2006). DME and ROS1 were identified in genetic screens while DML2 and DML3 were identified based on sequence homology. Mutations in DME lead to endosperm overgrowth followed by seed abortion when inherited maternally, and mutations in ROS1 cause transcriptional gene silencing of a previously stable promoter:reporter gene fusion (Choi et al., 2002; Gong et al., 2002).
DME and ROS1 have been the most extensively biochemically characterized in vitro, although the details from different groups are not always identical. Purified, recombinant DME excises 5-methylcytosine, but not cytosine, from double-stranded oligonucleotides. 5-methylcytosine is released as a free base as a product of the reaction (Morales-Ruiz et al., 2006). Excision occurs in CG, CHG, and CHH sequence contexts, on either fully methylated or hemimethylated substrates, with the strongest activity observed on CG sites (Gehring et al., 2006; Morales-Ruiz et al., 2006). HhH-GPD DNA glycosylases are characterized by a conserved aspartic acid residue and an invariant lysine. When either of these residues is mutated in recombinant DME, 5-methylcytosine DNA glycosylase activity is lost (Gehring et al., 2006; Morales-Ruiz et al., 2006).
Similar biochemical results were obtained for purified recombinant ROS1. One group found that ROS1 had in vitro preference for methylated CHG sites, whereas another found the highest activity against CG (Agius et al., 2006; Morales-Ruiz et al., 2006). This discrepancy might be due to the different sequences of the methylated oligos used in these studies. A comparison of the kinetics of glycosylase action on 5-methylcytosine in different sequence contexts found that for both DME and ROS1, meC in the CAG context is removed much more efficiently than meC at the outer cytosine in the CCG context (Morales-Ruiz et al., 2006). CCG is also the least likely CHG site to be methylated in vivo (Cokus et al., 2008). DML2 and DML3 activities have also been investigated. DML3 removes all types of cytosine methylation from methylated oligonucleotides (Penterman et al., 2007c). DML2 also does so, but far more weakly than the other glycosylases and not in all sequence contexts (Penterman et al., 2007c).
The idea that DNA glycosylases serve as demethylases by recognizing and removing 5-methylcytosine is not a new one and has some precedence in the animal literature (Cortazar et al., 2007). One persistent criticism of this idea has been that removal of symmetrically methylated cytosines on opposite strands of the DNA could lead to double-stranded DNA breaks, a side effect that would be disastrous. It appears, however, that inhibition of the enzyme by nearby abasic sites solves this problem. DME activity on meCG is greatly reduced when it is opposite an abasic site. Moving the abasic site farther away from the methylated CG relieves the inhibition (Gehring et al., 2006).
HhH-GPD DNA glycosylases can be monofunctional or bi-functional (Fromme and Verdine, 2004). Monofunctional glycosylases attack the base using water and leave the DNA backbone intact. Bifunctional DNA glycosylases (or glycosylases/lyases as they are also known) use nucleophillic attack to remove the damaged base and nick the DNA 3′ of the base. The bifunctional glycosylase pathway acts only on the single base being repaired while the monofunctional glycosylase pathway can lead to long-patch repair, in which the DNA strand is displaced and several bases replaced by polymerase. Monofunctional and bifunctional DNA glycosylases are distinguished based on the different elimination products generated by the two reactions and on the fact that bifunctional DNA glycosylases form a covalent intermediate with the DNA that can be trapped with sodium borohydride. All of the Arabidopsis enzymes are bifunctional DNA glycosylases, releasing characteristic beta and delta elimination products (Agius et al., 2006; Gehring et al., 2006; Morales-Ruiz et al., 2006; Penterman et al., 2007c) and DME and ROS1 can be trapped by sodium borohydride (Agius et al., 2006; Gehring et al., 2006; Morales-Ruiz et al., 2006). This means that the enzymes only affect a single base at a time and reduces the chance that they could cause double strand breaks during 5-methylcytosine removal.
Photosynthetic organisms are subject to continual assault on their DNA from the Sun and photosynthesis byproducts. Plants contain a large number of DNA glycosylases, which might have allowed additional functional specialization of these enzymes (Britt, 2002). While HhH-GPD DNA glycosylases are the largest class of glycosylases among all organisms, the DME family appears to be unique to the plant lineage. The chromatin database (www.chromdb.org) identifies members in all of the sequenced or partially sequenced plant genomes, down to the unicellular algae Ostreococcus luminarias. In most organisms, HhH-GPD glycosylases range in size from 18–40 kDa, with the glycosylase domain itself being about 200 amino acids. In contrast, the Arabidopsis DME family of enzymes are between 1100 and 1800 amino acids long. The only other glycosylase that approaches this size is the Drosophila MUG (mismatch uracil glycosylase) protein Thd1, which is 191 kDa and processes U:G mismatches (Cortazar et al., 2007).
It is possible that the DEMETER family of enzymes also plays a role in repair of spontaneous DNA damage. Although many organisms repair DNA damage caused by spontaneous deamination of methylated cytosines to thymines with a thymine-specific MUG DNA glycosylase, no MUG homologs are found in the Arabidopsis genome (Britt, 2002). This is especially surprising given the large amounts of 5-methylcytosine found in plant genomes and the fact that spontaneous deamination is not uncommon. The DEMETER family of enzymes might serve this function as well in Arabidopsis. Studies have shown that DME and ROS1 have substantial activity in vitro against T:G mismatches (results for DML2 and DML3 have not been reported) (Gehring et al., 2006; Morales-Ruiz et al., 2006). ros1 mutants are also more sensitive than wild type plants to the DNA alkylating agent MMS (methyl methanesulfonate) and the oxidizing agent hydrogen peroxide (Gong et al., 2002). It's unknown whether this is a direct or indirect effect. It would be unusual for a single glycosylase to repair so many diverse types of DNA damage.
How 5-methylcytosine DNA glycosylases recognize their substrate is a mystery. 5-methylcytosine is a “normal” base. Thymine DNA glycosylases, which recognize the normal base thymine, do so by also querying the complementary strand of DNA, so that only thymines paired to guanines are removed, not thymines paired to adenines (Cortazar et al., 2007). This sort of mechanisms does not seem sufficient for 5-methylcytosine DNA glycosylase activity. Solving the crystal structure of these enzymes in complex with DNA will be vital for understanding how they recognize and remove 5-methylcytosine.
