

Abstract
Mitochondrial dysfunction has been implicated in chemical toxicities. The present study used an in vitro model to investigate the differential expression of metabolic pathways during cellular stress in p53-efficient embryonic fibroblasts compared to p53-deficient cells. These cell lines differed with respect to NADH/NAD+ balance. This ratio constitutes a driving force for NAD- and NADH-dependent reactions and is inversed upon exposure to Rotenone (complex I inhibitor). Rotenone perturbed the structure of the elongated fibrillar tubulin network and decreased mRNA expression of tubulin genes both suggesting reprogramming and reorganization of the cytoskeleton in both cell lines. These changes were reflected in the abundance of specific mRNA and microRNA (miRNA) species as determined from genome-based analysis. Changes in mRNA and miRNA expression profiles reflected differences in energy utilizing pathways, consistent with the notion that the p53 pathway influences the cellular response to mitochondrial dysfunction and that at least some control may be embedded within specific mRNA/miRNA networks in embryonic cells.
Keywords: p53, mitochondria, rotenone, miRNA, embryo, MEF, mouse
1. Introduction
The p53 tumor suppressor protein is a key player in the response to cellular stress (Vousden and Lane, 2007). p53 is a tetrameric transcription factor that trans-activates a number of regulatory genes leading to cell cycle arrest and apoptosis (el-Deiry et al., 1992; Harper et al., 1993; Jin and Levine, 2001). Although p53 has been widely studied in tumor development, it impacts developmentally regulated changes in the embryo. Inactivation of p53 is associated with depletion of mitochondrial DNA (mtDNA), neural tube defects and differential teratogenic sensitivity in early embryos (Kulawiec et al., 2008; Ibrahim et al. 1998; Sah et al., 1995; Wubah et al., 1996).
p53 impacts mitochondrial homeostasis. An early differential display analysis of normal p53 (+/+) and mutant p53 (−/−) mouse embryos on gestation day 8 revealed differences in the relative expression of 16S rRNA, a mtDNA genomic locus, and cellular respiratory deficiency among p53-deficient embryos (Ibrahim et al. 1998). The p53 (−/−) mutant embryos also displayed decreased cytochrome c oxidase staining and reduced ATP content. In addition, p53-deficient mouse embryonic fibroblast cells exhibited significant disruption of cellular ROS homeostasis (Lebedeva et al. 2009). Possible mechanisms of p53 action that may have led to these changes in mitochondrial biogenesis and/or oxidative metabolism include a direct action on the mitochondrion (Donahue et al. 2001), biophysical interaction with mtDNA and mitochondrial transcription factor A (Wong et al., 2009; Yoshida et al., 2003), and nuclear transactivation of specific genes imported to the mitochondrial respiratory chain (Matoba et al., 2006). Although it is well established that p53 activity supports mitochondrial biogenesis, less is known about definitive p53 cellular signaling pathways regulating the mitochondria. Conversely, genetic defects in mtDNA cause embryonic lethality, disease and cancer that directly involve p53. Therefore, the relationship between p53 and mitochondria was examined.
The electron transport chain (ETC) passes electrons through an electrochemical proton gradient that produces chemical energy in the form of ATP. The ETC consists of electron transfer complexes I-IV embedded in the inner mitochondrial membrane. These complexes pass electrons from reduced electron carriers such as NADH and FADH2 to ultimately drive ATP synthesis at ATP-synthase (complex V). NADH-ubiquinone oxidoreductase (complex I) catalyzes the first step in the respiratory chain. Structural and functional defects in complex I are characteristic of mitochondrial dysfunction and disease (DiMauro and Hirano, 2005). Maintenance of proper regulatory functions in the respiratory chain of mitochondria must be tightly coupled with the demand in cellular metabolism, oxygen homeostasis and ROS balance that ultimately leads to mitochondrial dysfunction. Therefore, a p53 response may occur when electron transfer is reduced in the mitochondrial respiratory chain.
We used a well-known environmental pollutant, Rotenone, as a highly-selective inhibitor of complex I (Chance et al. 1963) to assess the impact of p53 on programmed and inducible responses to mitochondrial dysfunction. Rotenone concentrations (0.1–100μM) showed inhibition of complex I-dependent respiration in NIH/3T3 cells (Yang et al. 2010; Trifunovic et al. 2005; MacKenzie et al. 2008). Micromolar ranges of Rotenone were used throughout these experiments in NIH/3T3 cells without severe cell damage. Compton et al. 2011 not only inhibited respiration at sub-micromolar levels, but also showed Rotenone-induced mitochondrial dysfunction suppressed p53 expression and function reversibly in human embryonic kidney cells. Recently, the EPA’s ToxCast™ project identified mitochondrial dysfunction and cytotoxicity as critical effects of Rotenone in a high-content screening in human hepatocyte cells. In addition, p53 induction and microtubule disruption were both identified as targets of Rotenone cellular bioactivity (http://www.epa.gov/ncct/toxcast/). Other mechanisms of Rotenone toxic action include oxidative damage and destabilization of the cellular microtubule network (Brinkley et al., 1974; Li et al., 2003; Srivastava and Panda, 2007). Rotenone was chosen as a mitochondrial stressor in this study because the interaction between p53 and the mitochondria may be triggered by Rotenone-induced mitochondrial dysfunction.
Wild-type (p53+/+) and p53-deficient (p53−/−) mouse embryonic fibroblast cell lines were selected as a prototype to develop a model pathway based on our understanding of the link between p53 and mitochondrial metabolism during embryo development. Since early stage p53 (+/+) and p53 (−/−) mouse embryos displayed significant differences in mitochondrial biogenesis, we wanted to determine p53-dependent mechanisms directly involved in regulating mitochondrial energy transition such as that observed in p53-deficient embryos exhibiting low oxidative metabolism. As such, little is known about how mitochondrial stress influences cell signaling pathways that are sensitive to p53 function.
In our study, cellular responses to Rotenone were monitored at different levels, including transcriptomic profiling of mRNA and miRNA. The latter are transcripts that regulate the translation of multiple proteins in a pathway (Ambros, 2004; Rhodes and Chinnaiyan, 2005; Lim et al. 2003; Meister and Tuschi, 2004). Many miRNAs fluctuate developmentally and play diverse roles in developmental programming or developmental buffering in response to environmental change (Hornstein and Shomron, 2006; Li et al. 2009). This suggests a key role for miRNAs in the regulation of embryonic development. Evidence supporting the function of miRNAs in developmental processes include differential expression profiles, similarities in expression patterns of miRNAs during development in mammals, cell culture studies and analysis of dicer mouse mutants. The elucidation of the spatial and temporal patterns of their mRNA and mircoRNA expression is critical for understanding precise roles of the mammalian miRNAs in development, particularly those that target p53.
Here, we investigated pathway-level regulation of mRNA and miRNA expression in normal and p53-deficient embryonic 3T3 cells during Rotenone exposure. Our findings reveal p53-dependent and -independent signaling pathways that mediate cellular responses to Rotenone. This data supports the p53 impact on mitochondrial biogenesis. We also found that mitochondrial dysfunction influences p53 localization and function in embryonic fibroblast cells. An approach to develop a model based on our understanding of the link between p53, mitochondrial metabolism, gene regulation and embryo development aids in our understanding of molecular mechanisms that contribute to the manifestation of birth defects.
2. Materials and Methods
2.1. Cell culture and reagents
NIH/3T3 mouse embryonic fibroblast (MEF) cells containing wild-type p53 and p53-deficient mouse embryonic fibroblasts were obtained from the American Type Culture Collection (ATTC, Manassas, VA) and grown at 37°C in a humidified atmosphere of 5% CO2 in either 4-well chamber slides or T-75 flasks (Fisher Scientific, Pittsburgh, PA). Cells were harvested at approximately 80% confluence based on cell counts; all studies performed between 2 and 15 passages using multiple stocks with multiple passages and all studies were run, at least, in triplicate. Cells were passaged every three days and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco/Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 0.025% gentamicin sulfate. Rotenone (CAS Number 83-79-4, PubChem Substance ID 24278683, ≥95% purity) and tissue culture grade dimethylsulfoxide (DMSO, ≥99.9% purity) were purchased from (Sigma-Aldrich, St. Louis, MO). Rotenone concentrations (0–1000nM) concentrations of the known range of its inhibitory action on NIH/3T3 cells were used (Yang et al. 2010).
