

Abstract
Lamin B receptor (LBR) is a bifunctional nuclear membrane protein with N-terminal lamin B and chromatin binding domains plus a C-terminal sterol Δ14 reductase domain. LBR expression increases during neutrophil differentiation and deficient expression disrupts neutrophil nuclear lobulation characteristic of Pelger-Huët anomaly. Thus LBR plays a critical role in regulating myeloid differentiation, but how the two functional domains of LBR support this role is currently unclear. We previously identified abnormal proliferation and deficient functional maturation of promyelocytes (EPRO cells) derived from EML-ic/ic cells, a myeloid model of ichthyosis (ic) bone marrow that lacks Lbr expression. Here we provide new evidence that cholesterol biosynthesis is important to myeloid cell growth and is supported by the sterol reductase domain of Lbr. Cholesterol biosynthesis inhibitors caused growth inhibition of EML cells that increased in EPRO cells, whereas cells lacking Lbr exhibited complete growth arrest at both stages. Lipid production increased during wild-type neutrophil maturation, but ic/ic cells exhibited deficient levels of lipid and cholesterol production. Ectopic expression of a full length Lbr in EML-ic/ic cells rescued both nuclear lobulation and growth arrest in cholesterol starvation conditions. Lipid production also was rescued, and a deficient respiratory burst was corrected. Expression of just the C-terminal sterol reductase domain of Lbr in ic/ic cells also improved each of these phenotypes. Our data support the conclusion that the sterol Δ14 reductase domain of LBR plays a critical role in cholesterol biosynthesis, and that this process is essential to both myeloid cell growth and functional maturation.
Keywords: Lamin B receptor, Cholesterol biosynthesis, Neutrophil development
INTRODUCTION
Neutrophils are the most abundant white blood cells in circulation and present the first line of defense against invading pathogens. As critical mediators of innate immunity, neutrophils mount an impressive barrier to infectious microbes by undergoing two critical processes during their development in the bone marrow: rapid proliferation of myeloid progenitors that produces large numbers of circulating mature cells, and dramatic changes in morphology and protein expression that support functional responses (1, 2). Myeloid progenitor proliferation is caused by signalling pathways activated by certain hematopoietic cytokines released during inflammatory responses, including granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) (3, 4). Following proliferation, neutrophil progenitors then undergo morphologic maturation that includes lobulation of the nucleus, a process thought to facilitate the escape of mature neutrophils from capillary beds as they undergo chemotaxis toward sites of infection. Upon contact with bacteria, the neutrophils engulf the microbes by phagocytosis and then kill them via the production of proteolytic enzymes and reactive oxygen during the respiratory burst. The importance of high neutrophil numbers and their correct function is revealed by the consequences of human disorders that either block neutrophil development (cyclic or severe congenital neutropenia) or cause abnormal neutrophil functions (chronic granulomatous disease) (recently reviewed in (5, 6)). Studies aimed at identifying the molecular causes of these disorders have helped to unravel the complex transcriptional mechanisms that regulate neutrophil differentiation. However, there is increasing evidence that cholesterol biosynthesis also plays a significant role in supporting myeloid progenitor proliferation and functional maturation. For example, treatment of neutrophils with the cholesterol synthesis inhibitor lovastatin, the lanosterol 14α-demethylase inhibitor SSKF 104976, or the cholesterol sequestering agent methyl-β-cyclodextrin (MβCD) dramatically inhibited multiple neutrophil functions, including adhesion and rolling, chemotaxis, phagocytosis and activation of the respiratory burst (7-11). In each case, cholesterol levels were shown to influence properties of membrane microdomains termed lipid rafts. These lipid platforms facilitate the assembly of protein complexes in plasma or phagosome membranes, including the membrane-bound proteins of NADPH oxidase (11, 12). Treatment of neutrophils with cholesterol inhibitors therefore caused deficient membrane cholesterol levels, thereby disrupting the formation of membrane protein complexes critical to normal functional responses. Cholesterol bioavailability and its synthesis in myeloid progenitors is also important to proliferative responses; treatment of myeloblastic HL-60 and acute myeloid leukemia (AML) cells with lipoprotein deficient serum (LPDS) and/or inhibitors of cholesterol biosynthesis severely diminished their proliferation (13-18). Together these data indicate that levels of cholesterol directly influence both myeloid cell proliferation and functional maturation, but whether the metabolic mechanisms that drive cholesterol biosynthesis are critical to neutrophil differentiation is currently unknown.
The cholesterol biosynthesis pathway is part of the larger isoprenoid biosynthetic system and its end product is not only an important structural lipid but also a starting metabolite for the synthesis of multiple metabolically active compounds (reviewed in (19)). A major intermediate in cholesterol biosynthesis from the starting compound acetyl-CoA is lanosterol, which must undergo a series of enzymatic modifications including reductions of C7-, C14- and C24-unsaturated bonds. Each of these reductions is catalyzed by a member of the highly related sterol reductase family. A possible association between the activities of one of these enzymes and myeloid development was recently revealed when mutations of the lamin B receptor (LBR) gene were found to cause Pelger-Huët anomaly, a disorder of neutrophil morphologic maturation characterized by mononuclear or bilobed nuclei (often referred to as pince-nez cells) (20, 21). LBR encodes a bifunctional protein with an N-terminal domain localized to the nucleoplasm that interacts with B-type lamins, heterochromatin and chromatin binding proteins (e.g. HP1), and a C-terminal domain with 8 predicted transmembrane segments that anchor LBR to the inner nuclear membrane (INM) (22-28). The combined functions of these domains, which tether chromatin and components of the nuclear lamina to the INM, are thought to mediate reformation of the nuclear envelope after mitosis and perhaps orchestrate localization of chromatin to peripheral regions of the nucleus at distinct stages of the cell cycle. In addition to these identified domains, the C-terminal ~407 amino acids of LBR are 58% identical to the 3β-hydroxysterol Δ14 reductase TM7SF2 (also known as DHCR14 or SR-1), recently identified in bovine, human and mouse tissues (29-32). This high level of homology indicates that both LBR and TM7SF2 can function as sterol reductases, which is supported by studies that demonstrated LBR can complement C14 sterol reductase mutants of Saccharomyces cerevisia and Neurospora crassa, and TM7SF2 exhibits sterol reductase activity when overexpressed in COS cells (30, 33, 34). Which of these two proteins is essential to cholesterol biosynthesis is unclear, but homozygous mutations of Tm7sf2 in mice cause no apparent abnormality and there is no evidence to date that the gene is associated with human disorders (31, 35). In contrast, homozygous mutations in LBR cause not only severe PHA but also a fatal disorder in humans termed Hydrops-Ectopic calcification-Moth-eaten skeletal dysplasia (Greenberg/HEM dysplasia) and the ichthyosis (ic) phenotype in mice (20, 36). Furthermore, a stillborn fetus with Greenberg/HEM dysplasia exhibited increased levels of the precursors cholesta-8,14-dien-3β-ol and cholesta-8,14,24-trien-3β-ol, indicating that the loss of sterol reductase activity provided by LBR may be the underlying cause of the disease (37). Despite these data, views differ as to whether disorders caused by loss of LBR expression are more likely laminopathies, i.e. caused by a disruption of normal interactions between LBR and nuclear lamins (e.g. lamin B), or due to deficient cholesterol biosynthesis. For example, studies of mice deficient for both Tm7sf2 and Lbr indicated that these two proteins play redundant roles and that a disruption of the interactions between lamin B and chromatin with the INM underlies the cause of Greenberg/HEM dysplasia and ichthyosis (35). This data is contrasted by the recent discovery that certain missense mutations within the C-terminal sterol reductase domain of LBR caused Greenberg/HEM dysplasia with no apparent effect on neutrophil nuclear maturation (38). Thus the importance of the two disparate functional domains of LBR during early mammalian development or adult blood cell differentiation remains to be resolved.
To further investigate the functions of LBR in myeloid differentiation, we previously used bone marrow of a homozygous ic mouse that lacked Lbr expression to generate an immortalized cell line that can be induced to differentiate into mature neutrophils. The cell line was generated by expressing a dominant negative form of the retinoic acid receptor alpha (RARα), which previously was shown to block differentiation of bone marrow stem cells at either an early progenitor stage (erythroid, myeloid and lymphoid, or EML cells) or at the promyelocyte stage (EML-derived promyelocytes, or EPRO cells); further differentiation of either cell type can be induced by superphysiologic levels of all-trans retinoic acid (ATRA) (39). We previously discovered that derived EML-ic/ic and EPRO-ic/ic cells exhibited aberrant proliferative responses and upon ATRA-induced differentiation displayed severe nuclear hypolobulation, loss of chemotaxis and a deficient respiratory burst (40). We speculated that the loss of chemotaxis could be explained by the lack of lobulation, which may have impeded the migration of cells through the transwell apparatus used in our assays. However, causes for the loss of growth and respiratory burst were unresolved – although expression of gp91phox, a critical component of NADPH oxidase, was reduced in ic/ic cells as compared to +/ic cells, significant expression was nonetheless identified and this did not explain the dramatic effects on respiratory burst induced by opsinized zymosan. Here we tested our hypothesis that loss of the sterol reductase domain of Lbr may contribute to the growth and respiratory burst defects in the ic/ic cells, and that cholesterol biosynthesis supported by Lbr is critical to myelopoiesis. Both wild type (+/+) and ic/ic cells were assayed for their capacity to produce lipids and their growth in lipid deprived conditions. We found that loss of Lbr expression severely affected EML and EPRO responses in both assays, and caused a loss of overall cholesterol synthesis in either normal or lipid deprived medium. Expression of a full length version of mouse Lbr rescued both lipid production and the respiratory burst phenotypes. Moreover, truncated versions of Lbr that lacked N-terminal lamin B and chromatin binding domains also improved the responses. These studies indicate that cholesterol biosynthesis is a critical feature of myeloid cell differentiation that influences progenitor cell growth and their maturation into functional neutrophils, and that expression of the sterol Δ14 reductase domain of Lbr plays an essential role in myeloid cell cholesterol biosynthesis.
MATERIALS AND METHODS
Cell lines
All EML cells lines were cultured in IMDM (HyClone, Logan, UT) supplemented with 20% horse serum (HS, Invitrogen, Carlsbad, CA), and 15% BHK-MKL conditioned medium as a source of Stem Cell Factor (SCF). EPRO cells from each genotype were derived from EML cells as previously described (41) and maintained in IMDM supplemented with 20% horse serum and 20% BHK-HM5-conditioned medium as a source of GM-CSF. EPRO cell inductions were performed using 10 μM all-trans retinoic acid (ATRA, Sigma-Aldrich, St. Louis, MO) for 3 to 5 days. Generation of EML-Tm7sf2-knockout (KO) and littermate control cells was performed as described for EML-+/ic and -ic/ic cells (40), using bone marrow from knockout mice that were previously described (32). Human Embryonic Kidney (HEK) 293 and Phoenix retrovirus producer cells were maintained in DMEM (HyClone) supplemented with 10% FBS (Gemini, Aukland, New Zealand). Media were supplemented with 5 Units/mL penicillin, 5 μg/mL streptomycin sulfate and 0.25 μg/mL amphotericin B; all cells were maintained at 37°C in 5% CO2.
Chemicals
For cholesterol depletion studies, lovastatin (Sigma-Aldrich) was dissolved in 100% ethanol to make a 10 mg/mL stock solution, and methyl-β cyclodextrin (MβCD) was dissolved in sterile water to make a 500 mg/mL stock solution. Lipoprotein deficient serum (LPDS) was generated by the addition of 2% (weight/volume) Cab-O-Sil (Arcos, Morris Plains, NJ) to horse serum followed by overnight incubation at 4°C and centrifugation at 9000 x g for 1 h. The cholesterol content in regular serum vs. LPDS was determined using the Amplex Red cholesterol assay kit according to the manufacturer’s instructions (Invitrogen). For staining total lipids in cells, 1 mg/mL Nile red stock solution (Sigma-Aldrich) was prepared in acetone. Both LXR agonists T0901317 and GW3965 (Sigma) were dissolved in DMSO to yield final concentrations of 10 and 8 mM, respectively, each of which was added to growth medium of EML and EPRO cells to yield the final concentrations indicated for each assay.