In vivo evidence also supports the designation of these enzymes as DNA demethylases and indicates that their glycosylase activity is required for function in planta. When DME with a missense mutation in the critical aspartic acid residue is transformed back into plants, it no longer complements the dme phenotype (Choi et al., 2004). The MEA Polycomb group gene, defined genetically as a target of DME in the female gametophyte, becomes hypermethylated when dme is mutated (Gehring et al., 2006). Similarly, the endogenous RDR29A promoter and RDR29A promoter transgene become hypermethylated and transcriptionally silent in a ros1 background (Gong et al., 2002).
Two different approaches have been undertaken to search for endogenous targets of ROS1 and the other DEMETER-LIKE genes. One group compared gene expression in wild type plants and ros1 plants, the idea being that ros1-induced hypermethylation could lead to gene silencing (Zhu et al., 2007). Three genes that decrease in expression in a ros1 mutant showed increases in DNA methylation at the promoter region. They also looked at several well-studied transposons and identified increases in non-CG methylation at a subset of these (CG methylation was already near 100% in wild type and could not be expected to go much higher).
Targets of ROS1 and the DML2 and DML3 enzymes have been directly mapped using genome-wide methylation profiling. Methylation patterns in wild type and ros1 dml2 dml3 triple mutant whole plants were compared by isolating methylated DNA from each genotype and hybridizing it to tiling microarrays (Penterman et al., 2007c). By subtracting one methylation pattern from the other, about 200 regions that were hypermethylated in the mutant were identified. Detailed bisulfite analysis at a subset of these sequences indicated that hypermethylation occurred both at sites where there was no methylation in wild type, or where some was already present. Overall, hypermethylation of cytosines was found in all sequence contexts, although this varied depending on the locus. Most of the hypermethylation occurred in genic sequences. Among genes that were not methylated in wild type but became methylated in the triple mutant, methylation accumulated predominantly at the 5′ and 3′ ends of genes. This is the opposite of the genic methylation pattern found in wild type, and are the regions potentially the most deleterious to gene function if methylated (Figure 2). However, of the 13 targets tested, very few showed an expression change between wild type and mutant, indicating that hypermethylation is not necessarily immediately deleterious to gene expression. The methylation removed by the DEMETER family of enzymes is deposited by RdDM and targets of the DEMETER family are enriched in siRNAs (Penterman et al., 2007a). This suggests that one of the functions of the demethylases is to “clean-up” after a methylation system that robustly recognizes and silences parasitic transposable elements. In this way, genes would be protected from methylation that can potentially silence normal gene expression.
At present, nothing is known about how the glycosylases are targeted to their substrates. How, for example, are the ends of genes distinguished? One possibility is that chromatin differences mark the ends of genes, and that this is recognized by ROS1, DML2 and DML3. How any glycosylase finds its target among millions of base pairs of DNA condensed into chromatin is still an open question. One possibility is that glycosylases look for their substrates by constantly scanning the DNA. The endogenous RDR29A gene promoter is not a target for ROS1 in wild type plants, but becomes so when a siRNA-producing RDR29A trans-gene is introduced (Gong et al., 2002). This suggests that ROS1 can immediately recognize new genic targets and remove methylation. The mechanism of 5-methylcytosine DNA glycosylase targeting is likely to be a fruitful area of future research, as will be identifying interacting factors.
Finally, evidence for cross-talk and potential coordination between the methylation, RNA silencing, and demethylation pathways is emerging. Expression of ROS1 is reduced in mutants of the RdDM pathway and in met1 (Huettel et al., 2006; Mathieu et al., 2007; Penterman et al., 2007a) and ROS1 target loci become hypermethylated in rdr2 and drm2 mutants (Penterman et al., 2007a). Reduction of DNA demethylase function in RdDM and methyltransferase mutants complicates the analysis of methylation in these backgrounds.
Methylation, Demethylation, and Regulation
Methylation has been ascribed many roles in Arabidopsis; it is widely reported to regulate transposons, regulate genes, and regulate development. The word regulate has a tendency to be bandied about rather loosely in scientific discourse and applied to any situation in which a molecular or morphological phenotype emerges: mutation of factor X causes phenotype y to emerge, therefore Factor X regulates process Y. Mutations in HUELLEN-LOS (HLL) lead to a striking developmental phenotype. Ovule growth is arrested and the pistil does not elongate. HLL encodes a mitochondrial ribosomal gene (Skinner et al., 2001). Does this mean that ribosomal proteins regulate development? Clearly not. With regard to methylation, it might be helpful to be more precise with our use of the word regulate. Methylation and demethylation activities appear to function primarily in genome housekeeping, with regulation limited to, at this point, the expression of imprinted genes (Figure 4).
Figure 4.

Housekeeping and regulatory functions of DNA methylation.
A) Housekeeping function of DNA methylation. DNA methyltransferases (DMT) maintain DNA methylation at transposons, keeping them from being transcribed. DNA glycosylases (DNG) remove methylation that accumulates near genes.
B) Regulatory function of DNA methylation. The DNA glycosylase DME removes 5-methylcytosine from imprinted genes in the central cell of the female gametophyte, allowing expression (the status of siRNAs in the central cell has not been experimentally addressed). After fertilization only the maternal allele is expressed in the endosperm. (Red lollipops = 5-methylcytosine, short squiggly lines = siRNAs).
For the majority of methylated sequences found in the Arabidopsis genome, like TEs and centromeres, “methylated” is the default state and methylation is propagated indefinitely. Unlike mammals, the plant methylome does not appear to be reset during gametogenesis. There is little evidence that the status of methylated sequences changes during growth and development of the organism. Methylation certainly impacts repeated sequences, as a dramatic effect is observed when methylation is removed, but does not appear to dynamically regulate them. Similarly, DNA demethylation serves a housekeeping function by removing genic RNA-directed DNA methylation (Gong et al., 2002; Penterman et al., 2007a). In these ways transposable elements are kept silent and genes are protected from methylation-induced silencing (Figure 4A).
A similar housekeeping scenario may apply to the clusters of CG methylation found in genes. In met1 mutants, genes that are body-methylated in wild type have a slight overall increase in expression compared to genes that are not methylated in wild type (Zilberman et al., 2007). This suggests that methylation in the body of genes dampens their expression, but does not suggest that it does so in a regulatory manner.
