

Abstract
Lasting B-cell persistence depends on survival signals that are transduced by cell surface receptors. Here, we describe a novel biological mechanism essential for survival and homeostasis of normal peripheral mature B cells and chronic lymphocytic leukemia (CLL) cells, regulated by the heparin-binding cytokine, midkine (MK), and its proteoglycan receptor, the receptor-type tyrosine phosphatase zeta (RPTPζ). We demonstrate that MK initiates a signaling cascade leading to B cell survival, by binding to RPTPζ. In mice lacking PTPRZ, the proportion and number of the mature B cell population is reduced. Our results emphasize a unique and critical function for MK signaling in the previously described MIF/CD74 induced survival pathway. Stimulation of CD74 with MIF leads to c-Met activation, resulting in elevation of MK expression in both normal mouse splenic B and CLL cells. Our results indicate that MK and RPTPζ are important regulators of the B cell repertoire. These findings could pave the way towards understanding the mechanisms shaping B cell survival, and suggest novel therapeutic strategies based on the blockade of the midkine/RPTPζ-dependent survival pathway.
Introduction
Adaptive immunity depends on the production and maintenance of a pool of mature peripheral lymphocytes throughout life. In normal individuals, the pool of peripheral lymphocytes is constant in size. The control of lymphoid homeostasis is the result of a very fine balance between lymphocyte production, survival, and proliferation. Survival factors have been shown to play a critical role in maintaining lymphocyte homeostasis.
For many years, the maintenance of peripheral B-cell homeostasis was thought to rely only on two key elements, BCR tonic signals (e.g., Igα and Syk), and the B cell activating factor, a member of the TNF family (BAFF; BLyS/TALL-1/THANK/zTNF4) (1). Conditional deletion of the BCR on mature B cells results in their apoptosis (2, 3). Similarly, ablation of BAFF or its receptor, BAFFR, in mice by either genetic inactivation or using treatments designed to block their action, dramatically reduces the mature B cell pool (1).
An additional mechanism that regulates mature B cell survival, and depends on CD74 (invariant chain, Ii) and its ligand, macrophage migration inhibitory factor (MIF), was recently described. CD74 is a non-polymorphic type II integral membrane protein that is expressed on antigen presenting cells, including macrophages and B cells. The CD74 chain was initially thought to function mainly intracellularly as an MHC class II chaperone. A small proportion of CD74 is modified by the addition of chondroitin sulfate (CD74-CS), and this form of CD74 is expressed on the surface of antigen presenting as well as other cell types. CD74 is expressed as a cell surface complex with CD44 on macrophages (4), B cells (5), and epithelial cells (6). This complex is directly involved in shaping the B cell repertoire (7–11) by regulating mature B cell survival (5, 12, 13). It was previously shown that MIF binds to the CD74 extracellular domain on macrophages (14) and on B cells (5). Moreover, we recently demonstrated that CD74 stimulation by MIF recruits the tyrosine kinase receptor, c-Met, to the CD74/CD44 complex and thereby enables the induction of its signaling cascade within the cell (13). This leads to NF-κB activation, entry to the cell cycle, and augmented expression of anti-apoptotic proteins in a CD44-dependent manner. These findings indicate that surface CD74 functions as a survival receptor (5, 12, 13). In addition, the survival of re-circulating B cells was shown to require the presence of BM-resident dendritic cells (bmDC), which produce MIF in the perivascular BM niches (15). These compartments are organized into unique perivascular clusters enveloping selected blood vessels and seeded with mature B and T lymphocytes.
Interestingly, both MIF, CD74, and CD44 have been associated with tumor progression and metastasis (16–18). CD74 expression has been suggested to serve as a prognostic factor in many cancers, with higher relative expression of CD74 behaving as a marker of tumor progression (19). Chronic lymphocytic leukemia (CLL) is characterized by the progressive accumulation of small, mature CD5+ B-lymphocytes in the peripheral blood, lymphoid organs and bone marrow. In CLL cells, binding of MIF to CD74 initiates a similar signaling cascade that induces NF-κB activation and upregulation of TAp63 expression, resulting in the secretion of interleukin 8 (IL-8), which in turn promotes cell survival (20, 21). Thus, CD74 expressed on the surface of CLL cells plays a critical role in regulating the survival of these cells.
To determine the identities of the molecules whose expression is modulated by CD74, thereby regulating B cell survival, we investigated CD74 target genes. One striking example that we identify here, whose expression is strongly regulated by CD74, is the cytokine, midkine (MK).
MK is an heparin-binding cytokine, whose activities include anti-apoptotic, mitogenic, transforming, angiogenic, and chemotactic functions (22). MK is a basic, cysteine-rich polypeptide with a molecular mass of 13 kDa (23). It is composed of two domains, the N- and C-terminal domains. The domain corresponding to the C-terminal half domain of vertebrate MK is evolutionally conserved, and is important in binding heparin and chondroitin sulfate proteoglycans (24–26). MK tends to form dimers via spontaneous association, and the dimers are stabilized by cross-linking with transglutaminase. This dimerization appears to be required for MK activity (27). MK is expressed in adult peripheral lymphocytes (28) and specifically, in normal splenic B cells (29).
The expression of MK is significantly up regulated in various malignant tumors and plays crucial roles in carcinogenesis. Its expression has been shown to be strongly correlated with poor prognosis in patients with neuroblastomas, astrocytomas, pancreatic head carcinomas, or gastrointestinal stromal tumors. In accordance with its high expression in various malignant tumors, MK exerts cancer-related activities in the process of carcinogenesis, including transformation, fibrinolysis, cell migration, cell survival, anti-apoptosis, and angiogenesis (22, 30, 31).
Several cell-surface receptors were shown to bind to MK, including members of syndecan family, syndecan-1, -3, and -4 (32), a proteoglycan receptor-type tyrosine phosphatase ζ (RPTPζ) (26), a transmembrane low density lipoprotein (LDL), a receptor-related protein (LRP) (33), the anaplastic lymphoma kinase (ALK) (34), and the α4β1 and α6β1-integrins (35). Following MK stimulation, activation of the PI3 kinase/Akt and MAP kinase signaling pathways takes place, resulting in suppression of caspase activation (36). The signaling cascade involved in this pathway is not well understood, although it likely involves Src, since PPI, an inhibitor of Src, inhibits MK activity (22).
Little is known about the role of MK in B cells; however, it was recently shown that transfection of MK cDNA into the IL-3-dependent pro-B cell line Ba/F3 promotes cell cycle progression and partially inhibits apoptosis of these cells (37).
In the current study, we followed the role of MK and its receptor, RPTPζ, in normal and CLL B cells. We show that MK and RPTPζ are expressed in normal B and CLL cells, and play an important role in the MIF/CD74 induced survival cascade. These findings establish a key and novel role for MK and RPTPζ in the regulation of B cell survival during health and disease.
Materials and Methods
Cells
Murine B cells
B cells were obtained from spleen, lymph nodes, bone marrow and peritoneal cavity of C57BL/6, CD74−/− (38), 129SvEv and PTPζ (39) mice, at 6 to 8 weeks of age. All animal procedures were approved by the Animal Research Committee at the Weizmann Institute. Splenic B cells were purified using CD45R beads (BD Biosciences). The purity of the isolated cells (between 96–99%) was confirmed by FACS (using the B220+ marker) following each experiment.
Human Cells
B lymphocytes were taken from the peripheral blood of both healthy subjects (control), and CLL patients at varying stages of the disease; blood samples were provided by the Hematology Institute of Kaplan Medical Center, in accordance with the IRB of the hospital, as previously described (20). The diagnosis of CLL was based on standard criteria. Patients were staged according to the Rai staging system (40). B lymphocytes were purified using a RosettSep Ab mixture (StemCell, Vancouver, British Columbia, Canada), as described previously (20).