2.2. Transient Transfection
Plasmids used in transfection studies were purified with HiSpeed Midi and Maxi Kits (Qiagen, Valencia CA). Dominant negative mitochondrial p53 mutant (m53DD) was previously described (Donahue et al, 2001). This vector encodes a mitochondrial-driven p53 dominant deletion protein that lacks its DNA transactivation domain while maintaining a capacity to interact with other proteins (Bowman et al., 1996). Mitochondrially targeted EGFP (mEGFP) or mitochondrially targeted wild-type p53 (m53-EGFP) vector contains the same mitochondria peptide leader sequence, cloned from the murine cytochrome c oxidase subunit Cox8l gene ligated to cytosolic p53-EGFP fragment. The pEGFP-N2 plasmid (Clontech) was the vector backbone for the m53-EGFP construct used for transfection. Plasmids (1:1 ratio, 3 μg), serum-free DMEM, and SuperFect Transfection Reagent (1 DNA: 5 SuperFect ratio) (Qiagen) were incubated for 10 min and complete DMEM was added to the suspension. Cells were incubated in transfection solution for 3 h; the solution was aspirated and replaced with complete DMEM. These vectors were transfected into the p53(−/−) cells that were then selected with gentamycin (Sigma-Aldrich, St. Louis, MO). Selected cells were treated with DMSO or Rotenone 500nM for 3 h before harvesting and analyzing NADH levels. Sham experiments used cultures with transfection solution, but without plasmid DNA. Gel purification allowed visualization and isolation of the vector band.
2.3. MTT assay
Cell viability was assessed in p53(+/+) and p53(−/−) MEF cells using the colorimetric reagent, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT, Sigma, St. Louis, MO). Cells were seeded in triplicate at 1 × 105 per well in 24-well plates. A stock solution of the dye was prepared, filter-sterilized and stored at −20°C. Cells were either exposed to Rotenone (0, 20, 100, 500, 1000nM) or 0.01% DMSO for 0, 3, 24 or 48 h. In a separate experiment, cells were treated with 500nM rotenone for 3 h and media replenished until 24 or 48 h. After treatments, MTT (2 mg/ml) was added to each well, and the incubation was continued for another 4 h. The supernatant was removed and the blue formazan product was solubilized with acidic isopropanol (0.04 M HCl in absolute isopropanol). Reduced MTT was measured at a wavelength of 560 nm using UltraSpec 3100 (GE Healthcare Bio-sciences, Piscataway, NJ). Relative formazan product was graphed.
2.4. NADH analysis
p53(+/+) and p53(−/−) MEF cells were either treated with DMSO vehicle or Rotenone (100, 500, or 1000 nM, 3 h). Cells were extracted with 60% methanol at −20°C and the supernatant was dried in a Savant Speedvac Concentrator System (Key Scientific, Mt. Prospect, IL). Samples were resuspended in mobile phase (50 mM ammonium phosphate monobasic, 2 mM tetrabutyl ammonium hydroxide, pH 6.5 with ammonium hydroxide and 5% acetonitrile) and analyzed by reverse-phase HPLC with ion-pairing using a Xterra RP18 5μm column (Waters, Milford, MA). Samples were monitored with UV wavelength collection between 249nm and 285nm (NAD+) and with in-line fluorescence excitation of 340nm and emission of 465nm (NADH) and compared to standards. Samples were also standardized by measuring their protein contents using the BCA method (Pierce, Rockford, IL) and bovine serum albumin standards were used to generate a calibration curve.
2.5. Reverse transcription/polymerase chain reaction (RT-PCR) Analysis
Total RNA was isolated from p53 +/+ and p53−/− cells using RNeasy (Qiagen, Valencia, CA) for four biological replicates. RNA was quantitated spectrophotometrically. After an 1 hour RNase-free DNase digestion, 2 μg of RNA was reversed transcribed with Superscript II Reverse transcriptase (Gibco-BRL, Gaithersburg, MD) at 42°C in the presence of dithiothreitol (DTT), random primers, and 0.2mM each dNTP. In each tube of Ready-to-go PCR beads (GE Healthcare Biosciences, Piscataway, NJ), 500 ng of cDNA and 2.5 μL of each primer were added, with a total volume of 25 μL. PCR cycles were 95°C (1 minute), 64°C (1 minute), and 72°C (1 minute). Reference gene (β-actin) and glycolytic target genes (LDH-A and LDH-B,) reactions used 27 cycles. The primers were: 5′-TAC CAC AGG CAT TGT GAT GG-3′ and 5′-AAT AGT GAT GAC CTG GCC GT-3′ for β-actin (310 bp), 5′-TGA CGC AGA CAA GGA GCA GTG-3′ and 5′-TCC CCA CAC CAT CTC AAC ACC-3′ for LDH-A (455 bp), 5′-GAT TCT GCT AGA TTT CGC TAC-3′ and 5′-CCA CAT TCA CAC CAC TCC-3′ for LDH-B (124 bp). PCR products were loaded on the DNA 1000 LabChip for quantification by the Agilent Bioanalyzer 2100. This application of the Bioanalyzer sizes and measures PCR double-stranded DNA fragments ranging from 25–1000 bp. Gel images are digitally created to replicate electrophoresis results.
2.6. Immunofluorescence for p53, cytochrome c, and tubulin
Cells were fixed treated with Rotenone (100nM for p53 staining and 500nM for tubulin staining) in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked with 10% BSA in phosphate-buffered saline (PBS), and incubated overnight with either polyclonal anti-p53, monoclonal anti-cytochrome c, or monoclonal anti-α-tubulin (1:1000; 1:200 dilution in 1% BSA/PBS) antibodies (Molecular Probes/Invitrogen, Carlsbad, CA) at 4°C in a humidified chamber. Slides were rinsed in PBS and later incubated with either AlexaFluor 488-tagged goat anti-rabbit IgG secondary antibody or AlexaFluor 594-tagged goat anti-mouse IgG (1:2000 dilution, Molecular Probes). Finally, the cells were counterstained with Hoechst 33342. Slides were mounted with ProLong Anti-fade (Molecular Probes). Fluorescence was observed with a Nikon E600 microscope for each individual channel and imaged using ACT software and Adobe Acrobat to superimpose three-color images. For p53 immunostaining, samples were scored for either nuclear or mitochondrial p53 localization using MetaMorph software. Localization of p53 was based on co-localization with compartmental markers Hoechst (nuclear) or MitoTracker Red (mitochondria). A total of two hundred cells were scored in 5–6 fields of view containing approximately 50 cells per field for each time point (0, 30, 60, 120 min) during Rotenone treatment (100 nM) or DMSO vehicle control, or after UV irradiation for 4 min as a positive reference control. Four biological replicates were run for each study.
2.7. Total RNA isolation
Cells were either exposed to Rotenone (500nM) or DMSO for 3 h for all PCR and microarray experiments. To isolate intact RNA and preserve mature miRNAs (<200 bp), total cellular RNA was isolated from MEF cells using Trizol Reagent (Invitrogen, Carlsbad, CA). Cells were scraped, homogenized and RNA extracted by acid-phenolchloroform method using mirVANA kit according to manufacturer’s protocol (Applied Biosystems/Ambion, Austin, TX). Total RNA was eluted with RNAse-free water (Applied Biosystems) and assessed for QA/QC using RNA 6000 Nano-LabChip on the Agilent 2100 Bioanalyzer. The NanoDrop ND-1000 (NanoDrop Technologies) was used for accurate measurement of concentrations (A260), protein contamination (ratio A260/280) or contamination with organic compounds (ratio A260/230). Each RNA sample passing QA/QC standards was divided for mRNA (Agilent) and miRNA (Exiqon) profiling. Less than 5-μg high-quality total RNA was needed.