Expression plasmids and transductions in EML cells
Coding sequences for all plasmids were polymerase chain reaction (PCR) amplified from cDNA generated from ATRA-induced EPRO cells using Superscript II Reverse Transcriptase (Invitrogen). The FLAG-tagged mouse Lbr expression plasmid was generated by amplifying the Lbr cDNA using a forward primer that included a BamHI site (underlined) adjacent to the second amino acid (5′-CGGGATCCCAAGTAGGAAGTTTG-3′) and reverse primers with a XhoI site directly downstream from the stop codon (5′-CGCTCGAGGTCAGTAAATGTAGGGG-3′), which was ligated into the pCMV-Tag2C vector (Stratagene, La Jolla, CA). The FLAG-Lbr fragment was then excised with NotI (located upstream of the FLAG sequences) and XhoI, overhangs filled in with Klenow Fragment (New England Biolabs, Ipswich, MA) and then the fragment was cloned into pMSCVpuro at the HpaI site. The Lbr71-626 and Lbr145-626 mutant sequences were generated using the forward primers 5′-CGGGATCCTGTCTCCCTCTCGACGC-3′ and 5′-CGGGATCCTGGAGCCCGAACACATG-3′, respectively, combined with the reverse primer 5′-CGGAATTCTCAGTAAATGTAGGGGAA-3′, and the fragments were cloned into the pMSCV-FLAG-Lbr expression vector using the BamHI (located downstream from the FLAG sequences) and an EcoRI site located downstream from the stop site. All PCR amplifications were carried out with a Px2 thermocycler (Thermo Electron Corporation, Milford, MA); PCR for wild-type Lbr was carried out for 30 cycles using 94°C × 1 min, 60°C × 1 min and 72°C × 1 min, whereas amplifications of the mutant Lbr fragments employed a touch-down amplification using the following conditions: 94°C, 58°C, 72°C for 30 sec each, 6 cycles; 94°C, 58→46°C gradient, 72°C for 30 sec each, 14 cycles; 94°C, 45°C, 72°C for 30 sec each, 15 cycles. The expression vector pMSCVpuro-Tm7sf2 was constructed by excising the mouse Tm7sf2 cDNA from clone MMM1013-7510962 (Open Biosystems, Huntsville, AL) via SalI and XhoI sites, and then ligating the cDNA into the XhoI site of the pMSCV-puro vector. Correct sequences for all inserted fragments were confirmed by double-stranded sequencing reactions (Beckman Coulter Genomics, Danvers, MA). Each plasmid was transduced into EML-+/ic or -ic/ic cells by first transiently transfecting Phoenix producer cells using FuGENE 6 transfection reagent (Roche Diagnostics, Mannheim, Germany) following the manufacturer’s instructions. After 24 hours, EML cells were added to the adherent cells, allowed to incubate in the presence of polybrene (8 mg/mL) for 24 hours, and then harvested and cultured for 24 h. Cells positive for each expression vector were selected for 3 weeks in 1 μg/mL puromycin (Sigma-Aldrich).
Proliferation assays
For each assay, 2 × 105 cells/mL were plated in triplicate wells and growth analysis was performed over a period of either 3 or 5 days. Viable cells were identified using trypan blue exclusion and cells were quantified using a hemacytometer. Proliferation responses to the cholesterol depleting drugs lovastatin and MβCD also were determined using the Non Radioactive Cell Proliferation assay (Promega, Madison, WI), which is a colorimetric assay that identifies metabolic activity in viable cells. Briefly, 2 × 105 cells/mL were cultured in triplicate wells in a 96-well plate in medium containing increasing concentrations of the appropriate cholesterol depleting drug (concentration range from 3.125 μM to 100 μM of lovastatin and 0.3125 to 10 mM of MβCD). After 3 days of growth, reagents were added according to the manufacturer’s instructions and the absorbance in each well was measured at 490 nm using a Spectramax M2 spectrophotometer (Molecular Devices, Sunnyvale, CA). The differences in absorbance at levels shown to cause growth inhibition but not complete cell death were then graphed.
Nile red staining of total lipids
Cells were cultured in growth medium supplemented with 10% oleic acid-albumin (Sigma-Aldrich) for 3 days, then were fixed with 4% paraformaldehyde and dried onto superfrosted microscope slides for 15 min. Slides were washed with PBS and stained with light-sensitive Nile Red solution (1μg/mL, Sigma-Aldrich) for 20 min. The slides then were rinsed three times in PBS and lipid droplets were visualized using an Olympus CKX41 microscope (Optical Analysis Corporation) at 40X magnification and fluorescent light.
Gas chromatography and mass spectrometry analyses
Methods used to extract sterols from cells and for GC/MS analysis were adapted from previously described procedures (42, 43). Briefly, cells were saponified for 1 h with 4% KOH in ethanol at 60°C. Stigmasterol was added as an internal standard. The samples were extracted with an equal volume of hexane by vortexing for 1 min. Once the samples separated into two phases, the organic layers were transferred to fresh tubes and dried under nitrogen. Dried extracts were reacted with BSTFA at 55°C for 45 min to form the trimethylsilyl derivative, which were then diluted with cyclohexane prior to loading one microliter into an Agilent 7890A GC system with 5975C inert MSD triple axis detector and a DB-5 column (J& W Scientific, Folsom, CA, USA, 30 m × 0.250 mm × 0.25 μm) for sterol separation. High purity helium gas was used as the carrier gas at a flow rate of 1.2 mL/min. The GC temperature was ramped from 150 to 260°C at 10°C/min and held for 25 min. Injector and ion source were kept at 260 and 230°C, respectively. The mass spectrometer was operated in SIM (selective ion monitoring) mode. Ions 368, 253 and 394 were used to identify cholesterol, desmosterol and stigmasterol, respectively.
Morphology
Cytospins were performed on terminally differentiated EPRO cells and the morphological maturation was examined by Wright-Giemsa staining of the cell using an Olympus BX41 microscope at 60X magnification under bright field optics.
Immunohistochemistry
For examining expression of Lbr mutants in HEK293 cells, transient transfections were performed using cells cultured on Lab-Tek chamber slides (Nalge Nunc International, Rochester, NY), which were then fixed with 4% paraformaldehyde in PBS for 30 min, followed by a PBS wash. Cells were then permeablized in 0.2% Triton X-100 in TBS (20 mM Tris-Cl, 1.4 mM CaCl2, 1% BSA adjusted to pH 7.4) for 20 min at room temperature. Cells were then rinsed in TBS and blocked with TBS + 5% FBS for 30 min at 37°C. This was followed by incubation with Anti-FLAG primary antibody (Stratagene) at 1:1000 dilution in blocking buffer, for 30 min at 37°C. Slides were rinsed three times with 0.1% Tween in PBS, 5 min each, then incubated with Fluorescein Isothiocyanate (FITC)-conjugated goat anti-mouse secondary antibody ((Millipore, Danvers, MA) at 1:5000 dilution in blocking buffer for 30 min at 37°C. Slides were washed three times with 0.1% Tween in PBS for 5 min each, and then viewed at 40X magnification and fluorescent light using an Olympus BX41RF-5 microscope; images were taken using an Olympus DP71 digital camera with imaging software.
Western blot analyses
Phosphorylation of STAT5 and Erk1/2 was analyzed in EPRO cells by first serum starving the cells in IMDM plus 2% HS for 60 min and then stimulating the cells with 10 ng/mL of recombinant murine GM-CSF (Peprotech) for 15 min. For all Western blot analyses, cells (1 × 107) were collected, washed with PBS supplemented with Complete proteinase inhibitors (Roche Applied Science) and Phenyl Methyl Sulfonyl Fluoride (1 mM, Sigma-Aldrich), and then lysed in 1X Laemmli buffer as described previously (44). Equal amounts (~5 μg) of each lysate were gel electrophoresed using 4% to 12% precast gradient gels (Invitrogen) and electroblotted onto polyvinylidene difluoride membranes (Millipore). Membranes were probed using the following primary antibodies: Anti-STAT5, anti-phospho-STAT5, anti-Erk1/2 and anti-phospho-Erk1/2 (Cell Signaling Technologies, Beverly, MA), each at 1:1000 dilution; Anti-Akt and anti-phospho-Akt (Millipore) at 1:1000 dilution, Anti-Flag (Stratagene) at 1:1000 dilution; Anti-tubulin (Sigma-Aldrich) at 1:2000 dilution; mouse monoclonal anti-lamin A/C at a dilution of 1:5 (45), and guinea pig serum against LBR at 1:1000 dilution (kindly provided by Prof. Harald Herrmann, German Cancer Research Center, Heidelberg, Germany). Horse radish peroxidase (HRP)-conjugated secondary antibodies were used to detect primary antibody binding, and chemiluminescence was detected using Immobilon Western HRP substrate solutions (Millipore).
Respiratory burst assays
Terminally differentiated cells (1 × 107) were washed in PBS, suspended in 400 μL of Hank’s Balanced Salt Solution (HBSS, 135 mM NaCl, 5.4 mM KCl, 0.3 mM Na2HPO4.7H2O, 0.4 mM KH2PO4, 4.2 mM NaHCO3, 1 mM MgCl2, 1.3 mM CaCl2, 0.6 mM MgSO4.7H2O; Sigma-Aldrich) with 0.1% glucose plus 100 μL of the cellular luminescence enhancement reagent Diogenes (National Diagnostics, Atlanta, GA) and incubated for 15 min at 37°C. Cells then were stimulated with Phorbol 12-myristate 13-acetate (PMA, 3.2 μM; Sigma Aldrich) and chemiluminescence emission was recorded at minute intervals for a period of five min. For assays of cells stimulated with opsonized zymosan, the washed cells were resuspended in 300 μL of HBSS plus 100 μL of Diogenes, incubated for 15 min at 37°C, and then stimulated with zymosan particles (4 mg/mL final concentration, Sigma) that were previously opsonized for 30 min at 37°C with fresh mouse serum; chemiluminescence emission was recorded at 2 min intervals over 60 min.
Northern blot analyses
Total RNA was isolated from 1 × 107 cells using TRIzol reagent (Life Technologies, Rockville, MD) according to the manufacturer’s specifications. For Northern blot analyses, approximately 5 μg of total RNA was electrophoresed and blotted on Nytran Plus membranes as performed previously (40). Radionucleotide-labeled probes were generated from cDNAs encoding mouse Lbr (amplified from EPRO cells), Tm7sf2 (clone MMM1013-7510962, Open Biosystems, Huntsville, AL), lactoferrin, neutrophil collagenase, neutrophil gelatinase or β-actin.
Staining of cells with CTB-FITC
EPRO cells of each genotype (+/+, +/ic, ic/ic and ic/ic-FLAG-Lbr) were induced with ATRA (10 μM) for three days and then 5 μL of cells at 2 × 106 cells/mL were dried onto chambered microscope slides coated with 0.01% poly-L-Lysine (Sigma-Aldrich) by incubating the slides at 37°C for 20 min. Cells were then stained with 5 μl of 50 μg/mL CTB-FITC or FITC (Sigma-Aldrich) for 30 min in the dark and on ice. The slides were dipped in 1X PBS and cells were fixed using 2% paraformaldehyde in 1X PBS for 10 min followed by dipping the slides in 1X PBS. The chambers were covered with a cover slip and the cells were imaged using fluorescence microscopy at 400x final magnification.
Statistical Analyses
All statistical analyses for calculations of standard deviations were performed using Excel software (Microsoft, Redmond, WA, USA) and the Student’s two sample t-test where significance (p value) is indicated.