Stimulation of cells
MIF stimulation
Recombinant murine MIF was prepared in its native sequence form and purified from an expression system, as previously described (14). Contaminating endotoxin was removed by C8 affinity chromatography prior to protein renaturation, and the experimental preparations had an activity of 1800 EU/mg MIF. For MIF stimulation, 1 × 107 primary B cells of CLL cells were incubated in ISCOV’s medium containing 0.1% (v/v) FCS at 37°C for 1 hr. Next, cells were re-suspended in medium containing recombinant MIF (600 ng/ml). The cells were then incubated at 37°C for the indicated periods.
HGF stimulation
1×107 primary B cells were incubated in ISCOVEs medium containing 10% FCS with 20 ng/ml of recombinant HGF (R&D systems) and incubated at 37° C for the indicated periods.
MK stimulation
1 × 107 primary B cells or CLL cells were incubated in ISCOVEs medium containing 10% FCS with 100 ng/ml of recombinant MK (Peprotech; endotoxin level is < 1 ng/μg), and maintained at 37° C for the indicated periods.
MK and MIF injections
MIF injections
C57BL/6 mice were intraperitoneally injected with 1200 ng of MIF or PBS. After 24 hrs, spleens were collected and splenocytes analyzed for their MK expression.
MK injections
C57BL/6 mice were intraperitoneally injected daily with 400 ng of MK or PBS for 2 days. Spleens were collected and splenocytes were analyzed for their B cell repertoire and survival.
HGF, c-Met and RPTPζ blocking
For HGF blocking, 1 × 107 B cells were incubated in 1 ml of RPMI medium containing 1–3% (v/v) FCS in the presence of 5μg/ml anti-murine HGF (R&D systems) or 5 μg/ml anti-isotype control antibody for 16h at 37°C. For c-Met blocking, 1 × 107 B cells were incubated in 1ml of RPMI medium containing 10% (v/v) FCS in the presence of 0.1 ng/ml c-Met inhibitor, PH-665752 (a kind gift from Pfizer) at 37°C. For RPTPζ blocking, 1 × 107 CLL cells were incubated in 1 ml of RPMI medium containing 10% (v/v) FCS in the presence of anti-human RPTPζ (2 μg/ml; Santa Cruz; rabbit IgG raised against extracellular domain residues 141–440) or an isotype control antibody (2 μg/ml, Jackson Laboratories) for 24h at 37°C.
RNA isolation and reverse transcription
Total RNA was isolated from cells using the Tri Reagent kit (Sigma). Reverse transcription was carried out using Superscript II RT (Gibco-BRL). Primers used included:
HPRT: 5′GAGGGTAGGCTGGCCTATGGCT
3′ GTTGGATACAGGCCAGACTTTGTT
MK: 5′CCCCCAGCAGCAAGGACTG 3′GGGCCTGTGGGAGAGGAGGG
RPTPζ:5′CACATGGGGACTAAATACAATGAA
3′ GGATCCTTTAGTGATTCCTCTGAA
Real-time reverse transcription–PCR analysis
Levels of mRNA of MK and Bcl-2 were analyzed by quantitative real-time RT-PCR using a Light-Cycler instrument (Roche Diagnostics, Mannheim, Germany). The reaction volume (10 μl) contained 3 mM MgCl2, LightCycler HotStart DNA SYBR Green I mix (Roche Diagnostics), specific primer pairs, and 2.5 μl of cDNA. β-Actin levels were used to normalize samples for calculation of the relative expression levels of all of the genes. Primer sequences were as follows:
Actin: 5′ GTGACGTTGACATCCG 3′ CAGTAACAGTCCGCCT
Bcl-2 (murine): 5′ GCTACCGTCGTGACTT 3′ GCCGGTTCAGGTACTC
MK 5′ CCAGGAGACCATCCGCG 3′ TCCTTTTCCTTTCTTGGCTTTG
RP2 5′ GTGCGGCTGCTTCCATAA 3′
Bcl-2 (human) 5′GCCAACTGAGCAGAGTCTC 3′
Preparation of cell extracts
Stimulated cells were lysed in buffer containing: 25 mM Tris, pH 7.4; 2 mM Vanadate; 75 mM β glycophosphate, pH 7.2; 2 mM EDTA; 2 mM EGTA; 10 mM NaPPi; and 0.5% NP-40 in the presence of the following protease inhibitors: 10μg/ml Leupeptin, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 10 μg/ml chymostatin (Roche), 1mM PMSF (Sigma), and 20 mM N-ethyl-meleimide (Sigma Aldrich).
Immunoprecipitation
Protein-G Sepharose beads (Pharmacia) were conjugated to Tyr(P) (Santa Cruz) mAb for 2 hours at 40°C, followed by three washes in PBS. Beads were added to the cell lysates, and Tyr(P) proteins were immunoprecipitated overnight. The protein G bound material was washed three times with PBS containing 0.1% SDS and 0.5% NP40. Immunoprecipitates were separated by 10% (w/v) SDS-PAGE.
Western blot analysis
To detect changes in protein phosphorylation, lysates or immunoprecipitates were separated by 8–12% (w/v) SDS-PAGE. The proteins were transferred onto a nitrocellulose membrane and probed with anti p-Tyr (pTyr99; Santa Cruz), anti-p-Akt (Cell Signaling Technology), anti-Syk (LR; Santa Cruz Biotechnology), anti-RPTPζ (directed against the C- terminal domain of the protein Santa Cruz), and anti–Bcl-2 (C-2; Santa Cruz Biotechnology) followed by 1 h incubation with horseradish peroxidase-conjugated anti-mouse (Jackson Laboratories), anti-rabbit (Jackson Lab) or anti-goat (Santa Cruz), and peroxidase visualization by enhanced chemiluminescence (Amersham). The membrane was then stripped and reprobed with anti-tubulin antibody (Sigma), followed by peroxidase-conjugated anti-mouse (Jackson Labs).
Flow cytometry
Magic Red apoptosis detection kit
Cells were incubated with Magic Red (Immunochemistry Technology) according to the manufacturer’s instructions, at 37 °C for 1 h. Magic Red staining was measured by FACS analysis. The positive population was identified by comparison of the staining to that of the negative stained population.
Annexin/PI staining
Cells were washed and stained with Annexin (BD Biosciences) and PI (Bender MedSystems, Vienna, Austria) for 30 min at 4°C. Annexin and PI staining were analyzed by FACS. The positive population was identified by comparison of the staining to that of the negative stained population.
Phenotypic characterization of B cells
Cells were stained for anti-CD45R/B220, anti-CD21 (CR2/CR), anti-CD24 (heat stable Ag), and anti-CD23, anti-IgM, anti-IgD, anti-Mac-1 and anti-CD5 (all from eBioscience) and analyzed by FACS.
Intracellular staining for MK
Cells were permeabilized with 4% of PFA for 20 min at 4°C and washed twice with 0.1% saponin buffer. They were then stained for 30 min on ice with anti–MDK Ab (Santa Cruz Biotechnology) or anti-isotype control, followed by 30 min staining with PE-anti-rabbit (Jackson ImmunoResearch Laboratories). Cell staining was assessed in an FACS Calibur flow cytometer (BD Biosciences).
Staining of PTPζ
Human B cells were stained for anti-CD19 (Biolegend) and anti-PTPζ (Santa Cruz), and analyzed by FACS.
ELISA
MK in the serum of CLL patients and in conditioned media of stimulated human B cells was determined by ELISA according to the manufacturer’s instructions (Peprotech).
Statistical analysis
Comparisons between groups were evaluated by Student’s t test. Data are expressed as mean ± SD, and were considered statistically significant if p values were ≤0.05.