2.8. Real-time RT- PCR
TaqMan miRNA assays were used to quantify miRNA abundance. In miRNA profiling experiments, each reverse transcriptase (RT) reaction contained 10 ng of purified total RNA, 50 nM stem–loop RT primer, 10XRT buffer, 100mM dNTPs, 50U/μl MultiScribe reverse transcriptase and 20U/μl RNase inhibitor (Applied Biosystems, Foster City, CA). The reactions were incubated for 30 min at 16°C, 30 min at 42°C, and 5 min at 85°C. Real-time PCR reactions for each miRNA (25 μl volume) were performed in quadruplicate, and each 25 μl reaction mixture included l.66 μl of diluted RT product (1:20 dilution), 12.5 μl of TaqMan Universal PCR Master Mix, 1.25 μl TaqMan miRNA Assay primer (mature sequences for snoRNA202: GCTGTACTGACTTGAT GAAAGTACTTTTGAACCCTTTTCCATCTGATG, hsa-miR34a: CAAUCAGCAAGUAU ACUGCCCU and mmu-miR34c; AGGCAGUGUAGUUAGCUGAUUGC), and 9.59 μl nuclease-free water, respectively (Applied Biosystems). Reactions were incubated in an Applied Biosystems 7900HT Fast Real-Time PCR system in 96-well plates at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 sec and 60 °C for 1 min. The threshold cycle (Ct) is defined as the fractional cycle number at which the fluorescence exceeds the fixed threshold of 0.2. The ratios of RNA species in each sample were determined in triplicate with the use of an ABI 7900HT TaqMan sequence detector following the standard curve method. snoRNA202 was used as internal control. Fold-change values were determined according to the relationship: fold-change = 2-ΔΔCt, where Ct is the threshold value for real-time PCR amplification detection, ΔCt is the difference in Ct for the same probe:primer pair and ΔΔCt = ΔCt, sample minus ΔCt (Livak and Schmittgen, 2001).
2.9. mRNA labeling and hybridization
For mRNA analysis, total RNA was shipped on dry-ice to COGENICS (Morrisville, NC, http://www.cogenics.com) for processing. A concentration of 500 ng of RNA was converted to labeled cRNA with oligo-dT-T7-promoter primer and T7 RNA polymerase in the presence of nucleotides coupled to a fluorescent dye (Cy5 test-sample, Cy3 reference-sample) using the Agilent Low RNA Input Linear Amp Kit (#5184-3523). After limited hydrolysis, labeled cRNA (750 ng) was hybridized to Agilent Whole Mouse Genome Oligonucleotide Microarrays, a comprehensive array with 44K probe features to 18K genes. Data were collected using Feature Extraction v9.1 (Agilent Technologies).
3.0. miRNA labeling and hybridization
For miRNA analysis, total RNA was shipped to Exiqon A/S services (Vedbaek, Denmark) for processing. The samples (2-μg) were labeled using the miRCURY Hy3/Hy5 labeling kit (Exiqon). Hybridization was based on locked-nucleic acid (LNA) modified oligos and does not require size fractionation. The miRCURY LNA Arrays (Exiqon, v 8.1) capture 2.6K vertebrate miRNAs in quadruplicate, of which 359 are annotated mouse miRNAs. Signals on the scanned slides were assessed by Exiqon to confirm capture of all control spike-in oligo-nucleotides.
3.1. Data Preprocessing
Various QA/QC metrics for mRNA (Agilent) and miRNA (Exiqon) profiling included internal positive (spike-in) and negative (unexpressed) controls. For variance modeling, datasets were subjected to multiple ANOVA using Partek Genomics Solution Software (http://www.partek.com/). A ‘source of variance’ plot mapped each sample and characterized the biological noise and procedural variance for the different parameters. Partek software containing a sophisticated de-noising algorithm, removed batch-effect variance associated with each array, dye channel, and array-dye interaction. The optimal normalization schema was selected based on plots of values of M (difference of log intensities) versus A (average of log intensities). Normalized datasets for the 48 mRNA arrays and 48 miRNA arrays were transformed to logarithm base 2 (log2). Quantified signals were adjusted in R using global Lowess (LOcally WEighted Scatterplot Smoothing) regression. The resultant data were scaled for each microarray to median = 0.00 and STDEV = 0.5.
3.2. Statistical analysis
For NADH quantitation and Relative and real-time-PCR analysis, differences among the treatment groups were analyzed by either Student’s Paired T-test or One-way ANOVA and p values less than 0.05 were taken as significant. Post hoc analysis was performed using the Dunnett’s test. The software package Prism 5.0d (GraphPad Software, Inc., La Jolla, CA) was used for these analyses. For p53 transfection experiments, the data was graphed in MS Excel 2003 and analyzed using Statistical Package for the Social Sciences version 16.0.
For microarray analysis, the data were normalized and analyzed using GeneSpring GX* 7.3 (Agilent Technologies) and Partek. Precision was based on variance components analysis (VCA) that calculates the standard error and P-value. VCA uses cross-replica and cross-gene error variations to give a mean expression. ANOVA was run with Benjamini and Hochberg False Discovery Rate as a beta correction. Summary statistics are run at a range of alpha levels (P= 0.001, 0.005, 0.01, 0.05) to achieve the desired sensitivity-specificity.
3.3. Predicting miRNA targets
The miRBase database (http://www.mirbase.org/) was utilized to make predictions of miRNA targets. The database represents a predicted hairpin portion of a miRNA transcript (termed mir in the database), with information on the location and sequence of the mature miRNA sequence (termed miR). The retrieved targets were then annotated using Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation tool (http://david.abcc.ncifcrf.gov/) to analyze the predicted miRNA targets.
3. Results
3.1. Alterations in cellular metabolism are p53-dependent
Previous studies in our laboratory have shown that p53 activity in the early embryo is necessary for developmental programming of mitochondrial biogenesis and/or oxidative metabolism (Ibrahim et al. 1998). Furthermore, the effect of p53 disruption is evident in NAD metabolism, because NADH content is lower in p53-deficient embryos compared to p53-competent counterparts. Rotenone concentrations of 100 and 500nM at 3 h were chosen for this study based on cell viability with the MTT assay (Supplementary Figure 1). Wild-type p53 and null embryonic fibroblasts also had significant difference in NADH content that simulated what had been observed in the embryo (Figure 1). Furthermore, when the fibroblasts were treated with Rotenone (500 nM, 3 h), p53 wild-type cells showed a dose-dependent decrease in NADH concentrations. This could be explained during inhibition of complex I inhibition by shunting of NADH to other metabolic reactions. On the other hand, p53-deficient cells showed the opposite response - an increase in cellular NADH levels, during Rotenone block (Figure 1A). This implies a bottleneck of substrate oxidation due to inhibition of complex I activity by Rotenone. The differential response was reproducible in independent assays based on ANOVA (n=3, *p<0.05) (Figure 1B). Therefore, the two cell lines responded quite differently to Rotenone block in terms of changes in NADH levels.
Fig. 1. Differences in metabolic markers correlate with p53 genotype.

A: Cells from wild-type p53 (+/+) mice treated with rotenone show a dose-dependent decrease in NADH content assessed by HPLC analysis. p53 (−/−) cells show an opposite effect with increased NADH levels upon rotenone (0, 100nM and 1000nM) treatment. B: Quantitative measurements show a remarkable inverted response to 500nM rotenone. Data were expressed as means ± SEM. Asterisk (*) indicates statistically significant *p ≤0.05 vs. p53 −/− control based on One-way ANOVA analysis. Post-hoc analysis was performed using the Dunnett’s test. C: Virtual gel reconstructed from electropherograms of PCR amplicon assay on the Agilent 2100 Bioanaylzer. mRNA gene expression levels were determined by relative RT-PCR for lactate dehydrogenase alpha and beta (ldha, ldhb) and actin (actb) control.
To further characterize the metabolic programming between the two cell lines, lactate dehydrogenase (LDH) mRNA levels were monitored using relative RT-PCR (Figure 1C and Supplementary Figure 2). Whereas LDH-A (Ldha) catalyzes the conversion of pyruvate to lactate by using NAD+ as a coenzyme, LDH-B (Ldhb) catalyzes the reverse reaction. The A-type subunit predominates in anaerobic tissues because NAD+ ion substitutes for oxygen as an electron acceptor. The B-type subunit predominates in aerobic tissues where NADH is regenerated from NAD+. The functional importance of LDH isozyme shifts is generally attributed to a need to increase A-subunit, which can derive more energy from pyruvate by reduction to lactate. PCR analysis indeed showed a greater level of Ldha transcripts in the p53-deficient cells compared to levels found in wild-type cells. Quantitative measurements comparing Ldha/Actb ratios between the two cell lines revealed a significant difference in gene expression by Student’s paired t-test (n=3, *p<0.005) (Supplementary Figure 2). These data support the notion that p53-deficient MEFs primarily utilize anaerobic metabolism under similar conditions as wild-type cells.
