

Background: The heterogeneity of substrains of Bacillus Calmette-Guérin (BCG), the only available tuberculosis vaccine, was studied.
Results: BCG Tokyo 172 types I and II were different in morphology and lipid composition.
Conclusion: Type II lacks phenolic glycolipid/phthiocerol dimycocerosate due to a ppsA mutation, and its phenotype was implicated in host responses.
Significance: The lipid phenotype is important to evaluate the efficacy of BCG vaccines.
Keywords: Lipids, Mycobacterium tuberculosis, Toll-like Receptors (TLR), Tuberculosis, Vaccines, Bacillus Calmette-Guérin (BCG), Phenolic Glycolipid, Phthiocerol Dimycocerosate, Tuberculosis, Vaccine
Abstract
Bacillus Calmette-Guérin (BCG) Tokyo 172 is a predominant World Health Organization Reference Reagent for the BCG vaccine. Recently, the BCG Tokyo 172 substrain was reported to consist of two subpopulations with different colony morphologies, smooth and rough. Smooth colonies had a characteristic 22-bp deletion in Rv3405c of the region of difference (RD) 16 (type I), and rough colonies were complete in this region (type II). We hypothesized that the morphological difference is related to lipid phenotype and affects their antigenicity. We determined the lipid compositions and biosynthesis of types I and II. Scanning electron microscopy showed that type I was 1.5 times longer than type II. Phenolic glycolipid (PGL) and phthiocerol dimycocerosate (PDIM) were found only in type I. Although it has been reported that the RD16 is involved in the expression of PGL, type II did not possess PGL/PDIM. We examined the ppsA-E gene responsible for PGL/PDIM biosynthesis and found that the existence of PGL/PDIM in types I and II is caused by a ppsA gene mutation not regulated by the RD16. PGL suppressed the host recognition of total lipids via Toll-like receptor 2, and this suggests that PGL is antigenic and involved in host responses, acting as a cell wall component. This is the first report to show the difference between lipid phenotypes of types I and II. It is important to clarify the heterogeneity of BCG vaccine substrains to discuss and evaluate the quality, safety, and efficacy of the BCG vaccine.
Introduction
Tuberculosis (TB)2 is a major public health problem in the world. The World Health Organization (WHO) estimates that one-third of the world's population is infected with Mycobacterium tuberculosis and annually reports the global burden of the disease caused by TB (1). Moreover, the global spread of drug-resistant mycobacteria and the number of immunocompromised hosts, including victims of the human immunodeficiency virus (HIV) epidemic, are important problems. The Mycobacterium bovis Bacillus Calmette-Guérin (BCG) vaccine is the only available TB vaccine licensed under the WHO Expanded Program on Immunization. The BCG vaccine was first introduced in 1921 and has been used to prevent TB for nearly a century. The BCG strain was disseminated and maintained in different laboratories until the introduction of archival seed lots in the 1960s.
Currently, more than 14 BCG substrains are used throughout the world, and the heterogeneity of their genomics has been studied. These strains are different from each other and the original BCG strain and possibly affect the efficacy of the vaccine (2–5). All BCG substrains lack the region of difference (RD) 1. The BCG substrains are divided into early (represented by BCG Tokyo, Birkhaug, Sweden, and Russia) and late (including BCG Pasteur, Danish, Glaxo, and Prague) strains by the deletions and insertions of genes plus some single nucleotide polymorphisms (SNPs) of RDs (6). The first WHO International Reference Preparation for the BCG vaccine was established in 1965, and this International Reference Preparation was produced using BCG Tokyo 172. Based on collective international expert opinions from three WHO consultation meetings on the BCG vaccine in 2005, there is a demand for replacement of the first WHO International Reference Preparation for the BCG vaccine with substrain-specific BCG Reference Reagents. Three different substrains of BCG, Tokyo 172-1, Russian BCG-I, and Danish 1331, were identified and represent the predominant substrains used for BCG vaccine production and distribution worldwide (7). BCG Tokyo 172 strain has kept the position of International Reference Preparation or Reference Reagent for over 40 years, and the quality of this vaccine preparation is the best estimation of the viability and stability in the three Reference Reagent substrains (8–10).
Bedwell et al. (11) showed two genotypes in the BCG Tokyo 172 preparation that either have or do not have a characteristic 22-bp deletion in RD16. Honda et al. (12) identified two types of colony morphology, smooth and rough. In most cases, the smooth colonies showed a 22-bp deletion in Rv3405c of RD16 (type I), and rough colonies were complete in this region (type II). Both genotypes and colony morphologies have been found in common BCG Tokyo 172 vaccine preparations, but the major population was always type I.
For global BCG vaccination policy and practice, it is important to understand the heterogeneity in the morphology and genetics of BCG substrains (4, 13). The most common characteristic of acid-fast bacteria is that they are rich in lipids, and this may be reflected in the morphology (14, 15). The differences in the nature of the BCG Tokyo 172 subpopulation types I and II and vaccine efficacy were not fully understood except for the mutation in RD16. In this study, we demonstrate the heterogeneity of lipid components in types I and II and clarify the lipid phenotypes, their biosynthesis, and their functions.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
BCG Tokyo 172 subpopulation types I and II were isolated on Middlebrook 7H10 agar medium (Difco) with 0.5% glycerol and 10% Middlebrook oleic acid-albumin-dextrose-catalase enrichment (Difco) at 37 °C for 2 weeks (12). BCG Moreau, Connaught (ATCC 35745), and Pasteur (ATCC 35734) substrains and parent M. bovis Ushi 10 were used as reference strains. The identities and history were described previously (3).
Rv3405c of RD16
Bacterial cells were disrupted mechanically, and genomic DNA was extracted with phenol-chloroform followed by precipitation with ethanol. To examine Rv3405c of the RD16, PCR was performed with primers RD16-F (5′-GGCTGGTGTTTCGTCACTTC-3′) and RD16-R (5′-ACATTGGGAAATCGCTGTTG-3′).