Results
The MIF/CD74 induced cascade elevates MK mRNA and protein expression
CD74 stimulation by its ligand MIF induces cell survival (5, 12). To further follow the CD74 induced survival cascade, we sought to identify genes whose expression is modulated by CD74 and are involved in regulation of B cell survival. Hence, gene expression profiles of cells treated in the presence or absence of MIF were compared using the Affymetrix GeneChip® expression analysis system (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33352; GSE33352). Many genes were found to be differentially expressed in these two populations. One striking example was Midkine (MK), whose expression was markedly elevated in the MIF stimulated cells. In order to confirm this result, splenic B cells were stimulated with or without MIF for 8 h, and MK mRNA levels were analyzed by RT-PCR (Fig 1A) and quantitative Real-time reverse transcription-PCR (qRT-PCR; Fig 1B). As shown in Fig 1A and B, MK mRNA levels were elevated following stimulation with MIF. No elevation in MK mRNA levels was observed in CD74 deficient B cells, indicating that this elevation was specific for MIF binding to its receptor, CD74 (Fig 1C). We next followed MK intracellular protein levels following MIF stimulation, in control or CD74 deficient B cells, by intracellular staining (Fig 1D). A specific elevation in MK protein was observed in wild type B cells following MIF stimulation, while no change was observed in CD74-deficient cells. MK protein was further followed in vivo following MIF injection to C57BL/6 mice. As demonstrated in Fig 1E, a significant elevation in MK protein levels was detected in B cells derived from MIF injected compared to PBS-treated mice. Thus, MIF binding to CD74 induces transcription and expression of MK in vitro and in vivo.
Figure 1. MIF elevates MK expression in B cells in a CD74-dependent manner.

(A–C) B cells derived from C57BL/6 (A–B) or CD74 deficient (C) mice were incubated in the presence or absence of MIF (600 ng/ml) for 8h. Total RNA was then isolated. (A) RT-PCR using primers for MK or HPRT, was performed. The intensity of the MK band after each treatment was normalized by dividing by the measured intensity of the HPRT band from the same treatment. The fold activation ratio in the absence of any treatment was normalized to 1, and the ratio for each treatment was calculated as the intensity of the treatment sample relative to 1. The results presented are representative of at least six different experiments. (B–C) Quantitative Real Time PCR was performed using primers for MK and β-actin. β-actin levels were used to normalize samples for calculation of the relative expression levels of MK. Results are expressed as fold change in MK expression in stimulated cells compared to non-stimulated cells, which was defined as 1. The results presented are representative of at least five different experiments. (D) B220+ cells derived from C57BL/6 and CD74−/−mice were incubated in the presence or absence of MIF (600 ng/ml) for 16 hr, and intracellular MK protein levels were analyzed by intracellular staining as described in Materials and Methods. Grey line- isotype control; Black line- MK staining. The graphs show an average of five independent experiments. (E) Control mice were injected with MIF (1.2 μg) or PBS. After 24 h, mice were sacrificed, splenic B cells were purified, and intracellular MK protein levels were analyzed by intracellular staining as described in Materials and Methods. Graph shows an average of three independent experiments. Grey line- isotype control; Black line- MK staining.
The expression of the c-Met receptor and its ligand, HGF, was recently demonstrated to be regulated by MIF and CD74. Both c-Met and HGF were shown to be involved in the CD74 induced survival cascade (13). We therefore examined whether the modulation of MK expression by CD74 is HGF/c-Met dependent. Wild type and CD74 deficient B cells were stimulated with HGF for 8 hours, and MK expression levels were analyzed. Stimulation of c-Met with its ligand elevated MK mRNA (Fig 2A) and protein (Fig 2B) levels in both wild type and CD74 deficient B cells. Moreover, blocking the c-Met activity using the c-Met inhibitor, PHA-665752, a selective small molecule, active-site inhibitor of the catalytic activity of c-Met kinase (Ki 4 nM) that competes with its ATP binding (41), or with anti-HGF blocking antibody (13), decreased intracellular protein levels of MK (Fig 2C). Thus, c-Met activation by its ligand, HGF, elevates MK expression. To determine whether c-Met activation is essential for the MIF-induced expression of MK, we next stimulated B cells with MIF in the presence or absence of anti-HGF blocking antibody or an isotype control antibody, and MK levels were analyzed by intracellular staining. As shown in Fig. 2D, treatment with HGF blocking antibody significantly reduced the MIF-induced elevation of MK expression. Together, these results suggest that MIF elevates MK expression in a c-Met/HGF dependent manner.
Figure 2. Binding of HGF to c-Met induces MK expression in B cells.

(A) B cells derived from C57BL/6 and CD74 deficient mice were incubated in the presence or absence of HGF (20 ng/ml) for 8 hr, and quantitative Real Time PCR was performed using primers for MK and β-actin. β-actin levels were used to normalize samples for calculation of the relative MK expression levels. Results are expressed as fold change in MK expression in stimulated cells compared to non-stimulated cells, which was defined as 1. The results presented are representative of at least five different experiments. (B–C) B cells derived from C57BL/6 or CD74 deficient mice were incubated in the presence or absence of HGF (20 ng/ml) (B), or the c-Met inhibitor, PHA-665752 (0.3 ng/ml) (C) for 16hr, and MK intracellular protein levels were analyzed by intracellular staining as described in Materials and Methods. Grey line- isotype control; black line- MK staining. The graphs show an average of five (B) or three (C) independent experiments. (D) B cells derived from C57BL/6 mice were incubated with or without MIF (600 ng/ml) in the presence of anti-HGF (5 μg/ml) or isotype control (5 μg/ml) antibodies for 16h. MK protein levels were analyzed by intracellular staining as described in Materials and Methods. Grey line- isotype control; Black line- MK staining. The graph summarizes the results of three independent experiments.
MK induces survival cascades in B cells
In order to follow the role of MK in B cells, we analyzed the MK-induced signaling cascade. Syk and Akt phosphorylation were shown to be involved in the B cell survival cascade (42, 43) and in the MIF induced signaling cascade (5, 12). We therefore followed the phosphorylation levels of Syk and Akt upon MK stimulation. As shown in Fig 3A and B, MK stimulation induced Akt and Syk phosphorylation in B cells.
Figure 3. MK induced cascade in B cells.

B cells derived from C57BL/6 mice were incubated in the presence or absence of MK (100 ng/ml) for various periods. Immediately after stimulation, cells were washed and fast frozen in liquid N2. The cells were lysed as described in Materials and Methods (A) Lysates were separated on 10% (w/v) SDS-PAGE, and proteins were blotted with anti-p-Akt or anti-Tubulin antibodies. The results presented are representative of at least four different experiments. (B) An aliquot from the lysates was reserved for total Syk analysis. Phosphorylated proteins from the remaining lysate were immunoprecipitated (IP) with an anti-Tyr(P) antibody. Immunoprecipitates and total lysate proteins were separated on 10% (w/v) SDS-PAGE and blotted with an anti-Syk antibody. The results presented are representative of at least three different experiments. (C) RNA was purified from control B cells after stimulation with PBS or MK (100ng/ml) for 6h. Quantitative Real time PCR was performed using primers for Bcl-2 and β-actin. β-actin levels were used to normalize samples for calculation of the relative expression levels of Bcl-2. Results are expressed as a fold change in MK expression in stimulated cells compared to non-stimulated cells, which was defined as 1. Results shown represent an average of at least eight separate experiments. (D) B220+ B cells derived from C57BL/6 mice were incubated in the presence or absence of MK (100ng/ml) for 6h. Cells were lysed, and levels of Bcl-2 and Tubulin were analyzed by western blot analysis. The results presented are representative of at least four different experiments.
We further studied the survival cascade by following the expression of the anti-apoptotic protein, Bcl-2, in MK stimulated or control B cells. As shown in Fig 3, MK stimulation elevated Bcl-2 mRNA (Fig 3C) and protein (Fig 3D) levels.