3.2. Comparative mRNA and miRNA profiles
Gene expression profiling was carried out using Agilent Whole Mouse Genome Oligonucleotide Microarrays in 4 pack format on a set of 12 total RNA samples (3 exposed and 3 vehicle control from each cell line). Profiling miRNA was carried out with miRCURY LNA Arrays that captured 2600 vertebrate miRNAs, of which 359 are annotated mouse miRNAs and over 640 human miRNAs. The normalized gene expression data were analyzed by two-way ANOVA for all 12 chips. Of 18K genes this identified 5712 differentially expressed at a corrected alpha cutoff (p-value) of 0.001. We observed the major source of variance between cell lines (5645 genes), whereas Rotenone treatment had only minor effect (67 genes), and generally in the same relative pattern displayed by the control cell lines. We observed no significant treatment × cell line interaction at alpha 0.001, although lowering the stringency of the summary statistics to alpha 0.05 yielded 164 genes that passed for a significant treatment × cell line interaction. Cluster analysis of these genes again showed the effects of Rotenone to be overshadowed by the cross-cell line differences. We then applied a less stringent statistical filter on either cell line independently in order to focus on the differential effects of Rotenone. This analysis detected 470 differentially expressed genes in p53 wild-type MEFs (alpha 0.005, no multi-comparison correction) and 1424 differentially expressed genes in the p53-deficient MEFs (alpha 0.005, correction applied).
Bioinformatic analysis (Table 1) showed enrichment of the Biocarta p53 signaling pathway, as might be expected, in p53 wild type cells. Other biological themes enriched in these cells included glutathione metabolism, TCA cycle, adipocytokines, ECM-receptor interaction, propanoate metabolism, and pentose phosphate pathway. By contrast, biological themes enriched in p53-deficient cells included adherens junction, focal adhesion, gap junction, tight junction, actin cytoskeleton, cell cycle, MAPK signaling, WNT signaling, and TGF-β signaling. These findings reflect marked differences inherent to p53(+/+) and p53(−/−) embryonic cell lines.
Table 1. Biological themes over-represented between mRNA (Agilent) and microRNA (Exiqon) microarray gene profiles.
mRNA Agilent Whole Mouse Genome Oligonucleotide Microarrays contain 44K probe features to 18K genes. The miRCURY LNA Arrays (Exiqon, v 8.1) capture 2.6K vertebrate miRNAs in quadruplicate, of which 359 are annotated mouse miRNAs. Normalized datasets were analyzed using GeneSpring GX* 7.3 (Agilent Technologies). For data analysis, ANOVA was run with Benjamini and Hochberg False Discovery Rate as a beta correction and summary statistics are run at a range of alpha levels (P= 0.05) to achieve the desired sensitivity-specificity. We scaled each microarray to median = 0.00 and STDEV = 0.5. Wild-type p53 (+/+) cells display 2263 mRNA genes and 2759 miRNAs involved in major metabolic pathways throughout the murine genome, whereas; p53 (−/−) cells display 1787 mRNA genes and 2805 miRNAs involved in major cellular morphology pathways, suggesting differences in pathway utilization.
KEGG pathways differentiating between p53 (+/+) and p53 (−/−) MEFs
| KEGG pathway | Derived by mRNA (Agilent) profiling | Translated by microRNA (Exiqon) profiling | ||
|---|---|---|---|---|
| gene count | p value | gene count | p value | |
| Wild-type > p53 null | 2263 genes (of 4085), 5 KEGGs of 14 | 2759 genes from 11 miRs, 5 KEGGs of 13 | ||
| glutathione metabolism | 16 | <0.001 | 16 | 0.002 |
| ribosome | 21 | 0.019 | 24 | 0.015 |
| axon guidance | 24 | 0.067 | 30 | 0.018 |
| pentose phosphate pathway | 8 | 0.032 | 9 | 0.026 |
| biosynthesis of steroids | 6 | 0.044 | 7 | 0.024 |
| p53 null > Wild-type | 1787 genes (of 4085), 4 KEGGs of 13 | 2805 genes from 8 miRs, 4 KEGGs of 11 | ||
| adherens junction | 22 | <0.001 | 17 | 0.042 |
| MAPK signaling pathway | 41 | 0.007 | 50 | 0.013 |
| cell cycle | 22 | 0.019 | 28 | 0.056 |
| GAG (N-glycan) degradation | 5 | 0.059 | 5 | 0.066 |
Enrichment of the genes altered by Rotenone showed a striking down-regulation of many tubulin genes, both the alpha and beta family (Figure 2). To confirm this effect at the cellular level, we stained MEF cells with anti-alpha tubulin antibody (Figure 2 and Supplementary Figure 3). The tubulin network was normally fibrous and expansive in the control cells. Rotenone treatment caused a disruption of the microtubule network and cellular detachment and rounding within 3 h, preceding mitotic failure and polyploidy (Supplementary Figure 3 and 4). However, the early effect on cell rounding and disruption of the microtubular network at 3 h coincided with down-regulation of tubulin mRNAs. These features were ultimately shown to be p53-independent because the same effects were observed in both p53(+/+) and p53(−/−) MEF cell lines. Rotenone-induced microtubule destabilization was reversed when treated media was replaced with fresh complete media at 3 h and cells further analyzed at 24 h (Supplementary Figure 3).
Fig. 2. Rotenone-induced changes in cytoskeletal morphology.

Tubulin genes were selected from the total array. A: mRNA gene expression analysis shows rotenone down-regulated tubulin genes in both strains. B, C: Immunofluorescence microscopy was used to detect cytoskeletal changes using monoclonal anti-alpha-tubulin observed with secondary anti-rabbit antibody in p53 (+/+) cells treated with either DMSO (B) or 500nM rotenone (C) for 3 h.
We condensed the tubulin-responsive genes to model a computational gene network for Rotenone using the ResNet Database (Pathway Architect). The central hubs in this network were the MAPK (MAP3K1) and NFKB (Figure 3). These pathways were elevated in the p53-deficient line (see Table 1) and may be a focal point for the cell rounding following mitochondrial dysfunction; however, this effect is p53-independent, because both cell strains were sensitive to Rotenone-induced tubulin disruption.
Fig. 3. Computational gene network prediction based on genes associated with the significant KEGG pathways.

Gene records for the significantly altered genes in the derived KEGG pathways were portrayed as an interacting network using Pathway Architect v1.20 (Stratagene). Gene products are coded in red and small molecules applied to this study (rotenone, active oxygen, H2O2) in green. Lines indicate curated interactions in the ResNet database for direct binding (purple), direct regulation (blue), and direct modulation (gray).
3.3. Identification of p53-dependent and independent miRNA transcripts
The normalized miRNA data were also analyzed by Two-way ANOVA to identify miRNA components of the p53 pathways. We compared miRNA expression profiles of p53 wild-type and p53-deficient cells (Figure 4). Transcripts were denoted as either having high expression levels (red blocks), neutral expression (black blocks) or low expression levels (green blocks). Although our analysis found 19 miRNAs differentially expressed by the two cell lines, 11 miRNAs were highly up-regulated (red blocks) in the p53(+/+) line compared to the down-regulated (green blocks) transcripts in the p53(−/−) line. Of these 11 miRNAs, 6652 potential gene targets were identified. Conversely, 8 miRNAs were highly regulated in the p53(−/−) line compared to the p53(+/+) line. These 8 miRNAs showed 4352 potential gene targets. Due to the large numbers of potential targets for cross-regulation by miRNA species, we performed a comparative pathway analysis from mRNA and miRNA targets to find common pathways being either up-regulated or down-regulated by Rotenone for each genotype. These pathways are listed in Table 1.
Fig. 4. Expression of miR-34 is correlated with p53 status and rotenone exposure in MEFs.

A: Heat-map of miRNA gene expression between wild-type p53(+/+) and p53(−/−) mouse embryonic fibroblast cells. GeneSpring v 7.0 analysis shows red denotes (up-regulated), black (neutral) and green (down-regulated) gene expression clusters. Twenty differentially regulated miRNAs clusters were amongst the two cell lines. B: miR34b and miR34c expression profiles were derived from Exiqon miRNA arrays in either p53 (+/+) (white columns) or p53 (−/−) (black columns) MEFs treated with DMSO or rotenone (500 nM, 3 h). C: Mature miR34a and miR34c levels were measured by Taqman assays in a similar manner. Results are presented with respect to DMSO treated controls. Data were expressed as means ± SEM (n=3). Statistically significant by Student’s Paired t-test (*p<0.05) (**p<0.005) differences in miR34 fold changes between p53 genotypes treated with rotenone. p53 status correlates with miR34 a,b and c expression levels.