Colony Morphology and Scanning Electron Microscopy
Colony morphology was observed using an optical microscope after incubation for 2 weeks on 7H10 agar plates. To prepare the bacteria for scanning electron microscopy, types I and II were grown in 7H9 broth with shaking to the midlog phase. The bacteria were collected, passed through a 5-μm membrane filter to remove aggregates, and then put on coverslips (Thermanox Plastic, 13-mm diameter; Thermo Fisher Scientific, Rochester, NY) coated with 0.1% (w/v) poly-l-lysine solution. The bacteria were fixed with 2.5% glutaraldehyde (TAAB Laboratories Equipment Ltd., West Berkshire, UK) in 0.1 m phosphate buffer (pH 7.2) for 2 h at room temperature, washed twice with 0.1 m phosphate buffer, and postfixed with 1% osmium tetroxide (TAAB Laboratories Equipment Ltd.) in 0.1 m phosphate buffer for 1 h at room temperature. The bacteria were then dehydrated in a graded ethanol series. After permutation with 3-methylbutyl acetate, critical point drying was performed using a Hitachi HCP-2 Critical Point Dryer (Hitachi High-Technologies Corp., Tokyo, Japan). Scanning electron microscopy was performed at ×5,000 in a Hitachi FE-SEM S-4700 instrument (Hitachi High-Technologies Corp.) using an accelerating voltage of 10 kV (16).
Analysis of Mycolic Acid Compositions
Mycolic acids (MAs) were analyzed as described previously (17). In brief, MAs of each subpopulation were liberated from the bacteria by alkaline hydrolysis using 10% potassium hydroxide in methanol at 90 °C for 2 h followed by extraction with n-hexane. After treatment with diazomethane, the methyl ester derivatives of total MAs were detected by thin-layer chromatography (TLC) on Silicagel G (Uniplate; 20 × 20 cm, 250 μm; Analtech, Inc., Newark, DE), which was developed with n-hexane-diethyl ether (90:15, v/v; three runs) and visualized by spraying with 20% sulfuric acid in ethanol and heating at 180 °C for 3 min. The ratio of each MA subclass was measured and calculated with ImageJ software. For each MA subclass, α-, methoxy-, and keto-MA was purified by preparative TLC until a single spot was obtained. The molecular species of the MA methyl esters were detected by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS).
Comparisons of Fatty Acids and Carbohydrates
To determine the bacterial fatty acid (FA) and carbohydrate compositions, gas chromatography-mass spectrometry (GC/MS) of FA methyl ester, trimethylsilyl ester, and alditol acetate derivatives was performed (18, 19). The bacteria were sonicated, and total lipids were extracted with chloroform-methanol (2:1, v/v). The lipids were partitioned by a two-layer system with chloroform-methanol (2:1, v/v) and water. The organic phase was evaporated. Bacterial cells and total lipids were dried followed by the alkaline hydrolysis with 15% sodium hydroxide in 50% methanol at 100 °C for 72 h. After acidification by 6 m hydrochloric acid, FAs were extracted with n-hexane. FAs were methylated with diazomethane or trimethylsilylated with N,O-bis(trimethylsilyl)trifluoroacetamide (Supelco, Bellefonte, PA). Alditol acetate derivatives of carbohydrates were obtained by hydrolysis with 2 m trifluoroacetic acid at 120 °C for 2 h, reduction with 10 mg/ml sodium borohydride at 25 °C for 2 h, and acetylation with acetic anhydride at 100 °C for 1 h (20, 21).
Analysis and Conditions of Mass Spectrometry
MALDI-TOF MS was performed by using a SpiralTOF mass spectrometer (JMS-S3000; JEOL Ltd., Tokyo, Japan) for MA methyl esters and an Ultraflex II mass spectrometer (Bruker Daltonics, Billerica, MA) for glycolipids. The matrix was 10 mg/ml 2,5-dihydroxybenzoic acid in chloroform-methanol (1:1, v/v), and it was analyzed in the reflectron mode with an accelerating voltage operating in positive mode at 20 kV. GC/MS was performed by using a quadrupole mass spectrometer (GCMS-QP2010 Plus; Shimadzu Corp., Kyoto, Japan) equipped with a fused capillary column (SP-2380 and Equity-1; 30 m, 0.25-mm inner diameter; Supelco). Helium was used as the carrier gas. The temperature program for FA methyl esters was started at 60 °C, held for 1 min, increased 30 °C/min to 190 °C followed by an increase of 6 °C/min to 320 °C, and held for 3 min. The temperature program for FA trimethylsilyl esters was started at 60 °C, held for 1 min, increased 30 °C/min to 200 °C, increased 8 °C/min to 280 °C, and held for 16 min. The program for alditol acetate derivatives was started at 60 °C, increased 40 °C/min to 220 °C, held for 5 min followed by an increase of 10 °C/min to 260 °C, and held for 10 min. The molecular separator and ion source energies were 70 eV, and the accelerating voltage was 8 kV (20, 21).
Analysis of Total Lipids by Two-dimensional TLC
Two-dimensional TLC of total lipids was performed. The TLC plates were developed with three systems: first with chloroform-methanol-water (65:25:4, v/v/v) and second with chloroform-methanol-acetic acid-water (80:12:15:4, v/v/v/v) for the low hydrophobic fraction; first with chloroform-methanol-acetone-acetic acid (90:10:6:1, v/v/v/v) and second with chloroform-methanol-water (90:10:1, v/v/v) for the middle hydrophobic fraction; and first with n-hexane-ethyl acetate (98:2, v/v, three runs) and second with n-hexane-acetone (98:2, v/v) for the high hydrophobic fraction (22, 23). To detect phenolic glycolipids (PGLs), the TLC plates were developed first with chloroform-methanol (96:4, v/v) and second with toluene-acetone (80:20, v/v). The TLC plates were sprayed with 20% sulfuric acid in ethanol and heated at 180 °C for 3 min.
Sequencing of ppsA Region
The ppsA region of types I and II was amplified by PCR using primers ppsA-F (5′-CATATGACGGGAAGCATCAGTGGTGAAG-3′) and ppsA-R (5′-AAGCTTTCACACCGACCTCTCGGCCTCAG-3′). The amplified fragment was sequenced using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) and a sequence analyzer (ABI3130xl; Applied Biosystems) (21).
Transformation of Type II with Type I ppsA
The ppsA fragments from type I (type I ppsA) and type II (type II ppsA) were amplified and cloned into pVV16, an expression plasmid vector for mycobacteria, downstream of the hsp60 promoter. Types I and II were transformed by incorporation of pVV16-type I ppsA and pVV16-type II ppsA by electroporation, and kanamycin-resistant colonies were isolated (21). Total lipids were prepared from heat-killed transformants, and the lipids produced were checked by TLC.
