

Background: CuII(atsm) [(diacetylbis(N(4)-methylthiosemicarbazonato) copper(II)] was orally administrated to transgenic SOD1G93A mice.
Results: Treatment significantly prolonged lifespan with preservation of motor neurons. Reduced protein oxidation, attenuated astrocyte, and microglial activation also resulted from treatment.
Conclusion: CuII(atsm) is neuroprotective in this model even when treatment begins after the onset of disease symptoms.
Significance: The drug has therapeutic potential for amyotrophic lateral sclerosis.
Keywords: Amyotrophic Lateral Sclerosis, Neurodegenerative Diseases, Oxidative Stress, Superoxide Dismutase (SOD), Transgenic Mice, CuII(atsm), SODG93A, TDP-43, Peroxynitrite
Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive paralyzing disease characterized by tissue oxidative damage and motor neuron degeneration. This study investigated the in vivo effect of diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)), which is an orally bioavailable, blood-brain barrier-permeable complex. In vitro the compound inhibits the action of peroxynitrite on Cu,Zn-superoxide dismutase (SOD1) and subsequent nitration of cellular proteins. Oral treatment of transgenic SOD1G93A mice with CuII(atsm) at presymptomatic and symptomatic ages was performed. The mice were examined for improvement in lifespan and motor function, as well as histological and biochemical changes to key disease markers. Systemic treatment of SOD1G93A mice significantly delayed onset of paralysis and prolonged lifespan, even when administered to symptomatic animals. Consistent with the properties of this compound, treated mice had reduced protein nitration and carbonylation, as well as increased antioxidant activity in spinal cord. Treatment also significantly preserved motor neurons and attenuated astrocyte and microglial activation in mice. Furthermore, CuII(atsm) prevented the accumulation of abnormally phosphorylated and fragmented TAR DNA-binding protein-43 (TDP-43) in spinal cord, a protein pivotal to the development of ALS. CuII(atsm) therefore represents a potential new class of neuroprotective agents targeting multiple major disease pathways of motor neurons with therapeutic potential for ALS.
Introduction
Amyotrophic lateral sclerosis (ALS)3 is a fatal neurodegenerative disorder characterized by selective loss of cortical, brainstem, and spinal motor neurons leading to paralysis within 2–5 years of diagnosis. Although the majority of ALS cases are sporadic (sALS), ∼10% of ALS is familial (fALS) in which dominant mutations in superoxide dismutase 1 (SOD1) and TAR DNA-binding protein-43 (TDP-43) are the most common cause (1, 2). Both sALS and fALS affect the same neurons with similar clinical characteristics, implying common disease mechanisms. Because most SOD1 mutants retain normal antioxidant activity and SOD1 knock-out mice do not develop spontaneous ALS-like symptoms, these mutations may lead to a toxic gain of function rather than loss of superoxide scavenging ability (3, 4). One such mechanism is oxidative or nitrosative stress resulting from aberrant catalysis conferred by SOD1 mutations (5–7). In contrast to wild-type enzyme, mutant SOD1 can promote nitration of tyrosine residues on target proteins in the presence of peroxynitrite formed by superoxide and nitric oxide radicals (8). Both peroxynitrite and 3-nitrotyrosine have been shown to induce apoptosis in cultured motor neurons with evidence of protein nitration (9, 10), whereas increased levels of 3-nitrotyrosine were reported in ALS spinal cord (11). Transgenic rodents expressing human mutant SOD1, especially the SOD1G93A model, develop pathology consistent with ALS patients and have been studied widely for pathogenic mechanisms and testing therapeutic strategies in ALS (3, 12, 13). Of particular relevance is the finding that nitrated proteins accumulate in spinal cords of mice months before symptom onset (14, 15), implicating a causal role of nitrosative stress in motor neuron degeneration.
Diacetylbis(N(4)-methylthiosemicarbazonato)copper(II) (CuII(atsm)) (see Fig. 1A) is a neutral, lipophilic, thermodynamically stable complex that is membrane-permeable and readily crosses the blood-brain barrier (16, 17). CuII(atsm) prepared with radioactive 64Cu is currently undergoing clinical trials as a positron emission tomography tracer for hypoxia imaging (18, 19). It was reported that CuII(atsm) possesses a SOD1-like antioxidant activity as CuII(atsm) treatment in a model of ischemic reperfusion injury lowered lipid peroxidation, suggesting that its protective effects were linked to amelioration of radical-mediated tissue damage (16, 20).
FIGURE 1.