Analysis of the miRNA microarray dataset also revealed that p53(+/+) MEFs had greater miRNA basal levels compared to that of p53(−/−) MEFs. In addition, treatment with Rotenone further elevated miR-34b and miR-34c in p53 (+/+) MEFs, but further decreased the transcript levels in the p53-deficient cells. To validate these data by a non-array method, we performed RT-PCR with hsa-miR-34a and mmu-miR-34c primers, since the human miR-34b primer available was not homologous to miR-34b in our murine system (Figure 4 B and C). As expected, in Rotenone-treated wild-type samples, miR34a expression showed a four-fold increase in gene expression compared to DMSO-vehicle controls. Conversely, a two-fold decrease in gene expression occurred in the p53-deficient samples for miR34a. The results were similar for miR-34c. The effect that Rotenone exposure had on miR34a and miR34c were significant for both normal and p53-deficient MEF cells by Student’s Paired t-test (*p<0.005).
Taking these data further, we utilized the miRbase to predict miR-34 target genes. This extrapolated 380 possible gene targets of miR34a-c. In order to identify whether a common biological theme existed amongst the collective miR34 target genes in our list, the gene list was annotated by DAVID to elevate KEGG pathways with the highest ranked biological themes. Many of the over-represented pathways from the predicted targets were involved in the p53-signaling pathway (cell-cycle) or cytoskeletal (cellular adhesion molecules) related. Representation of the p53 pathway in the list bolsters confidence in the data. In an attempt to identify a common underlying disease related to the genes in the dataset, the only OMIM (Online Mendelian Inheritance in Man) pathway mapped to the list was Parkinson’s disease (Table 2).
Table 2. Biological themes over-represented by miR34a-c targets.
miRbase revealed 380 miR34 gene targets. The Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation tool was then used to analyze the predicted miR34 targets for biological meaning behind the large list of targets. Genes are visualized by KEGG pathway maps. The database identified that most of the over-represented pathways from the predicted targets were cancer related, involved in the p53-signaling pathway (cell-cycle) or cytoskeletal (cellular adhesion molecules) related.
KEGG pathways identified by miR34 targets using miRNA classifier
| KEGG pathway | Derived by microRNA (Exiqon) profiling | Common OMIM disease target | |
|---|---|---|---|
| gene count | p value | ||
| miR34a-c | 380 possible gene targets, 21 KEGGs | ||
| prostate cancer | 8 | 0.006 | Parkinson’s disease |
| non small-cell lung cancer | 6 | 0.009 | |
| pancreatic cancer | 7 | 0.009 | |
| glioma | 6 | 0.017 | |
| axon guidance | 9 | 0.017 | |
| bladder cancer | 5 | 0.017 | |
| colorectal cancer | 7 | 0.019 | |
| focal adhesion | 11 | 0.027 | |
| p53 signaling | 6 | 0.027 | |
| melanoma | 6 | 0.029 | |
| notch signaling | 5 | 0.029 | |
| glycan structure | 8 | 0.030 | |
| biosynthesis 1 | 8 | 0.030 | |
| melanogenesis | 7 | 0.036 | |
| adherens junction | 6 | 0.037 | |
| inositol phosphate | 5 | 0.038 | |
| phosphatidyl inositol | 6 | 0.041 | |
| small-cell lung cancer | 6 | 0.067 | |
| Wnt signaling | 8 | 0.084 | |
| N-glycan biosynthesis | 4 | 0.088 | |
| Gap junction | 6 | 0.090 | |
3.4. Complex I inhibition induces p53 to localize to the mitochondrion
Immunofluorescence staining of p53 in wild-type MEFs (Figure 5) showed a weak p53 signal inherent to the mitochondrion at culture time 0 h as shown by co-localization with MitoTracker red. Since changes in cell morphology occurred with Rotenone (500nM) exposure, we chose a lesser concentration of Rotenone (100nM) for the p53 localization experiment. Following 100 nM Rotenone treatment, p53 levels increased in mitochondria within 1 h and further increased by 2 h (Figure 5). In contrast to Rotenone exposure, a UV light stimulus induced the expected nuclear p53 accumulation. This implies that upstream activating signals leading to mitochondrial translocation of the p53 protein differed from a classical nuclear DNA damage response. However, this was not accompanied by excessive apoptosis monitored by cytochrome c release from the mitochondrion (Supplementary Figure 5).
Fig. 5. Localization of endogenous p53.

A: Immunofluorescence staining of p53+/+ MEF cells were grown in 4-well chamber slides and either unexposed or exposed to 100 nM rotenone for 0, 30, 60, or 120 min, or UV light for 4 min. Green, p53 staining; red, MitoTracker Red (mitochondria); blue, Hoechst (nucleus). B: Cells were scored for either p53 localization in either mitochondria (white bars) or nuclear (black bars) compartments. C: Quantitative measurements of NADH in p53 (−/−) cells treated with rotenone 500nM for 3 h. Cells were transfected with the m53DD or m53-EGFP vectors. The results were standardized against each control treated with DMSO. Data were expressed as means ± SEM. Statistically significant (*p ≤0.05 vs. p53 −/−) based on One-way ANOVA analysis. Post-hoc analysis was performed using the Bonferroni correction for multiple comparisons.
To test whether mitochondrial p53 might mediate the NADH response to Rotenone block (e.g., decreased in wild-type cells, increased in p53-deficient cells), we generated a mitochondrion-specific wild-type p53 construct (m53-EGFP) (Supplementary Figure 6) and a mutant m53DD control construct (Donahue et al., 2001). Transfecting p53(−/−) cells with m53-EGFP drove the recombinant wild-type p53 protein to the mitochondrion. This significantly reversed the cellular NADH response during Rotenone, meaning that introducing wild-type p53 to the mitochondrion significantly reversed accumulation of NADH in the p53-deficient state (Figure 5C; p<0.05) based on One-way ANOVA analysis. We similarly transfected p53(−/−) cells with the dominant deletion mutant of p53, thus driving m53DD to the mitochondrion. The m53DD protein, unlike its wild-type counterpart, did not significantly restore the cellular NADH response to Rotenone block (Figure 5C; p=0.136). This implies that Rotenone-induced accumulation of NADH could be directly or indirectly linked with mitochondrial p53 function.
4. Discussion
We previously demonstrated a connection between p53 and activities encoded by the mtDNA genome in the early embryo (Ibrahim et al. 1998) and mitochondrial compartmentation in mouse embryonic fibroblast cells (Donahue et al. 2001). The experiments described further demonstrate that mitochondrial compartmentation of p53 has a differential effect on cellular NADH metabolism during mitochondrial dysfunction induced with Rotenone. We found that intermediary metabolism in mouse embryonic fibroblasts may be at least partly influenced by p53 based on the differential response in NADH content and LDHA expression in comparing wild-type and p53-deficient MEF lines. It is, of course possible that other factors may explain these differences since the cell lines tested here have different genetic backgrounds. We also found several differentially expressed miRNA transcripts dependent on the p53 background, including miR34 known to be p53 dependent (Bommer et al. 2007; Chang et al. 2007; He et al. 2007; Raver-Shapira et al. 2007; Tarasov et al. 2007). Recent studies showed that the silent information regulator (SIRT1), that functions as an NAD+-dependent p53 deacetylase and maintains metabolic homeostasis, can be inhibited by miR34 in a feedback loop (Yamakuchi and Lowenstein, 2009). SIRT1 deficient mice demonstrate embryonic developmental defects (Lee and Kemper, 2010; Tarantino et al. 2010; Saunders et al. 2011). Therefore, the pathways inferred from miRNA targets suggest a direct link to p53 regulated metabolism and embryonic health and development.
Evidence for p53 as a key player in the balance between aerobic respiration and glycolysis comes from a number of studies. For example, p53-deficient mouse embryos are at increased risk for exencephaly (Sah et al., 1995), a defect in neural tube closure that occurs during the critical time period when the embryo transitions from anaerobic (glycolytic) to aerobic (oxidative) bioenergy metabolism (Ibrahim et al. 1998). Results from a number of studies, independent of those focused on the embryo, showed that changes in glycolysis are a secondary effect of p53-regulated mitochondrial respiration and were previously reviewed by Ma et al. (2007). p53 is a central hub to this oxidative transition which was further supported by the work of Park et al. (2009). They showed that p53 promotes aerobic metabolism during exercise and this increases mitochondrial DNA content in mouse skeletal muscle. In addition, cellular insults that alter mitochondrial function can have important consequences on p53 activity. Supporting this notion is the finding that endogenous p53 in unstressed HCT116 colorectal carcinoma cells regulates the steady-state level of SCO2, a protein required for the assembly of cytochrome c oxidase, to facilitate aerobic respiration and in turn caused oxygen consumption to be lower in a preparation of liver mitochondria from p53-deficient mice (Matoba et al., 2006). Furthermore, that study showed a general correlation between p53 protein induction and an increased abundance of mtDNA-coded subunits of cytochrome c oxidase (COI and COII).