RESULTS
Cytokine signalling in EPRO-ic/ic cells
The model EML cell lines derived from +/+, +/ic and ic/ic bone marrow are dependent on SCF and can be induced toward EPRO cells by culturing in ATRA plus interleukin-3 (IL-3) followed by selection for GM-CSF-dependent cells; EPRO cells can then be induced to mature neutrophils by a second dose of ATRA, allowing analysis at essentially three stages of neutrophil development (see Fig. 1A). We previously showed that Lbr-deficient EML-ic/ic cells exhibited a small decrease in growth response to SCF, but that EPRO-ic/ic cells displayed an approximately 50% decrease in proliferation as compared to heterozygous EPRO-+/ic cells (40). To further investigate the cause of defective growth of EPRO-ic/ic cells, we determined whether proliferative signals activated by the GM-CSF receptor might be disrupted in these cells. Activation of the GM-CSF receptor results in the phosphorylation of several transcription factors critical to myeloid cell proliferation, including signal transducer and activator of transcription 5 (STAT5), the extracellular signal-related kinase (ERK) proteins Erk1/2, and the phosphoinositide 3-kinase/Akt pathway (3, 46, 47). We therefore analyzed the phosphorylation states of STAT5, Erk1/2 and Akt in EPRO-+/+ vs. -+/ic and -ic/ic cells, each of which were cultured in the absence of GM-CSF and then induced with GM-CSF for 15 min; cell lysates for Western blot analyses were recovered before and after the induction. The results showed that GM-CSF caused rapid phosphorylation of STAT5 in all three cell types (Fig. 1B). Furthermore, increased levels of Erk1/2 phosphorylation were also detected in lysates from all three genotypes (Fig. 1C), whereas levels of phosphorylated Akt remained unchanged (Fig. 1D). Lysates of EML cells initially starved and then stimulated with SCF showed similar results: phosphorylation of Erk1/2 and Akt, both known targets of the SCF signalling pathway (48), were equivalent in all three genotypes (data not shown). Together these data indicate that signalling pathways downstream of either GM-CSF or SCF are not affected by the loss of Lbr expression in either +/ic or ic/ic cells.
FIGURE 1.

Targets of GM-CSF receptor activation in EPRO cells are not affected by the loss of Lbr expression. A, Depicted is the procedure for generating GM-CSF-dependent EPRO cells and mature neutrophils from SCF-dependent EML progenitors. B, Phosphorylation of STAT5 was assayed by culturing cells for 1 hour in the absence of GM-CSF followed by addition of GM-CSF (10 ng/mL). Proteins from whole cell lysates were electrophoresed, blotted and then probed for phospho-STAT5 and STAT5. C, Levels of phosphorylated Erk 1/2 were assayed using the same blot that was stripped and probed using anti-phospho-Erk1/2 and anti-Erk1/2 antibodies. D. A new blot was generated from starved and stimulated EPRO cells, which was then probed for anti-Akt and anti-phospho-Akt; levels of phospho-STAT5 in the same cell lysates are shown for comparison.
Effects of sterol deprivation on the growth of wild-type vs. Lbr-deficient myeloid cells
To determine if the decreased growth of ic/ic cells may be associated with disrupted cholesterol biosynthesis, we next subjected each of the +/+, +/ic or ic/ic cell lines at both EML and EPRO stages to sterol starvation conditions. Treatments were based on previous studies performed with human HL-60 cells in which combinations of lipoprotein deficient serum (LPDS) plus the 3-HMG CoA reductase inhibitor lovastatin or the cholesterol binding agent methyl β-cyclodextrin (MβCD) were used to induce sterol starvation (16, 17). We began by first comparing the effects of LPDS (confirmed to contain 0.85 μg/mL of cholesterol compared to 23 μg/mL in normal serum) vs. two concentrations of lovastatin (5 μM and 10 μM), and two concentrations of MβCD (5 mM and 10 mM) on wild-type EML cell growth. The results indicated that expansion of the number of EML-+/+ cells by 3 days of culture decreased by 18% when treated in LPDS and 12% when treated with 5 μM lovastatin, whereas cell numbers decreased by approximately 40% when treated with 10 μM lovastatin or 5 mM MβCD (see Figs. 2A-C for typical growth profiles of EML-+/+ cells in each condition; all differences were significant with p < 0.01). Treatment with 10 mM MβCD induced rapid cell death and therefore was not further utilized. We next tested the proliferation of EML-ic/ic and +/ic cells vs. +/+ cells in regular medium or medium with LPDS. As graphed in Fig. 2A, both EML-+/ic and –ic/ic cells exhibited growth profiles in regular medium similar to that of +/+ cells by three days of culture, although growth of EML-ic/ic cells lagged behind that of the other two cell types (the difference between ic/ic vs. +/+ were significant, with p = 0.001). This result was consistent with our previous studies showing that EML-ic/ic cells exhibit a small decrease in proliferation compared to EML-+/ic cells (40). Growth profiles of EML-+/+ vs. -+/ic cells in LPDS, lovastatin and MβCD were similar: both cell lines exhibited decreased proliferation in each condition. In contrast, each treatment either blocked proliferation of EML-ic/ic cells or caused cell death (Figs. 2B and 2C).
FIGURE 2.
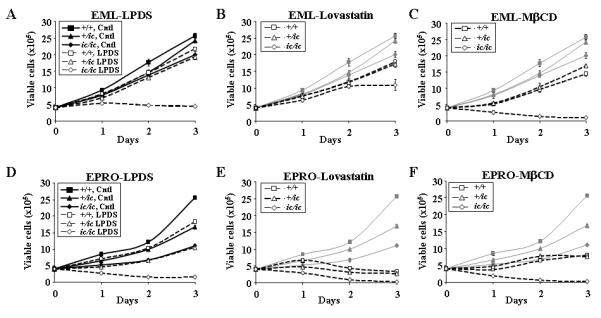
Loss of Lbr expression in EML and EPRO-+/ic and –ic/ic cells suppresses growth responses in regular medium or under cholesterol deprivation conditions. EML cells were plated at 2 × 105 cells/ml in 2 ml medium that contained (A) normal serum (Ctrl) or lipoprotein deficient serum (LPDS), (B) normal serum plus 10μM lovastatin, or (C) normal serum plus 5mM Methyl β-Cyclodextrin (MβCD), and daily counts of viable cells were performed. Data shown are from multiple assays performed simultaneously, in which cells were plated in 2 ml of medium with each condition using triplicate plates per assay; the data from cells grown in regular medium are displayed in (A) as black lines and then shown as grey lines in (B) and (C) to highlight the changes of growth in cholesterol depleted conditions. Similar studies were performed with EPRO cells derived from the EML cells of each genotype, using (D) normal serum vs. LPDS, (E) lovastatin or (F) MβCD. Viable cells were identified by trypan blue exclusion and increases in total cell numbers was determined manually using a hemacytometer at 24-hour intervals for three days. Data represent results from at least two independent assays.
Growth of each genotype at the EPRO stage was next tested using either regular medium or the three different cholesterol stressed conditions, which yielded very different results as compared to EML cells. First, EPRO-ic/ic cells exhibited much slower growth in regular medium as compared to EPRO-+/ic cells, as was previously observed (Fig. 2D and (40), p < 0.001). We also discovered that differences between the growth of EPRO-+/ic cells vs. +/+ cells were greater than differences observed at the EML stage, indicating that loss of one genomic copy of Lbr has a greater affect on proliferation at this later stage of differentiation. Second, growth of EPRO-+/ic cells in LPDS medium was substantially less than that of +/+ cells (after three days, total cell numbers of EPRO-+/ic cells were approximately half that of +/+ cells). As previously observed for EML-ic/ic cells, each treatment completely inhibited growth of EPRO-ic/ic cells, many of which stained positive for trypan blue by day 2 indicating cell death. Third, all cells treated with either 10 μM lovastatin or 5 mM MβCD ceased to proliferate, again with the most dramatic affect on ic/ic cells (Figs. 2E, F). Finally, a nonradioactive proliferation assay for metabolic activity (the MTS assay) confirmed growth inhibitory effects of cholesterol inhibitors on each cell type at the EML or EPRO stages: specifically, EML–ic/ic cells exhibited little change in absorbances as compared to +/ic or +/+ cells in regular medium but significant differences in medium treated with lovastatin, and EPRO-ic/ic cells treated with either lovastatin or MβCD exhibited a dramatic loss of metabolic activity while EPRO-+/ic cells exhibited an intermediate phenotype as compared to +/+ cells (data not shown). Altogether these data indicate that EPRO cells are more susceptible to the growth inhibitory effects of lipid starvation as compared to EML cells, deficient Lbr expression inhibits the growth of EML cells under sterol deprivation conditions, and deficient Lbr expression in EPRO cells inhibits their growth in either regular medium or sterol deprivation conditions.
Production of lipids and cholesterol during neutrophil development is dependent on Lbr expression
Neutrophils are dependent on cholesterol synthesis for the formation of lipid rafts in membrane microdomains, which support multiple functional responses (7, 8, 11). Based on this observation and previous studies demonstrating that LBR expression increases during neutrophil differentiation, we reasoned that as myeloid cells differentiate toward mature neutrophils they may exhibit increasing cholesterol production during differentiation. Moreover, this cholesterol production may be disrupted due to loss of Lbr expression in EPRO-+/ic or -ic/ic cells. Previous studies have shown that certain fatty acids, including oleic acid, can modulate neutrophil functions, some of which may be mediated by the accumulation of lipid droplets in the cytoplasm or cell membranes(49-51). We previously observed that Nile Red effectively stained membrane lipids that accumulate in oleic acid-stimulated liver PLC cells (Zwerger and Herrmann, unpublished observations). Based on these results, we tested EML, EPRO and ATRA-induced EPRO cells for oleic acid-induced production of lipid droplets, visualized by staining the cells with Nile Red. We found that neither EML nor EPRO cells exhibited detectable lipid staining when cultured in normal medium (data not shown). However, induction of these myeloid cells with oleic acid caused lipid production that could be easily visualized under fluorescence microscopy. As shown in Fig. 3A, oleic acid induced lipid production at all 3 stages of differentiation, but levels increased as EML cells differentiated into mature neutrophils. By comparison, the number of cells positive for lipid production was reduced in heterozygous +/ic or homozygous ic/ic cells, with only 20% of ATRA-induced EPRO-ic/ic cells showing lipid production as compared to >80% of mature +/+ neutrophils (Fig. 3B). Furthermore, the average number of lipid droplets identified per cell also increased as EML cells differentiated toward mature neutrophils, but this increase was less pronounced in +/ic cells compared to +/+ cells and no change was observed during differentiation in ic/ic cells (Fig. 3C). These results indicated that the capacity of neutrophils to synthesize lipids increased during their differentiation from progenitor EML cells toward mature neutrophils, and this capacity was dependent on the expression of Lbr.
FIGURE 3.

Lipid production in oleic acid-induced cells and cholesterol biosynthesis is disrupted by loss of Lbr expression. (A-C) EML, EPRO and ATRA-induced EPRO cells from each genotype were cultured in medium that contained 10% oleic acid for 3 days, then fixed with 4% paraformaldehyde and stained with Nile Red. A, Cells stained for Nile red were photographed under fluorescence to visualize lipids (original magnification, x 400). The numbers of yellow droplets indicate the amount of lipids synthesized in each cell. Graphed are the percentage of +/+, +/ic and ic/ic cells positively stained for lipids (B), and the average number of individual droplets that could be counted per cell (C). Data are the average counts of approximately 100 analyzed cells from each induction ± SD. D, GC/MS analysis of EML cells of each genotype cultured in regular medium (upper panels) or LPDS medium (lower panels) reveals changes in sterol levels. Shown are chromatographs that reveal the relative abundance of extracted sterols with respect to stigmasterol loaded as an internal control. Labeled peaks correspond to cholesterol (peak 1, confirmed by analyses of cholesterol alone), two precursors (peaks 2 and 3), and stigmasterol (peak 4).
To confirm that differences in lipid staining induced by oleic acid in EML and EPRO cells reflects the ability of these cells to drive cholesterol biosynthesis, we also performed gas chromatography/mass spectrometry (GC/MS) analyses on cells of each genotype, using either proliferating EML cells or EML cells stressed with medium that contained LPDS. The addition of stigmasterol provided an internal control. Assays of both +/+ and +/ic cells in regular medium revealed a major peak at approximately 16 min retention time that was consistent with cholesterol, as determined by a control run with pure cholesterol (Fig. 3D, upper panels, peak 1). However, this peak was dramatically reduced in ic/ic cells, whereas two additional peaks were observed (peaks 2 and 3). Since standards for identifying where cholesta-8,14-dien-3β-ol or cholesta-8,14,24-trien-3β-ol (previously identified in mice tissues that lacked DHCR14)(35) are not available, it remains unclear as to the identity of these two peaks, but the retention time of peak 2 was very near that of desmosterol (17 min, data not shown). Interestingly, all cell types produced this same peak when stressed by growth in LPDS medium (Fig. 3D, bottom panels), and ic/ic cells also exhibited less relative levels of cholesterol (peak 1). Combined with the oleic acid studies, these data indicate that both overall lipid levels and cholesterol biosynthesis are impaired by loss of Lbr, and that lipid deprived conditions produce a similar defect in cholesterol biosynthesis.