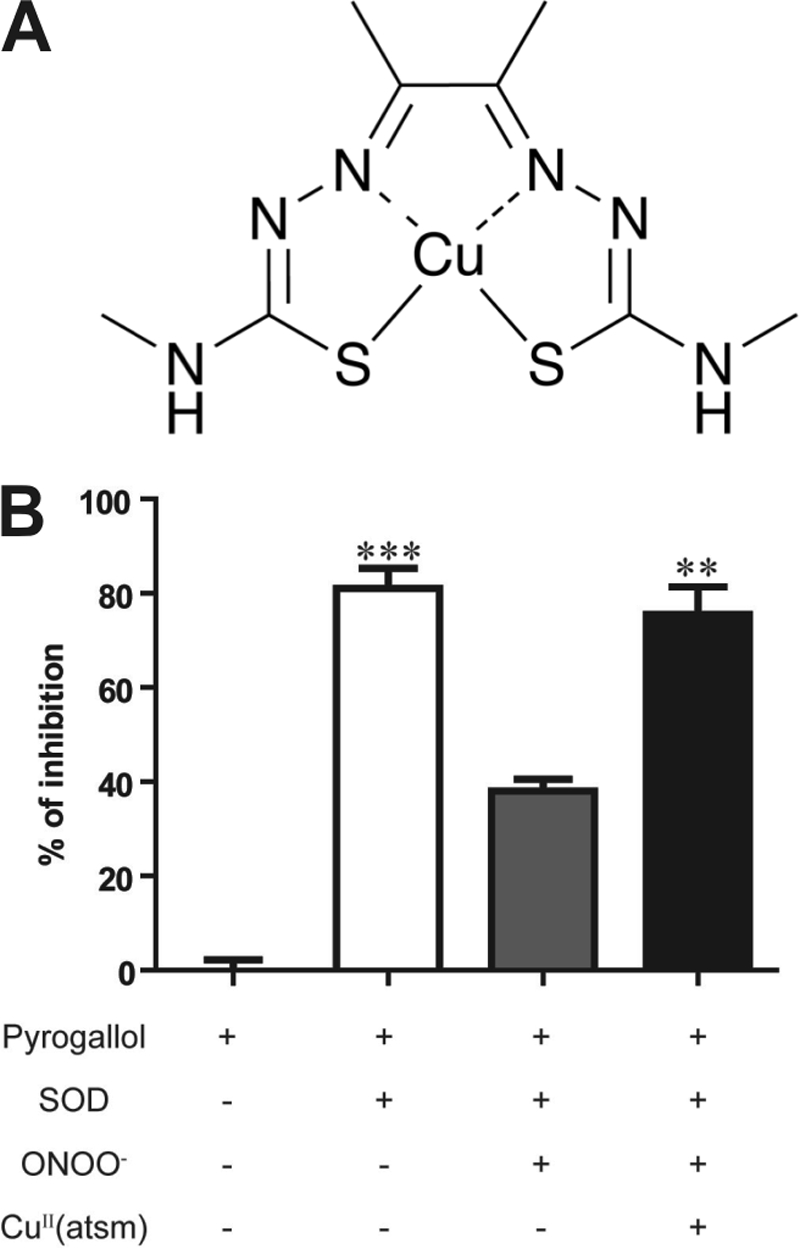
CuII(atsm) action on peroxynitrite. A, molecular structure of CuII(atsm). B, SOD activity was measured as the ability to inhibit pyrogallol oxidation determined by absorbance at 420 nm after a 1-h incubation. 10 units/ml SOD1 was able to inhibit ∼80% of pyrogallol oxidation. SOD1 activity was inhibited by the addition of 1 μm peroxynitrite (ONOO−) and rescued by 1 μm CuII(atsm). Data shown are an average of three experiments and expressed as mean ± S.E. One-way analysis of variance with Dunnett's post hoc test was used for statistical analysis, **, p < 0.01, ***, p < 0.001 difference when compared with SOD1 + ONOO− treatment.
Here, we uncover that CuII(atsm) is an effective scavenger of peroxynitrite. In the present study, we used a well characterized mouse model with low expression of mutant SOD1G93A (21, 22) to test the therapeutic effects of CuII(atsm) on symptom onset and progression of disease. Treatment significantly enhanced animal survival, correlating with reduced neurodegeneration, gliosis, and peroxynitrite-induced oxidative stress in the spinal cords of mice. In addition, we also identified prominent TDP-43 pathology in this model, which is not present in mice with high expression of mutant SOD1 transgenes (23, 24). CuII(atsm) also inhibited the phosphorylation and fragmentation of TDP-43. Overall, the beneficial therapeutic effect of CuII(atsm) may be due to its ability to inhibit peroxynitrite activity.
EXPERIMENTAL PROCEDURES
Chemical Synthesis
CuII(atsm) was prepared according to published procedures (25, 26).
SOD Activity Assay
SOD1 activity was measured using pyrogallol oxidation, as described previously (27). Briefly, pyrogallol (Sigma-Aldrich) was made up to 0.2 mm in 50 mm Tris-HCl (pH 8.2) containing 1 mm diethylenetriaminepentaacetic acid. 10 units/ml SOD1 (isolated from human erythrocytes; Sigma-Aldrich) was preincubated with 1 μm CuII(atsm) made up in acetonitrile. SOD was then nitrated by 1 mm of ONOO−. SOD activity was measured as the ability to inhibit the oxidation of pyrogallol as determined by increased absorbance at 420 nm after a 1-h incubation.
Transgenic SOD1G93A Mice
All animal protocols conformed to the Australian National Health and Medical Research Council published code of practice for animal research and conduct. Experimental procedures carried out with mice were approved by the Animal Ethics Committee of The University of Melbourne. Transgenic SOD1G93A low copy mice used in this study were originally derived from the TgN[SOD1-G93A]1Gur line from The Jackson Laboratory (Bar Harbor, ME) and develop a delayed phenotype due to a stable reduction in transgene copy number (28). Hemizygous carriers were backcrossed to C57BL6/J for >15 generations to produce a congenic line (21). Transgene analysis was carried out at weaning by DNA extraction from tail biopsies using the Extract-N-Amp tissue PCR kit (Sigma).
Treatment Regime
SOD1G93A mice were orally administered CuII(atsm) at 30 mg/kg starting at 140 days old (presymptomatic with no clinical symptoms or motor impairments) or 200 days old (symptomatic). The compound was resuspended in standard suspension vehicle (SSV) containing 0.9% (w/v) NaCl, 0.5% (w/v) sodium carboxymethylcellulose, 0.5% (v/v) benzyl alcohol, 0.4% (v/v) Tween 80. Treatment of mice from 140 days with CuII(atsm) or stand suspension vehicle was carried out five times per week via gavage until 270–300 days (end stage, survival study). Cohorts used for cell counts and biochemical analysis were treated in parallel with survival groups, but animals were killed at 200 days (n = 5 per treatment) for histology or 230 days (wild-type vehicle n = 9, CuII(atsm) n = 7; SOD1G93A vehicle n = 9 CuII(atsm) n = 14) for histological and biochemical analyses. Non-transgenic littermates were used as wild-type controls. Experimental mouse groups are shown in supplemental Table S1. The investigators were blinded for mouse genotype when performing all the procedures.
Treatment of mice of 200 days old (defined as symptom onset with body weight loss prior to motor impairment (22)) was performed daily. Biochemical and histological studies were not carried out for these mice. In both studies, no gender effects were observed with and between treatment groups. Therefore, sexes were combined for analyses. Quantitative real-time PCR assessment confirmed equivalent mutant SOD1 transgene copies in experimental and control mice (14 copies, data not shown).
Motor Performance Test (Rotarod and Stride Length Tests)
The rotarod apparatus was used to monitor disease progression and motor function deficit in mice by recording the latency to fall from a rotating rod (Rota MK2, Monash Uni) within 180 s at a constant speed of 16 rpm as described before (29). Data are presented as mean ± S.E.
Stride length was assessed by the footprint test using an ∼60-cm runway strip. An average of six steps per mouse was used per calculation. Both rotarod and stride tests were performed every fortnight beginning at 140 days of age followed by weekly testing at 200 days of age and subsequently twice per week from 240 days onwards. Body weight was recorded weekly from 140 days of age and then twice per week from symptomatic stage (200 days).