The fact that drm2 cmt3 plants exhibit reproducible recessive phenotypes has been interpreted to mean that non-CG methylation regulates development (Chan et al., 2006). The molecular basis of the phenotypes has not been elucidated and no target genes are known. As of yet, there is no evidence that non-CG methylation actively regulates genes.
Regulation by methylation describes a change in the methylation status of a sequence associated with a change in function. In mammals, tissue specific differences in methylation have been discovered. There is evidence that germline-specific genes become methylated as they are no longer expressed in differentiated tissues (Weber et al., 2007). These questions have not been fully experimentally addressed in plants. Several plant studies have found differences in methylation between tissue types, although the responsible sequences have not, for the most part, been identified (Gehring and Henikoff, 2007). These must be viewed with a degree of caution as most rely on detecting differences by methods that utilize methylation-sensitive enzymes. Methylation can be lost at individual cytosines even though the region remains methylated overall (Tran et al., 2005a). A recent study profiled DNA methylation and histone modifications in young rice shoots and cultured cells using McrBC digestion followed by hybridization to high density tiling microarrays. DNA methylation differences were observed at several genes and transposons (Li et al., 2008b). The question of whether there are tissue-specific patterns of DNA methylation in Arabidopsis is now ready to be addressed with the new methods available for isolating DNA from specific tissues and for determining high resolution methylation patterns. It might be particularly interesting to compare undifferentiated and differentiated tissue types (i.e. meristems and leaves). For example, the PHB gene is less methylated and more highly expressed in inflorescence meristems than in mature tissues. However, expression levels are under miRNA control, and methylation appears to play no role in regulating mRNA. Determining whether tissue-specific methylation differences are biologically significant will be a challenge.
Despite caveats concerning the possible regulatory roles of most plant DNA methylation, imprinted genes are clear examples in which DNA demethylation has been harnessed to regulate gene expression. Alleles of imprinted genes are expressed differently depending on whether they are inherited from the male or female parent. There is good evidence that the methylation of expressed alleles of imprinted genes changes during female gametophyte development (Figure 4) (Gehring and Henikoff, 2007; Huh et al., 2007; Penterman et al., 2007b). In Arabidopsis we know of four imprinted genes, MEDEA, FWA, FIS2, and PHERES. The first three are expressed maternally and silenced paternally in the endosperm. The endosperm is one of the products of double fertilization (the embryo being the other) and arises when a haploid sperm fertilizes the diploid central cell of the female gametophyte. The triploid endosperm nourishes the embryo during seed development. Expression of maternal MEDEA and FIS2 alleles is essential for normal endosperm development and seed viability (Grossniklaus et al., 1998; Kiyosue et al., 1999; Luo et al., 1999). These genes encode components of Polycomb group complexes, which generally function to maintain gene repression. Maternal inheritance of mea and fis2 alleles causes endosperm overproliferation and embryo arrest. The role of FWA during endosperm development is unknown.
MEA, FWA, and FIS2 are all associated with DNA methylation that is maintained by MET1 (Soppe et al., 2000; Xiao et al., 2003; Kinoshita et al., 2004; Jullien et al., 2006a). Maternal expression of MEA, FWA, and FIS2 in the female gametophyte and endosperm depends on inheritance of a wild type maternal allele of the 5-methylcytosine DNA glycosylase DME (Choi et al., 2002; Kinoshita et al., 2004; Jullien et al., 2006a). DME exhibits a restricted pattern of expression during reproductive development; it is expressed in the polar nuclei (which fuse to form the central cell nucleus) and central cell nucleus, but not in the egg cell, which gives rise to the embryo (Choi et al., 2002).
The simplest imprinting scenario (or so it seems at present) is represented by FWA (Figure 4B). In the sporophyte, FWA is highly methylated at tandem repeats in the promoter and first exon and is silenced. When methylation is lost, as in met1 mutants, the gene is expressed (Soppe et al., 2000). In wild type endosperm, but not embryo, FWA is relatively hypomethylated and is expressed. Expression depends on DME, suggesting that DME removes 5-methylcytosine from the 5′ region of FWA (Kinoshita et al., 2004). Since the endosperm is a terminally differentiated tissue, the hypomethylated allele is never inherited, thus obviating the need for methylation resetting. Regulation of MEA expression is slightly more complicated, but the same basic features are present. MEA is methylated at tandem direct repeats 3′ of the gene and, in some ecotypes, at a CG cluster 5′ of the gene (Xiao et al., 2003; Gehring et al., 2006). The expressed maternal MEA allele is hypomethylated in the endosperm compared to the silent paternal allele and the expressed embryo alleles. If a mutant maternal dme allele is inherited, the maternal MEA allele is hypermethylated in the endosperm (Gehring et al., 2006). Unlike FWA, loss of MEA paternal allele methylation via a met1 mutation does not lead to paternal allele expression in the endosperm. Polycomb group (PcG) genes, including maternal MEA, repress paternal MEA expression (Baroux et al., 2006; Gehring et al., 2006; Jullien et al., 2006b). For both FWA and MEA (and FIS2, although endosperm methylation data has not yet been reported), the presumptive site of demethylation is in the central cell of the female gametophyte, where DME is expressed (Choi et al., 2002). Work on maize imprinted genes also supports this conclusion. A PcG gene that is maternally expressed and paternally silent in the endosperm is hypomethylated in the central cell compared to the egg cell and sperm cells (Gutierrez-Marcos et al., 2006). The restricted expression pattern of the DME demethylase during reproductive development allows imprinting to be established. Thus far, the methylation at the FWA and MEA genes is the only example of methylation changing during the growth and development of the plant and actively regulating gene expression.
OUTLOOK
The pace of methylation and demethylation research in Arabidopsis is expected to remain brisk. A little over a year after the first high-resolution methylation map for Arabidopsis (or any organism) was published (Zhang et al., 2006), a map with single base resolution is now available (Cokus et al., 2008). The challenge now is to make sense of all this information. Both genetic and genomic experiments will be required to tease apart the impact of methylation and demethylation on the genome. How significant is methylation at a particular sequence? How dynamic is it? Our understanding of DNA demethylation is still relatively rudimentary and significant questions remain unanswered. With all of the molecular, genetic, and genomic tools available for studying these problems in Arabidopsis, it is expected that this chapter will need a significant update within just a few years.