Expression of Tm7sf2 does not change during neutrophil differentiation but increases in response to lipid deprivation
The observations that oleic acid-induced production of lipids increased upon ATRA induction in EPRO cells suggest that sterol biosynthesis may be upregulated during neutrophil differentiation. Since Tm7sf2 gene expression is controlled by cellular sterol levels via activation of the sterol-sensing transcriptional regulator SREBP-2 (52), and LBR transcription increases during neutrophil differentiation in both human and mouse myeloid cells, we next examined how expression levels of the endogenous Tm7sf2 and Lbr genes compared in differentiating EML cells and how they may be affected by cholesterol starvation. Lbr transcription increased during ATRA-induced differentiation of EML cells toward mature neutrophils in both +/+ and +/ic cells, with +/ic cells expressing approximately half the transcript levels of +/+ cells, whereas Lbr expression was completely lacking in ic/ic cells (Fig. 4A). By comparison, Tm7sf2 expression did not increase; rather, expression remained steady throughout neutrophil differentiation and may have actually decreased in ATRA-induced EPRO-ic/ic cells. Consistent with our previous findings (40), loss of Lbr expression did not affect the typical increased expression of multiple transcripts indicative of neutrophil differentiation, including lactoferrin, neutrophil gelatinase and neutrophil collagenase. Lipid stressed conditions also did not affect neutrophil-specific gene expression in ATRA-induced EPRO cells (Fig. 4B, EPRO cells were cultured in regular medium plus ATRA, LPDS plus ATRA, or pretreated in LPDS for three days and then induced in LPDS plus ATRA). Interestingly, expression of Tm7sf2 increased when all three EML genotypes were treated with lovastatin, whereas Lbr expression was not affected (Fig. 4C). Similar results were found for +/ic and +/+ cells treated with MβCD (ic/ic cells could not be analyzed due to rapid cell death caused by MβCD), and for human HL-60 cells treated with the LPDS or MβCD (data not shown). Thus during the differentiation of myeloid cells toward neutrophils, Lbr transcription is upregulated whereas Tm7sf2 expression remains in general unaffected. On the other hand, sterol deprivation caused activation of Tm7sf2 expression in both human and mouse myeloid progenitors, but did not affect the expression of Lbr nor did it disrupt transcriptional activation of neutrophil-specific genes.
FIGURE 4.
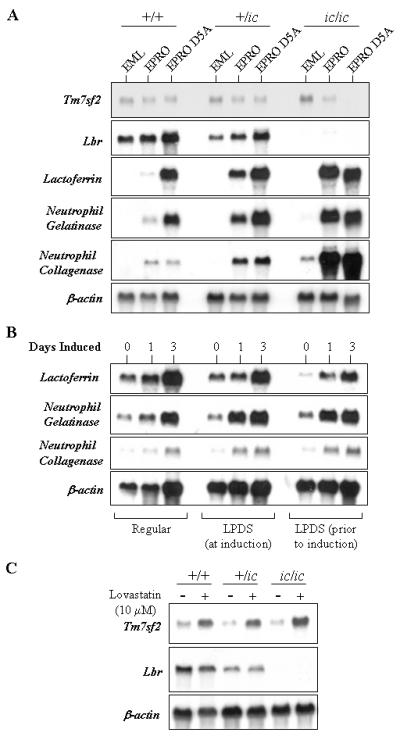
Gene expression profile during the differentiation of EML cells toward mature neutrophils and under lipid-stressed conditions. A, A Northern blot was generated using total RNA extracted from EML, EPRO and ATRA-induced EPRO cells from the three genotypes +/+, +/ic and ic/ic, which was sequentially probed with radioactive probes generated from mouse cDNAs for the genes Tm7sf2, Lbr, lactoferrin, neutriophil gelatinase and neutrophil collagenase. B, EPRO cells were a) cultured in regular medium and then induced with ATRA, b) transferred to medium with LPDS plus ATRA, or c) cultured in LPDS medium for three days prior to induction with ATRA, and total RNA was isolated just before ATRA induction and 1 or 3 days after ATRA induction. The Northern blot generated from the total RNA was probed for the expression of lactoferrin, neutrophil gelatinase and neutrophil collagenase to demonstrate normal expression patterns. C, Total RNA was isolated from EML cells of each genotype after incubation in either regular medium or medium with 10 μM lovastatin, and then used to generate a Northern blot that was sequentially probed for Tm7sf2 or Lbr gene expression. Shown below each blot is the result from hybridizations with a β-actin probe to demonstrate amount of RNA loaded in each lane.
Loss of Tm7sf2 expression does not affect the growth of EML cells
Previous studies demonstrated that loss of Tm7sf2 expression does not cause any observable phenotype in knockout mice, therefore we predicted that myeloid progenitors generated from these knockout mice will lack the same growth defect in lipid starved conditions as observed with EML-ic/ic cells. To investigate this possibility, we generated EML-like immortalized progenitors from bone marrow of a Tm7sf2 knockout mouse and from bone marrow of a littermate that was genotypically normal (32). After transduction with the dominant negative RARα and a prolonged growth in SCF, we identified populations of cells from each genotype that morphologically resembled EML cells (see Fig. 5A). Both of the resulting cell lines were able to form EPRO-like cells after culture in IL-3 plus ATRA, and to further differentiate into morphologically mature neutrophils upon ATRA induction. The EML-Tm7sf2-KO cells were confirmed to have the neomycin resistance cassette inserted in exon 5 of the Tm7sf2 gene (data not shown) and to lack Tm7sf2 gene expression (Fig. 5B). By comparison, Tm7sf2 expression in the littermate control cells was equivalent to that of the original EML/EPRO-+/+ cells. All lines examined expressed similar levels of Lbr and displayed activation of both lactoferrin and neutrophil gelatinase, each hallmarks of normal neutrophil maturation (41). We then tested the EML cells for growth in lipid deprivation conditions, and found that there was no significant difference between the growth of Tm7sf2 knockout cells compared to that of the littermate-derived cells (Fig. 5C). In other words, loss of Tm7sf2 expression did not cause the dramatic growth defect as observed for EML-ic/ic cells in lipid starved conditions. This data supports the notion that the combined sterol reductase activities provided by Lbr and Tm7sf2 are important for normal myeloid cell growth, but that Lbr may be a more critical regulator of cholesterol biosynthesis in myeloid cells as compared to Tm7sf2.
FIGURE 5.
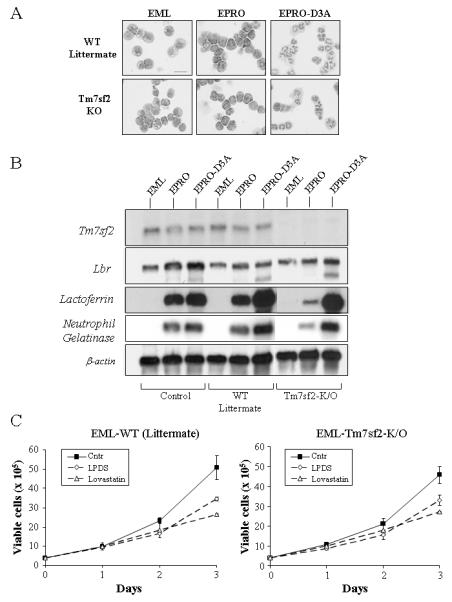
EML cells generated from Tm7sf2 knockout mouse bone marrow exhibit normal differentiation and growth in lipid stressed conditions. A, Morphologic maturation of EML cells derived from Tm7sf2 knockout bone marrow and a littermate control was examined by staining cytospun cells after ATRA-induced differentiation to EPRO cells and mature neutrophils. Scale bar in uninduced EML-WT cells represents 50 μm. B, A Northern blot was generated from total RNA isolated from control vs. Tm7sf2 knockout cells and then sequentially probed for expression of Tm7sf2, Lbr, mouse lactoferrin, mouse neutrophil gelatinase and finally β-actin to demonstrate amounts of RNA loaded into each lane. C, Growth profiles of control vs. Tm7sf2 knockout cells were analyzed in regular medium vs. medium with LPDS or lovastatin. Data shown are the average number of cells ± SD in triplicate wells with each condition, and are representative of three independent assays.
Phenotypes caused by loss of Lbr expression are not due to effects on the liver X Receptor nor can they be rescued by overexpression of Tm7sf2
Previous studies have demonstrated that loss of Tm7sf2 causes an increase in cellular levels of the cholesterol precursor cholesta-8,14,24-trien-3β-ol, which is also a ligand for the nuclear Liver X Receptor (LXR) (35, 56). LXR is similar to RARα, in that they both function by forming obligate heterodimers with the retinoid X receptors and bind to the promoters of genes that are critical to the proliferation and/or function of macrophages, which are highly related to neutrophils as professional phagocytes (57-60). Because we found cholesterol precursors in our GC/MS analyses of ic/ic cells, one of which may be cholesta-8,14,24-trien-3β-ol, it is conceivable that an abnormal accumulation of cholesta-8,14,24-trien-3β-ol levels in EML/EPRO-ic/ic cells contributes to the observed phenotypes via aberrant activation of the LXR signalling pathway. To test this possibility, we cultured wild-type EML and EPRO cells in different concentrations of the synthetic LXR ligands T0901317 and GW3965, both shown to affect macrophage proliferation or gene expression (57, 61, 62). Our results showed that neither LXR agonist consistently affected the proliferation of EPRO cells (Fig. 6A). Similar results were found with EML cell growth, and morphologic maturation of EPRO cells was normal in medium that contained the maximum concentration of either agonist (data not shown). Furthermore, the LXR antagonist fenofibrate failed to improve the lack of nuclear maturation or deficient growth profiles of EML/EPRO-ic/ic cells (data not shown). We also tested whether simply increasing the expression of Tm7sf2, which may decrease levels of cholesta-8,14,24-trien-3β-ol by increasing sterol Δ14 reductase activity, might rescue the growth or differentiation defects of EPRO-ic/ic cells. Using an expression vector with the full length Tm7sf2 cDNA (pMSCVpuro-Tm7sf2), EML-ic/ic cells that overexpress Tm7sf2 were derived, ectopic expression was confirmed (Fig. 6B), and the cells were cultured in regular or LPDS medium, or induced to differentiate with ATRA. As shown in Fig. 6C, the Tm7sf2 expression vector failed to affect deficient growth of EML-ic/ic in LPDS, whereas this was rescued by full-length FLAG-Lbr. In addition, the derived EPRO-ic/ic-Tm7sf2 cells exhibited severe hypolobulation upon ATRA induction and deficient respiratory burst responses similar to those observed with cells transduced with an empty vector (data not shown). These data indicate that the observed phenotypes in ic/ic cells are most likely due to the lack of unique functions provided by Lbr and not mediated by disruption to LXR activities or functions that can be readily provided by high-level expression of Tm7sf2.
FIGURE 6.

Analysis of EPRO cell growth in response to LXR agonists or overexpression of Tm7sf2. A, Wild-type EPRO cells were cultured at a starting concentration of 2 × 105 cells/mL in the presence of different concentrations of the LXR agonists T0901317 or GW3965, and cell numbers were counted after three days of culture. Shown are the average total number of cells ± SD from triplicate assays with the indicated final concentrations of each drug or DMSO as a control, and represent results from three independent assays. B, Total RNAs from EML-ic/ic cells transduced with either an empty vector, the full length FLAG-tagged Lbr or full-length Tm7sf2 were blotted and probed for Tm7sf2 expression. The blot is overexposed for expression of Tm7sf2 in the transduced cells, which was necessary to show the endogenous levels of Tm7sf2. Also shown are expression levels in EML-TM7sf2-WT and KO cells, and the ethidium bromide-stained gel prior to membrane transfer. C, Growth responses of EML-ic/ic cells transduced with the empty vector, FLAG-Lbr or Tm7sf2 are shown from cultures that used regular medium (left panel) vs. LPDS medium (right panel). Data shown are average total cell numbers from daily cell counts ± SD, each from triplicate assays.