Clinical Assessments
Symptom onset was defined as age following first decline from peak body weight. Disease onset was defined either by age of decreased stride length or by rotarod activity from peak performance. Disease progression was confirmed by the presence of hindlimb resting tremor and increased hyporeflexia followed by unilateral or bilateral paralysis. End stage was defined by our ethical requirements as 15–20% body weight loss and <8 s of rotarod performance.
Mouse Tissue Collection
Mice were euthanized with carbon dioxide and transcardially perfused with butylated hydroxytoluene (Sigma-Aldrich) and phosphatase and protease inhibitor mixtures (Sigma-Aldrich) in PBS. Spinal cord tissues were removed and frozen with dry ice before storing in −80 °C for subsequent biochemical analysis.
Motor Neuron Assessment
Mice killed at 200 days were perfused with 4% paraformaldehyde in PBS, and lumbar spinal cords were dissected out. For mice killed at 230 days, lumbar spinal cords were dissected out and immersion-fixed in 10% neutral buffered formalin. Spinal cords were dehydrated in increasing alcohol concentrations, before embedding in paraffin. For motor neuron counts, 20 serial horizontal sections (6 μm thick) per mouse were cut using a microtome as described previously (22) and mounted onto slides. Cresyl violet staining was performed on the 1st, 9th, and 18th serial sections (∼50-μm interval between each section), and motor neurons in both ventral horns on each section were counted within an area defined by a horizontal line through the central canal.
Spinal Cord Fractionation
Spinal cord fractionation was performed according to published protocols with modification (30). Briefly, flash-frozen tissue was homogenized gently (7–8 times) using a Dounce homogenizer in 200 μl of chilled homogenizing buffer containing 0.25 m sucrose, 5 mm MgCl2, 10 mm Tris-HCl, pH 7.4, 1:100 dilution of butylated hydroxytoluene, (0.5 m) and protease inhibitor mixture and phosphatase inhibitor mixture (Sigma-Aldrich). Homogenates were collected, and another 200 μl of homogenizing buffer was used to extract residual tissue suspensions. Homogenates were centrifuged at 100 × g for 5 min, and supernatants were centrifuged at 600 × g for 10 min to collect pellets designated as the nuclear fraction. Supernatants were clarified by ultracentrifugation at 80,000 × g for 80 min at 5 °C to collect cytoplasmic fractions.
Mouse Pharmacokinetic and Brain Uptake Studies
CuII(atsm) was administered orally by gavage at a dose of 30 mg/kg to adult male C57BL/6 or SOD1G93A mice. At 2 h after dose, mice were anesthetized with sodium pentobarbitone (100 mg/kg, Lethobarb, Jurox, Rutherford, NSW, Australia), and ∼500 μl of blood was withdrawn by terminal cardiac puncture and transferred to heparinized tubes. Blood samples were immediately centrifuged, and duplicate 50-μl aliquots of plasma were collected for analysis. Immediately postmortem, brains and spinal cord were removed, blotted to remove residual blood, weighed, and placed into tubes that were snap frozen on dry ice. Plasma and tissue samples were stored frozen at −80 °C until analysis by LC-MS.
Bioanalysis of Plasma and Brain Samples
Quantitation of CuII(atsm) in plasma and brain samples was performed using LC-MS (Waters Xevo TQ mass spectrometer coupled to a Waters ACQUITY UPLC). Chromatographic separation was conducted employing a Supelco Ascentis Express RP amide 50 × 2.1-mm column eluting with methanol-water gradient with 0.05% formic acid. Elution of CuII(atsm) and the internal standard, diazepam, was monitored in positive electrospray ionization multiple-reaction monitoring mode using the transition of 322.0 > 248.8 and 285.2 > 154.1, respectively. Whole brains were homogenized in three parts (by weight) of water on ice prior to precipitation. CuII(atsm) concentrations were quantified in plasma and brain homogenate against calibration standards prepared in blank plasma or brain homogenate, as appropriate. Both samples and standards were prepared by precipitation with acetonitrile followed by centrifugation and analysis of the supernatant. The analytical lower limit of quantitation was 5 ng/ml in mouse plasma and 5 ng/g in mouse brain homogenate.
SOD1 Gel Zymography
SOD1 enzymatic activity was determined according to methods previously described (31). Briefly, spinal cord cytoplasmic protein fractions from transgenic mice (2 μg) and wild-type mice (10 μg) were separated using 7.5% native PAGE. Gels were incubated in 2.45 mm nitro blue tetrazolium (Sigma-Aldrich) solution for 20 min and soaked in developer solution (28 mm tetramethylethylenediamine, 28 μm riboflavin in 36 mm KH2PO4) for 15 min in the dark with constant agitation. Gels were illuminated for 15 min until sufficient contrast between the achromatic zones (dismutation) and blue background was achieved. Gel images were captured using an Epson transmission scanner. SOD1 activities were measured by quantifying absorbance of the clear bands using the Gene Tools analysis software (Syngene, Cambridge, UK).
SDS-PAGE and Western Blotting
Cytoplasmic protein fractions (20 μg) were separated using NuPAGE® Novex 4–12% Bis-Tris midi gel electrophoresis (Invitrogen). Proteins were transferred to nitrocellulose membranes, blocked in 0.5% casein in PBS, and incubated for 1 h with the sheep SOD1 (1:1000) (Calbiochem), mouse GFAP (1:20,000) (Dako), rabbit Iba1 (1:1000) (Wako), rabbit TDP-43 (1:1500) (Proteintech), mouse phospho-TDP-43 (1:2000) (Cosmobio), neurofilament (1:1000) (Sigma), or β-actin (1:2000) (Sigma) antibodies. HRP-conjugated secondary antibodies were used accordingly and incubated for 1 h, and membranes were developed using ECL Advance Lumigen TMA-6 chemiluminescence (GE Healthcare, Rydalmere, Australia). The optical density of target protein bands normalized to actin was quantitated using the GeneGnome chemiluminescence image system (Syngene). Graphs represent analysis of data collected from two midi gels (26 wells) showing data across the whole cohort.
Protein Carbonyl Measurement
Protein oxidation assessed by carbonyl levels in cytoplasmic fractions was measured using the OxyELISATM oxidized protein quantitation kit (Millipore, S7250) according to the manufacturer's instructions.