Indication of a functional link between cellular metabolism and mitochondrial p53 status was also demonstrated in cells containing recombinant and endogenous p53. Given the potential variability between cell lines derived from different p53 gene backgrounds, we evaluated the inherent response of the p53-deficient cells to recombinant p53, driven to the mitochondrion by signals for the respiratory chain component COVIIIL (cytochrome c oxidase subunit VIII-L). Transfection of mitochondrially targeted p53 reversed accumulation of NADH observed in untransfected p53-deficient cells. Rotenone promoted mitochondrial compartmentation of endogenous p53 in wild-type cells whereas, UV irradiation which promoted nuclear compartmentation. These findings also support the notion that p53 may vary the cellular response to mitochondrial dysfunction through a direct effect on mitochondrial metabolism.
Rotenone-induced p53 translocation to the mitochondria suggests a novel connection that involves key alterations in nicotinamide adenine dinucleotide (NAD) metabolism. Previous studies showed that preferential utilization of NADH by LDHA in the cytosol impedes mitochondrial respiration (Fantin et al., 2006). In addition, it has been hypothesized that in oxygen-depleted cells, mitochondria are depolarized and the F1, F0-proton ATPase catalyzes net ATP hydrolysis when the cells exhibit the signs of an aerobic-to-anaerobic metabolic transition, such as increased lactate formation and decline in high-energy phosphate reserves (Rouslin et al., 1990). Differences in how p53 wild-type and deficient embryonic cells adapt to changes in oxygen utilization may be correlated with altered metabolism. Our findings are consistent with the notion that p53-deficient cells may utilize glycolysis compared to p53 wild-type cells that utilize mitochondrial metabolism. Rotenone inhibits respiratory complex I (NADH:ubiquinone dehydrogenase) and may disrupt the flux of NAD/NADH. Chemical-induced p53 can bind NAD(P)H:quinone oxidoreductase1, a reductase that catalyzes the reduction of substrates and utilizes NADH and NADPH as reducing cofactors. Binding activates and stabilizes p53 because it is protected from 20S proteosomal degradation (Gong et al., 2007). Accumulation of NADH in p53-deficient cells treated with Rotenone, and the rescue of this effect by mitochondrial transfection with wild-type p53, suggest that loss of p53 function may impair the cellular adaptation to mitochondrial dysfunction. This could also reflect uncoupled respiration in p53-deficient cells (Ricquier, 2005), or less likely, a reversal of electron flow (to NADH) as in some anaerobic systems (Heinen et al., 2007). Khutornenko et al. (2010) showed that a deficiency in pyrimidine biosynthesis was responsible for activation of p53 in human A549 lung adenocarcinoma cells and HCT116 colorectal cancer cells. However, Rotenone induced p53 to a lesser extent than the complex III inhibitor, Myxothiazol.
We identified a number of mRNAs that participate in pathways that could mediate cellular adaptive responses to mitochondrial dysfunction independent of p53 status. Besides complex I inhibition, another mechanism of Rotenone toxic action is to depolymerize microtubules (Brinkley et al., 1974), and the present study captures this. Although the expression network showed a Rotenone-induced response, tubulin disruption linked with mitochondrial dysfunction was not apparent. Rotenone exposure mediated down-regulation of tubulin gene expression. Although we did not observe any significant changes in cellular proliferation or viability between the two cell-lines treated with Rotenone, we cannot rule out that these changes would not have occurred at higher concentrations. When we condensed the genes associated with tubulin to build a computational gene network for mitochondrial dysfunction, the most proximate nodes were the Mitogen-activated protein kinases, originally known as microtubule-associated protein 2 kinases (Rossomondo et al., 1989). Specifically, MAP3K1 and NFKB were elevated from the dataset as potentially important focal points in the response to mitochondrial dysfunction. p53 can regulate NFKB-dependent cellular responses (Ravi et al. 1998). Knocking down MAPK kinase (MKK) blocks MAPK activation in midbrain dopaminergic neurons and significantly reduced the toxicity of rotenone or colchicine (Ren et al. 2009). In addition, these authors found that over-expression of MAPKs caused marked toxicities that were significantly attenuated by the Parkin protein, whose mutations are linked to Parkinson’s disease (Ren et al., 2009). Parkin is a protein-ubiquitin isopeptide ligase (E3) that binds and stabilizes alpha/beta tubulin heterodimers and microtubules (Yang et al., 2005). These data suggest that MAPK activation may play an important role in signaling events related to stabilization of microtubules and morphological adaptations of the cell.
The miRNA microarray data reported here suggest that changes in gene expression, such as cell-cycle transcript levels, are mediated by p53. For instance, miR-34 data analysis predicted a number of downstream cell-cycle targets, such as cyclin D1 and cyclin E2. Our data suggest that several sets of miRNAs may work in concert to intricately regulate cell-cycle progression, and therefore may not depend solely on one single miR transcript. Nonetheless, these data lead us to speculate that the Rotenone pathway to mitochondrial dysfunction may be embedded within miRNA networks.
In summary, we modeled these results for p53-dependent and independent effects during Rotenone exposure (Figure 6). Data from p53(+/+) and p53(−/−) mouse embryonic fibroblasts mirror our previous findings in the early embryo during a period in development when rapid growth depends on mitochondrial biogenesis. Based on previous results from our lab, we expected that the mere presence of p53 in the cell would change its metabolic profile. Here, we aimed to identify signaling pathways that mediate this p53-dependent response. Interestingly, intricate interactions with mRNAs and miRNAs emerged as metabolic regulators and in turn, such precise cellular processes are necessary for proper embryonic growth and development. Our data not only showed p53 loss influences mitochondrial activity (metabolism), but conversely, mitochondrial stress directly influences p53 localization and function. Together this information suggests that the p53 molecule has an intimate relationship with the mitochondrial organelle such that their interaction allows the cell to compensate and adapt to stress in order to continue healthy growth and development of the whole embryo. The conceptual model also supports the notion that mitochondrial dysfunction and redistribution depends upon an intact microtubule network (Richter-Landsberg 2001) that is independent of p53 and may be mediated by MAP kinase signaling. A large-scale mitochondrial screening compendium uncovered a link between microtubule disruption and an increase in the expression of genes for mitochondrial oxidative phosphorylation while suppressing ROS (Wagner et al. 2008). Further understanding of pathways regulated by specific mRNA and miRNA interactions may give us insight into how mitochondrial dysfunction influences p53 activity in developing embryonic tissues.
Fig. 6. Model for rotenone exposure in mouse embryonic fibroblast cells.

Differences between p53 (+/+) and p53 (−/−) cells show sensitivity to NADH imbalance and gene expression, but not their modes of action upon rotenone exposure, suggesting a role for p53 in cellular programming.
Supplementary Material
MTT assay was used to measure cell viability in p53 (+/+) and p53 (−/−) MEFS in a rotenone dose-response and time-course manner. A: MEF cells were treated with varying concentrations of rotenone (0–1000nM) over a 24 and 48 h period. B: MEF cells were treated with 500nM rotenone for 3 h. MTT assay was assessed 24 and 48 h after media replenished at 3 h.
{kind=link}
Virtual gels reconstructed from electrophorograms of PCR amplicon assay on the Agilent 2100 Bioanaylzer. Panel A: β-Actin, LDH-A, and LDH-B. Ladder (15–1500 bp) found in first lane of all three gels. Lanes 1–2 or 9–10 are untreated p53+/+ MEFs and Lanes 3–4 or 11–12 are p53−/− MEFs. Panel B: Relative gene expression level for LDH-A was normalized to control (β-actin) gene expression. LDH-A expression was significantly increased in p53(−/−) MEFs compared to p53 (+/+) based on Student’s Paired T-Test (n=4, *p ≤0.05).