Full length and carboxy-terminal domains of Lbr rescue morphologic maturation and lipid starved growth of EML-ic/ic cells
In order to determine which domains of Lbr support growth of EML or EPRO cells in lipid starved conditions, we aimed to express truncated variants of Lbr that contained either the N-terminal chromatin/lamin B binding domains or the C-terminal sterol reductase domain in the EML-ic/ic model cells, and then examine their responses to differentiation and sterol deprivation. We began by transducing a full-length version of Lbr into EML-ic/ic cells to demonstrate that expression of wild-type Lbr proteins can rescue the observed phenotypes. For expression of full-length Lbr, a mouse cDNA that encodes a FLAG-tagged version of Lbr was cloned into the pMSCV-puro retroviral expression vector, previously shown to drive abundant protein expression in the EML cell line (53). Both EML-+/ic and -ic/ic cells were transduced with the expression vector or an empty vector. Expression in each cell type was confirmed by Western blot analysis of whole cell lysates using a commercially available anti-FLAG antibody (Fig. 7A). To compare levels expressed from pMSCV-FLAG-Lbr to endogenous Lbr levels in +/+ versus +/ic and ic/ic cells, protein lysates also were probed with an anti-LBR antibody, which demonstrated that ectopic expression in transduced cells was similar to endogenous expression in +/+ cells (Fig 7B). Importantly, ATRA-induced EPRO-ic/ic cells expressing FLAG-Lbr exhibited normal nuclear lobulation, with total numbers of cells with multilobed or ring-shaped nuclei equivalent to that of differentiated +/ic cells (Figs. 7C and D). In contrast, very few ic/ic cells transfected with the empty pMSCV vector showed any signs of lobulation (see Fig. 7D for compiled data). These data indicated that the full length mouse Lbr completely rescued the defective nuclear lobulation phenotype of differentiated EPRO-ic/ic cells.
FIGURE 7.
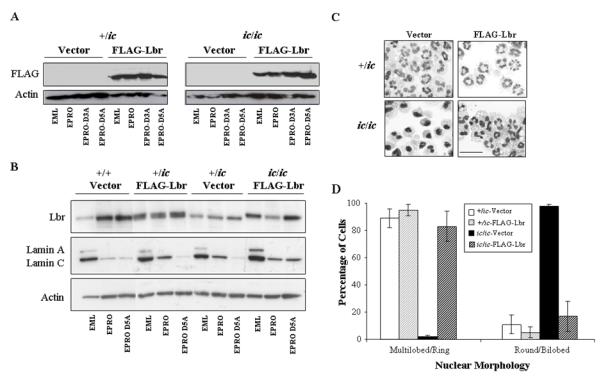
Expression of a FLAG-tagged Lbr in EPRO-ic/ic cells rescues nuclear lobulation in differentiated neutrophils. A, EML-+/ic and –ic/ic cells were transduced with a FLAG-Lbr expressing retroviral vector or an empty vector as a control, and total proteins from derived EML, EPRO and ATRA-induced EPRO cells were electrophoresed and blotted onto a PVDF membrane that was probed using antibodies to the FLAG epitope or actin. B, Expression of Lbr, lamin A/C and actin were assessed in cells transduced with the FLAG-Lbr expression vector vs. an empty vector control by immunoblotting of electrophoresed total proteins. C, ATRA-induced EPRO-+/ic and –ic/ic cells were derived from EML cells of each genotype transduced with the FLAG-Lbr expression vector, stained with Wright-Giemsa and photographed to demonstrate nuclear lobulation. Scale bar represents 50 μm. D, Percentages of cell that contained either multilobed or ring shaped nuclei vs. round or bilobed nuclei were determined by analyzing >100 ATRA-induced cells from the indicated genotypes from at least three independent inductions. Values are averages ± SD; differences between +/ic, +/ic-FLAG-Lbr and ic/ic-FLAG-Lbr were not significant (p > 0.05).
We next generated two different expression vectors that encoded N-terminal domains of Lbr but that lacked portions of the C-terminal sterol reductase domain, specifically residues 1-387 and 1-544 (see Fig. 8A). Each were transfected into Phoenix retrovirus producer cells, but after co-growth with EML-+/ic or ic/ic cells, expression of either C-terminal truncated proteins were undetectable, despite successful selection in puromycin (data not shown). This suggested that ectopic expression of these N-terminal residues in the absence of the C-terminal region disrupted the survival or growth of EML cells. While performing these experiments, studies being conducted by our collaborators demonstrated that the corresponding human constructs caused severe disruptions to the nucleus in three different cell lines (54). We therefore focused on transducing EML-ic/ic cells with two truncated variants of Lbr that lacked N-terminal sequences, one that lacked residues critical to lamin B binding in the first 70 amino acids (Lbr71-626) and a second that lacked both this domain and important binding sites for HP1 and/or heterochromatin located between amino acids 71 and 145 (Lbr145-626, Fig. 8A) (25-27). Expression of both truncated proteins in transduced EML-ic/ic cells was confirmed using whole cell lysates in a Western blot and an anti-FLAG antibody (Fig. 8B). To confirm nuclear expression, we also analyzed nuclear vs. cytoplasmic lysates from the transduced EML-ic/ic cells, but the analysis failed to identify proteins at the correct size for Lbr (data not shown). This problem was previously observed in earlier studies with these cells (Donald and Ada Olins, personal communication), and may suggest that myeloid cells express enough proteases to cause excessive protein degradation in this particular assay, despite the use of protease inhibitors. We therefore isolated nuclear and cytoplasmic proteins from HEK293 cells transfected with each construct, and successfully identified expression of each protein, including the full-length FLAG-Lbr, in the nuclear lysates (Fig. 8C, left panels). Expression of each variant was also detected in the cytoplasm. This result was consistent with immunohistochemistry analyses performed with transfected HEK293 cells, where expression was identified in both the cytoplasm and the nuclear rim (Fig. 8C, right panels). Nuclear morphology was then examined in differentiated EPRO cells derived from the transduced EML-ic/ic cells (Fig. 8D). A small but significant number of cells transduced with either the Lbr71-626 or Lbr145-626 mutant proteins exhibited morphologic maturation (see Fig. 8E for summary of percentages in each category of nuclear morphology; asterisks indicate that differences between the cells expressing mutant Lbr proteins vs. cells expressing the empty vector are significant, with p < 0.01). This result was surprising, given that the interactions of LBR with lamin B and/or chromatin are thought to be critical to the functions of LBR in mediating neutrophil nuclear lobulation (55).
FIGURE 8.
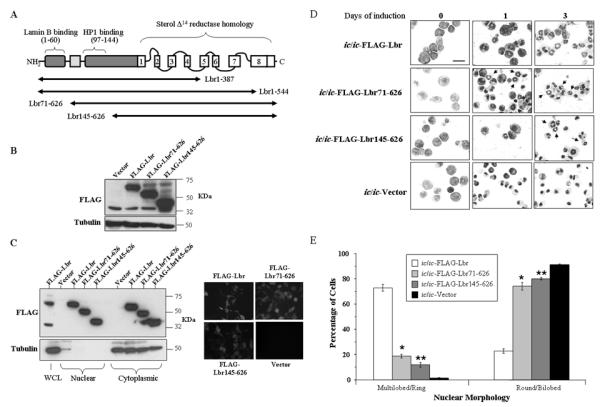
Expression of C-terminal domains of Lbr restores nuclear lobulation to a significant population of ic/ic neutrophils. A, Depicted is a diagram of the Lbr protein with the N-terminal binding domains and the C-terminal membrane spanning region that is homologous to sterol Δ14 reductase Tm7sf2 (adapted from Hoffmann et al, 20). The black horizontal arrows represent the length of mutant variants of Lbr that were transduced into EML-ic/ic cells via the pMSCV-puro retroviral expression vector. B, An immunoblot was generated using whole cell lysates from EML-ic/ic cells transduced with FLAG-Lbr, FLAG-Lbr71-626, FLAG-Lbr145-626 expression vectors or the empty vector, and probed with anti-FLAG antibody to identify expression of each mutant construct. C, Expression of the C-terminal variants of Lbr was assessed in transfected HEK293 cells by immunoblotting nuclear vs. cytoplasmic lysates and probing with anti-FLAG antibodies (left panels), or by immunohistochemistry using a FITC-conjugated secondary antibody (right panels). D, Morphologic maturation of differentiated EPRO-ic/ic cells expressing FLAG-Lbr, FLAG-Lbr71-626 and FLAG-Lbr145-626 was assessed by analyzing cytospun cells with Wright-Giemsa stain. Pictures shown indicate cells with the normal ring-shaped nuclei (arrows) in mature neutrophils. Scale bar represents 50 μm. E, Percentages of ATRA-induced EPRO-ic/ic cells expressing FLAG-Lbr, FLAG-Lbr71-626 and FLAG-Lbr145-626 were calculated by examining >100 cells from at least three independent inductions. Values shown are percentages of cells with the indicated nuclear morphology ± SD; differences between EPRO-ic/ic cells transduced with the empty vector vs. * FLAG-Lbr71-626 and ** FLAG-Lbr145-626 were significant, (p < 0.01).
The proliferation profiles of +/ic and ic/ic cells expressing the full-length FLAG-Lbr in sterol starvation conditions were then analyzed. EML-ic/ic cells transduced with the empty vector proliferated in regular medium at rates similar to that of parental EML-ic/ic cells (Fig. 9A; note that the analyses were performed over 5 days to accentuate the differences in growth profiles). As expected, these cells failed to proliferate in medium with LPDS. Importantly, EML-ic/ic cells transduced with FLAG-Lbr proliferated in LPDS at levels equivalent to that of cells grown in regular medium (p = 0.2 for differences at Day 2), indicating that expression of the full-length Lbr protein in the ichthyosis genetic background rescued not only morphologic maturation but also the growth of EML-ic/ic cells under lipid starved conditions. Expression of FLAG-Lbrhad essentially no effect on the growth of EML-+/ic cells, which were not dramatically influenced by use of LPDS (Fig. 9B). Despite these results, expression of the full-length FLAG-Lbr was unable to rescue growth of either +/ic or ic/ic cells when induced to the EPRO stage and cultured in LPDS (Figs. 9C and D). This result suggested that lack of Lbr expression caused changes to the hematopoietic stem cells in the ichthyosis mouse that can not be reversed once they are harvested and used to derive EML/EPRO cell lines.
FIGURE 9.
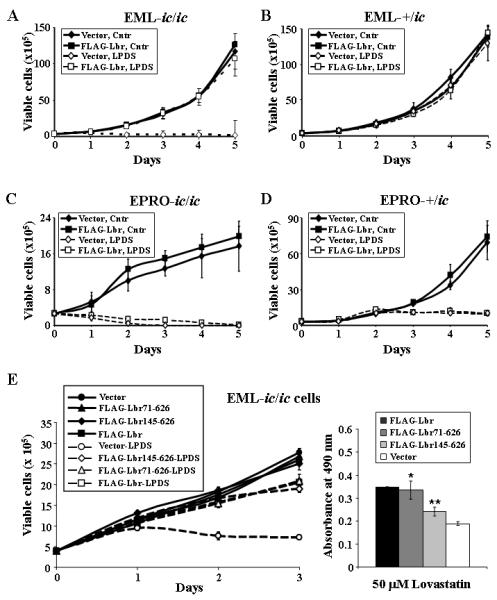
Expression of either full length or C-terminal domains of Lbr rescues growth of EML-ic/ic in lipid deprived conditions, but does not improve the growth of EPRO-ic/ic cells. A and B, EML-ic/ic cells transduced with ether an empty vector or the full length FLAG-Lbr were started at a concentration of 2 × 105 cells/mL and then cultured in medium with normal serum vs. LPDS, and daily cell counts were performed to assess their proliferation over 5 days. C and D, EPRO-ic/ic cells that express either the empty vector or FLAG-Lbr were assayed for growth in medium with normal serum vs LPDS. Shown in each graph is the total number of viable cells ± SD from three independent experiments. E, Proliferation assays were performed on EML-ic/ic cells transduced with FLAG-Lbr, FLAG-Lbr71-626, FLAG-Lbr145-626 or the empty vector, using either viability counts of cells plated at a starting concentration of 2 × 105 cells/mL in medium with regular serum versus LPDS (left panel), or MTS reduction assays of cells cultured for 3 days in regular media containing lovastatin (right panel). Data shown are averages ± SD from three independent assays; graphs with * and ** indicate statistical significance in differences compared to growth of EML-ic/ic-vector cells (p < 0.02).