3-Nitrotyrosine (3-NT) Enzyme-linked Immunosorbent Assay (ELISA)
3-NT levels were determined by ELISA. Briefly, ELISA was performed at 22 °C, and all antibodies were prepared in 0.1% casein in PBS, pH 7.4. Immunosorbent 96-well plates were incubated with cytoplasmic protein fractions (∼20 μg) diluted in 100 mm carbonate buffer, pH 9.6, for 2 h. After blocking with StabilCoat (Surmodics) for 1 h and washing with PBS containing 0.1% Tween 20, plates were incubated with rabbit anti-tyrosine (1:2000; Cell Signaling Technologies) antibodies for 2 h. Next, plates were incubated with HRP-conjugated secondary antibodies for 1 h and with TMB substrate (GE Healthcare) for 15 min, and the reaction was stopped by the addition of 10% HCl. Absorbance was measured at 450 nm. 3-NT levels were normalized to protein concentration and expressed as the percentage of vehicle-treated wild-type mice.
Immunohistochemistry
Immunostaining procedures were performed as described previously (21). Antigen retrieval was carried out with citrate buffer for Iba1 only. Sections were quenched with 3% hydrogen peroxide, blocked with the appropriate 20% serum, and incubated with primary antibodies (GFAP 1:500 and Iba1 1:1500) for 1 h. Sections were developed using the universal LSAB® + HRP kit (Dako) as described by the manufacturer. Diaminobenzidine tetrahydrochloride reagent (Dako) was applied to sections, before dehydration and mounting with DPX.
Statistical Analysis
Statistical analyses were performed using Kaplan-Meier and log rank tests (Mantel-Cox) for disease onset, duration, and survival (GraphPad Prism version 4 software). Optical density changes between Western and zymography bands were analyzed using SPSS version 17.0. Statistical significance was determined using a Student's t test or one-way analysis of variance with Tukey's honestly significant difference (HSD) test or Dunnett's post hoc analysis and defined as p < 0.05.
RESULTS
CuII(atsm) (Fig. 1A) is a neutral, lipophilic, thermodynamically stable complex that was reported to possess SOD1-like antioxidant activity (16, 20). We have found that CuII(atsm) has very little SOD1-like activity (supplemental Fig. S1C), but instead it specifically inhibits the action of peroxynitrite. This is exemplified by the ability of CuII(atsm) to rescue SOD1 activity in vitro in the presence of peroxynitrite, which inactivates SOD1 (Fig. 1B, supplemental Fig. S1D). SOD1 activity was measured as the ability to inhibit pyrogallol oxidation. 10 units/ml SOD1 was able to inhibit ∼80% of pyrogallol oxidation. This SOD1 activity was inhibited by the addition of 1 μm peroxynitrite (ONOO−) and was rescued by co-treatment with 1 μm CuII(atsm) (p < 0.01). This is likely due to the ability of CuII(atsm) to enhance the breakdown of peroxynitrite. ONOO− inhibited SOD1 activity in a dose-dependent manner and did not interfere with the absorbance of pyrogallol or pyrogallol oxidation. CuII(atsm) also had no any effect on pyrogallol oxidation (supplemental Fig. S1B).
To evaluate the potential therapeutic effect of CuII(atsm) for ALS, the drug was orally administered (30 mg/kg) to presymptomatic SOD1G93A mice (21, 22) at 140 days. Treatment significantly prolonged mean survival time from 263 ± 3 days (n = 18) to 300 ± 11 days (n = 14, p < 0.001), a 14% extension in lifespan (Fig. 2A). Treatment also significantly delayed the onset of locomotor deficits from 237 ± 2 days to 256 ± 4 days (Fig. 2, B and C, p < 0.001) and delayed onset of weight loss (182 ± 2 days versus 194 ± 4 days) (Fig. 2D, Table 1, p < 0.01). This led to a significant slowing of progression from onset of motor impairment to death by 70% (27 ± 2.4 days versus 45 ± 6.5 days), and weight loss onset to death was extended by 30% (79 ± 4.1 days to 103 ± 7.5 days) (Table 1). The dose at 30 mg/kg was well tolerated with no adverse effects observed in WT mice (supplemental Fig. S2). The in vivo concentration of CuII(atsm) 2 h after gavage in WT mice was 304 ng/ml (plasma), 69 ng/g (brain), and 19 ng/g (spinal cord). The concentrations achieved in SOD1G93A mice were 331 ng/ml (plasma), 67 ng/g (brain), and 15 ng/g (spinal cord) (supplemental Table S2).
FIGURE 2.

Effect of CuII(atsm) on symptom onset, progression, and survival in SOD1G93A mice. A, Kaplan-Meier survival analysis of CuII(atsm)- and vehicle-treated SOD1G93A mice dosed at presymptomatic (Pre-sym) age. CuII(atsm) extended lifespan of SOD1G93A mice from 263 ± 3.4 days to 300 ± 10.8 days (mean ± S.E.), p < 0.001. B and C, motor function assessed by rotarod was significantly improved in drug-treated mice when compared with controls with delayed onset at 256 ± 3.9 days versus 237 ± 1.7, p < 0.001. D, onset of weight loss determined by age of decline from peak body weight. E, Kaplan-Meier survival curves for CuII(atsm) treatment started at symptomatic (Sym) age showing a 25-day extension, p < 0.001. In either study, there were no sex-related differences in survival time.
TABLE 1.