Acknowledgments
We thank Roger Deal for comments on the manuscript. M.G. is a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation.
Footnotes
Citation: Gehring M., and Henikoff S. (2008) DNA Methylation and Demethylation in Arabidopsis. The Arabidopsis Book 6:e0102. doi:10.1199/tab.0102
elocation-id: e0102
Published on: May 23, 2008
REFERENCES
- Agius F., Kapoor A., Zhu J. K. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc. Natl. Acad. Sci. USA. 2006;1036(1):11796–11801. doi: 10.1073/pnas.0603563103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aufsatz W., Mette M. F., Matzke A. J., Matzke M. The role of MET1 in RNA-directed de novo and maintenance methylation of CG dinucleotides. Plant Mol. Biol. 2004;546(1):793–804. doi: 10.1007/s11103-004-0179-1. [DOI] [PubMed] [Google Scholar]
- Bao N., Lye K. W., Barton M. K. MicroRNA binding sites in Arabidopsis class III HD-ZIP mRNAs are required for methylation of the template chromosome. Dev. Cell. 2004;76(1):653–662. doi: 10.1016/j.devcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Baroux C., Gagliardini V., Page D. R., Grossniklaus U. Dynamic regulatory interactions of Polycomb group genes: MEDEA autoregulation is required for imprinted gene expression in Arabidopsis. Genes Dev. 2006;206(1):1081–1086. doi: 10.1101/gad.378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee L., Malagnac F., Bender J. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 2001;156(1):1753–1758. doi: 10.1101/gad.905701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender J. Chromatin-based silencing mechanisms. Curr. Opin. Plant Biol. 2004;76(1):521–526. doi: 10.1016/j.pbi.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Bostick M., Kim J. K., Esteve P. O., Clark A., Pradhan S., Jacobsen S. E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;3176(1):1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- Britt A. Repair of Damaged Bases. In: Somerville C., Meyerowitz E., editors. The Arabidopsis Book. American Society of Plant Biologists; Rockville, MD: April 4, 2002. doi: 10.1199/tab.0005, http://www.aspb.org/publications/arabidopsis/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X., Jacobsen S. E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 99 Suppl. 2002;46(1):16491–16498. doi: 10.1073/pnas.162371599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. W., Henderson I. R., Jacobsen S. E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005;66(1):351–360. doi: 10.1038/nrg1601. [DOI] [PubMed] [Google Scholar]
- Chan S. W., Zilberman D., Xie Z., Johansen L. K., Carrington J. C., Jacobsen S. E. RNA silencing genes control de novo DNA methylation. Science. 2004;3036(1):1336. doi: 10.1126/science.1095989. [DOI] [PubMed] [Google Scholar]
- Chan S. W., Henderson I. R., Zhang X., Shah G., Chien J. S., Jacobsen S. E. RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in arabidopsis. PLoS Genet. 2006;26(1):e83. doi: 10.1371/journal.pgen.0020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E. J., Carrington J. C. Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 2007;86(1):884–896. doi: 10.1038/nrg2179. [DOI] [PubMed] [Google Scholar]
- Chawla R., Nicholson S. J., Folta K. M., Srivastava V. Transgene-induced silencing of Arabidopsis phytochrome A gene via exonic methylation. Plant J. 2007;526(1):1105–1118. doi: 10.1111/j.1365-313X.2007.03301.x. [DOI] [PubMed] [Google Scholar]
- Choi Y., Harada J. J., Goldberg R. B., Fischer R. L. An invariant aspartic acid in the DNA glycosylase domain of DEME-TER is necessary for transcriptional activation of the imprinted MEDEA gene. Proc. Natl. Acad. Sci. USA. 2004;1016(1):7481–7486. doi: 10.1073/pnas.0402328101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y., Gehring M., Johnson L., Hannon M., Harada J. J., Goldberg R. B., Jacobsen S. E., Fischer R. L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in arabidopsis. Cell. 2002;1106(1):33–42. doi: 10.1016/s0092-8674(02)00807-3. [DOI] [PubMed] [Google Scholar]
- Citterio E., Papait R., Nicassio F., Vecchi M., Gomiero P., Mantovani R., Di Fiore P. P., Bonapace I. M. Np95 is a histone-binding protein endowed with ubiquitin ligase activity. Mol. Cell Biol. 2004;246(1):2526–2535. doi: 10.1128/MCB.24.6.2526-2535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S. J., Harrison J., Paul C. L., Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;226(1):2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus S. J., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C. D., Pradhan S., Nelson S. F., Pellegrini M., Jacobsen S. E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;4526(1):215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar D., Kunz C., Saito Y., Steinacher R., Schar P. The enigmatic thymine DNA glycosylase. DNA Repair (Amst) 2007;66(1):489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Ebbs M. L., Bender J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell. 2006;186(1):1166–1176. doi: 10.1105/tpc.106.041400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Shami M., Pontier D., Lahmy S., Braun L., Picart C., Vega D., Hakimi M. A., Jacobsen S. E., Cooke R., Lagrange T. Reiterated WG/GW motifs form functionally and evolutionarily conserved ARGONAUTE-binding platforms in RNAi-related components. Genes Dev. 2007;216(1):2539–2544. doi: 10.1101/gad.451207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan E. J., Peacock W. J., Dennis E. S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA. 1996;936(1):8449–8454. doi: 10.1073/pnas.93.16.8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan E. J., Kovac K. A. Plant DNA methyltransferases. Plant Mol. Biol. 2000;436(1):189–201. doi: 10.1023/a:1006427226972. [DOI] [PubMed] [Google Scholar]
- Fransz P., Soppe W., Schubert I. Heterochromatin in interphase nuclei of Arabidopsis thaliana. Chromosome Res. 2003;116(1):227–240. doi: 10.1023/a:1022835825899. [DOI] [PubMed] [Google Scholar]