MK was recently shown to act as an anti-apoptotic factor in the human hepatoma cell line, HepG2, by downregulation of caspase-3 activity (36). Therefore, caspase 3 and 7 activity was analyzed in B cells following MK stimulation, using Magic Red staining. As shown in Fig 4A, MK stimulation significantly reduced the activity of these caspases. This reduced caspase activity resulted in an elevation of the live population and reduced the apoptotic population, as revealed by Annexin/PI staining (Fig 4B). Furthermore, an MK induced survival effect was also observed in CD74−/− B cells (Fig 4A). Although the absence of CD74 results in elevated levels of apoptotic cells, MK rescued the survival of these B cells. These results suggest that MK can rescue the absence of CD74, and therefore acts downstream of the CD74 induced survival cascade. However, MK does not completely rescue the absence or inactivity of CD74, suggesting that other compounds might be involved in this CD74 induced survival cascade. To further demonstrate that MK activity occurs downstream to HGF/c-Met, c-Met activity was blocked with the PHA-665752 inhibitor. Caspase activity was analyzed by Magic Red staining. As seen in Fig 4C, MK stimulation reduced caspase activity in cells incubated either in the presence or absence of the c-Met inhibitor. Furthermore, MK was able to rescue B cells from apoptosis induced by the blocking anti-HGF antibody (Fig 4D). Thus, MK acts downstream to the MIF/CD74 and HGF/c-Met survival cascade.
Figure 4. MK stimulation induces cell survival in vitro.

(A) B220+ B cells derived from C57BL/6 or CD74 deficient mice were incubated in the presence or absence of MK (100ng/ml) for 16h. Caspase activity was analyzed by Magic Red apoptosis detection kit. The graph shows the average of ten independent experiments. (B) B220+ B cells derived from control mice were incubated in the presence or absence of MK (100 ng/ml) for 18 hr. Cell death was then analyzed by Annexin V and PI staining of intact cells. The graph summarizes the average apoptotic population in four independent experiments. (C–D) B220+ B cells derived from C57BL/6 were incubated in the presence or absence of MK (100 ng/ml) with or without the c-Met inhibitor, PHA-665752 (0.3 ng/ml) (C), or with anti-HGF (5 μg/ml) or an isotype control (5 μg/ml) antibody (D) for 16 hr, and caspase activity was analyzed by Magic Red. The graphs show an average of five (C), or three (D) independent experiments.
In order to determine whether MK has a physiological effect on B cell survival, PBS or MK were injected daily i.p. into control (C57BL6) and CD74 deficient mice. After 48 hr, live versus apoptotic B cell populations were analyzed by Annexin/PI staining. As demonstrated in Fig 5A, the percent of apoptotic B populations was reduced following MK injection, resulting in elevation of the live cell population, as compared with the PBS treated cells in both C57BL/6 and CD74 deficient mice (Fig 5A). Moreover, in order to determine whether the MK-induced survival cascade leads to elevation of the peripheral mature population, we next analyzed the splenic B cell populations in MK treated mice. As demonstrated in Fig 5B, treatment of C57BL6 and CD74 deficient mice with MK resulted in an elevation of the percent and number of mature B (CD21low CD24low) cells. The mature B cell population in mice lacking CD74 is markedly reduced (7). Our results show that the proportion of elevation in the mature population following MK stimulation in both strains is similar, suggesting that MK has a similar effect on control and CD74−/− B cells. These results suggest that treatment with MK induces a survival cascade, which enlarges the proportion of the mature B cell population in the spleen.
Figure 5. The MK-induced survival pathway regulates survival of the mature B population in vivo.

C57BL/6 or CD74 deficient mice were injected daily with MK (400 ng) or PBS for 2 days. (A) Cell survival was analyzed by Annexin V and PI staining of intact B220+ cells. The graphs show the average apoptotic population in five independent experiments. (B) B cell subpopulations were analyzed for CD21, CD24, and CD23 and B220+ expression. The graphs show the number of mature B cells in seven independent experiments.
Together, these results show that MK activation is a downstream event to the MIF/CD74 and HGF/c-Met induced survival cascade. MK induces a signaling cascade leading to the expression of Bcl-2, resulting in the suppression of apoptosis, cell survival, and elevation of the mature B cell population.
RPTPζ serves as the receptor for MK in B cells
We next wished to identify the receptor that binds MK and regulates B cell survival. RPTPζ has been recognized as one of the receptors of MK in many cell types (26), however, its expression in B cells has not been reported. In order to reveal whether RPTPζ serves as a receptor for MK in B cells, we initially examined its expression in these cells. Thus, B cells from control and RPTPζ knockout mice (39, 44) were isolated, and RPTPζ mRNA and protein were examined using RT-PCR and western blot analysis, respectively. As can be seen in Fig 6, RPTPζ mRNA was detected in the wild type, but not the RPTPζ deficient B cells (Fig 6A). RPTPζ protein levels were then evaluated using an antibody that recognizes the transmembrane isoform. As shown in Fig 6B, RPTPζ protein was detected in B cells derived from control mice, while they were not observed in the RPTPζ-deficient cells.
Figure 6. RPTPζ is the receptor for MK in B cells.
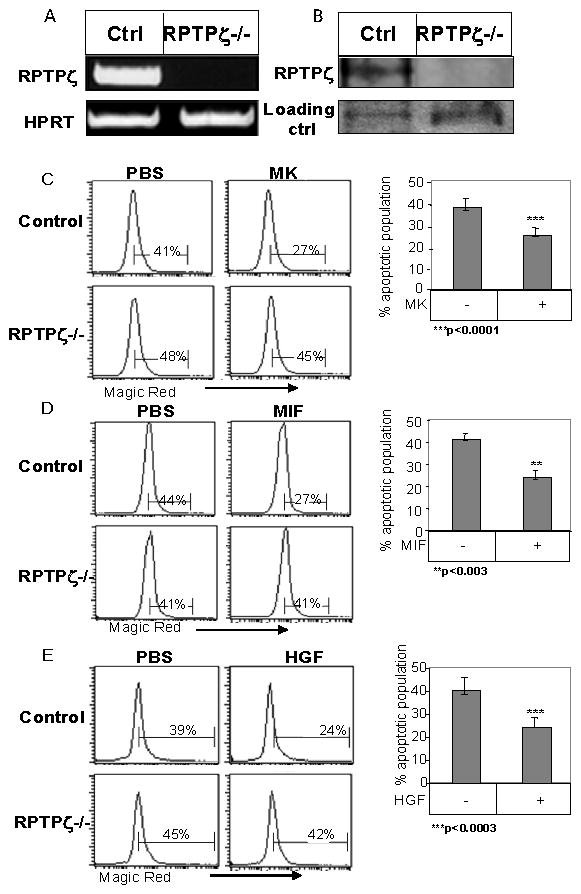
(A–B) B cells derived from control-129 SvEv mice and RPTPζ deficient mice were purified. (A) RT-PCR, was performed using primers for RPTPζ or HPRT. (B) RPTPζ expression was analyzed by western blot analysis. (C–E) B220+ B cells derived from control-129 SvEv or RPTPζ deficient mice were incubated in the presence or absence of MK (100 ng/ml) (C), MIF (600 ng/ml) (D), or HGF (20 ng/ml) (E) for 16h. Caspase activity was measured by Magic Red apoptosis detection kit. The graphs show an average of at least three independent experiments.
To test whether MK induces its survival cascade through RPTPζ, the survival of control and RPTPζ deficient B cells was analyzed in the presence or absence of MK. As seen in Fig 6C, MK stimulation reduced caspase 3 and 7 activities in control B cells, whereas cells lacking RPTPζ were unresponsive to MK. Moreover, in absence of RPTPζ, neither MIF nor HGF were able to elevate cell survival, demonstrating that RPTPζ is essential for the survival cascade induced by MIF/CD74 (Fig 6D) and HGF/c-Met (Fig 6E). These results indicate that RPTPζ serves as the receptor for MK in B cells and is involved in the survival cascade induced by MIF/CD74 and HGF/c-Met.