Effect of CuII(atsm) on onset of weight loss, gait abnormalities, motor impairment, and lifespan (days)
Disease onset was defined by age of first decline from peak body weight, stride length, or rotarod performance. Survival interval (SI) is the duration from onset to end point. Days (%) refers to percentage difference between CuII(atsm) and vehicle treated mice. ND, not determined; NA, not applicable as the weight of the mice has already decreased when the treatment started. *, p < 0.05, **, p < 0.01, ***, p < 0.001 difference between CuII(atsm)- and vehicle-treated groups.
| Presymptomatic |
Symptomatic |
|||||
|---|---|---|---|---|---|---|
| Vehicle | CuII (atsm) | Days (%) | Vehicle | CuII (atsm) | Days (%) | |
| Symptom onset (body weight loss) | 182 ± 2.3 | 194 ± 4.3** | 12 (6) | NA | NA | NA |
| Gait impairment onset (stride length) | 205 ± 2.0 | 229 ± 2.7*** | 24 (10) | ND | ND | ND |
| Motor impairment onset (rotarod) | 237 ± 1.7 | 256 ± 3.9*** | 19 (8) | 212 ± 1.1 | 216 ± 0.9* | 4 (1.8) |
| SI (body weight) | 79 ± 4.1 | 103 ± 7.5** | 24 (30) | NA | NA | NA |
| SI (stride length impairment) | 58 ± 3.4 | 72 ± 6.0* | 14 (24) | ND | ND | ND |
| SI (rotarod impairment) | 27 ± 2.4 | 45 ± 6.5 ** | 19 (70) | 37 ± 3.9 | 59 ± 3.0*** | 22 (59) |
| Lifespan | 263 ± 3.4 | 300 ± 10.8*** | 37 (14) | 250 ± 4.4 | 275 ± 2.9*** | 25 (10) |
Next, the more clinically relevant paradigm of postsymptomatic CuII(atsm) administration was evaluated. Treatment initiated at 200 days (after onset of muscle atrophy according to body weight decline) prolonged lifespan from 250 ± 4 days to 275 ± 3 days (p < 0.001, Fig. 2E), a 10% increase in survival. This was accompanied by a significant slowing of motor impairment (p < 0.05, Table 1).
In mice treated presymptomatically, the beneficial effects of CuII(atsm) were evidenced by significant reduction in several markers of protein oxidative damage in spinal cord. SOD1G93A mice showed elevated carbonyl levels when compared with wild-type animals, and CuII(atsm) treatment significantly reduced protein oxidation when compared with vehicle-treated SOD1G93A mice (p < 0.001, Fig. 3A). Furthermore, nitrated protein levels (3-NT) were significantly increased in SOD1G93A mice when compared with normal animals. CuII(atsm) treatment markedly reduced 3-NT levels when compared with vehicle-treated SOD1G93A mice (p < 0.05, Fig. 3B), whereas administration to wild-type mice did not affect 3-NT levels. Interestingly, CuII(atsm) treatment also significantly increased mutant SOD1 enzyme activity in spinal cords of SOD1G93A mice (∼11%, p < 0.05) (Fig. 3, C and D). Mutant SOD1 expression levels using a human-specific antibody were unchanged by treatment in SOD1G93A mice (supplemental Fig. S3). Interestingly, CuII(atsm) treatment also increased endogenous SOD1 activity, although this was not statistically significant.
FIGURE 3.

Impact of CuII(atsm) treatment on oxidative and nitrosative stress indices in SOD1G93A mouse spinal cord. A, protein carbonyl levels were determined using OxyELISA in 230-day-old mice and normalized to protein levels. The data are expressed as the percentage of change over vehicle (Veh)-treated wild-type mice. B, 3-NT levels were determined by ELISA and normalized to protein levels. The data are expressed as the percentage of change over vehicle-treated wild-type mice. 3-NT levels were increased in SOD1G93A mice when compared with wild types and reduced with CuII(atsm) treatment. C, SOD1 enzymatic activity gel analysis of spinal cords. An equal amount of protein was loaded for WT (10 μg) and SOD1G93A (2 μg) mice, respectively. D, quantitation of SOD1 activity expressed relative to respective vehicle-treated groups. CuII(atsm) treatment significantly increased SOD1 activity when compared with vehicle-treated transgenic mice. WT, non-transgenic (Non TG) littermate of the SOD1G93A mice. n is shown at bottom of bars (mean ± S.E.). One-way analysis of variance with Tukey's post hoc test, *, p < 0.05, **, p < 0.01, ***, p < 0.001 difference. ns, not significant.
The beneficial effects of CuII(atsm) were also demonstrated by significant sparing of lumbar spinal motor neurons in mice. At 200 and 230 days, there was a respective 25% (p < 0.05) and 21% (p < 0.01) rescue of motor neurons when compared with vehicle-treated SOD1G93A mice (Fig. 4, A and B). Consistent with preservation of motor neurons, neurofilament protein, which was depleted in vehicle-treated SOD1G93A mice, was also significantly maintained with CuII(atsm) treatment (p < 0.001) (Fig. 4, C and D).
FIGURE 4.

Effect of CuII(atsm) on motor neuron degeneration in SOD1G93A mice. A, micrographs of cresyl violet-stained lumbar spinal cord sections from wild-type (WT), vehicle (Veh)-, or CuII(atsm)-treated SOD1G93A mice. Higher magnification is shown in the insets. CuII(atsm)-treated mice exhibit larger, intact neuronal morphology when compared with vehicle-treated mice with atrophied and degenerating neurons. B, motor neuron numbers showing that CuII(atsm) treatment significantly attenuated neurodegeneration at 200 and 230 days when compared with vehicle group. Vehicle-treated SOD1G93A mice undergo significant motor neuron depletion at 200 and 230 days of disease when compared with wild-type mice. C and D, neurofilament expression determined by Western blot analysis of 230-day-old spinal cord showing restoration of neurofilament levels reflecting preservation of motor neurons with CuII(atsm) treatment, *, p < 0.05, **, p < 0.01, ***, p < 0.001. Error bars indicate mean ± S.E. The motor neuron counts were indistinguishable between vehicle- and CuII(atsm)-treated WT mice; therefore, these groups are pooled for the analysis.
Key cellular and molecular hallmarks of ALS pathology were next examined to investigate the mechanism of CuII(atsm)-mediated neuroprotection. Immunohistochemical analysis of astrocytic activation (GFAP) revealed widespread gliosis in spinal cords of SOD1G93A mice when compared with wild-type mice (Fig. 5A). CuII(atsm) treatment significantly reduced GFAP-positive cells at 200 and 230 days (Fig. 5A). This was confirmed by Western analysis of spinal cord lysates where GFAP induction in SOD1G93A mice was significantly inhibited by CuII(atsm) (Fig. 5, B and C). Numbers of Iba1-labeled activated microglia were also increased in spinal cords of control SOD1G93A mice when compared with normal mice (Fig. 5D), and these numbers were reduced upon CuII(atsm) treatment (Fig. 5D). This was in accordance with Western blot analysis showing reduction of Iba1 levels in CuII(atsm)-treated SOD1G93A mice to wild-type levels at 230 days (Fig. 5, E and F).
FIGURE 5.