- Fromme J. C., Verdine G. L. Base excision repair. Adv Protein Chem. 2004;696(1):1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- Fromme J. C., Banerjee A., Verdine G. L. DNA glycosylase recognition and catalysis. Curr. Opin. Struct. Biol. 2004;146(1):43–49. doi: 10.1016/j.sbi.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Frommer M., McDonald L. E., Millar D. S., Collis C. M., Watt F., Grigg G. W., Molloy P. L., Paul C. L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA. 1992;896(1):1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzani S., Lawrenson T., Woodward C., Headon D., Sablowski R. A link between mRNA turnover and RNA interference in Arabidopsis. Science. 2004;3066(1):1046–1048. doi: 10.1126/science.1101092. [DOI] [PubMed] [Google Scholar]
- Gehring M., Henikoff S. DNA methylation dynamics in plant genomes. Biochim. Biophys. Acta. 2007;17696(1):276–286. doi: 10.1016/j.bbaexp.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Gehring M., Huh J. H., Hsieh T. F., Penterman J., Choi Y., Harada J. J., Goldberg R. B., Fischer R. L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell. 2006;1246(1):495–506. doi: 10.1016/j.cell.2005.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll M. G., Bestor T. H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;746(1):481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Gong Z., Morales-Ruiz T., Ariza R. R., Roldan-Arjona T., David L., Zhu J. K. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell. 2002;1116(1):803–814. doi: 10.1016/s0092-8674(02)01133-9. [DOI] [PubMed] [Google Scholar]
- Grafi G., Zemach A., Pitto L. Methyl-CpG-binding domain (MBD) proteins in plants. Biochim. Biophys. Acta. 2007;17696(1):287–294. doi: 10.1016/j.bbaexp.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Grossniklaus U., Vielle-Calzada J. P., Hoeppner M. A., Gagliano W. B. Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science. 1998;2806(1):446–450. doi: 10.1126/science.280.5362.446. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Marcos J. F., Costa L. M., Dal Pra M., Scholten S., Kranz E., Perez P., Dickinson H. G. Epigenetic asymmetry of imprinted genes in plant gametes. Nat. Genet. 2006;386(1):876–878. doi: 10.1038/ng1828. [DOI] [PubMed] [Google Scholar]
- Henderson I. R., Zhang X., Lu C., Johnson L., Meyers B. C., Green P. J., Jacobsen S. E. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 2006;386(1):721–725. doi: 10.1038/ng1804. [DOI] [PubMed] [Google Scholar]
- Henikoff S., Comai L. A DNA methyltransferase homolog with a chromodomain exists in multiple polymorphic forms in Arabidopsis. Genetics. 1998;1496(1):307–318. doi: 10.1093/genetics/149.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr A. J., Jensen M. B., Dalmay T., Baulcombe D. C. RNA polymerase IV directs silencing of endogenous DNA. Science. 2005;3086(1):118–120. doi: 10.1126/science.1106910. [DOI] [PubMed] [Google Scholar]
- Hohn T., Corsten S., Rieke S., Muller M., Rothnie H. Methylation of coding region alone inhibits gene expression in plant protoplasts. Proc. Natl. Acad. Sci. USA. 1996;936(1):8334–8339. doi: 10.1073/pnas.93.16.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettel B., Kanno T., Daxinger L., Aufsatz W., Matzke A. J., Matzke M. Endogenous targets of RNA-directed DNA methylation and Pol IV in Arabidopsis. EMBO J. 2006;256(1):2828–2836. doi: 10.1038/sj.emboj.7601150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh J. H., Bauer M. J., Hsieh T. F., Fischer R. Endosperm gene imprinting and seed development. Curr. Opin. Genet. Dev. 2007;176(1):480–485. doi: 10.1016/j.gde.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson J. P., Lindroth A. M., Cao X., Jacobsen S. E. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;4166(1):556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- Jackson J. P., Johnson L., Jasencakova Z., Zhang X., PerezBurgos L., Singh P. B., Cheng X., Schubert I., Jenuwein T., Jacobsen S. E. Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana. Chromosoma. 2004;1126(1):308–315. doi: 10.1007/s00412-004-0275-7. [DOI] [PubMed] [Google Scholar]
- Jacobsen S. E., Sakai H., Finnegan E. J., Cao X., Meyerowitz E. M. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol. 2000;106(1):179–186. doi: 10.1016/s0960-9822(00)00324-9. [DOI] [PubMed] [Google Scholar]
- Jeddeloh J. A., Stokes T. L., Richards E. J. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet. 1999;226(1):94–97. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- Johnson L., Mollah S., Garcia B. A., Muratore T. L., Shabanowitz J., Hunt D. F., Jacobsen S. E. Mass spectrometry analysis of Arabidopsis histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Res. 2004;326(1):6511–6518. doi: 10.1093/nar/gkh992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L. M., Bostick M., Zhang X., Kraft E., Henderson I., Callis J., Jacobsen S. E. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 2007;176(1):379–384. doi: 10.1016/j.cub.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan N. D., West J. P., Bottley A., Sheikh M., Furner I. Transcript profiling of the hypomethylated hog1 mutant of Arabidopsis. Plant Mol. Biol. 2007;656(1):571–586. doi: 10.1007/s11103-007-9221-4. [DOI] [PubMed] [Google Scholar]
- Jullien P. E., Kinoshita T., Ohad N., Berger F. Maintenance of DNA methylation during the Arabidopsis life cycle is essential for parental imprinting. Plant Cell. 2006a;186(1):1360–1372. doi: 10.1105/tpc.106.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien P. E., Katz A., Oliva M., Ohad N., Berger F. Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr. Biol. 2006b;166(1):486–492. doi: 10.1016/j.cub.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Kankel M. W., Ramsey D. E., Stokes T. L., Flowers S. K., Haag J. R., Jeddeloh J. A., Riddle N. C., Verbsky M. L., Richards E. J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics. 2003;1636(1):1109–1122. doi: 10.1093/genetics/163.3.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T., Mette M. F., Kreil D. P., Aufsatz W., Matzke M., Matzke A. J. Involvement of putative SNF2 chromatin remodeling protein DRD1 in RNA-directed DNA methylation. Curr. Biol. 2004;146(1):801–805. doi: 10.1016/j.cub.2004.04.037. [DOI] [PubMed] [Google Scholar]