Host Responses to Total Lipids and Suppression by PGL
The host responses to total lipids were estimated by activations of murine bone marrow-derived macrophages (BMMs) of wild-type (C57BL/6), Toll-like receptor (TLR)2-KO, and TLR4-KO mice (Oriental Yeast Co., Ltd., Tokyo, Japan). The bone marrow cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS and 20% L929 cell supernatant at 37 °C in a 5% CO2 atmosphere for 7 days, and adherent cells were harvested as BMMs. The BMMs were seeded at a concentration of 5 × 105 cells/ml on 96-well flat bottom tissue culture plates and stimulated with the total lipids (21). The lipid samples were stored at a concentration of 1 mg/ml in PBS containing 0.025% Tween 80 and diluted to the optimal concentration in each experiment. After 24-h incubation, the production of tumor necrosis factor (TNF)-α in the supernatants was measured using a DuoSet ELISA development kit (R&D Systems, Inc., Minneapolis, MN) according to the manufacturer's instructions. As positive controls, LPS from Escherichia coli 055:B5 (Sigma-Aldrich) for TLR4 and Pam3CSK4 (InvivoGen, San Diego, CA) for TLR2 were used. Two independent experiments were performed and conducted in duplicate.
Nucleotide Sequence Accession Number
The nucleotide sequence of type II ppsA has been deposited in the NCBI GenBankTM database under accession number AB665170.
RESULTS
Colony Morphology and Genotypes of Types I and II
The colony morphologies of types I and II were different. The type I colonies were smooth, irregular, and flat, and those of type II were rough, raised, and convex (Fig. 1A). The scanning electron microscopy showed that the average bacterial length was 2.25 ± 0.11 μm for type I and 1.44 ± 0.05 μm for type II. Thus, the bacterial cell of type I was 1.5 times longer than that of type II, although there was no significant difference in their widths (Fig. 1, B and C).
FIGURE 1.

Genotypes and morphologies of BCG Tokyo 172 type I and II subpopulations. A, the colony morphology was observed by an optical microscope after incubation for 2 weeks on 7H10 agar plates. Type I was smooth, irregular, and flat, and type II was rough, raised, and convex. The magnification is ×4. Scanning electron microscopy (B) and a comparison of bacterial cell size (C) are shown. Types I and II were grown in 7H9 broth with shaking to the midlog phase. The bacteria were collected and filtered through a 5-μm membrane to remove aggregates. The average bacterial cell length was 2.25 ± 0.11 μm for type I and 1.44 ± 0.05 μm for type II. The magnification is ×5,000. The data are means ± standard deviations (SD) for 20 bacteria. *, p < 0.001. D, PCR of Rv3405c of the RD16. Type I showed a characteristic 22-bp deletion in Rv3405c, and type II was complete in this region. BCG Moreau, Connaught, and Pasteur substrains were used as reference strains.
Next, we examined the PCR products of the Rv3405c locus in RD16. Type I amplified a 183-bp band, whereas a 205-bp band was detected in type II that was identical to those of BCG Connaught and Pasteur. BCG Moreau showed the deletion of the specific band (Fig. 1D). These results clarified that type I had a 22-bp deletion in the Rv3405c locus and that of type II was complete.