Effect of CuII(atsm) treatment on reactive gliosis. A, GFAP expression shown by immunostaining of lumbar spinal cords from vehicle (Veh)- or CuII(atsm)-treated wild-type (WT) or SOD1G93A mice. B and C, Western blot quantitation of GFAP levels in 230-day-old mouse spinal cord. D, Iba1 expression revealed by immunostaining of spinal cords from mice. E and F, Western blot quantitation of Iba1 expression at 230 days in spinal cord. These results show reduction of astrogliosis and microgliosis with CuII(atsm) administration in mice. *, p < 0.05, **, p < 0.01, ***, p < 0.001. Error bars indicate mean ± S.E.
Phosphorylated and truncated TDP-43 species are characteristic of sALS pathology, some forms of mutant SOD1-linked fALS, and low transgene copy number strains of SOD1G93A mice (32, 33). In line with these studies, we showed an accumulation of abnormal TDP-43 species in our mouse model at 230 days. Levels of full-length phospho-TDP-43 (pTDP-43) were markedly increased in cytosolic fractions of spinal cord in control SOD1G93A mice (Fig. 6, A and B) when compared with wild-type animals (p < 0.01). Cytosolic full-length pTDP-43 species were significantly reduced by CuII(atsm) treatment (p < 0.01). C-terminal fragments of pTDP-43 (∼25 kDa) also implicated in toxicity (34, 35) were identified in cytosolic fractions of control SOD1G93A mice, and their production was abolished by CuII(atsm) in spinal cords (Fig. 6A).
FIGURE 6.

CuII(atsm) therapy prevents cytoplasmic TDP-43 pathology in SOD1G93A mice. A, Western blot of cytoplasmic pTDP-43. Veh, vehicle. B, quantitation of full-length cytoplasmic pTDP-43 levels in 230-day-old mouse spinal cord normalized to β-actin. Truncated C-terminal pTDP-43 was also detected in spinal cords. CuII(atsm) treatment significantly reduced full-length pTDP-43 accumulation as well as cytosolic C-terminal pTDP-43. Graphs represent the percentage of change when compared with WT vehicle controls. n is shown at bottoms of bars (mean ± S.E.). **, p < 0.01.
DISCUSSION
We demonstrate multiple and pronounced therapeutic effects of CuII(atsm) on clinical and pathological progression in this SOD1G93A mouse model of ALS. Oral administration of CuII(atsm) in the presymptomatic phase delayed disease onset and extended survival by 14%, whereas treatment of symptomatic mice still prolonged survival by 10%. Notably, there was a striking slowing of progression from onset of motor deficits to death, increasing the survival interval by 70 and 59% for presymptomatic and symptomatic interventions, respectively. Because muscle atrophy and spinal motor neuron loss are well underway in these mice at symptom onset (22), our data suggest that CuII(atsm) is effective even when initiated postsymptomatically, which is clinically relevant. These survival effects were linked to motor neuron rescue, reduced reactive gliosis, and decreased oxidative damage in spinal cords, consistent with effective blood-brain barrier penetration of CuII(atsm) as demonstrated in this study and others (17).
The beneficial effect of CuII(atsm) is likely due to its peroxynitrite scavenging ability, which reduces tissue accumulation of this radical. Peroxynitrite is a powerful oxidant formed by superoxide and nitric oxide; it can nitrate tyrosine or nitrosate cysteine residues and inactivate cellular targets including SOD1, and it has been shown to induce apoptosis in motor neurons and play an important role in ALS (8, 9, 36, 37). Although SOD1 mutations affect the scavenging of O2−, they also enhance SOD1-mediated peroxynitrite-induced nitration of many proteins (8). Nitrated products have been found to accumulate in spinal cords of SOD1G93A mice (14, 15) as confirmed here. Gene deletion of inducible nitric oxide synthase delayed disease onset and significantly extended the lifespan of SOD1G93A mice (38), supporting our findings that inhibiting nitrosative stress is protective in this model. We demonstrate that CuII(atsm) inhibits protein nitration in mice, consistent with diminished peroxynitrite accumulation in vivo. Our in vitro study also showed that peroxynitrite inhibited SOD1 activity, which was prevented by CuII(atsm) (Fig. 1B). Our animal data showed that SOD1 activity in spinal cord was enhanced in CuII(atsm)-treated mice when compared with vehicle-treated mice (Fig. 3C) independent of SOD1 synthesis. One consequence of elevated SOD1 activity in CuII(atsm)-treated mice would be reduction of protein carbonylation as observed. This suggests that CuII(atsm) is able to reduce oxidative and nitrosative damage in SOD1G93A mice, which could slow neurodegeneration. Because oxidative and nitrosative stress are also features of sALS (11), CuII(atsm) may have wider application for other common forms of ALS, not just mutant SOD1-linked disease. Our data also suggest that CuII(atsm) mediates activities other than scavenging peroxynitrite that warrant further study.
Attenuation of astrocyte and microglia activation in CuII(atsm)-treated mice is consistent with reduced inflammation. It is well accepted that neuroinflammatory cells may contribute to spinal motor neuron death in SOD1G93A transgenic mice and ALS patients (39–41). Astrocytes and microglia are strong determinants of disease progression in mutant SOD1 mice (42), and our data showing that CuII(atsm) extends survival interval even when administered postsymptomatically are consistent with modulation of glial activation and function. Further evidence for this proposal is that induction of astrocyte-derived pro-inflammatory metalloproteinase 9 activity in this model (22) was reduced in treated animals (data not shown). Peroxynitrite is known to stimulate astrocytic activation provoking motor neuron death (9), suggesting that glial cells are also likely targets for the CuII(atsm) action and neuroprotection observed.