To determine whether the survival cascade regulated by RPTPζ modifies the B cell repertoire, wild type (129 SvEv) and RPTPζ deficient peripheral B cell populations were compared. FACS analysis showed a severe deficit of B cells in the spleen, the bone marrow and the lymph nodes of RPTPζ deficient mice, compared to their numbers wild type mice. A significant reduction in the proportion and absolute numbers of mature splenic B cells (CD21low CD24low, IgMlow IgDhigh) was detected (Fig 7A–B respectively). In addition, lower absolute numbers and proportion of the mature B cell population was detected in the BM of PTPRζ deficient mice relative to control mice (Fig 7C). Moreover, while in the RPTPζ knockout mice, since mostly mature B cells can be detected in the lymph nodes, the percent of mature B cells in the this compartment was similar to its levels in control mice, however, the total number of mature B cells was about two fold lower (Fig 7D). Taken together, these results suggest that RPTPζ plays an important role in the survival and shaping of the mature B cell compartment. However, no significant difference in the B cell populations in the peritoneal cavity was observed (Fig 7E), suggesting that this receptor does not play a role in survival of B cells in this compartment.
Figure 7. RPTPζ is involved in shaping the mature B cell repertoire.

(A–B) Splenic cells derived from control-129 SvEv mice and RPTPζ deficient mice were purified. (A) Dot plots showing CD21 and CD24 expression on B220+ cells derived from 129SvEv and from RPTPζ deficient mice. Graphs show the average percent and numbers of the mature population in twelve mice. (B) Dot plots showing IgD and IgM expression on B220+ cells derived from the spleen of 129SvEv and RPTPζ deficient mice. (C) Dot plots showing IgD and IgM expression on B220+ cells derived from bone marrow of 129SvEv and RPTPζ deficient mice. The graphs show the average of the percent and numbers of the mature population in seven mice. (D) Dot plots showing IgD and IgM expression on B220+ cells derived from lymph nodes of 129SvEv and RPTPζ deficient mice. The graph summarizes the average number of mature B cells in lymph node derived from 129SvEv and RPTPζ deficient mice, with six mice in each group (E) Dot plots showing CD5 and MAC-1 expression on IgMhigh B220+ cells derived from the peritoneal cavity of 129SvEv and RPTPζ deficient mice.
MK regulates survival of chronic lymphocytic leukemia cells
In order to determine whether MK regulates survival of normal human B cells, we first analyzed the expression of the MK receptor on these cells. As shown in Fig 8A, normal human B cells express RPTPζ on their surface. We next followed MK secretion following MIF stimulation. Human B cells were incubated with or without MIF for 24 h and MK secretion was then analyzed by ELISA. As demonstrated in Fig 8B, MIF stimulation augmented MK secretion to levels in the range used in our in vitro experiments (174 ng/ml ± 56 ng/ml; n=3).
Figure 8. MK regulates survival of chronic lymphocytic leukemia cells.

(A) RPTPζ expression was analyzed on human primary CD19+ B cells by FACS analysis. (B) Human primary B cells were incubated with or without MIF (600 ng/ml). After 24 h, their conditioned medium was collected and analyzed by ELISA for human MK protein levels. (C) MK levels in serum derived from healthy early and advanced CLL patients. (D) B cells derived from early and advanced CLL patients were purified. Cells were incubated in the presence or absence of MIF (100 ng/ml). Quantitative Real time PCR was performed using primers for MK and Rp2. Rp2 levels were used to normalize samples for calculation of the relative expression levels of MK. Results are expressed as fold change in MK expression in stimulated compared to non-stimulated cells, which was defined as 1. Results shown in each graph summarize at least four CLL patients. (E–F) CLL cells were incubated in the presence or absence of MK (100ng/ml) (E) Quantitative Real time PCR was performed using primers for Bcl2 and Rp2. Rp2 levels were used to normalize samples for calculation of the relative expression levels of Bcl2. Results are expressed as fold change in Bcl-2 expression in stimulated cells compared to non-stimulated cells, which was defined as 1. Results shown in each graph summarize three different CLL patients. (F) Cells were lysed, and Bcl-2 and Tubulin levels were measured by western blot analysis. The results presented are representative of four CLL patients. (G) Caspase activity was analyzed by Magic Red apoptosis detection kit. The graph shows an average of five different patients. (H) CLL cells were incubated with or without MIF (600 ng/ml) in the presence of anti-RPTPζ (2 μg/ml) or isotype control (2 μg/ml) antibodies for 24h. Caspase activity was analyzed by Magic Red apoptosis detection kit. Graph summarizes the results of three independent experiments.
Numerous studies have reported that serum MK levels are increased in the blood of patients with several kinds of malignancy (45–47). However, little is known about circulating levels of MK in leukemia patients. Therefore, we next evaluated the levels of MK in sera derived from control versus early and advanced CLL patients utilizing an ELISA assay (Fig 8C). Higher levels of MK were detected in sera derived from both early and advanced CLL patients (Early CLL: mean =285.5ng/ml, n=12, Advanced CLL: mean= 400ng/ml, n=9, Normal: mean=79.5ng/ml, n=14), suggesting that MK can serve as a prognostic marker, even at early stages of the disease.
A similar survival pathway regulated by MIF and CD74 was shown to operate in CLL cells (12, 20). We therefore, wished to determine whether MK is involved in the cascade regulating CLL survival, as well. To determine whether MK is a target gene of the MIF/CD74 survival cascade in CLL cells, CLL cells from early and advanced-stage patients were stimulated with MIF for 18 hr, and MK gene expression was analyzed by qRT-PCR. As demonstrated in Fig 8D, an elevation in MK mRNA levels was detected in MIF-stimulated CLL cells regardless of the disease stage. To determine whether MK induces CLL survival, CLL cells were stimulated in the presence or absence of MK, and expression of Bcl-2 was analyzed by qRT-PCR and western blot analysis. MK elevated Bcl-2 mRNA (Fig 8E) and protein (Fig 8F) levels. This cascade led to a significant reduction in the level of caspase activity (Fig 8G). Thus, in analogy to its role in normal B cells, MK mediates the survival of CLL cells. To gain further insight into the role of RPTPζ in CLL survival, the receptor was inhibited using an anti-RPTPζ blocking antibody. To this end, CLL cells were treated with RPTPζ blocking or with an isotype-control antibody for 24 hr, and caspase activity was examined by Magic Red. As can be seen in Fig 8H, blocking of RPTPζ significantly increased the caspase activity. In addition, to confirm that MIF/CD74 induces a survival cascade that is RPTPζ dependent, CLL cells were stimulated with MIF in the presence or absence of RPTPζ blocking or isotype control antibodies. The MIF-induced survival was downregulated in cells whose RPTPζ receptor was blocked (Fig 8H). However, blocking RPTPζ did not completely abolish the MIF induced survival cascade, suggesting that there might be additional branches in the pathway controlled by CD74, or alternatively that the antibody did not entirely block RPTPζ function. All together, these results show that RPTPζ is involved in mediating the anti-apoptotic activity of MK and of the MIF/CD74 survival cascade in CLL cells.
Discussion
Adaptive immunity depends on the production and maintenance of a pool of mature peripheral lymphocytes throughout life. In healthy individuals, the pool of peripheral lymphocytes remains constant in size. The control of lymphoid homeostasis is the result of a very fine balance between lymphocyte production, survival, and proliferation. Survival factors have been shown to play a critical role in maintaining lymphocyte homeostasis (1). Moreover, resistance to apoptosis, leading to enhanced survival, is associated with initiation and progression of B cell malignancies such as CLL (48).