Profiles of Bacterial Components in Types I and II
Mycolic Acid Subclasses
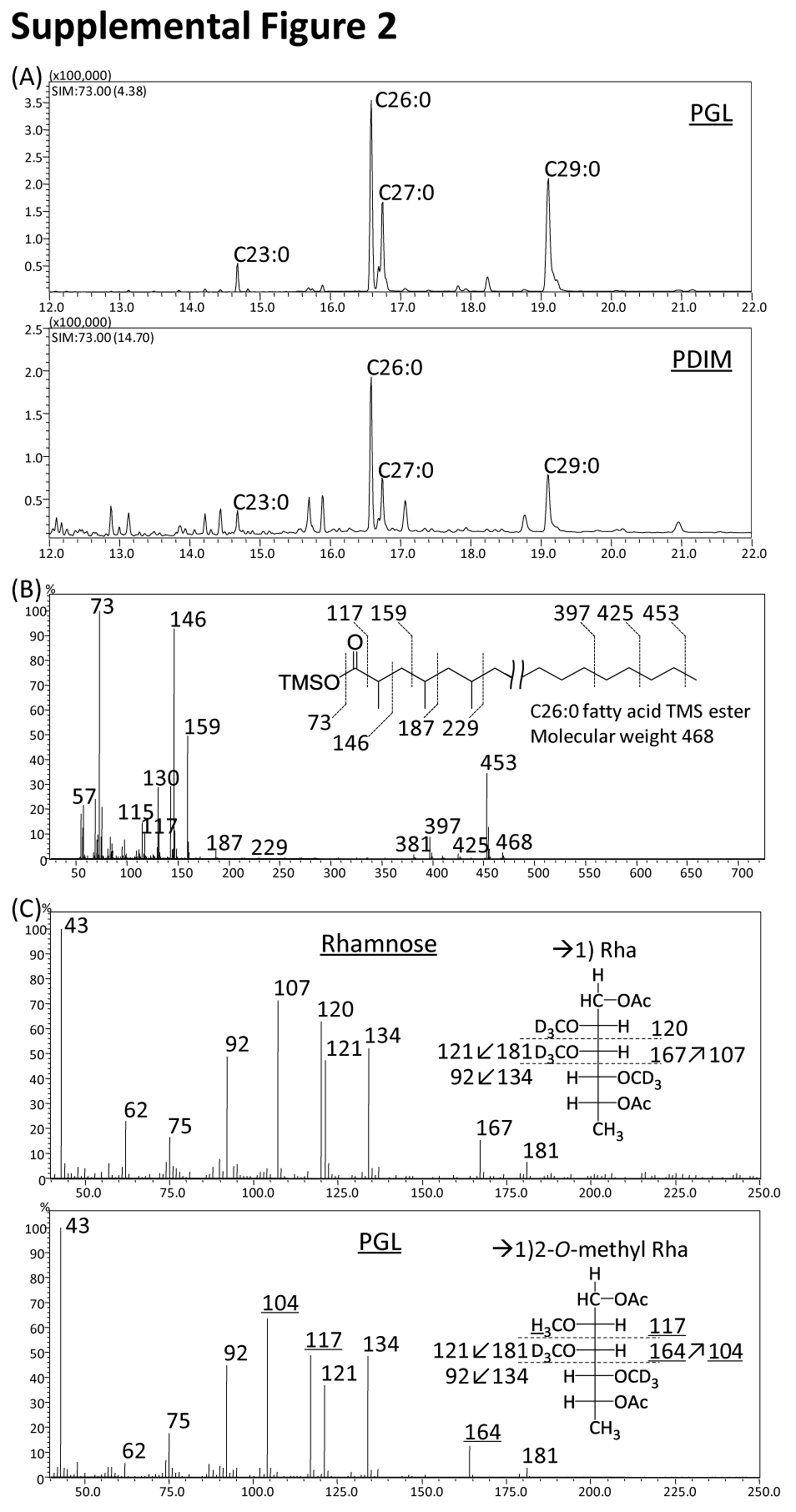
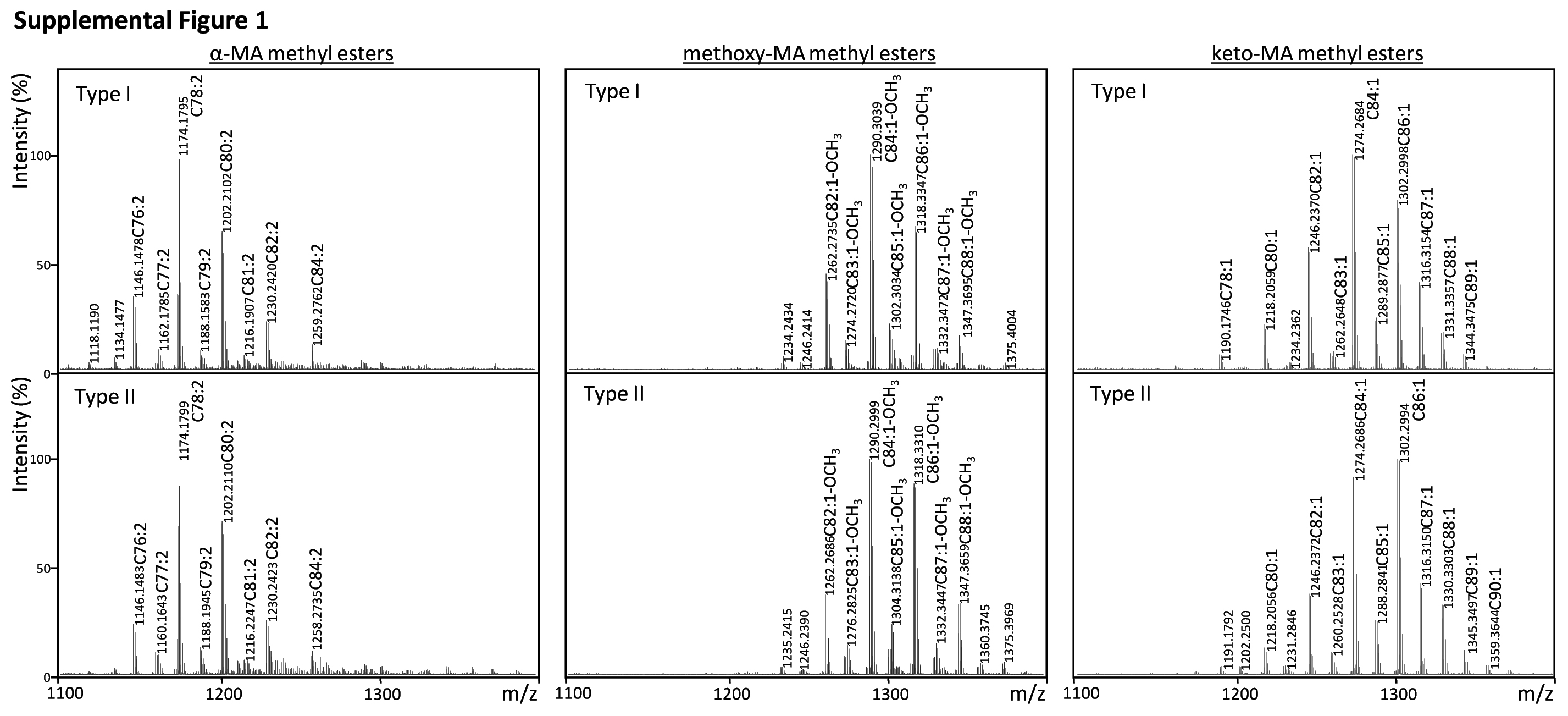
MAs are the most characteristic components of the acid-fast bacteria. The TLC of MA methyl esters showed the presence of three MA subclasses; α-, methoxy-, and keto-MA in both subpopulations (Fig. 2A). The composition of each subclass was keto- > methoxy- > α-MA. Keto-MA was over 50%, and the ratio of MA subclasses was significantly different from that of M. bovis Ushi 10 (Fig. 2B). The molecular species were assigned by MALDI-TOF MS, and the main species was C78:2 for α-MA, C84:1-OCH3 for methoxy-MA, and C84:1 for keto-MA, respectively (supplemental Fig. 1). These results imply that types I and II had identical distributions of MAs.
FIGURE 2.

Comparison of MA subclasses. A, the TLC plate was developed with n-hexane-diethyl ether (90:15, v/v; three runs) and visualized by spraying with 20% sulfuric acid in ethanol and heating at 180 °C for 3 min. B, the ratio of each MA subclass was measured and calculated with ImageJ software. The keto-MA was over 50%, and the ratio of MA subclasses was almost the same in types I and II but significantly different from that of the BCG parent strain M. bovis Ushi 10.
Fatty Acids
The FA compositions of bacterial cells and total lipids are summarized in Table 1. The major FA species were C16:0, C18:0, 10-methyl C18:0 (tuberculostearic acid), and C26:0. The ratio of each FA from bacterial cells was almost the same in both subpopulations. The bacterial cells contained much C18:0 and C26:0 FAs and less 10-methyl C18:0 FAs compared with total lipids. The C24:0 and C26:0 FAs may be components of triglycerol (24). The multiple methyl-branched C26:0 and C29:0 FAs in total lipids were much higher in type I compare with type II.
TABLE 1.
Fatty acid compositions in bacterial cells and total lipids from types I and II
| Fatty acidsa | Compositionb |
|||
|---|---|---|---|---|
| Bacterial cells |
Total lipids |
|||
| Type I | Type II | Type I | Type II | |
| % | ||||
| C14:0 | 1.5 | 2.3 | 1.4 | 1.9 |
| C15:0 | 0.5 | 0.3 | 0.2 | 0.8 |
| C16:1 | 0.5 | 0.8 | 0.8 | 1.3 |
| C16:0 | 28.3 | 30.4 | 27.5 | 38.4 |
| C17:0 | 0.9 | 1.7 | 1.0 | 2.6 |
| C18:1 | 2.4 | 2.2 | 5.1 | 8.2 |
| C18:0 | 17.3 | 18.0 | 12.1 | 15.8 |
| C20:0 | 0.6 | 1.3 | 1.8 | 1.0 |
| C22:0 | 1.1 | 0.8 | 0.8 | 0.4 |
| C24:0 | 3.2 | 2.2 | 3.0 | 1.6 |
| C25:0 | ND | 1.8 | 0.7 | 1.2 |
| C26:0 | 29.7 | 27.9 | 13.7 | 9.8 |
| C28:0 | ND | ND | 0.2 | 0.1 |
| 10-Methyl C16:0 | 1.2 | ND | 0.6 | 1.3 |
| 10-Methyl C17:0 | 0.2 | 0.5 | 0.1 | 0.9 |
| 10-Methyl C18:0 | 8.5 | 8.8 | 11.4 | 12.2 |
| 10-Methyl C19:0 | ND | ND | 0.1 | ND |
| br C23:0 | 0.1 | ND | 0.6 | ND |
| br C26:0 | 0.9 | ND | 4.4 | 0.4 |
| br C27:0 | 0.5 | ND | 1.7 | ND |
| br C29:0 | 1.3 | 0.2 | 8.7 | 0.1 |
| br C30:0 | 0.4 | ND | 1.8 | ND |
| br C32:0 | 0.6 | 0.2 | 1.4 | 0.1 |
| br C33:0 | ND | ND | 0.2 | 0.1 |
| Unidentified | 0.4 | 0.8 | 0.7 | 1.9 |
| Total | 100 | 100 | 100 | 100 |
a br, multiple methyl-branched.