The identification of mislocalized TDP-43 in all sALS cases and mutant TDP-43-linked fALS was a recent breakthrough toward understanding ALS pathogenesis (33, 43). Pathological TDP-43 species include nuclear TDP-43 mislocalized to the cytosol, a truncation producing ∼25-kDa C-terminal fragments, ubiquitination, and phosphorylation in affected tissues of patients (33). Previous studies with SOD1G93A low copy mice with a lifespan of 224–281 days reported mislocalization of neuronal TDP-43 by microscopy (32). This contrasts with mice highly expressing SOD1G93A where TDP-43 abnormalities were absent (23, 24) or rare (44). Here, we identify biochemical hallmarks of abnormal TDP-43 such as truncation and phosphorylation in our SOD1G93A mice with an average lifespan of 250–270 days (21, 22). Detection of cytosolic C-terminal pTDP-43 fragments here is also in line with observations in sALS tissues (33, 45, 46). This indicates that mice with low level accumulation of mutant SOD1 may recapitulate key pathological and biochemical features of TDP-43 proteinopathy as observed in ALS. The late onset and slow disease progression in these mice, unlike the aggressive disease course in other lines (23, 24), may therefore allow sufficient time for a more relevant disease model to develop as proposed by others (23, 32). CuII(atsm) treatment robustly reduced pathological forms of full-length as well as C-terminal fragmented pTDP-43, which may contribute to the neuroprotective effects and survival benefits seen. This is the first study showing drug modulation of TDP-43 pathology in vivo. Furthermore, given the mechanism of action of CuII(atsm), these data suggest a role for oxidative stress in the genesis of pathologic TDP-43. Indeed, recent findings suggest that TDP-43 is recruited to cytoplasmic stress granules under pro-oxidative conditions (47, 48). We have also recently shown that oxidative stress leads to the accumulation of TDP-43 in stress granules and that this may be a precursor for TDP-43 aggregation (49). Most relevant here was the demonstration that SIN1, a peroxynitrite donor, was able to induce abnormal cellular processing of TDP-43 resulting in its cytoplasmic accumulation (49). It remains to be determined whether CuII(atsm) can inhibit peroxynitrite-mediated TDP-43 pathology in these cell models. Although inhibition of peroxynitrite was the driving force behind the rationale to test CuII(atsm) in the ALS animal model here, our data do not exclude other possible mechanisms. For example, we have demonstrated that kinases can control TDP-43 cytosolic accumulation (49). However, more definitive evidence of how CuII(atsm) modulates aberrant TDP-43 metabolism will require further work.
In summary, our findings are that oral administration of the peroxynitrite scavenger CuII(atsm) reduces oxidative and nitrosative tissue burden, TDP-43 pathology, and motor neuron loss, leading to significant clinical improvement in ALS model mice. This indicates that this class of compound has promise as a therapeutic candidate for ALS and possibly other major forms of neurodegenerative disorders where oxidative and nitrosative stress are the driving force of disease progression.
Supplementary Material
This work was supported by the Australian National Health and Medical Research Council (NHMRC), C. J. Martin Fellowship 359269 (to B. J. T.), the Bethlehem Griffiths Research Foundation, the Motor Neurone Disease Research Institute of Australia, and the Australian Research Council.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1–S3.
- ALS
- amyotrophic lateral sclerosis
- sALS
- sporadic ALS
- fALS
- familial ALS
- CuII(atsm)
- diacetylbis(N(4)-methylthiosemicarbazonato)copper(II)
- TDP-43
- TAR DNA-binding protein-43
- pTDP-43
- phospho-TDP-43
- SOD1
- superoxide dismutase 1
- GFAP
- glial fibrillary acidic protein
- 3-NT
- 3-nitrotyrosine
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol
- TAR
- Transactive response.
REFERENCES
- 1. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X. (1993) Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 2. Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., Miller C. C., Nicholson G., Shaw C. E. (2008) Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turner B. J., Talbot K. (2008) Prog. Neurobiol. 85, 94–134 [DOI] [PubMed] [Google Scholar]
- 4. Boillée S., Vande Velde C., Cleveland D. W. (2006) Neuron 52, 39–59 [DOI] [PubMed] [Google Scholar]
- 5. Crow J. P. (2006) Expert Opin. Investig. Drugs 15, 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mariani E., Polidori M. C., Cherubini A., Mecocci P. (2005) J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 827, 65–75 [DOI] [PubMed] [Google Scholar]
- 7. Cozzolino M., Ferri A., Carrì M. T. (2008) Antioxid. Redox Signal. 10, 405–443 [DOI] [PubMed] [Google Scholar]
- 8. Beckman J. S., Carson M., Smith C. D., Koppenol W. H. (1993) Nature 364, 584. [DOI] [PubMed] [Google Scholar]
- 9. Cassina P., Peluffo H., Pehar M., Martinez-Palma L., Ressia A., Beckman J. S., Estévez A. G., Barbeito L. (2002) J. Neurosci. Res. 67, 21–29 [DOI] [PubMed] [Google Scholar]
- 10. Peluffo H., Shacka J. J., Ricart K., Bisig C. G., Martìnez-Palma L., Pritsch O., Kamaid A., Eiserich J. P., Crow J. P., Barbeito L., Estèvez A. G. (2004) J. Neurochem. 89, 602–612 [DOI] [PubMed] [Google Scholar]
- 11. Beal M. F., Ferrante R. J., Browne S. E., Matthews R. T., Kowall N. W., Brown R. H., Jr. (1997) Ann. Neurol. 42, 644–654 [DOI] [PubMed] [Google Scholar]
- 12. Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X., et al. (1994) Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 13. Scott S., Kranz J. E., Cole J., Lincecum J. M., Thompson K., Kelly N., Bostrom A., Theodoss J., Al-Nakhala B. M., Vieira F. G., Ramasubbu J., Heywood J. A. (2008) Amyotroph. Lateral Scler. 9, 4–15 [DOI] [PubMed] [Google Scholar]