{kind=link}
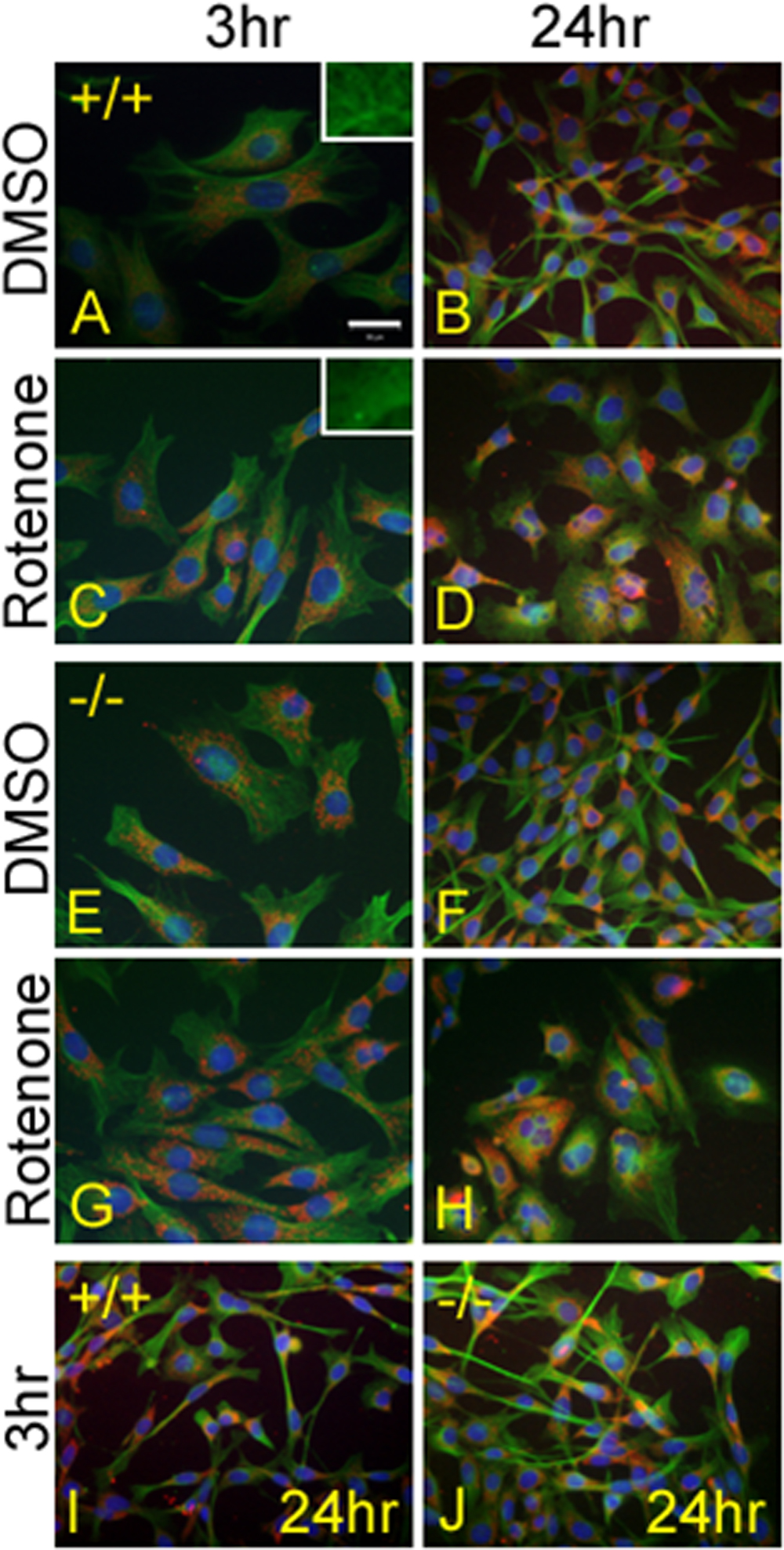
Images of p53 (+/+) (A-D) or p53 (−/−) cells (E-H) treated with either DMSO (A, B, E, F) or 500 nM rotenone (C, D, G, H) for 3 or 24 h. A, B, E, F. Cells appear to have a normal cell structure when treated with DMSO for 3 and 24 h in both p53 (+/+) and p53−/−cells. C, G. After 3 h of rotenone treatment the cells appear normal in structure. The microtubules are slightly fragmented and depolymerized, however; overall structure is not affected greatly (see insets). D, H. After 24 h rotenone treatment there are far fewer cells than in the respective DMSO treated slides. The cells have also failed to divide causing an increase in both nuclear and cytoplasmic size. Polyploidy of the nuclei can be seen following this extended exposure to rotenone treatment. I, J. Cells that were treated for 3 h followed by a 21 h incubation period in normal media appeared normal, indicating that the effects of rotenone can be reversed. Calibration mark: 50 microns.
{kind=link}
Cells were treated with 500nM rotenone for 24 h and then harvested by trypsinization. The analysis showed a substantial number of cells were arrested in the M/S interphase (Sub-G2) of cell division upon rotenone treatment thus increasing the amount of DNA found within each cell, explaining the polyploidy phenotype.
{kind=link}
Panel A: Immunofluorescent staining of mitochondrial localized cytochrome c (red) in rotenone exposed (100 nM, 1.5 hr) p53 null MEFs. Panel B: Mitochondrial marker mEGFP (green) in pre null transfected cells. Panel C: Colocalization of mitochondrial cytochrome c and mEGFP.
{kind=link}
Merged image of in rotenone exposed (500 nM, 1.5 hr) fibroblasts transfected with the m53-EGFP construct (green) and counterstained with MitoTracker Red to reveal mitochondria localization. Co-localization of p53 and the MitoTracker appears as yellow on the merged image.
{kind=link}
Acknowledgments
The authors would like to thank the members of Dr. Knudsen’s laboratory: Marjaneh Razmara, Nafeesa Owens, Reetu Singh and Jennifer Case for their contributions to this research. Special thanks to Dr. Robert MacPhail at EPA/NHEERL for helpful comments on the manuscript.
Funding
This research was supported by the NIH grants RO1-AA13205, RO-1ES09120 and T32-ES07282 and a pilot grant from the University of Louisville Center for Genetics and Molecular Medicine.
Footnotes
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Conflict of interest statement
None declared.