b Composition is given as the percentage of total integrated chromatographic areas. ND, not detected.
Carbohydrates
The alditol acetate derivatives of carbohydrates in bacterial cells and total lipids were detected by GC/MS. The ratio of each carbohydrate in bacterial cells was identical in both subpopulations. In total lipids, type I contained 11.2% 2-O-methylrhamnose (2-O-Me-Rha), although only a trace was detected in type II (Table 2). This result strongly implied the deletion of glycolipids containing 2-O-Me-Rha in type II.
TABLE 2.
Carbohydrate compositions in bacterial cells and total lipids from types I and II
| Carbohydrates | Compositiona |
|||
|---|---|---|---|---|
| Bacterial cells |
Total lipids |
|||
| Type I | Type II | Type I | Type II | |
| % | ||||
| 2-O-Methyl Rhamnose | 0.5 | tr | 11.2 | 0.4 |
| Rhamnose | 0.8 | 0.6 | 0.6 | 0.2 |
| Ribose | 1.6 | 1.2 | ND | ND |
| Arabinose | 15.7 | 14.8 | 0.8 | 1.0 |
| Xylose | 0.2 | 0.2 | ND | ND |
| Mannose | 12.8 | 13.3 | 42.7 | 47.5 |
| Galactose | 14.9 | 14.6 | 1.1 | 1.7 |
| Glucose | 45.8 | 47.2 | 10.6 | 12.9 |
| myo-inositol | 4.6 | 4.7 | 23.9 | 28.5 |
| Unidentified | 3.3 | 3.3 | 9.1 | 7.9 |
| Total | 100 | 100 | 100 | 100 |
a Composition is given as the percentage of total integrated chromatographic areas. tr, trace (less than 0.05%); ND, not detected.
Lipids
The heterogeneity of lipid components of types I and II was examined by two-dimensional TLC (Fig. 3). Total lipids were developed with three different solvent systems to detect low, middle, and high hydrophobic lipid components. In the low hydrophobic fraction, the TLC patterns were similar in both subpopulations, and phosphatidylinositol mannosides and phosphatidylethanolamine were detected as major components. The middle hydrophobic fraction mainly contained several mycoloyl glycolipids, cord factor (trehalose 6,6′-dimycolate), and trehalose 6-monomycolate. Interestingly, an unknown spot (X1) at the upper side of cord factor was detected as a yellow-brown spot only in type I. In the same manner, one more unknown spot (X2) expressed only in type I was detected in the high hydrophobic fraction.
FIGURE 3.

Two-dimensional TLC of total lipids extracted from types I and II. The TLC plates were developed with three systems: chloroform-methanol-water (65:25:4, v/v/v) followed by chloroform-methanol-acetic acid-water (80:12:15:4, v/v/v/v) for the low hydrophobic fraction (A), chloroform-methanol-acetone-acetic acid (90:10:6:1, v/v/v/v) followed by chloroform-methanol-water (90:10:1, v/v/v) for the middle hydrophobic fraction (B), and n-hexane-ethyl acetate (98:2, v/v; three runs) followed by n-hexane-acetone (98:2, v/v) for the high hydrophobic fraction (C). The TLC plates were sprayed with 20% sulfuric acid in ethanol and heated at 180 °C for 3 min. PIMs, phosphatidylinositol mannosides; PE, phosphatidylethanolamine; TMM, trehalose 6-monomycolate; TDM, cord factor (trehalose 6,6′-dimycolate).
We identified the precise structures of X1 and X2 by using mass spectrometry. The positions of X1 and X2 in TLCs developed with each solvent system suggested that X1 and X2 were PGL and phthiocerol dimycocerosate (PDIM), respectively (22). X1 and X2 were purified by preparative TLC. The MALDI-TOF MS spectrum of X1 showed m/z 1531 and other mass units at 14-Da intervals for [M + Na]+ as molecule-related ions in positive mode (Fig. 4A). In addition, the MS/MS spectrum showed fragment ion peaks m/z 1371 based on the elimination of methyl deoxysugar, m/z 1135 and 1093 based on the elimination of the C26:0 and C29:0 FAs, m/z 696 based on the elimination of both C26:0 and C29:0 FAs, and m/z 535 for the phenolic phthiocerol that lacked methyl deoxysugar and C26:0 and C29:0 FAs (Fig. 4B and supplemental Fig. 2A). Alkaline hydrolysis of X1 was performed, and the FA was extracted. A trimethylsilyl ester of the FA fraction was analyzed by GC/MS. The main peaks of C26:0 and C29:0 were detected, and fragmented ions m/z 146, 187, and 229 were methyl-branched at every other branch from the α-position (supplemental Fig. 2B). This result was supported by the compositions of FA in total lipids from types I and II (Table 1). The GC/MS of the partially deuteromethylated alditol acetate derivatives of the X1 sugar moiety were detected as 2-O-methyl-3,4-di-O-deuteromethyl-1,5-di-O-acetylrhamnitol and assigned to 2-O-Me-Rha (supplemental Fig. 2C). The MALDI-TOF MS and MS/MS of X2 showed m/z 1349 for [M + Na]+ as molecule-related ions (Fig. 4C) and m/z 952 and 910 based on the elimination of C26:0 and C29:0 FAs (Fig. 4D). As a result, X1 was identified as PGL, and X2 was identified as PDIM, which is composed of PGL. The proposed structure and fragment patterns of PGL/PDIM are shown in Fig. 4E. Analysis of the lipid distributions of types I and II clarified that type II did not produce PGL/PDIM, which contribute to the pathogenesis of M. tuberculosis and M. bovis (15).