Our previous studies have demonstrated that mature B cell survival is mediated by CD74 and its natural ligand, MIF. MIF is a ubiquitous protein that has a broad tissue distribution and is found in virtually all cells (49); thus, it undergoes tonic production in the spleen, as well. Engagement of the CD74/CD44 complex by MIF triggers recruitment of the tyrosine kinase receptor, c-Met, to this complex (13), resulting in induction of an anti-apoptotic Syk-PI3K/Akt signaling pathway and HGF secretion, which stimulates the survival of the mature B cell population in an autocrine manner (5, 12, 13, 50). Here, we identified two novel components of the CD74/CD44 induced B cell survival pathway, the heparin-binding cytokine, midkine, and its receptor, RPTPζ
Midkine is an heparin-binding cytokine, that promotes growth, survival, migration and other activities of its target cells (31). MK was shown to promote growth and suppress apoptosis through activation of Raf-1/MEK/ERK and PI3K/Akt kinase pathways in neurons, cardiomyocytes and some malignant neuroblastomas. The activated MAPK pathway was reported to downregulate caspase-3 activity (51, 52). In addition, MK-induced Akt phosphorylation promotes a series of anti-apoptotic pathways (53). In B cells, the PI 3-kinase/Akt pathway has a major role in determining cell fate and resting B-cell survival (42, 43). Akt is activated in a Syk-dependent pathway (54, 55). Disruption of Syk expression by genetic deletion indicates that it is essential for regulation of cell survival (56). Here, we show that exogenous MK stimulation triggers the Syk and Akt signaling cascade, which leads to B cell survival. Thus, MK appears to be necessary for the Syk and Akt phosphorylation induced by MIF.
Several reports have previously demonstrated that MK acts as an anti-apoptotic growth factor, at least in part, through the regulation of Bcl-2 expression (57, 58). Our present data show that the MK-induced signaling cascade enhances the expression of Bcl-2, and inhibits activity of caspases 3 and 7, leading to cell survival. Furthermore, MK injection to C57BL6 and CD74−/−mice leads to a survival cascade, which results in enlargement of the mature B cell compartment in the spleen. The MK induced survival cascade can partially bypass the lack of survival signals transmitted in CD74 deficient B cells. MK was also able to induce survival in cells in which c-Met activity was perturbed, indicating that MK is an important downstream inducer of the B cell survival cascade, which is regulated by the HGF/c-Met axis. Thus, MK is a crucial downstream component in the MIF/CD74 induced survival cascade. However, other downstream elements might be also involved in this induced pathway.
Several receptors are known to be activated by MK (31). Here, we report that RPTPζ is expressed in mature B cells and functions as a survival receptor in these cells. RPTPζ forms a cell surface complex with LRP (33), α4β1-integrin and α6β1-integrin (35), which also serve as MK receptors. MK, which acts as a dimer, binds to components of the receptor including glycosaminoglycan chains, and promotes formation of the receptor complex. It was previously shown in embryonic neurons that the survival-promoting signal of MK is induced by binding to a receptor complex containing RPTPζ (59). Similarly, our study demonstrated the importance of RPTPζ in reducing cell death in B cells. In addition, characterization of the peripheral B cell repertoire of PTPRζ deficient mice revealed a significant decrease in the proportion of mature B cells leading to a dramatic reduction in the number of mature B cells in PTPRζ deficient as compared to control mice. These results demonstrate the essential role of RPTPζ in controlling and shaping the B cell repertoire.
MIF/CD74 (6, 60) and HGF/c-Met (61) induced pathways were shown to regulate tumor progression and survival. In addition, in CLL, we previously showed that MIF and CD74 play a pivotal role in the regulation of CLL cell survival (20, 21). MK has also been associated with tumor progression (22). Moreover, significantly higher MK expression levels were detected in B-ALL, AML-M2, and AML-M3 than those in normal controls (62). However, expression and function of MK in leukemia cells was never characterized, although it was demonstrated that transfection of MK into the IL-3-dependent pro-B cell line, Ba/F3, promotes cell cycle progression and partially inhibits apoptosis in these cells (37).
Our study demonstrates that MK and its receptor RPTPζ play a major role in survival of CLL cells. MK suppresses CLL apoptosis by elevating the expression of Bcl-2 and inhibiting caspase 3 and 7 activities. Moreover, blocking RPTPζ in CLL cells, resulted in the induction of cell death and inhibition of the MIF/CD74-induced survival cascade. However, blocking RPTPζ did not completely abolish the MIF induced survival cascade, suggesting that there might be additional branches in the pathway controlled by CD74. We further show that MK serum levels are elevated in CLL patients compared to their levels in normal individuals, regardless of the disease stage. These findings are in agreement with other studies in which MK was significantly elevated in serum derived from cancer patients (45, 46). Since reliable predictors of prognosis at the early stages of CLL are lacking in the clinical workup of CLL patients (63), the identification of MK as a prognostic marker may contribute to improving the clinical management of CLL patients. To conclude, our findings establish a key and novel role for MK and its receptor, RPTPζ, in the regulation of B cell survival during health and disease, and may suggest novel therapeutic strategies aimed at interrupting this survival pathway in B cell malignancies.
Acknowledgments
The authors gratefully acknowledge members of the Shachar laboratory team for helpful discussions.
Footnotes
This research was supported by The Israel Science Foundation (Morasha), the Israel Science foundation and the Israel Cancer Association. I.S. is the incumbent of the Dr. Morton and Ann Kleiman Professorial Chair. R Bucala and L Leng are supported by NIH AR049610, AR050498, AI042310, and the Alliance for Lupus Research.
Author contribution: S.C.- Designed and performed most of the experiments, analyzed results, wrote the paper.
O.S.- Designed and performed some of the experiments, analyzed results.
E.Z.-T.- Designed and performed some of the experiments, analyzed results.
N.M.- Designed and performed some of the experiments, analyzed results.
I.B.-E. Designed and performed some of the experiments, analyzed results.
M.G.- Designed and performed some of the experiments, analyzed results.
I. H.H.- Provided reagents.
Y.H.- Provided reagents.
L.S.- Provided reagents.
M.H.- Provided reagents.
L.L.- Provided reagents.
R.B.- Provided reagents and participated in writing the paper.
S.H.- Provided reagents, participated in some of the experiments and in writing the paper.
I.S.- Designed and supervised the experiments, analyzed the results and wrote the paper.
References
- 1.Mackay F, Figgett WA, Saulep D, Lepage M, Hibbs ML. B cell stage and context dependent requirements for survival signals from BAFF and the B cell receptor. Immunol Rev. 2010;237:205–225. doi: 10.1111/j.1600-065X.2010.00944.x. [DOI] [PubMed] [Google Scholar]
- 2.Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 3.Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, Schmidt-Supprian M. Canonical NF-[kappa] B Activity, Dispensable for B Cell Development, Replaces BAFF-Receptor Signals and Promotes B Cell Proliferation upon Activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 Is the Signaling Component of the Macrophage Migration Inhibitory Factor-CD74 Receptor Complex. Immunity. 2006;25:595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gore Y, Starlets D, Maharshak N, Becker-Herman S, Kaneyuki U, Leng L, Bucala R, Shachar I. Macrophage migration inhibitory factor (MIF) induces B cell survival by activation of a CD74/CD44 receptor complex. J Biol Chem. 2008;283:2784–2792. doi: 10.1074/jbc.M703265200. [DOI] [PubMed] [Google Scholar]