References
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- Bowman T, Symonds H, Gu L, Yin C, Oren M, Van Dyke T. Tissue- specific inactivation of p53 tumor suppression in the mouse. Genes Dev. 1996;10(7):826–35. doi: 10.1101/gad.10.7.826. [DOI] [PubMed] [Google Scholar]
- Brinkley B, Barham S, Barranco S, et al. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Exp Cell Res. 1974;85(1):41–46. doi: 10.1016/0014-4827(74)90210-9. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR, Hollunger G. Inhibition of electron and energy transfer in mitochondria. I. Effects of Amytal, thiopental, rotenone, progesterone, and methylene glycol. J Biol Chem. 1963;238:418–31. [PubMed] [Google Scholar]
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton S, Kim C, Griner NB, et al. Mitochondrial dysfunction impairs tumor suppressor p53 expression/function. J Bio Chem. 2011;286(23):20797–20312. doi: 10.1074/jbc.M110.163063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Hirano M. Mitochondrial encephalomyopathies: an update. Neuromusculo Disord. 2005;15:276–286. doi: 10.1016/j.nmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Donahue R, Razmara M, Hoek J, et al. Direct influence of the p53 tumor suppressor on mitochondrial biogenesis and function. FASEB. 2001;15:635–644. doi: 10.1096/fj.00-0262com. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, et al. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- Fantin V, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Gong X, Kole L, Iskander K, Jaiswal AK. NRH:quinone oxidoreductase 2 and NAD(P)H:quinone oxidoreductase 1 protect tumor suppressor p53 against 20s proteasomal degradation leading to stabilization and activation of p53. Cancer Res. 2007;67(11):5380–5388. doi: 10.1158/0008-5472.CAN-07-0323. [DOI] [PubMed] [Google Scholar]
- Harper J, Adami G, Wei N, et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen A, Aldakkak M, Stowe D, et al. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am J Physiol Heart Circ Physiol. 2007;293:H1400–H1407. doi: 10.1152/ajpheart.00198.2007. [DOI] [PubMed] [Google Scholar]
- Hornstein E, Shomron N. Canalization of development by microRNAs. Nature Gen. 2006;38:S20–S24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- Ibrahim M, Razmara M, Nguyen D, et al. Altered expression of mitochondrial 16S ribosomal RNA in p53-deficient mouse embryos revealed by differential display. Biochem Biophys Acta. 1998;1403:254–264. doi: 10.1016/s0167-4889(98)00066-4. [DOI] [PubMed] [Google Scholar]
- Jin S, Levine AJ. The p53 functional circuit. J Cell Sci. 2001;114:4139–4120. doi: 10.1242/jcs.114.23.4139. [DOI] [PubMed] [Google Scholar]
- Khutornenko AA, Roudko VV, Chernyak BV, et al. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Nat Acad Sci. 2010;107(29):12828–33. doi: 10.1073/pnas.0910885107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulawiec M, Safina A, Desouki M, et al. Tumorigenic transformation of human breast epithelial cells induced by mitochondrial DNA depletion. Cancer Biol Ther. 2008;7(11):1732–1743. doi: 10.4161/cbt.7.11.6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta. 2009;1787(5):328–334. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kemper JK. Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging. 2010;2(8):527–534. doi: 10.18632/aging.100184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez J, Robinson J. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. A microRNA imparts robustness against environmental fluctuation during development. Cell. 2009;137:273–282. doi: 10.1016/j.cell.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Glasner ME, Yekta S. Vertebrate microRNA genes. Science. 2003;7;299(5612):1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma W, Sung HJ, Park JY, et al. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr. 2007;39(3):243–246. doi: 10.1007/s10863-007-9083-0. [DOI] [PubMed] [Google Scholar]
- MacKenzie EL, Ray PD, Tsuji Y. Role and regulation of ferritin H in rotenone-mediated mitochondrial oxidative stress. Free Radical Biology and Medicine. 2008;44(9):1762–1771. doi: 10.1016/j.freeradbiomed.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang J, Patino W, et al. p53 Regulates mitochondrial respiration. Science. 2006;312(16):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Meister B, Tuschi T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- Park JY, Wang PY, Matsumoto T, et al. P53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res. 2009;105(7):705–712. doi: 10.1161/CIRCRESAHA.109.205310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Ravi R, Mookerjee B, van Hensbergen Y, et al. p53-mediated repression of nuclear factor-kappaB RelA via the transcriptional integrator p300. Cancer Res. 1998;58(20):4531–4536. [PubMed] [Google Scholar]
- Ren Y, Jiang H, Yang F, et al. Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation. J Biol Chem. 2009;284(6):4009–4017. doi: 10.1074/jbc.M806245200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Chinnaiyan AM. Integrative analysis of the cancer transcriptome. Nat Genet. 2005;37(Suppl):S31–7. doi: 10.1038/ng1570. Review. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C. Organization and functional roles of the cytoskeleton in oligodendrocytes. Microsc Res Tech. 2001;53:628–636. doi: 10.1002/jemt.1047. [DOI] [PubMed] [Google Scholar]
- Ricquier D. Respiration uncoupling and metabolism in the control of energy expenditure. Proc Nutr Soc. 2005;64:47–52. doi: 10.1079/pns2004408. [DOI] [PubMed] [Google Scholar]
- Rouslin W, Broge CW, Grupp IL. ATP epletion and mitochondrial functional loss during ischemia in slow and fast heart-rate hearts. Am J Physiol. 1990;259(6 Pt 2):H1759–66. doi: 10.1152/ajpheart.1990.259.6.H1759. [DOI] [PubMed] [Google Scholar]
- Rossomando AJ, Payne DM, Weber MJ, Sturgill TW. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc Natl Acad Sci. 1989;86:6940–6943. doi: 10.1073/pnas.86.18.6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah V, Attardi L, Mulligan G, et al. A subset of p53-deficeint embryos exhibit exencephaly. Nat Genet. 1995;2:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- Saunders LR, Sharma AD, Tawney J, et al. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging. 2011;2(7):415–431. doi: 10.18632/aging.100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P, Panda D. Rotenone inhibits mammalian cell proliferation by inhibiting microtubule assembly through tubulin binding. FEBS J. 2007;274(18):4788–4801. doi: 10.1111/j.1742-4658.2007.06004.x. [DOI] [PubMed] [Google Scholar]
- Tarantino C, Paolella G, Cozzuto L, et al. miRNA 34a, 100, and 137 modulate differentiation of mouse embryonic stem cells. FASEB J. 2010;24(9):3255–3263. doi: 10.1096/fj.09-152207. [DOI] [PubMed] [Google Scholar]
- Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. PNAS. 2005;102(50):17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nature Rev. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Wagner B, Kitami T, Gilbert T, et al. Large-scale chemical dissection of mitochondrial function. Nature Biotechnology. 2008;26(3):343–351. doi: 10.1038/nbt1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong T, Rajagopalan S, Townsley F, et al. Physical and functional interactions between human mitochondrial single-stranded DNA binding protein and tumour suppressor p53. Nucleic Acid Res. 2009;37(2):568–81. doi: 10.1093/nar/gkn974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wubah J, Ibrahim M, Gao X, et al. Teratogen-induced eye defects mediated by p53-dependent apoptosis. Curr Biol. 1996;6(1):60–69. doi: 10.1016/s0960-9822(02)00422-0. [DOI] [PubMed] [Google Scholar]
- Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009;8(5):712–715. doi: 10.4161/cc.8.5.7753. [DOI] [PubMed] [Google Scholar]
- Yang F, Jiang Q, Zhao J, et al. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J Biol Chem. 2005;280:17154–17162. doi: 10.1074/jbc.M500843200. [DOI] [PubMed] [Google Scholar]
- Yang D, Wang MT, Tang Y, et al. Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RAS(Q61L) Cancer Biol Ther. 2010;9(2):122–133. doi: 10.4161/cbt.9.2.10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Izumi H, Torigoe T, et al. p53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res. 2003;63:3729–3734. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MTT assay was used to measure cell viability in p53 (+/+) and p53 (−/−) MEFS in a rotenone dose-response and time-course manner. A: MEF cells were treated with varying concentrations of rotenone (0–1000nM) over a 24 and 48 h period. B: MEF cells were treated with 500nM rotenone for 3 h. MTT assay was assessed 24 and 48 h after media replenished at 3 h.
Virtual gels reconstructed from electrophorograms of PCR amplicon assay on the Agilent 2100 Bioanaylzer. Panel A: β-Actin, LDH-A, and LDH-B. Ladder (15–1500 bp) found in first lane of all three gels. Lanes 1–2 or 9–10 are untreated p53+/+ MEFs and Lanes 3–4 or 11–12 are p53−/− MEFs. Panel B: Relative gene expression level for LDH-A was normalized to control (β-actin) gene expression. LDH-A expression was significantly increased in p53(−/−) MEFs compared to p53 (+/+) based on Student’s Paired T-Test (n=4, *p ≤0.05).
Images of p53 (+/+) (A-D) or p53 (−/−) cells (E-H) treated with either DMSO (A, B, E, F) or 500 nM rotenone (C, D, G, H) for 3 or 24 h. A, B, E, F. Cells appear to have a normal cell structure when treated with DMSO for 3 and 24 h in both p53 (+/+) and p53−/−cells. C, G. After 3 h of rotenone treatment the cells appear normal in structure. The microtubules are slightly fragmented and depolymerized, however; overall structure is not affected greatly (see insets). D, H. After 24 h rotenone treatment there are far fewer cells than in the respective DMSO treated slides. The cells have also failed to divide causing an increase in both nuclear and cytoplasmic size. Polyploidy of the nuclei can be seen following this extended exposure to rotenone treatment. I, J. Cells that were treated for 3 h followed by a 21 h incubation period in normal media appeared normal, indicating that the effects of rotenone can be reversed. Calibration mark: 50 microns.
Cells were treated with 500nM rotenone for 24 h and then harvested by trypsinization. The analysis showed a substantial number of cells were arrested in the M/S interphase (Sub-G2) of cell division upon rotenone treatment thus increasing the amount of DNA found within each cell, explaining the polyploidy phenotype.
Panel A: Immunofluorescent staining of mitochondrial localized cytochrome c (red) in rotenone exposed (100 nM, 1.5 hr) p53 null MEFs. Panel B: Mitochondrial marker mEGFP (green) in pre null transfected cells. Panel C: Colocalization of mitochondrial cytochrome c and mEGFP.
Merged image of in rotenone exposed (500 nM, 1.5 hr) fibroblasts transfected with the m53-EGFP construct (green) and counterstained with MitoTracker Red to reveal mitochondria localization. Co-localization of p53 and the MitoTracker appears as yellow on the merged image.