FIGURE 4.

Structural analyses of PGL/PDIM by mass spectrometry. A, MALDI-TOF MS of PGL from type I showed the sodium adduct ion [M + Na]+ with a major peak at m/z 1531 as a molecule-related ion. B, MALDI-TOF MS/MS of m/z 1531 showed four major fragment ions (m/z 696, 1093, 1135, and 1371) based on elimination of FAs and/or monomethylrhamnose from PGL. C, MALDI-TOF MS of PDIM from type I showed the sodium adduct ion [M + Na]+ with a major peak at m/z 1349. D, MALDI-TOF MS/MS of m/z 1349 showed two minor fragment ions (m/z 910 and 952) based on elimination of FAs from PDIM. E, proposed structures and fragment patterns of PGL/PDIM from type I. Intens., intensity; a.u., arbitrary units.
Genetic Analysis of PGL/PDIM Biosynthesis
The ppsA-E, drrA-C, papA5, and mas genes are reported to be responsible for PGL/PDIM biosynthesis (25). We compared the sequences of the ppsA gene of types I and II. Type II ppsA had a single base insertion at nucleotide 379 and two single base substitutions at nucleotides 275 and 2415 compared with type I ppsA (Fig. 5A). As a result, a frameshift due to the insertion of this adenine relocated the stop codon from codon 1877 to 129, and an abnormal truncated protein was synthesized. To check the activity of the type II ppsA, we constructed type I and II transformants by inserting pVV16-type I ppsA and pVV16-type II ppsA. The type II transformant with type I ppsA restored the production of PGL (Fig. 5, B and C). These results imply that the type II ppsA is not functional and that the PGL/PDIM deletion in type II was caused by mutation of the ppsA gene.
FIGURE 5.

Genetic organization of PGL/PDIM locus (25) and PGL expression. A, the nucleotide sequence of ppsA genes showed that the type II ppsA had a single base insertion and two single base substitutions. The frameshift due to the insertion of this adenine relocated the stop codon from codon 1877 to 129, and an abnormal truncated protein was synthesized. B and C, total lipids extracted from type I (B) and II (C) transformants with type I ppsA or type II ppsA inserted. The manifestation of PGL was confirmed by insertion of the type I ppsA into the type II transformant. The TLC plates were developed with chloroform-methanol (96:4, v/v) first and then toluene-acetone (80:20, v/v). GroMM, glycerol monomycolate.
Suppression of Host Responses by PGL
To explore the antigenicity of the lipids, BMMs derived from wild-type mice were stimulated with total lipids of types I and II. The BMMs were activated by the total lipids and released TNF-α into the supernatant in a dose-dependent manner but did not respond to the PGL purified from type I. BMMs derived from TLR4-KO mice responded to the total lipids, but BMMs from TLR2-KO mice did not (Fig. 6A). These results imply that components of the total lipids stimulated BMMs via TLR2. Next, stimulations were performed using a mixture of total lipids and PGL at ratios of 10:1, 10:5, and 10:10. The TNF-α release was decreased in a concentration-dependent manner by the addition of PGL. This result implies that activation of the TLR2 pathway via total lipids was suppressed by PGL (Fig. 6, B and C).
FIGURE 6.

Inhibition of proinflammatory cytokine release from murine macrophages by PGL. A, BMMs were seeded at a concentration of 5 × 105 cells/ml on 96-well flat bottom tissue culture plates and stimulated with the purified PGL and total lipids. After 24-h incubation, the TNF-α production in the supernatants was measured. B, as positive controls, LPS (0.5 μg/ml) from E. coli 055:B5 was used for TLR4, and Pam3CSK4 (0.5 μg/ml) was used for TLR2. C, the BMMs were stimulated with type II total lipids (25 μg/ml) in the presence of increasing amounts of type I PGL (0, 2.5, 12.5, and 25 μg/ml). The TNF-α release was decreased in a PGL ratio-dependent manner. The data are means ± standard deviations (SD) for two independent experiments done in duplicate.
DISCUSSION
BCG was introduced as a live attenuated vaccine for TB using the original BCG in 1921. Since then, the BCG substrains have undergone spontaneous mutations in the laboratory. To produce a stable and safe vaccine, it is important to clarify the differences of each substrain and estimate whether BCG substrains have similar protective potency. Of the representative phenotypes, BCG substrains obtained from the Pasteur Institute before 1927 produced methoxy-MA, but those obtained after 1931 failed to synthesize methoxy-MA (2). A point mutation in the mma3 gene is responsible for this phenotype (26, 27). MAs, the major cell wall component, play a key role in the pathogenesis of TB and thus affect host responses. Recently, the genomes of the BCG Pasteur, Tokyo 172, and Moreau substrains were sequenced, and the genome of BCG Tokyo 172 had 20 insertion or deletion mutations of less than 20 bp and 68 SNPs compared with that of BCG Pasteur (4, 28, 29). The heterogeneity of BCG substrains is being clarified. BCG Tokyo 172 is important as a standard substrain of the BCG vaccine used by the WHO (8). It has been noted that BCG Tokyo 172 is composed of two subpopulations with different morphologies (12). Therefore, the relationships between bacterial phenotypes and genotypes should be evaluated in detail to validate the quality and protective effects of the BCG vaccine.
First, we characterized the lipids in types I and II biochemically because we suppose that mycobacterial lipids may affect colony morphology (smooth and rough; Refs. 30–32). The type I bacterial cells were significantly longer than those of type II (Fig. 1C). Scherman et al. (33) have reported that the mutant cells of M. tuberculosis with H37Rv disrupting the Rv3779 gene are significantly shorter than the wild-type parent strain. Rv3779 is responsible for the direct synthesis of polyprenoyl-phosphomannopyranose and indirectly for lipoarabinomannan, lipomannan, and phosphatidylinositol mannosides. It is considered that disruption of the Rv3779 gene induces decreases of lipomannan, lipoarabinomannan, and phosphatidylinositol mannosides and affects the cell growth and shape (33). Based on that report, we examined the Rv3779 gene of types I and II. The Rv3779 sequences were completely identical in both types I and II (data not shown), and thus, this gene cluster is not associated with cell length. The shorter cells of type II may be linked with slow growth, disruption of PGL/PDIM, and a change of morphology.
Next, we found that only type II lacked PGL/PDIM. The distribution of various species of glycolipids and phospholipids in the cell envelope determines the cell surface properties, cell wall permeability, and bacterial phenotype. For example, the glycopeptidolipids produced by several non-tuberculous mycobacteria are associated with smooth/rough colony morphology, biofilm formation, and sliding motility (30–32). PGL/PDIM play critical roles in the cell wall permeability and in the killing activity of host macrophages (15, 34, 35).