- 6.Meyer-Siegler KL, Leifheit EC, Vera PL. Inhibition of macrophage migration inhibitory factor decreases proliferation and cytokine expression in bladder cancer cells. BMC Cancer. 2004;4:34–45. doi: 10.1186/1471-2407-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shachar I, Flavell RA. Requirement for invariant chain in B cell maturation and function. Science. 1996;274:106–108. doi: 10.1126/science.274.5284.106. [DOI] [PubMed] [Google Scholar]
- 8.Matza D, Wolstein O, Dikstein R, Shachar I. Invariant Chain Induces B Cell Maturation by Activating TAFII105-NF-kB Dependent Transcription Program. J Biol Chem. 2001;276:27203–27206. doi: 10.1074/jbc.M104684200. [DOI] [PubMed] [Google Scholar]
- 9.Matza D, Lantner D, Bogoch Y, Flaishon L, Hershkoviz R, Shachar I. Invariant chain induces B cell maturation in a process which is independent of its chaperonic activity. Proc Natl Acad Sci USA. 2002;99:3018–3023. doi: 10.1073/pnas.052703299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matza D, Kerem A, Lantner F, Shachar I. Invariant chain induced B cell differentiation requires intramembrane - proteolytic release of the cytosolic domain. Immunity. 2002;17:549–560. doi: 10.1016/s1074-7613(02)00455-7. [DOI] [PubMed] [Google Scholar]
- 11.Matza D, Kerem A, Shachar I. Invariant chain, a chain of command. Trends Immunol. 2003;24:246–248. doi: 10.1016/s1471-4906(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 12.Starlets D, Gore Y, Binsky I, Haran M, Harpaz N, Shvidel L, Becker-Herman S, Berrebi A, Shachar I. Cell Surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood. 2006;107:4807–4816. doi: 10.1182/blood-2005-11-4334. [DOI] [PubMed] [Google Scholar]
- 13.Gordin M, Tesio M, Cohen S, Gore Y, Lantner F, Leng L, Bucala R, Shachar I. c-Met and Its Ligand Hepatocyte Growth Factor/Scatter Factor Regulate Mature B Cell Survival in a Pathway Induced by CD74. Journal of Immunology. 2010;185:2020–2031. doi: 10.4049/jimmunol.0902566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF Signal Transduction Initiated by Binding to CD74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sapoznikov A, Pewzner-Jung Y, Kalchenko V, Krauthgamer R, Shachar I, Jung S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat Immunol. 2008;9:388–395. doi: 10.1038/ni1571. [DOI] [PubMed] [Google Scholar]
- 16.Meyer-Siegler K, Hudson PB. Enhanced expression of macrophage migration inhibitory factor in prostatic adenocarcinoma metastases. Urology. 1996;48:448–452. doi: 10.1016/S0090-4295(96)00207-5. [DOI] [PubMed] [Google Scholar]
- 17.Bando H, Matsumoto G, Bando M, Muta M, Ogawa T, Funata N, Nishihira J, Koike M, Toi M. Expression of macrophage migration inhibitory factor in human breast cancer: association with nodal spread. Jpn J Cancer Res. 2002;93:389–396. doi: 10.1111/j.1349-7006.2002.tb01269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishihira J, Ishibashi T, Fukushima T, Sun B, Sato Y, Todo S. Macrophage migration inhibitory factor (MIF): Its potential role in tumor growth and tumor-associated angiogenesis. Ann N Y Acad Sci. 2003;995:171–182. doi: 10.1111/j.1749-6632.2003.tb03220.x. [DOI] [PubMed] [Google Scholar]
- 19.Mizue Y, Nishihira J, Miyazaki T, Fujiwara S, Chida M, Nakamura K, Kikuchi K, Mukai M. Quantitation of macrophage migration inhibitory factor (MIF) using the one-step sandwich enzyme immunosorbent assay: elevated serum MIF concentrations in patients with autoimmune diseases and identification of MIF in erythrocytes. Int J Mol Med. 2000;5:397–403. doi: 10.3892/ijmm.5.4.397. [DOI] [PubMed] [Google Scholar]
- 20.Binsky I, Haran M, Starlets D, Gore Y, Lantner F, Harpaz N, Leng L, Goldenberg DM, Shvidel L, Berrebi A, Bucala R, Shachar I. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. Proc Natl Acad Sci U S A. 2007;104:13408–13413. doi: 10.1073/pnas.0701553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Binsky I, Lantner F, Grabovsky V, Harpaz N, Shvidel L, Berrebi A, Goldenberg DM, Leng L, Bucala R, Alon R, Haran M, Shachar I. TAp63 regulates VLA-4 expression and CLL cell migration to the BM in a CD74 dependent manner. J Immunol. 2010;184:4761–4769. doi: 10.4049/jimmunol.0904149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadomatsu KJ, Muramatsu T. Midkine and pleiotrophin in neural development and cancer. Cancer Letters. 2004;204:127–143. doi: 10.1016/S0304-3835(03)00450-6. [DOI] [PubMed] [Google Scholar]
- 23.Muramatsu T. The Midkine Family of Growth/Differentiation Factors. Development Growth & Differentiation. 1994;36:1–8. doi: 10.1111/j.1440-169X.1994.00001.x. [DOI] [PubMed] [Google Scholar]
- 24.Asai T, Watanabe K, IchiharaTanaka K, Kaneda N, Kojima S, Iguchi A, Inagaki F, Muramatsu T. Identification of heparin-binding sites in midkine and their role in neurite-promotion. Biochemical and Biophysical Research Communications. 1997;236:66–70. doi: 10.1006/bbrc.1997.6905. [DOI] [PubMed] [Google Scholar]
- 25.Akhter S, Ichihara-Tanaka K, Kojima S, Muramatsu H, Inui T, Kimura T, Kaneda N, Talukder AH, Kadomatsu K, Inagaki F, Muramatsu T. Clusters of basic amino acids in midkine: Roles in neurite-promoting activity and plasminogen activator-enhancing activity. Journal of Biochemistry. 1998;123:1127–1136. doi: 10.1093/oxfordjournals.jbchem.a022052. [DOI] [PubMed] [Google Scholar]
- 26.Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. A receptor-like protein-tyrosine phosphatase PTP zeta/RPTP beta binds a heparin-binding growth factor midkine - Involvement of arginine 78 of midkine in the high affinity binding to PTP zeta. Journal of Biological Chemistry. 1999;274:12474–12479. doi: 10.1074/jbc.274.18.12474. [DOI] [PubMed] [Google Scholar]
- 27.Iwasaki W, Nagata K, Hatanaka H, Inui T, Kimura T, Muramatsu T, Yoshida K, Tasumi M, Inagaki F. Solution structure of midkine, a new heparin-binding growth factor. Embo J. 1997;16:6936–6946. doi: 10.1093/emboj/16.23.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hovanessian AG. Midkine, a cytokine that inhibits HIV infection by binding to the cell surface expressed nucleolin. Cell Research. 2006;16:174–181. doi: 10.1038/sj.cr.7310024. [DOI] [PubMed] [Google Scholar]
- 29.Kato H, Watanabe K, Murari M, Isogai C, Kinoshita T, Nagai H, Ohashi H, Nagasaka T, Kadomatsu K, Muramatsu H, Muramatsu T, Saito H, Mori N, Murate T. Midkine expression in Reed-Sternberg cells of Hodgkin’s disease. Leukemia & Lymphoma. 2000;37:415–424. doi: 10.3109/10428190009089442. [DOI] [PubMed] [Google Scholar]
- 30.Muramatsu T. Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J Biochem. 2002;132:359–371. doi: 10.1093/oxfordjournals.jbchem.a003231. [DOI] [PubMed] [Google Scholar]
- 31.Kadomatsu K. Midkine Regulation of the Renin-Angiotensin System. Current Hypertension Reports. 2010;12:74–79. doi: 10.1007/s11906-010-0092-8. [DOI] [PubMed] [Google Scholar]
- 32.Nakanishi T, Kadomatsu K, Okamoto T, Ichihara-Tanaka K, Kojima T, Saito H, Tomoda Y, Muramatsu T. Expression of syndecan-1 and-3 during embryogenesis of the central nervous system in relation to binding with midkine. J Biochem. 1997;121:197. [PubMed] [Google Scholar]
- 33.Muramatsu H, Zou K, Sakaguchi N, Ikematsu S, Sakuma S, Muramatsu T. LDL receptor-related protein as a component of the midkine receptor. Biochemical and Biophysical Research Communications. 2000;270:936–941. doi: 10.1006/bbrc.2000.2549. [DOI] [PubMed] [Google Scholar]