Based on the capacity of full length Lbr to rescue the growth of EML-ic/ic cells in LPDS, we examined the growth profiles of EML-ic/ic cells transduced with the truncated Lbr proteins, FLAG-Lbr71-626 and FLAG-Lbr145-626. As shown in Fig. 9E, both of the truncated proteins were able to improve the growth of EML-ic/ic cells in LPDS (left panel), and increase metabolic activity in MTS assays when cells were treated with lovastatin (right panel; the asterisks indicate that differences between responses by EML-ic/ic cells expressing either FLAG-Lbr71-626 or FLAG-Lbr145-626 versus vector control cells were significant, with p < 0.02). Finally, expression of either the FLAG-Lbr71-626 or FLAG-Lbr145-626 proteins in EML-ic/ic cells caused an increase in oleic acid-induced lipid production at either the EPRO stage or in ATRA-induced cells (Figs. 10A and B; the asterisks above values for cells transduced with the FLAG-Lbr145-626 indicate statistically significant differences as compared to cells with the empty vector, with p < 0.01 for both sets of graphs). It is also interesting to note that the average number of droplets produced by ATRA-induced EPRO-ic/ic cells expressing FLAG-Lbr was greater than that produced by +/ic cells. Together these data indicate that the sterol reductase domain of Lbr plays a critical role in supporting both the growth and lipid production of EML cells when cultured under cholesterol stressed conditions, and this domain alone can support morphologic maturation of EML-derived neutrophils.
FIGURE 10.

Lipids produced by oleic acid-induced EPRO-ic/ic cells increased by ectopic expression of full length or C-terminal variants of Lbr. A, EPRO cells expressing the three variants of Lbr were cultured in 10% oleic acid before or during ATRA induction, stained with Nile Red and photographed under fluorescence (original magnification x 400). B, Percentages of cells with positive lipid staining were calculated by counting >100 cells from at least three independent inductions with oleic acid. The average number of droplets per cell was also calculated. * Differences between FLAG-Lbr145 and vector expressing cells were significant (p < 0.01).
The sterol reductase domain of Lbr supports respiratory burst activity in mature neutrophils
During neutrophil maturation, components of the NADPH oxidase must assemble at both the plasma membrane and membranes that surround intracellular phagosomes. Once activated, the NADPH oxidases produce oxygen radicals that play essential roles in bacterial destruction. Recent studies have demonstrated that the two membrane-bound components gp91phox and p22phox, in addition to small GTPases Rac (1 or 2) and Rap1a, become localized due to the formation of lipid rafts, a process that facilitates assembly of functional NADPH oxidase complexes (11, 12). Furthermore, membrane cholesterol levels play an integral role in facilitating the formation of lipid rafts, depletion of which can dramatically affect activation of the respiratory burst (11, 63). In our previous studies, we found that differentiated ic/ic cells exhibited a severe decrease in the respiratory burst, which we reasoned may be partly explained by the modest decrease in the expression of gp91phox. An alternative explanation is that the phenotype is due to loss of sterol reductase activity provided by Lbr that reduces the required amount of membrane cholesterol for lipid raft formation. To investigate this possibility, we began by confirming that lipid deprivation inhibited the respiratory burst of ATRA-induced EPRO-+/+ cells. The cells were cultured in a) regular medium with ATRA, b) medium with LPDS plus ATRA, or c) first cultured in LPDS for 3 days and then in LPDS plus ATRA. As shown in Fig. 11A (top panel), cells cultured in LPDS during the 3 days of ATRA induction and then stimulated with phorbol myristate acetate (PMA) produced approximately half the level of respiratory burst compared to cells grown in regular medium, and cells pre-cultured in LPDS produced only 35% of the normal respiratory burst response. Similar results were found with cells cultured in LPDS and stimulated with opsonised zymasan (OZ) (Fig. 11B). Furthermore, cells cultured in regular medium in the presence of 5 μM lovastatin exhibited little to no respiratory burst activity in either PMA (data not shown) or OZ. Similar results were obtained from the related mouse promyelocytic cell line, MPRO (41), which also exhibited decreased respiratory burst responses in cells cultured in LPDS or lovastatin and stimulated with either PMA or OZ (data not shown). Importantly, expression of FLAG-Lbr in EPRO-ic/ic cells significantly increased levels of respiratory burst to near that produced by heterozygous cells (Fig. 11C). Furthermore, both of the C-terminal variants of Lbr also increased respiratory burst responses, albeit to levels less than that observed from differentiated ic/ic cells expressing the full length Lbr. Assays that used OZ to stimulate the cells also indicated that expression of either the full-length Lbr or C-terminal variants increased the respiratory burst response (specifically, FLAG-Lbr generated a 2.3 ± 0.2 fold increase over cells expressing the empty vector, and FLAG-Lbr71-626 or FLAG-Lbr145-626 each generated approximately 1.5 ± 0.2 fold increases; data not shown). Together these data indicate that ectopic expression of Lbr is sufficient to improve the activation of NADPH oxidases that is compromised in ic/ic cells, and part of this capacity is mediated by the C-terminal region that encodes the sterol Δ14 reductase homology domain.
FIGURE 11.
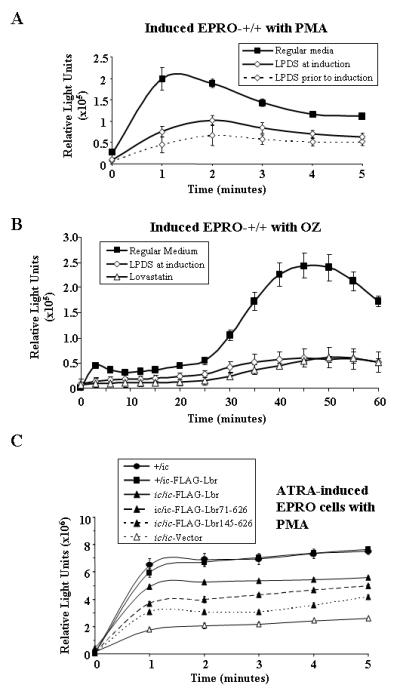
Respiratory burst responses of ATRA-induced EPRO cells treated with LPDS vs. ATRA-induced EPRO-ic/ic cells transduced with full-length or truncated variants of Lbr. A, EPRO-+/+ cells were cultured in either regular medium with ATRA, medium with LPDS plus ATRA, or pre-cultured for 3 days in medium with LPDS and then induced in medium with LPDS plus ATRA. Respiratory burst responses were measured after 5 days of induction and stimulation with 3.2μM phorbol myristate acetate (PMA) using luminol-enhanced chemiluminescence at one minute intervals. B, EPRO-+/+ cells cultured in regular medium, medium with LPDS or medium with lovastatin during ATRA induction were tested for respiratory burst responses caused by opsonized zymosan (OZ). C, Respiratory burst responses to PMA after 5 days of ATRA induction in regular medium were measured from EPRO cells of the indicated genotypes. Values of relative light units in each graph are averages ± SD from triplicate assays performed in parallel.
To test whether the observed loss of respiratory burst responses in cells that lack Lbr expression may be in part due to deficient formation of lipid rafts, we next analyzed differentiated EPRO-ic/ic cells for evidence of membrane lipid raft formation as compared to EPRO-+/ic and +/+ cells. This was performed by using a fluorescein-conjugated form of CTB, which is a ligand for the ganglioside raft marker GM-1 and therefore will identify concentrations of GM-1 expression in differentiated neutrophils that are associated with lipid rafts (63, 64). As shown in Fig. 12A, differentiated EPRO-ic/ic cells showed some background fluorescence but not the staining foci identified in either +/ic or +/+ cells. Furthermore, the percentage of cells showing staining foci increased in ic/ic cells tranduced with the FLAG-Lbr expression vector, as summarized in Fig. 12B. Use of FITC alone showed background staining but not the staining foci found with CTB-FITC (data not shown). Combined with the effects that deficient Lbr expression has on total respiratory burst responses, these data indicate that Lbr plays an important function in mediating the activation of NADPH oxidases, perhaps by maintaining critical levels of cholesterol required for membrane lipid raft formation during neutrophil differentiation.
Figure 12.
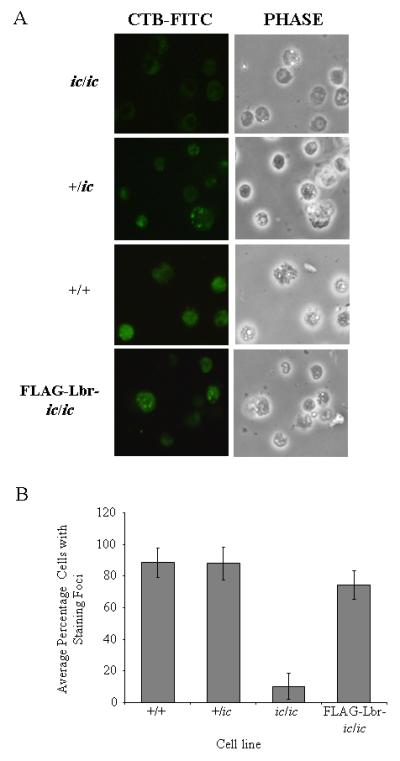
CTB-FITC staining identifies lipid rafts on differentiated EPRO cells. Cells of each genotype were induced with ATRA for three days and then stained for CTB-FITC, which identifies the raft marker GM-1. A, Cells were imaged using the fluorescence mode (left panels) and then under phase contrast (right panels), each shown in the same field of view (original magnification, x 400). B, Percentages of cells with staining foci were calculated by examining 5-6 fields per slide from multiple staining assays, which were then plotted for the indicated EPRO cells. Data shown are averages ± standard deviations.
DISCUSSION
A common feature of the enzymes involved in the conversion of lanosterol to cholesterol is that loss of their expression causes severe malformation syndromes in humans that include defects in skeletal and internal organ development, and in some cases skin abnormalities. This is clearly evident in Smith-Lemli-Opitz syndrome (SLOS) and the SLOS-related disorder desmosterolosis, caused by mutations in the gene DHCR7 and DHCR24, respectively (65-67). Homozygous mutations of the LBR gene cause symptoms similar to these inborn errors of cholesterol biosynthesis in both humans (Greenberg/HEM dysplasia) and mice (ichthyosis), which include post-axial polydactyly, short stature, skin defects and prenatal death (20, 36, 68). However, whether loss of sterol reductase activity provided by the membrane spanning C-terminus of LBR causes these developmental defects, or they are due to loss of lamin B/heterochromatin binding domains provided by N-terminal sequences, is still unclear (35, 38). We hypothesize that both domains are critical to normal development, but that sterol reductase activity provided by LBR may be particularly important to the growth and differentiation of certain cells that are unusually sensitive to levels of cholesterol biosynthesis. To demonstrate that myeloid cells are in this category, we utilized our EML/EPRO cells that lack Lbr expression to test for possible effects that cholesterol depletion may have on their growth, the results of which are summarized in Table I. We also tested whether the C-terminal domain of Lbr alone could rescue identified defects in the growth of EML-ic/ic cells under lipid stressed conditions.
Table I.