Chen et al. (22) compared the PGL/PDIM production in 12 BCG substrains and showed a deficiency of PGL/PDIM in three substrains, BCG Japan, Moreau, and Glaxo. They have suggested that Rv3405c encodes a transcription factor of the TetR family and is responsible for the biosynthesis of PGL/PDIM in BCG Japan and Moreau strains but not in BCG Glaxo. They also reported that insertion of the transformant of pRv3405c into BCG Japan partially restored the synthesis of PGLs but not PDIM (22). Our results are inconsistent with the report by Chen et al. (22). Although the Rv3405c gene was intact in type II, the production of PGL/PDIM was inhibited (Fig. 3). The type II ppsA had a single base insertion and two single base substitutions compared with type I ppsA. Moreover, the type II transformant with type I ppsA inserted recovered PGL/PDIM production. These results suggest that mutation of the ppsA gene is responsible for the lack of PGL/PDIM production. We constructed a type II transformant by inserting Rv3405c or Rv3405c-Rv3408 of RD16 in addition to type II ppsA, but no transformation restored the production of PGL/PDIM (data not shown). This result implies that Rv3405c and Rv3405c-Rv3408 do not participate in the biosynthesis of PGL/PDIM in type II. The previous report of Chen et al. (22) did not describe the origin of the BCG Japan substrain, and it is unclear whether the BCG Japan they used was the same as the BCG Tokyo 172 strain used in this study. We examined the BCG Japan ATCC 35737 substrain deposited in the American Type Culture Collection (Manassas, VA) and confirmed the 22-bp deletion in Rv3405c and the production of PGL/PDIM (data not shown). A recent report of the genome sequence of the BCG Moreau substrain clarified that fadD26-ppsA (976 bp) and RD16 (7,608 bp) were deleted (28, 36). The BCG Moreau substrain is incapable of producing PGL/PDIM like BCG Tokyo 172 type II (22), and the deletion of fadD26-ppsA in BCG Moreau is consistent with our results. Taken together, the BCG Tokyo 172 substrain may have been divided into at least two subpopulations, types I and II, in a very early stage after it was distributed from the Pasteur Institute.
Finally, the immune effect of PGL derived from the BCG Tokyo 172 substrain was studied because type I could produce PGL but type II could not. A previous report by Reed et al. (37) demonstrated that the PGL of M. tuberculosis (PGL-tb) inhibits the innate immune response. The loss of PGL-tb was responsible for an increase in the release of TNF-α and interleukins 6 and 12 in vitro, and the PGL-tb-deficient mutant showed a phenotype with low virulence/pathology (37). The PGL produced by BCG substrains is the so-called mycoside B, and its sugar moiety is different from that of PGL-tb. The PGL produced by BCG substrains has only a 2-O-Me-Rha branch elongated from the phenol moiety, although PGL-tb has three sugar residues elongated from the phenol moiety (38). The composition of the PDIM in PGL is similar in both species. The total lipids derived from types I and II activated BMMs via TLR2 (Fig. 6). Several lipid components containing phosphatidylinositol mannosides and mycoloyl glycolipids can induce the host response via TLR2 (39, 40). Although purified PGL molecules by themselves had no effect on the activation of macrophages in vitro, we found that PGL suppressed the activation of BMMs elicited by total lipids. It is considered that the PGL may have a competitive inhibitory effect or may mask the active site of other TLR2 agonistic lipid components and decrease their activity. The localization of PGL/PDIM in the cell envelope of BCG substrains is critical to their biological effects.
Our present study has demonstrated differences in the lipids and biosynthesis gene cluster of PGL/PDIM of two BCG Tokyo subpopulations and their effects on the host innate immune response. Type II lacks PGL/PDIM due to a ppsA mutation, and this phenotype is implicated in host responses. We propose that the lipid composition in the cell envelope is important for the efficacy of a BCG vaccine. These findings shed light on the quality, safety, and efficacy of BCG, which is the only vaccine currently available against TB.
Supplementary Material
Acknowledgments
We are grateful to Reina Yamamoto and Hideki Nakagawa (Osaka City University Graduate School of Medicine) and Dr. Kanae Teramoto and Dr. Takafumi Sato (JEOL Ltd.) for providing technical assistance.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology in Japan; and the Japan Health Sciences Foundation (Research on Publicly Essential Drugs and Medical Devices), the Ministry of Health, Labour and Welfare in Japan.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) AB665170.
- TB
- tuberculosis
- BCG
- Bacillus Calmette-Guérin
- PGL
- phenolic glycolipid
- PDIM
- phthiocerol dimycocerosate
- RD
- region of difference
- MA
- mycolic acid
- FA
- fatty acid
- BMM
- bone marrow-derived macrophage
- TLR
- Toll-like receptor
- 2-O-Me-Rha
- 2-O-methylrhamnose
- PGL-tb
- PGL of M. tuberculosis
- WHO
- World Health Organization.