- Kanno T., Huettel B., Mette M. F., Aufsatz W., Jaligot E., Daxinger L., Kreil D. P., Matzke M., Matzke A. J. Atypical RNA polymerase subunits required for RNA-directed DNA methylation. Nat. Genet. 2005;376(1):761–765. doi: 10.1038/ng1580. [DOI] [PubMed] [Google Scholar]
- Kasschau K. D., Fahlgren N., Chapman E. J., Sullivan C. M., Cumbie J. S., Givan S. A., Carrington J. C. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;56(1):e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Miura A., Bender J., Jacobsen S. E., Kakutani T. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr. Biol. 2003;136(1):421–426. doi: 10.1016/s0960-9822(03)00106-4. [DOI] [PubMed] [Google Scholar]
- Kinoshita T., Miura A., Choi Y., Kinoshita Y., Cao X., Jacobsen S. E., Fischer R. L., Kakutani T. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science. 2004;3036(1):521–523. doi: 10.1126/science.1089835. [DOI] [PubMed] [Google Scholar]
- Kiyosue T., Ohad N., Yadegari R., Hannon M., Dinneny J., Wells D., Katz A., Margossian L., Harada J. J., Goldberg R. B., Fischer R. L. Control of fertilization-independent endosperm development by the MEDEA polycomb gene in Arabidopsis. Proc. Natl. Acad. Sci. USA. 1999;966(1):4186–4191. doi: 10.1073/pnas.96.7.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R. J., Kallin E. M., Zhang Y. JmjC-domaincontaining proteins and histone demethylation. Nat. Rev. Genet. 2006;76(1):715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- Laird C. D., Pleasant N. D., Clark A. D., Sneeden J. L., Hassan K. M., Manley N. C., Vary J. C., Jr., Morgan T., Hansen R. S., Stoger R. Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc. Natl. Acad. Sci. USA. 2004;1016(1):204–209. doi: 10.1073/pnas.2536758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. F., Henderson I. R., Song L., Fedoroff N., Lagrange T., Jacobsen S. E. Dynamic Regulation of ARGONAUTE4 within multiple nuclear bodies in Arabidopsis thaliana. PLoS Genet. 2008a;46(1):e27. doi: 10.1371/journal.pgen.0040027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. F., Pontes O., El-Shami M., Henderson I. R., Bernatavichute Y. V., Chan S. W., Lagrange T., Pikaard C. S., Jacobsen S. E. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell. 2006;1266(1):93–106. doi: 10.1016/j.cell.2006.05.032. [DOI] [PubMed] [Google Scholar]
- Li X., Wang X., He K., Ma Y., Su N., He H., Stolc V., Tongprasit W., Jin W., Jiang J., Terzaghi W., Li S., Deng X. W. High-Resolution Mapping of Epigenetic Modifications of the Rice Genome Uncovers Interplay between DNA Methylation, Histone Methylation, and Gene Expression. Plant Cell. 2008b;206(1):259–276. doi: 10.1105/tpc.107.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroth A. M., Cao X., Jackson J. P., Zilberman D., McCallum C. M., Henikoff S., Jacobsen S. E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science. 2001;2926(1):2077–2080. doi: 10.1126/science.1059745. [DOI] [PubMed] [Google Scholar]
- Lindroth A. M., Shultis D., Jasencakova Z., Fuchs J., Johnson L., Schubert D., Patnaik D., Pradhan S., Goodrich J., Schubert I., Jenuwein T., Khorasanizadeh S., Jacobsen S. E. Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO. J. 2004;236(1):4286–4296. doi: 10.1038/sj.emboj.7600430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z., May B., Yordan C., Singer T., Martienssen R. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol. 2003;16(1):E67. doi: 10.1371/journal.pbio.0000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z., Gendrel A. V., Black M., Vaughn M. W., Dedhia N., McCombie W. R., Lavine K., Mittal V., May B., Kasschau K. D., Carrington J. C., Doerge R. W., Colot V., Martienssen R. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;4306(1):471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- Lorincz M. C., Dickerson D. R., Schmitt M., Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 2004;116(1):1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- Lu C., Kulkarni K., Souret F. F., MuthuValliappan R., Tej S. S., Poethig R. S., Henderson I. R., Jacobsen S. E., Wang W., Green P. J., Meyers B. C. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res. 2006;166(1):1276–1288. doi: 10.1101/gr.5530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M., Bilodeau P., Koltunow A., Dennis E. S., Peacock W. J., Chaudhury A. M. Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA. 1999;966(1):296–301. doi: 10.1073/pnas.96.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Chen Z. Improperly terminated, unpolyadenylated mRNA of sense transgenes is targeted by RDR6-mediated RNA silencing in Arabidopsis. Plant Cell. 2007;196(1):943–958. doi: 10.1105/tpc.106.045724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O., Reinders J., Caikovski M., Smathajitt C., Paszkowski J. Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell. 2007;1306(1):851–862. doi: 10.1016/j.cell.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Matzke M. A., Birchler J. A. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 2005;66(1):24–35. doi: 10.1038/nrg1500. [DOI] [PubMed] [Google Scholar]
- Morales-Ruiz T., Ortega-Galisteo A. P., Ponferrada-Marin M. I., Martinez-Macias M. I., Ariza R. R., Roldan-Arjona T. DEMETER and REPRESSOR OF SILENCING 1 encode 5-methylcytosine DNA glycosylases. Proc. Natl. Acad. Sci. USA. 2006;1036(1):6853–6858. doi: 10.1073/pnas.0601109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mull L., Ebbs M. L., Bender J. A histone methylation-dependent DNA methylation pathway is uniquely impaired by deficiency in Arabidopsis S-adenosylhomocysteine hydrolase. Genetics. 2006;1746(1):1161–1171. doi: 10.1534/genetics.106.063974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera Y., Haag J. R., Ream T., Nunes P. C., Pontes O., Pikaard C. S. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell. 2005;1206(1):613–622. doi: 10.1016/j.cell.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Penterman J., Uzawa R., Fischer R. L. Genetic interactions between DNA demethylation and methylation in Arabidopsis. Plant Physiol. 2007a;1456(1):1549–1557. doi: 10.1104/pp.107.107730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penterman J., Huh J. H., Hsieh T. F., Fischer R. L. Genomic imprinting in Arabidopsis thaliana and Zea mays. In: Olsen O. L., editor. Endosperm—Development and Molecular Biology. 1. Vol. 6. Heidelberg: Springer Berlin; 2007b. pp. 219–239. [Google Scholar]