Comparison of growth responses by derived EML and EPRO cells in lipid stressed conditions.
| Stage- Condition |
Genotype |
ic/ic |
||||
|---|---|---|---|---|---|---|
| +/+ | +/ic | ic/ic | Lbr | Lbr71-626 | Lbr145-626 | |
| EML- Regular Medium |
+++++ | +++++ | ++++ | +++++ | +++++ | +++++ |
| LPDS | ++++ | ++++ | − | ++++ | ++++ | ++++ |
| Lovastatin | +++ | +++ | + | +++ | +++ | ++ |
| EPRO- Regular Medium |
+++++ | +++ | ++ | ++ | ++ | ++ |
| LPDS | +++ | ++ | − | − | − | − |
| Lovastatin | + | + | − | − | ND | ND |
All data represent the percentage of growth compared to that of wild-type cells in regular medium for 3 days, with +++++ = 80-100%, ++++ = 70-80%, +++ = 50-70%; ++ = 30-50%, + = 10-30%, − = <10%.
ND = no data
By utilizing three different sterol starvation conditions, we were able to test the capacity of early progenitor EML cells vs. more differentiated EPRO cells to tolerate either loss of bioavailable cholesterol in serum (LPDS), inhibition of cholesterol synthesis (lovastatin), or sequestering of membrane-associated cholesterol (MβCD). In each condition, cells that are particularly sensitive to cholesterol levels would be expected to exhibit proliferation defects. We found that growth of EML cells was only modestly affected by culturing in LPDS but was inhibited by the addition of lovastatin or MβCD to the normal growth medium. This indicated that growth of these early progenitors depends on intracellular cholesterol biosynthesis, which is sufficient to support growth in the absence of serum-supplied cholesterol but insufficient to overcome lipid deprivation caused by the two drugs. By comparison, EPRO cells exhibited significant growth inhibition in LPDS and ceased to proliferate when treated with lovastatin or MβCD. Moreover, EML cells could be induced to produce lipid droplets, but this capacity greatly increased in EPRO cells and further increased in differentiated neutrophils. These results are consistent with the view that cholesterol levels are important to the proliferation of myeloid progenitors, as was previously shown for HL-60 and AML cells (13, 14, 16-18), and their dependence on cholesterol increases as the cells differentiate into mature neutrophils; this increased demand may support the observed increased lipid production that is required for functional responses in terminally differentiated cells.
How myeloid cells might meet the increased demand for cholesterol was addressed by testing EML- and EPRO-ic/ic cell growth in regular medium vs. treatment with the same three sterol starvation conditions. We first found that heterozygosity for Lbr at the EML stage was sufficient to support cell growth under sterol starved conditions, whereas homozygous mutations of Lbr in EML-ic/ic cells caused severe growth inhibition or cell death. In contrast, heterozygous or homozygous mutations of Lbr inhibited growth at the EPRO stage when cells were cultured in either regular medium or lipid deprived conditions, with the most severe affect observed in ic/ic cells. These results indicate that even an approximately 50% decrease in Lbr expression observed in +/ic cells (see Fig. 4 and (44)) was enough to disrupt normal cell growth at the EPRO stage. This effect was most likely not due to disrupted cytokine signalling pathways, because GM-CSF-induced phosphorylation levels of STAT5, Erk1/2 and Akt were normal in both genotypes as EPRO cells (Fig. 1). In addition, the percentage of cells positive for lipid production was reduced to < 50% in differentiated EPRO-+/ic cells and only 20% in induced EPRO-ic/ic cells; similar reductions in the average number of droplets per cell were also observed (Fig. 3). These results were consistent with our cholesterol assays by GC/MS: EML-ic/ic cells showed reduced overall cholesterol levels when cultured in either regular medium or in LPDS medium (Fig. 3D), and expressed a precursor that was observed in both wild-type and heterozygous cells treated with LPDS. The increase in lipid production during neutrophil differentiation could not be explained by an increase in Tm7sf2 expression, which was found to remain unchanged as EML cells differentiated toward mature neutrophils (Fig. 4A). On the other hand, sterol stressed conditions induced Tm7sf2 gene expression at all stages of differentiation (Fig. 4C), as previously observed in two different hepatoma cell lines and in human skin fibroblasts (31, 35). Despite this increase, both +/ic and ic/ic cells were dramatically affected by the lipid stressed conditions. Loss of Tm7sf2 expression did not accentuate the growth inhibitory effects of lipid starvation in EML cells, and differentiation was both morphologically and biochemically normal (Fig. 5). Thus myeloid cells are capable of activating the feedback mechanism(s) involved in TM7SF2 gene activation, perhaps via SREBP-2 (52), but this mechanism appears to provide insufficient levels of sterol Δ14 reductase activity required to support lipid-stressed growth of early progenitors that lack Lbr expression (e.g. EML-ic/ic cells), or even normal or lipid-stressed growth of promyelocytes heterozygous for Lbr (EPRO-+/ic cells). These data indicate that Lbr plays a major role in supporting the growth of myeloid progenitors, most likely by providing critical sterol reductase activity in addition to that supplied by Tm7sf2. Furthermore, activities of Lbr alone in early myeloid progenitors (e.g. EML cells) are sufficient to support growth under sterol stressed conditions, whereas those of Tm7sf2 alone are insufficient.
The vast majority of studies on LBR functions have concentrated on identifying different roles for the N-terminal domains of LBR in directing the localization of chromatin to the nuclear periphery or tethering lamins to the INM. The results of these studies were the basis of the postulated role for LBR in facilitating nuclear lobulation during neutrophil differentiation (42, 44, 47, 53, 55, 69). However, such studies did not determine whether sterol reductase activity provided by the C-terminus of LBR may also play an important role in supporting nuclear maturation. With this in mind, we determined how differentiation or growth of +/ic and ic/ic cells would be affected by ectopic expression of either a full-length version of mouse Lbr, two different C-terminal truncated forms (encoding residues 1-387 and 1-544) or two different N-terminal truncated forms (encoding residues 71-626 or 145-626). Expression of the full-length Lbr in ic/ic cells was demonstrated and shown to (a) provide for normal nuclear morphologic maturation, (b) significantly improve growth responses at the EML stage in sterol starved conditions, and (c) dramatically increase oleic acid-induced lipid production in mature cells. These results confirmed that the nuclear maturation, lipid-starved growth and lipid production defects in ic/ic cells were indeed due to loss of Lbr expression, which could be rescued by re-establishing full length Lbr expression in EML cells.
Our attempt to express mutant forms of Lbr that lack the C-terminal membrane spanning domains were unsuccessful, implicating that expression of such mutant Lbr proteins may severely disrupt nuclear morphology and therefore cause cell death, as demonstrated in previous studies by Zwerger et al (54). We therefore turned our attention to analyzing the roles of the N-terminal domains, and successfully expressed the two different N-terminal truncated forms in ic/ic cells. Importantly, we were able to identify ic/ic cells transduced with either construct that exhibited nuclear lobulation upon ATRA-induced differentiation, with ~19% of FLAG-Lbr71- 626 expressing cells and ~12% of FLAG-Lbr145-626 expressing cells showing lobulation as compared to 1.4% of ic/ic cells expressing the empty vector. Since these numbers are substantially less than that of differentiated ic/ic cells expressing the full length protein (~95% show lobulation), they reveal the importance of the first 71 amino acids of Lbr to nuclear lobulation, which encode the lamin B binding domain. The HP1 and heterochromatin binding domains may enhance the capacity for nuclear lobulation, because inclusion of these domains further increased the numbers of lobulated cells. These findings at least suggest that the first 145 amino acids of Lbr may be important to nuclear lobulation but not absolutely necessary. Moreover, domains provided by the N-terminus also help to support both survival in lipid-stressed conditions (Fig. 9E), and lipid production in oleic acid-induced cells (Fig. 10A and B). It is also interesting to note that we found abundant expression of all three forms of Lbr in both the nucleus and cytoplasm in HEK293 cells, suggesting that a fraction or pool of Lbr that resides in the ER may be responsible for the sterol reductase activities required by differentiating neutrophils. These results are consistent with a very recent publication demonstrated that three different antibodies to human LBR all identified its expression in the ER in certain cell types (38). This study also revealed that two different missense mutations in the sterol reductase domain of LBR caused Greenberg/HEM dysplasia, whereas nuclear maturation remained normal. Thus the features of LBR thought to drive nuclear maturation can be uncoupled from its role in cholesterol biosynthesis. The speculated role of LBR in membrane growth may therefore rely on both its sterol reductase domain and its capacity to bind nuclear components essential to regulating the nuclear architecture, e.g. lamin B and heterochromatin, perhaps as part of functional oligomers that have been indentified to form in distinct microdomains of the nuclear envelope (23).
Despite the capacity of either wild-type or truncated forms of Lbr to rescue lipid-starved growth of EML-ic/ic cells, we found that their expression failed to rescue growth of derived EPRO-ic/ic or -+/ic cells in lipid deprived conditions (Figs. 9C and D). We identified expression in the cytoplasm, but several studies have now demonstrated that LBR is expressed in both the nucleus and cytoplasm, so the lack of rescue is most likely not a result of aberrant localization of the FLAG-tagged Lbr proteins (38, 54). One plausible explanation is that deficient Lbr expression in the bone marrow stem cells used to generate EML-+/ic or EML-ic/ic cells caused irreversible changes, such that the differentiation programs of the derived cells were altered in a way that cannot be reversed by reintroducing wild-type Lbr expression. This explanation is based on recent findings that have identified interactions between proteins of the nuclear lamina and genetic loci that can change specific gene expression patterns during differentiation (70-73). In the “higher-order chromatin organization” model proposed from these studies, direct contacts between specific regions of the genome and nuclear lamina proteins, including B-type lamins, can lock genes into inactive states, but these interactions are remodelled during differentiation. This was shown to affect the expression of multiple “stemness” genes (e.g. Oct4, Klf and Nanog) in embryonic stem cells as they differentiated into neural precursor cells (73). Conversely, differentiation-inducing genes can be released from the nuclear lamina, thereby activating lineage specificity. In the case of our EML-ic/ic cells, lack of Lbr expression may have disrupted the anchoring of specific gene loci that are important to late stage growth of EPRO cells, but this affect does not disrupt the growth of EML cells. Therefore by re-establishing Lbr expression in the EML-ic/ic cells, sterol reductase activities were restored but this was probably too late to affect the aberrant expression patterns required for normal growth of EPRO cells in either regular medium or lipid deprived conditions. It is also important to note that our previous studies demonstrated that chromatin becomes localized to the nuclear periphery in +/ic neutrophils, but this failed to occur in ic/ic neutrophils, thus EPRO cells may already have chromatin localization patterns that cannot be reversed (40). Finally, EML-+/+ cells were generated from a different genetic background as compared to the Jackson Laboratory ichthyosis mouse and have been maintained for many more generations as compared to our EML-+/ic or ic/ic cells, which may also influence the levels of responses between these different cell types (39).
Membrane cholesterol levels facilitate the formation of membrane lipid rafts, which are comprised of different amounts of shingolipids and cholesterol packed with phospholipids, with cholesterol comprising ~25% of the mixture (74). The lipid-ordering nature of rafts draws signalling or enzymatic proteins embedded in membranes to close proximity, thereby facilitating protein-protein interactions and complex formation. The importance of this process to neutrophil function was clearly demonstrated when detergent insoluble membrane fractions from activated HL-60 and Ra2 cells were found to contain components of the NADPH oxidase, including the membrane bound subunits gp91phox and p22phox, and because activated HL-60 and Ra2 cells treated with cholesterol synthesis inhibitors displayed reduced respiratory burst activities (11). Our results showing that both lipid deprivation and loss of Lbr expression inhibited levels of oxidative bursts from mature EPRO cells indicated that the two phenotypes may be related. Moreover, expression of full-length Lbr dramatically improved the respiratory burst and improved the formation of CTB-FITC-stained loci in EPRO-ic/ic cells. We also showed that both C-terminal constructs improved reactive oxygen production, with cells expressing Lbr71-626 producing levels near that of those expressing the wild-type form. It is important to note that the expression of the C-terminal portions of Lbr only partially restored nuclear maturation, therefore the levels of respiratory burst responses in these cells may primarily be generated by those cells that exhibit complete nuclear maturation; this notion lends further support to the importance of neutrophil nuclear maturation to its functional responses. These results therefore support our suggestion that sterol reductase activity provided by the C-terminal sequences of LBR are important to cholesterol biosynthesis, which facilitates the formation of lipid rafts and thereby improves the activation of NADPH oxidase complexes.