- 14. Basso M., Samengo G., Nardo G., Massignan T., D'Alessandro G., Tartari S., Cantoni L., Marino M., Cheroni C., De Biasi S., Giordana M. T., Strong M. J., Estevez A. G., Salmona M., Bendotti C., Bonetto V. (2009) PLoS One 4, e8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casoni F., Basso M., Massignan T., Gianazza E., Cheroni C., Salmona M., Bendotti C., Bonetto V. (2005) J. Biol. Chem. 280, 16295–16304 [DOI] [PubMed] [Google Scholar]
- 16. Wada K., Fujibayashi Y., Tajima N., Yokoyama A. (1994) Biol. Pharm. Bull. 17, 701–704 [DOI] [PubMed] [Google Scholar]
- 17. Fodero-Tavoletti M. T., Villemagne V. L., Paterson B. M., White A. R., Li Q. X., Camakaris J., O'Keefe G., Cappai R., Barnham K. J., Donnelly P. S. (2010) J. Alzheimers Dis. 20, 49–55 [DOI] [PubMed] [Google Scholar]
- 18. Fujibayashi Y., Taniuchi H., Yonekura Y., Ohtani H., Konishi J., Yokoyama A. (1997) J. Nucl. Med. 38, 1155–1160 [PubMed] [Google Scholar]
- 19. Lewis J. S., Laforest R., Dehdashti F., Grigsby P. W., Welch M. J., Siegel B. A. (2008) J. Nucl. Med. 49, 1177–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wada K., Fujibayashi Y., Yokoyama A. (1994) Arch. Biochem. Biophys. 310, 1–5 [DOI] [PubMed] [Google Scholar]
- 21. Li Q. X., Mok S. S., Laughton K. M., McLean C. A., Volitakis I., Cherny R. A., Cheung N. S., White A. R., Masters C. L. (2006) Aging Cell 5, 153–165 [DOI] [PubMed] [Google Scholar]
- 22. Soon C. P., Crouch P. J., Turner B. J., McLean C. A., Laughton K. M., Atkin J. D., Masters C. L., White A. R., Li Q. X. (2010) Neuromuscul. Disord. 20, 260–266 [DOI] [PubMed] [Google Scholar]
- 23. Robertson J., Sanelli T., Xiao S., Yang W., Horne P., Hammond R., Pioro E. P., Strong M. J. (2007) Neurosci. Lett. 420, 128–132 [DOI] [PubMed] [Google Scholar]
- 24. Turner B. J., Bäumer D., Parkinson N. J., Scaber J., Ansorge O., Talbot K. (2008) BMC Neurosci. 9, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gingras B. A., Suprunchuk T., Bayley C. H. (1962) Can. J. Chem. 40, 1053 [Google Scholar]
- 26. Blower P. J., Castle T. C., Cowley A. R., Dilworth J. R., Donnelly P. S., Labisbal E., Sowrey F. E., Teat S. J., Went M. J. (2003) Dalton Trans. 4416–4425 [Google Scholar]
- 27. Marklund S., Marklund G. (1974) Eur. J. Biochem. 47, 469–474 [DOI] [PubMed] [Google Scholar]
- 28. Feeney S. J., McKelvie P. A., Austin L., Jean-Francois M. J., Kapsa R., Tombs S. M., Byrne E. (2001) Muscle Nerve 24, 1510–1519 [DOI] [PubMed] [Google Scholar]
- 29. Turner B. J., Parkinson N. J., Davies K. E., Talbot K. (2009) Neurobiol. Dis. 34, 511–517 [DOI] [PubMed] [Google Scholar]
- 30. Graham J. M., Rickwood D. (1997) Subcellular Fractionation: A Practical Approach, pp. 71–78, IRL Press, New York [Google Scholar]
- 31. Beauchamp C., Fridovich I. (1971) Anal. Biochem. 44, 276–287 [DOI] [PubMed] [Google Scholar]
- 32. Sumi H., Kato S., Mochimaru Y., Fujimura H., Etoh M., Sakoda S. (2009) J. Neuropathol. Exp. Neurol. 68, 37–47 [DOI] [PubMed] [Google Scholar]
- 33. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 34. Caccamo A., Majumder S., Deng J. J., Bai Y., Thornton F. B., Oddo S. (2009) J. Biol. Chem. 284, 27416–27424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y. J., Xu Y. F., Cook C., Gendron T. F., Roettges P., Link C. D., Lin W. L., Tong J., Castanedes-Casey M., Ash P., Gass J., Rangachari V., Buratti E., Baralle F., Golde T. E., Dickson D. W., Petrucelli L. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beckman J. S., Estévez A. G., Crow J. P., Barbeito L. (2001) Trends Neurosci. 24, S15–S20 [DOI] [PubMed] [Google Scholar]
- 37. Beal M. F. (2002) Free Radic. Biol. Med. 32, 797–803 [DOI] [PubMed] [Google Scholar]
- 38. Martin L. J., Liu Z., Chen K., Price A. C., Pan Y., Swaby J. A., Golden W. C. (2007) J. Comp. Neurol. 500, 20–46 [DOI] [PubMed] [Google Scholar]
- 39. Almer G., Guégan C., Teismann P., Naini A., Rosoklija G., Hays A. P., Chen C., Przedborski S. (2001) Ann. Neurol. 49, 176–185 [PubMed] [Google Scholar]
- 40. Kiaei M., Petri S., Kipiani K., Gardian G., Choi D. K., Chen J., Calingasan N. Y., Schafer P., Muller G. W., Stewart C., Hensley K., Beal M. F. (2006) J. Neurosci. 26, 2467–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hensley K., Fedynyshyn J., Ferrell S., Floyd R. A., Gordon B., Grammas P., Hamdheydari L., Mhatre M., Mou S., Pye Q. N., Stewart C., West M., West S., Williamson K. S. (2003) Neurobiol. Dis. 14, 74–80 [DOI] [PubMed] [Google Scholar]
- 42. Boillée S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A., Kassiotis G., Kollias G., Cleveland D. W. (2006) Science 312, 1389–1392 [DOI] [PubMed] [Google Scholar]
- 43. Lagier-Tourenne C., Polymenidou M., Cleveland D. W. (2010) Hum. Mol. Genet. 19, R46–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shan X., Vocadlo D., Krieger C. (2009) Neurosci. Lett. 458, 70–74 [DOI] [PubMed] [Google Scholar]
- 45. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., Oda T. (2006) Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 46. Igaz L. M., Kwong L. K., Xu Y., Truax A. C., Uryu K., Neumann M., Clark C. M., Elman L. B., Miller B. L., Grossman M., McCluskey L. F., Trojanowski J. Q., Lee V. M. (2008) Am. J. Pathol. 173, 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dewey C. M., Cenik B., Sephton C. F., Dries D. R., Mayer P., 3rd, Good S. K., Johnson B. A., Herz J., Yu G. (2011) Mol. Cell. Biol. 31, 1098–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., Buratti E., Silani V., Ratti A. (2009) J. Neurochem. 111, 1051–1061 [DOI] [PubMed] [Google Scholar]
- 49. Meyerowitz J., Parker S. J., Vella L. J., Ng DCh, Price K. A., Liddell J. R., Caragounis A., Li Q. X., Masters C. L., Nonaka T., Hasegawa M., Bogoyevitch M. A., Kanninen K. M., Crouch P. J., White A. R. (2011) Mol. Neurodegener. 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.