- Penterman J., Zilberman D., Huh J. H., Ballinger T., Henikoff S., Fischer R. L. DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. USA. 2007c;1046(1):6752–6757. doi: 10.1073/pnas.0701861104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikaard C. S. Transcription and tyranny in the nucleolus: the organization, activation, dominance and repression of ribosomal RNA genes. In: Somerville C., Meyerowitz E., editors. The Arabidopsis Book. American Society of Plant Biologists; Rockville, MD: August 12, 2002. doi: 10.1199/tab.0083, http://www.aspb.org/publications/arabidopsis/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes O., Li C. F., Nunes P. C., Haag J., Ream T., Vitins A., Jacobsen S. E., Pikaard C. S. The Arabidopsis chromatin-modifying nuclear siRNA pathway involves a nucleolar RNA processing center. Cell. 2006;1266(1):79–92. doi: 10.1016/j.cell.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Pontier D., Yahubyan G., Vega D., Bulski A., Saez-Vasquez J., Hakimi M. A., Lerbs-Mache S., Colot V., Lagrange T. Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes Dev. 2005;196(1):2030–2040. doi: 10.1101/gad.348405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., He X., Wang X. J., Kohany O., Jurka J., Hannon G. J. Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature. 2006;4436(1):1008–1012. doi: 10.1038/nature05198. [DOI] [PubMed] [Google Scholar]
- Rangwala S. H., Richards E. J. The value-added genome: building and maintaining genomic cytosine methylation landscapes. Curr. Opin. Genet. Dev. 2004;146(1):686–691. doi: 10.1016/j.gde.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Reinders J., Delucinge Vivier C., Theiler G., Chollet D., Descombes P., Paszkowski J. Genome-wide, high-resolution DNA methylation profiling using bisulfite-mediated cytosine conversion. Genome Res. 2008;186(1):469–476. doi: 10.1101/gr.7073008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha P. S., Sheikh M., Melchiorre R., Fagard M., Boutet S., Loach R., Moffatt B., Wagner C., Vaucheret H., Furner I. The Arabidopsis HOMOLOGY-DEPENDENT GENE SI-LENCING1 gene codes for an S-adenosyl-L-homocysteine hydrolase required for DNA methylation-dependent gene silencing. Plant Cell. 2005;176(1):404–417. doi: 10.1105/tpc.104.028332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H., Kakutani T. Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. Embo J. 2007;266(1):3641–3652. doi: 10.1038/sj.emboj.7601788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H., Mittelsten Scheid O., Paszkowski J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet. 2003;346(1):65–69. doi: 10.1038/ng1138. [DOI] [PubMed] [Google Scholar]
- Saze H., Shiraishi A., Miura A., Kakutani T. Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science. 2008;3196(1):462–465. doi: 10.1126/science.1150987. [DOI] [PubMed] [Google Scholar]
- Scharer O. D., Jiricny J. Recent progress in the biology, chemistry and structural biology of DNA glycosylases. Bioessays. 2001;236(1):270–281. doi: 10.1002/1521-1878(200103)23:3<270::AID-BIES1037>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T. A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K., Tajima S., Mitsuya K., Okano M., Koseki H. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;4506(1):908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- Skinner D. J., Baker S. C., Meister R. J., Broadhvest J., Schneitz K., Gasser C. S. The Arabidopsis HUELLENLOS gene, which is essential for normal ovule development, encodes a mitochondrial ribosomal protein. Plant Cell. 2001;136(1):2719–2730. doi: 10.1105/tpc.010323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin R. K., Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007;86(1):272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- Soppe W. J., Jacobsen S. E., Alonso-Blanco C., Jackson J. P., Kakutani T., Koornneef M., Peeters A. J. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell. 2000;66(1):791–802. doi: 10.1016/s1097-2765(05)00090-0. [DOI] [PubMed] [Google Scholar]
- Soppe W. J., Jasencakova Z., Houben A., Kakutani T., Meister A., Huang M. S., Jacobsen S. E., Schubert I., Fransz P. F. DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis. EMBO. J. 2002;216(1):6549–6559. doi: 10.1093/emboj/cdf657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar V. V., Kapoor A., Zhang K., Zhu J., Zhou T., Hasegawa P. M., Bressan R. A., Zhu J. K. Control of DNA methylation and heterochromatic silencing by histone H2B deubiquitination. Nature. 2007;4476(1):735–738. doi: 10.1038/nature05864. [DOI] [PubMed] [Google Scholar]
- Suck D. DNA-protein interactions. Flip out and modify. Curr. Biol. 1994;46(1):252–255. doi: 10.1016/s0960-9822(00)00057-9. [DOI] [PubMed] [Google Scholar]
- Sutherland E., Coe L., Raleigh E. A. McrBC: A multi-subunit GTP-dependent restriction endonuclease. J. Mol. Biol. 1992;2256(1):327–348. doi: 10.1016/0022-2836(92)90925-a. [DOI] [PubMed] [Google Scholar]
- Tamaru H., Selker E. U. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001;4146(1):277–283. doi: 10.1038/35104508. [DOI] [PubMed] [Google Scholar]
- Tanaka H., Masuta C., Uehara K., Kataoka J., Koiwai A., Noma M. Morphological changes and hypomethylation of DNA in transgenic tobacco expressing antisense RNA of the S-adenosyl-L-homocysteine hydrolase gene. Plant Mol. Biol. 1997;356(1):981–986. doi: 10.1023/a:1005896711321. [DOI] [PubMed] [Google Scholar]
- Tariq M., Saze H., Probst A. V., Lichota J., Habu Y., Paszkowski J. Erasure of CpG methylation in Arabidopsis alters patterns of histone H3 methylation in heterochromatin. Proc. Natl. Acad. Sci. USA. 2003;1006(1):8823–8827. doi: 10.1073/pnas.1432939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa R., McCallum C. M., Delrow J., Henikoff J. G., van Steensel B., Henikoff S. Genome-wide profiling of DNA methylation reveals transposon targets of CHRO-MOMETHYLASE3. Curr. Biol. 2002;126(1):65–68. doi: 10.1016/s0960-9822(01)00622-4. [DOI] [PubMed] [Google Scholar]