- 34.Stoica GE, Kuo A, Powers C, Bowden ET, Sale EB, Riegel AT, Wellstein A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem. 2002;277:35990–35998. doi: 10.1074/jbc.M205749200. [DOI] [PubMed] [Google Scholar]
- 35.Muramatsu H, Zou P, Suzuki H, Oda Y, Chen GY, Sakaguchi N, Sakuma S, Maeda N, Noda M, Takada Y, Muramatsu T. {alpha} 4 {beta} 1-and {alpha} 6 {beta} 1-integrins are functional receptors for midkine, a heparin-binding growth factor. J Cell Sci. 2004;117:5405–5415. doi: 10.1242/jcs.01423. [DOI] [PubMed] [Google Scholar]
- 36.Ohuchida T, Okamoto K, Akahane K, Higure A, Todoroki H, Abe Y, Kikuchi M, Ikematsu S, Muramatsu T, Itoh H. Midkine protects hepatocellular carcinoma cells against TRAIL-mediated apoptosis through down-regulation of caspase-3 activity. Cancer. 2004;100:2430–2436. doi: 10.1002/cncr.20266. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Xing H, Tian Z, Tang K, Wang J, Xu Z, Rao Q, Wang M, Wang J. Overexpression of Midkine promotes the viability of BA/F3 cells. Biochem Biophys Res Commun. 2009;384:341–346. doi: 10.1016/j.bbrc.2009.04.119. [DOI] [PubMed] [Google Scholar]
- 38.Elliott EA, Drake JR, Amigorena S, Elsemore J, Webster P, Mellman I, Flavell RA. The invariant chain is required for intracellular transport and function of major histocompatibility complex class II molecules. J Exp Med. 1994;179:681–694. doi: 10.1084/jem.179.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harroch S, Palmeri M, Rosenbluth J, Custer A, Okigaki M, Shrager P, Blum M, Buxbaum JD, Schlessinger J. No obvious abnormality in mice deficient in receptor protein tyrosine phosphatase beta. Molecular and Cellular Biology. 2000;20:7706–7715. doi: 10.1128/mcb.20.20.7706-7715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O’Brien S, Rai KR. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87:4990–4997. [PubMed] [Google Scholar]
- 41.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, Hansen M, Schaefer E, Naoki K, Lader A, Richards W, Sugarbaker D, Husain AN, Christensen JG, Salgia R. Functional expression and mutations of c-met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Research. 2005;65:1479–1488. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 42.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2:945–956. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- 43.Pogue SL, Kurosaki T, Bolen J, Herbst R. B cell antigen receptor-induced activation of Akt promotes B cell survival and is dependent on Syk kinase. J Immunol. 2000;165:1300–1306. doi: 10.4049/jimmunol.165.3.1300. [DOI] [PubMed] [Google Scholar]
- 44.Harroch S, Furtado GC, Brueck W, Rosenbluth J, Lafaille J, Chao M, Buxbaum JD, Schlessinger J. A critical role for the protein tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nature Genetics. 2002;32:411–414. doi: 10.1038/ng1004. [DOI] [PubMed] [Google Scholar]
- 45.Shimada H, Nabeya Y, Okazumi SI, Matsubara H, Kadomatsu K, Muramatsu T, Ikematsu S, Sakuma S, Ochiai T. Increased serum midkine concentration as a possible tumor marker in patients with superficial esophageal cancer. Oncol Rep. 2003;10:411–414. [PubMed] [Google Scholar]
- 46.Obata Y, Kikuchi S, Lin Y, Yagyu K, Muramatsu T, Kumai H. Serum midkine concentrations and gastric cancer. Cancer Sci. 2005;96:54–56. doi: 10.1111/j.1349-7006.2005.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ikematsu S, Yano A, Aridome K, Kikuchi M, Kumai H, Nagano H, Okamoto K, Oda M, Sakuma S, Aikou T, Muramatsu H, Kadomatsu K, Muramatsu T. Serum midkine levels are increased in patients with various types of carcinomas. British Journal of Cancer. 2000;83:701–706. doi: 10.1054/bjoc.2000.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352:8040815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 49.Calandra T, Froidevaux W, Martin C, Roger T. Macrophage migration inhibitory factor and host innate immune defenses against bacterial sepsis. Journal of Infectious Diseases. 2003;187:S385–S390. doi: 10.1086/374752. [DOI] [PubMed] [Google Scholar]
- 50.Lantner F, Starlets D, Gore Y, Flaishon L, Yamit-Hezi A, Dikstein R, Leng L, Bucala R, Machluf Y, Oren M, Shachar I. CD74 induces TAp63 expression leading to B cell survival. Blood. 2007;110:4303–4311. doi: 10.1182/blood-2007-04-087486. [DOI] [PubMed] [Google Scholar]
- 51.Sandra F, Harada H, Nakamura N, Ohishi M. Midkine induced growth of ameloblastoma through MAPK and Akt pathways. Oral Oncol. 2004;40:274–280. doi: 10.1016/j.oraloncology.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 52.Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, Michikawa M. Midkine inhibits caspase dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. Journal of Neurochemistry. 1999;73:2084–2092. [PubMed] [Google Scholar]
- 53.Dai LC, Wang X, Yao X, Lu YL, Ping JL, He JF. Antisense oligonucleotides targeting midkine induced apoptosis and increased chemosensitivity in hepatocellular carcinoma cells. Acta Pharmacologica Sinica. 2006;27:1630–1636. doi: 10.1111/j.1745-7254.2006.00459.x. [DOI] [PubMed] [Google Scholar]
- 54.Li HL, Davis WW, Whiteman EL, Birnbaum MJ, Pure E. The tyrosine kinases Syk and Lyn exert opposing effects on the activation of protein kinase Akt/PKB in B lymphocytes. Proc Natl Acad Sci U S A. 1999;96:6890–6895. doi: 10.1073/pnas.96.12.6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Astoul E, Watton S, Cantrell D. The dynamics of protein kinase B regulation during B cell antigen receptor engagement. J Cell Biol. 1999;145:1511–1520. doi: 10.1083/jcb.145.7.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geahlen RL. Syk and pTyr’d: Signaling through the B cell antigen receptor. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbamcr.2009.03.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horiba M, Kadomatsu K, Yasui K, Lee JK, Takenaka H, Sumida A, Kamiya K, Chen S, Sakuma S, Muramatsu T, Kodama I. Midkine plays a protective role against cardiac ischemia/reperfusion injury through a reduction of apoptotic reaction. Circulation. 2006;114:1713–1720. doi: 10.1161/CIRCULATIONAHA.106.632273. [DOI] [PubMed] [Google Scholar]
- 58.Qi MS, Ikematsu S, Ichihara-Tanaka K, Sakuma S, Muramatsu T, Kadomatsu K. Midkine rescues Wilms’ tumor cells from cisplatin-induced apoptosis: Regulation of Bcl-2 expression by midkine. Journal of Biochemistry. 2000;127:269–277. doi: 10.1093/oxfordjournals.jbchem.a022604. [DOI] [PubMed] [Google Scholar]
- 59.Sakaguchi N, Muramatsu H, Ichihara-Tanaka K, Madea N, Noda M, Yamamoto T, Michikawa M, Ikematsu S, Sakuma S, Muramatsu T. Receptor-type protein tyrosine phosphatase zeta as a component of the signaling receptor complex for midkine-dependent survival of embryonic neurons. Neuroscience Research. 2003;45:219–224. doi: 10.1016/s0168-0102(02)00226-2. [DOI] [PubMed] [Google Scholar]
- 60.Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Iwashige H, Aridome K, Hokita S, Aikou T. Invariant chain expression in gastric cancer. Cancer Lett. 2001;168:87–91. doi: 10.1016/s0304-3835(01)00503-1. [DOI] [PubMed] [Google Scholar]
- 61.Migliore C, Giordano S. Molecular cancer therapy: Can our expectation be MET? European Journal of Cancer. 2008;44:641–651. doi: 10.1016/j.ejca.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 62.Hidaka H, Yagasaki H, Takahashi Y, Hama A, Nishio N, Tanaka M, Yoshida N, Villalobos IB, Wang Y, Xu YY. Increased midkine gene expression in childhood B-precursor acute lymphoblastic leukemia. Leuk Res. 2007;31:1045–1051. doi: 10.1016/j.leukres.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 63.Van Bockstaele F, Verhasselt B, Philippe J. Prognostic markers in chronic lymphocytic leukemia: A comprehensive review. Blood Reviews. 2009;23:25–47. doi: 10.1016/j.blre.2008.05.003. [DOI] [PubMed] [Google Scholar]