REFERENCES
- 1. World Health Organization (2011) Global Tuberculosis Control 2010, WHO Press, World Health Organization, Geneva [Google Scholar]
- 2. Behr M. A. (2002) Lancet Infect. Dis. 2, 86–92 [DOI] [PubMed] [Google Scholar]
- 3. Behr M. A., Wilson M. A., Gill W. P., Salamon H., Schoolnik G. K., Rane S., Small P. M. (1999) Science 284, 1520–1523 [DOI] [PubMed] [Google Scholar]
- 4. Brosch R., Gordon S. V., Garnier T., Eiglmeier K., Frigui W., Valenti P., Dos Santos S., Duthoy S., Lacroix C., Garcia-Pelayo C., Inwald J. K., Golby P., Garcia J. N., Hewinson R. G., Behr M. A., Quail M. A., Churcher C., Barrell B. G., Parkhill J., Cole S. T. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5596–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zwerling A., Behr M. A., Verma A., Brewer T. F., Menzies D., Pai M. (2011) PLoS Med. 8, e1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brosch R., Behr M. A. (2005) in Tuberculosis and the Tubercule Bacillus (Cole S. T., Eisenach D. S., McMurray D. N., Jacobs W. R., Jr. eds) Chapter 10, pp. 155–164, ASM Press, Washington, D. C. [Google Scholar]
- 7. Knezevic I., Corbel M. J. (2006) Vaccine 24, 3874–3877 [DOI] [PubMed] [Google Scholar]
- 8. Ho M. M., Markey K., Rigsby P., Hockley J., Corbel M. J. (2011) Vaccine 29, 512–518 [DOI] [PubMed] [Google Scholar]
- 9. Ho M. M., Markey K., Rigsby P., Jensen S. E., Gairola S., Seki M., Castello-Branco L. R., López-Vidal Y., Knezevic I., Corbel M. J. (2008) Vaccine 26, 4754–4757 [DOI] [PubMed] [Google Scholar]
- 10. Markey K., Ho M. M., Choudhury B., Seki M., Ju L., Castello-Branco L. R., Gairola S., Zhao A., Shibayama K., Andre M., Corbel M. J. (2010) Vaccine 28, 6964–6969 [DOI] [PubMed] [Google Scholar]
- 11. Bedwell J., Kairo S. K., Behr M. A., Bygraves J. A. (2001) Vaccine 19, 2146–2151 [DOI] [PubMed] [Google Scholar]
- 12. Honda I., Seki M., Ikeda N., Yamamoto S., Yano I., Koyama A., Toida I. (2006) Vaccine 24, 4969–4974 [DOI] [PubMed] [Google Scholar]
- 13. Hayashi D., Takii T., Fujiwara N., Fujita Y., Yano I., Yamamoto S., Kondo M., Yasuda E., Inagaki E., Kanai K., Fujiwara A., Kawarazaki A., Chiba T., Onozaki K. (2009) FEMS Immunol. Med. Microbiol. 56, 116–128 [DOI] [PubMed] [Google Scholar]
- 14. Brennan P. J., Nikaido H. (1995) Annu. Rev. Biochem. 64, 29–63 [DOI] [PubMed] [Google Scholar]
- 15. Guenin-Macé L., Siméone R., Demangel C. (2009) Transbound. Emerg. Dis. 56, 255–268 [DOI] [PubMed] [Google Scholar]
- 16. Kremer L., de Chastellier C., Dobson G., Gibson K. J., Bifani P., Balor S., Gorvel J. P., Locht C., Minnikin D. E., Besra G. S. (2005) Mol. Microbiol. 57, 1113–1126 [DOI] [PubMed] [Google Scholar]
- 17. Bhatt A., Fujiwara N., Bhatt K., Gurcha S. S., Kremer L., Chen B., Chan J., Porcelli S. A., Kobayashi K., Besra G. S., Jacobs W. R., Jr. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5157–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gilbart J., Fox A., Morgan S. L. (1987) Eur. J. Clin. Microbiol. 6, 715–723 [DOI] [PubMed] [Google Scholar]
- 19. Miller L. T. (1982) J. Clin. Microbiol. 16, 584–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujiwara N., Nakata N., Naka T., Yano I., Doe M., Chatterjee D., McNeil M., Brennan P. J., Kobayashi K., Makino M., Matsumoto S., Ogura H., Maeda S. (2008) J. Bacteriol. 190, 3613–3621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Naka T., Nakata N., Maeda S., Yamamoto R., Doe M., Mizuno S., Niki M., Kobayashi K., Ogura H., Makino M., Fujiwara N. (2011) J. Bacteriol. 193, 5766–5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J. M., Islam S. T., Ren H., Liu J. (2007) Vaccine 25, 8114–8122 [DOI] [PubMed] [Google Scholar]
- 23. Fujiwara N., Kobayashi K. (2008) in Glycolipids: New Research (Sasaki M. ed) Chapter IV, pp. 99–116, Nova Science Publishers, Inc., New York [Google Scholar]
- 24. Sirakova T. D., Dubey V. S., Deb C., Daniel J., Korotkova T. A., Abomoelak B., Kolattukudy P. E. (2006) Microbiology 152, 2717–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guilhot C., Chlut C., Daffé M. (2008) in The Mycobacterial Cell Envelope (Daffé M., Reyrat J.-M. eds) Chapter 17, pp. 273–288, ASM Press, Washington, D. C. [Google Scholar]
- 26. Behr M. A., Schroeder B. G., Brinkman J. N., Slayden R. A., Barry C. E., 3rd (2000) J. Bacteriol. 182, 3394–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan Y., Zhu Y., Crane D. D., Barry C. E., 3rd (1998) Mol. Microbiol. 29, 1449–1458 [DOI] [PubMed] [Google Scholar]
- 28. Gomes L. H., Otto T. D., Vasconcellos E. A., Ferrão P. M., Maia R. M., Moreira A. S., Ferreira M. A., Castello-Branco L. R., Degrave W. M., Mendonça-Lima L. (2011) J. Bacteriol. 193, 5600–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seki M., Honda I., Fujita I., Yano I., Yamamoto S., Koyama A. (2009) Vaccine 27, 1710–1716 [DOI] [PubMed] [Google Scholar]
- 30. Barrow W. W., Brennan P. J. (1982) J. Bacteriol. 150, 381–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Etienne G., Laval F., Villeneuve C., Dinadayala P., Abouwarda A., Zerbib D., Galamba A., Daffé M. (2005) Microbiology 151, 2075–2086 [DOI] [PubMed] [Google Scholar]
- 32. Martínez A., Torello S., Kolter R. (1999) J. Bacteriol. 181, 7331–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scherman H., Kaur D., Pham H., Skovierová H., Jackson M., Brennan P. J. (2009) J. Bacteriol. 191, 6769–6772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Camacho L. R., Constant P., Raynaud C., Laneelle M. A., Triccas J. A., Gicquel B., Daffe M., Guilhot C. (2001) J. Biol. Chem. 276, 19845–19854 [DOI] [PubMed] [Google Scholar]
- 35. Rousseau C., Winter N., Pivert E., Bordat Y., Neyrolles O., Avé P., Huerre M., Gicquel B., Jackson M. (2004) Cell. Microbiol. 6, 277–287 [DOI] [PubMed] [Google Scholar]
- 36. Leung A. S., Tran V., Wu Z., Yu X., Alexander D. C., Gao G. F., Zhu B., Liu J. (2008) BMC Genomics 9, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reed M. B., Domenech P., Manca C., Su H., Barczak A. K., Kreiswirth B. N., Kaplan G., Barry C. E., 3rd (2004) Nature 431, 84–87 [DOI] [PubMed] [Google Scholar]
- 38. Siméone R., Léger M., Constant P., Malaga W., Marrakchi H., Daffé M., Guilhot C., Chalut C. (2010) FEBS J. 277, 2715–2725 [DOI] [PubMed] [Google Scholar]
- 39. Geisel R. E., Sakamoto K., Russell D. G., Rhoades E. R. (2005) J. Immunol. 174, 5007–5015 [DOI] [PubMed] [Google Scholar]
- 40. Gilleron M., Quesniaux V. F., Puzo G. (2003) J. Biol. Chem. 278, 29880–29889 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}