- Tost J., Gut I. G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007;26(1):2265–2275. doi: 10.1038/nprot.2007.314. [DOI] [PubMed] [Google Scholar]
- Tran R. K., Henikoff J. G., Zilberman D., Ditt R. F., Jacobsen S. E., Henikoff S. DNA methylation profiling identifies CG methylation clusters in Arabidopsis genes. Curr. Biol. 2005a;156(1):154–159. doi: 10.1016/j.cub.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Tran R. K., Zilberman D., de Bustos C., Ditt R. F., Henikoff J. G., Lindroth A. M., Delrow J., Boyle T., Kwong S., Bryson T. D., Jacobsen S. E., Henikoff S. Chromatin and siRNA pathways cooperate to maintain DNA methylation of small transposable elements in Arabidopsis. Genome Biol. 2005b;66(1):R90. doi: 10.1186/gb-2005-6-11-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki M., Nishidate T., Nakamura Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. 2004;236(1):7601–7610. doi: 10.1038/sj.onc.1208053. [DOI] [PubMed] [Google Scholar]
- Vaucheret H. Post-transcriptional small RNA pathways in plants: mechanisms and regulations. Genes Dev. 2006;206(1):759–771. doi: 10.1101/gad.1410506. [DOI] [PubMed] [Google Scholar]
- Vaughn M. W., Martienssen R. A. Finding the right template: RNA Pol IV, a plant-specific RNA polymerase. Mol. Cell. 2005;176(1):754–756. doi: 10.1016/j.molcel.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Vaughn M. W., Tanurd Ic M., Lippman Z., Jiang H., Carrasquillo R., Rabinowicz P. D., Dedhia N., McCombie W. R., Agier N., Bulski A., Colot V., Doerge R. W., Martienssen R. A. Epigenetic Natural Variation in Arabidopsis thaliana. PLoS Biol. 2007;56(1):e174. doi: 10.1371/journal.pbio.0050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y., Ohya H., Yamaguchi Y., Koizumi N., Sano H. Preferential de novo methylation of cytosine residues in non-CpG sequences by a domains rearranged DNA methyltransferase from tobacco plants. J. Biol. Chem. 2003;2786(1):42386–42393. doi: 10.1074/jbc.M303892200. [DOI] [PubMed] [Google Scholar]
- Wade P. A. Methyl-CpG binding proteins and transcriptional repression. Bioessays. 2001;236(1):1131–1137. doi: 10.1002/bies.10008. [DOI] [PubMed] [Google Scholar]
- Warnecke P. M., Stirzaker C., Song J., Grunau C., Melki J. R., Clark S. J. Identification and resolution of artifacts in bisulfite sequencing. Methods. 2002;276(1):101–107. doi: 10.1016/s1046-2023(02)00060-9. [DOI] [PubMed] [Google Scholar]
- Weber M., Hellmann I., Stadler M. B., Ramos L., Paabo S., Rebhan M., Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;396(1):457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- Woo H. R., Pontes O., Pikaard C. S., Richards E. J. VIM1, a methylcytosine-binding protein required for centromeric heterochromatinization. Genes Dev. 2007;216(1):267–277. doi: 10.1101/gad.1512007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W., Custard K. D., Brown R. C., Lemmon B. E., Harada J. J., Goldberg R. B., Fischer R. L. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell. 2006;186(1):805–814. doi: 10.1105/tpc.105.038836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W., Gehring M., Choi Y., Margossian L., Pu H., Harada J. J., Goldberg R. B., Pennell R. I., Fischer R. L. Imprinting of the MEA Polycomb gene is controlled by antagonism between MET1 methyltransferase and DME glycosylase. Dev. Cell. 2003;56(1):891–901. doi: 10.1016/s1534-5807(03)00361-7. [DOI] [PubMed] [Google Scholar]
- Xie Z., Johansen L. K., Gustafson A. M., Kasschau K. D., Lellis A. D., Zilberman D., Jacobsen S. E., Carrington J. C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004;26(1):E104. doi: 10.1371/journal.pbio.0020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki J., Gregory B. D., Ecker J. R. Mapping the genome landscape using tiling array technology. Curr. Opin. Plant Biol. 2007;106(1):534–542. doi: 10.1016/j.pbi.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A., Grafi G. Characterization of Arabidopsis thaliana methyl-CpG-binding domain (MBD) proteins. Plant J. 2003;346(1):565–572. doi: 10.1046/j.1365-313x.2003.01756.x. [DOI] [PubMed] [Google Scholar]
- Zemach A., Li Y., Wayburn B., Ben-Meir H., Kiss V., Avivi Y., Kalchenko V., Jacobsen S. E., Grafi G. DDM1 binds Arabidopsis methyl-CpG binding domain proteins and affects their subnuclear localization. Plant Cell. 2005;176(1):1549–1558. doi: 10.1105/tpc.105.031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Jacobsen S. E. Genetic analyses of DNA methyltransferases in Arabidopsis thaliana. Cold Spring Harb. Symp. Quant. Biol. 2006;716(1):439–447. doi: 10.1101/sqb.2006.71.047. [DOI] [PubMed] [Google Scholar]
- Zhang X., Henderson I. R., Lu C., Green P. J., Jacobsen S. E. Role of RNA polymerase IV in plant small RNA metabolism. Proc. Natl. Acad. Sci. USA. 2007;1046(1):4536–4541. doi: 10.1073/pnas.0611456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yazaki J., Sundaresan A., Cokus S., Chan S. W., Chen H., Henderson I. R., Shinn P., Pellegrini M., Jacobsen S. E., Ecker J. R. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;1266(1):1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Zheng X., Zhu J., Kapoor A., Zhu J. K. Role of Arabidopsis AGO6 in siRNA accumulation, DNA methylation and transcriptional gene silencing. EMBO J. 2007;266(1):1691–1701. doi: 10.1038/sj.emboj.7601603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Kapoor A., Sridhar V. V., Agius F., Zhu J. K. The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr. Biol. 2007;176(1):54–59. doi: 10.1016/j.cub.2006.10.059. [DOI] [PubMed] [Google Scholar]
- Zilberman D., Henikoff S. Silencing of transposons in plant genomes: kick them when they're down. Genome Biol. 2004;56(1):249. doi: 10.1186/gb-2004-5-12-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D., Henikoff S. Genome-wide analysis of DNA methylation patterns. Development. 2007;1346(1):3959–3965. doi: 10.1242/dev.001131. [DOI] [PubMed] [Google Scholar]
- Zilberman D., Gehring M., Tran R. K., Ballinger T., Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007;396(1):61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]