We propose that our studies establish the importance of the C-terminal sterol reductase region of LBR in myeloid cell growth and differentiation, but our results raise important questions regarding the evolution of LBR vs. other sterol reductases, why LBR is particularly important to myeloid cell differentiation, and how cholesterol biosynthesis in general may be related to disorders of myeloid differentiation. Clearly LBR is not required for cholesterol synthesis in all tissues since embryogenesis can proceed to the fetal stage in humans with homozygous knockout mutations of LBR, and some ichthyosis mice do survive. However, the lack of phenotypes associated with mouse Tm7sf2 knockout mutations as compared to the dramatic developmental defects caused by homozygous LBR mutations indicates that the proliferative or differentiation capacities of certain cells depend more on the activities provided by LBR as compared to TM7SF2. Such cells may have evolved unique cholesterol requirements that depend on sterol reductase activities of LBR but also require the N-terminal functions of LBR in tethering lamins and chromatin to the nuclear envelope, perhaps as a mechanism to orchestrate chromatin remodelling that drives differential gene expression. We propose that several properties of myeloid cells indicate that they are in this category: (i) mature neutrophils require increased cholesterol to mediate the assembly of membrane proteins critical to functional responses, (ii) their growth can be inhibited by drugs that disrupt membrane cholesterol levels, (iii) nuclear maturation is coincident with a relocation of chromatin to the nuclear periphery, and (iv) loss of LBR expression disrupts functional responses, myeloid cell growth and their capacity to synthesize cholesterol. Future studies aimed at defining whether additional components of the cholesterol biosynthetic pathway are important to neutrophil differentiation, and whether additional cell differentiation processes depend on LBR for increased cholesterol biosynthesis, may further define the importance of LBR as an authentic C14 sterol reductase. We are especially interested in testing the Tm7sf2 knockout cells for growth defects at the EPRO cell stage, and whether loss of Tm7sf2 expression in mature neutrophils will affect the respiratory burst; such studies are the focus of our current analyses of this new cell line. These ongoing studies may also reveal important associations between molecular mechanisms that regulate cellular cholesterol levels and hematopoietic cell growth, as was recently discovered for the ATP-binding cassette (ABC) transporters that promote cellular cholesterol efflux, which suppressed the proliferation of myeloid progenitors and hematopoietic stem cells (75). Finally, our cell line that lacks Lbr expression provides a model for studying additional variants of Lbr and their functions in either cell growth or differentiation, including forms that mimic the human variants found to cause Greenberg/HEM dysplasia but not PHA (38).
Acknowledgements
We thank Dr. Harald Herrmann for providing antibodies to LBR and for providing support and advice on analyzing its expression, and Michaela Hergt (DKFZ Heidelberg) for her advice concerning lipid induction and staining. We also thank Emmanuelle Paiva for constructing the pMSCV-puro-Tm7sf2 expression vector.
This work was supported in part by the National Heart, Lung and Blood Institute (AREA grant Nos. 1R15HL89933 and 1R15HL104593 to P.G.). M.Z. was supported by the European Union’s FP6 Life Science, Genomics and Biotechnology for Health area (LSHM-CT-2005-018690).
Abbreviations used in this paper
- ATRA
all-trans retinoic acid
- EML cells
erythroid, myeloid, and lymphoid cells
- EPRO cells
EML-derived promyelocytes
- GM-CSF
granulocyte-macrophage colony stimulating factor
- IL-3
interleukin-3
- ic
ichthyosis
- LBR
lamin B receptor
- LPDS
lipoprotein deficient serum
- MβCD
methyl-beta-cyclodextrin
- SCF
stem cell factor
Footnotes
Disclosures The authors have no financial conflicts of interest
REFERENCES
- 1.Gaines P, Berliner N. In: Granulocytopoiesis. Young NS, Aebi U, Gerson SL, High KA, editors. Mosby Elsevier; Pennsylvania: 2006. p. 61. [Google Scholar]
- 2.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 3.Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Dev. Comp. Immunol. 2004;28:509–554. doi: 10.1016/j.dci.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Hill AD, Naama HA, Calvano SE, Daly JM. The effect of granulocyte-macrophage colony-stimulating factor on myeloid cells and its clinical applications. J. Leukoc. Biol. 1995;58:634–642. doi: 10.1002/jlb.58.6.634. [DOI] [PubMed] [Google Scholar]
- 5.Berliner N. Lessons from congenital neutropenia: 50 years of progress in understanding myelopoiesis. Blood. 2008;111:5427–5432. doi: 10.1182/blood-2007-10-077396. [DOI] [PubMed] [Google Scholar]
- 6.Hager M, Cowland JB, Borregaard N. Neutrophil granules in health and disease. J. Intern. Med. 2010;268:25–34. doi: 10.1111/j.1365-2796.2010.02237.x. [DOI] [PubMed] [Google Scholar]
- 7.Oh H, Mohler ER, 3rd, Tian A, Baumgart T, Diamond SL. Membrane cholesterol is a biomechanical regulator of neutrophil adhesion. Arterioscler. Thromb. Vasc. Biol. 2009;29:1290–1297. doi: 10.1161/ATVBAHA.109.189571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barabe F, Pare G, Fernandes MJ, Bourgoin SG, Naccache PH. Cholesterol-modulating agents selectively inhibit calcium influx induced by chemoattractants in human neutrophils. J. Biol. Chem. 2002;277:13473–13478. doi: 10.1074/jbc.M112149200. [DOI] [PubMed] [Google Scholar]
- 9.Pierini LM, Eddy RJ, Fuortes M, Seveau S, Casulo C, Maxfield FR. Membrane lipid organization is critical for human neutrophil polarization. J. Biol. Chem. 2003;278:10831–10841. doi: 10.1074/jbc.M212386200. [DOI] [PubMed] [Google Scholar]
- 10.Peyron P, Bordier C, N’Diaye EN, Maridonneau-Parini I. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J. Immunol. 2000;165:5186–5191. doi: 10.4049/jimmunol.165.9.5186. [DOI] [PubMed] [Google Scholar]
- 11.Vilhardt F, van Deurs B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 2004;23:739–748. doi: 10.1038/sj.emboj.7600066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guichard C, Pedruzzi E, Dewas C, Fay M, Pouzet C, Bens M, Vandewalle A, Ogier-Denis E, Gougerot-Pocidalo MA, Elbim C. Interleukin-8-induced priming of neutrophil oxidative burst requires sequential recruitment of NADPH oxidase components into lipid rafts. J. Biol. Chem. 2005;280:37021–37032. doi: 10.1074/jbc.M506594200. [DOI] [PubMed] [Google Scholar]
- 13.Newman A, Clutterbuck RD, Powles RL, Catovsky D, Millar JL. A comparison of the effect of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors simvastatin, lovastatin and pravastatin on leukaemic and normal bone marrow progenitors. Leuk. Lymphoma. 1997;24:533–537. doi: 10.3109/10428199709055590. [DOI] [PubMed] [Google Scholar]
- 14.Newman A, Clutterbuck RD, Powles RL, Millar JL. Selective inhibition of primary acute myeloid leukaemia cell growth by lovastatin. Leukemia. 1994;8:274–280. [PubMed] [Google Scholar]
- 15.Clutterbuck RD, Millar BC, Powles RL, Newman A, Catovsky D, Jarman M, Millar JL. Inhibitory effect of simvastatin on the proliferation of human myeloid leukaemia cells in severe combined immunodeficient (SCID) mice. Br. J. Haematol. 1998;102:522–527. doi: 10.1046/j.1365-2141.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Botas J, Ferruelo AJ, Suarez Y, Fernandez C, Gomez-Coronado D, Lasuncion MA. Dose-dependent effects of lovastatin on cell cycle progression. Distinct requirement of cholesterol and non-sterol mevalonate derivatives. Biochim. Biophys. Acta. 2001;1532:185–194. doi: 10.1016/s1388-1981(01)00125-1. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez C, Mdel V. Lobo Md, Gomez-Coronado D, Lasuncion MA. Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp. Cell Res. 2004;300:109–120. doi: 10.1016/j.yexcr.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez C, Martin M, Gomez-Coronado D, Lasuncion MA. Effects of distal cholesterol biosynthesis inhibitors on cell proliferation and cell cycle progression. J. Lipid Res. 2005;46:920–929. doi: 10.1194/jlr.M400407-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu. Rev. Genomics Hum. Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- 20.Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Muller D, Vaya A, Aznar J, Ware RE, Cruz N. Sotelo, Lindner TH, Herrmann H, Reis A, Sperling K. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly) Nat. Genet. 2002;31:410–414. doi: 10.1038/ng925. [DOI] [PubMed] [Google Scholar]
- 21.Cunningham JM, Patnaik MM, Hammerschmidt DE, Vercellotti GM. Historical perspective and clinical implications of the Pelger-Huet cell. Am. J. Hematol. 2009;84:116–119. doi: 10.1002/ajh.21320. [DOI] [PubMed] [Google Scholar]
- 22.Duband-Goulet I, Courvalin JC, Buendia B. LBR, a chromatin and lamin binding protein from the inner nuclear membrane, is proteolyzed at late stages of apoptosis. J. Cell. Sci. 1998;111(Pt 10):1441–1451. doi: 10.1242/jcs.111.10.1441. [DOI] [PubMed] [Google Scholar]
- 23.Makatsori D, Kourmouli N, Polioudaki H, Shultz LD, McLean K, Theodoropoulos PA, Singh PB, Georgatos SD. The inner nuclear membrane protein lamin B receptor forms distinct microdomains and links epigenetically marked chromatin to the nuclear envelope. J. Biol. Chem. 2004;279:25567–25573. doi: 10.1074/jbc.M313606200. [DOI] [PubMed] [Google Scholar]
- 24.Meier J, Georgatos SD. Type B lamins remain associated with the integral nuclear envelope protein p58 during mitosis: implications for nuclear reassembly. EMBO J. 1994;13:1888–1898. doi: 10.1002/j.1460-2075.1994.tb06458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pyrpasopoulou A, Meier J, Maison C, Simos G, Georgatos SD. The lamin B receptor (LBR) provides essential chromatin docking sites at the nuclear envelope. EMBO J. 1996;15:7108–7119. [PMC free article] [PubMed] [Google Scholar]
- 26.Simos G, Georgatos SD. The inner nuclear membrane protein p58 associates in vivo with a p58 kinase and the nuclear lamins. EMBO J. 1992;11:4027–4036. doi: 10.1002/j.1460-2075.1992.tb05496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J. Biol. Chem. 1996;271:14653–14656. doi: 10.1074/jbc.271.25.14653. [DOI] [PubMed] [Google Scholar]
- 28.Ye Q, Callebaut I, Pezhman A, Courvalin JC, Worman HJ. Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J. Biol. Chem. 1997;272:14983–14989. doi: 10.1074/jbc.272.23.14983. [DOI] [PubMed] [Google Scholar]
- 29.Holmer L, Pezhman A, Worman HJ. The human lamin B receptor/sterol reductase multigene family. Genomics. 1998;54:469–476. doi: 10.1006/geno.1998.5615. [DOI] [PubMed] [Google Scholar]
- 30.Roberti R, Bennati AM, Galli G, Caruso D, Maras B, Aisa C, Beccari T, Della Fazia MA, Servillo G. Cloning and expression of sterol Delta 14-reductase from bovine liver. Eur. J. Biochem. 2002;269:283–290. doi: 10.1046/j.0014-2956.2001.02646.x. [DOI] [PubMed] [Google Scholar]
- 31.Bennati AM, Castelli M, Della Fazia MA, Beccari T, Caruso D, Servillo G, Roberti R. Sterol dependent regulation of human TM7SF2 gene expression: role of the encoded 3beta-hydroxysterol Delta14-reductase in human cholesterol biosynthesis. Biochim. Biophys. Acta. 2006;1761:677–685. doi: 10.1016/j.bbalip.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 32.Bennati AM, Schiavoni G, Franken S, Piobbico D, Della Fazia MA, Caruso D, De Fabiani E, Benedetti L, De Angelis M. G. Cusella, Gieselmann V, Servillo G, Beccari T, Roberti R. Disruption of the gene encoding 3beta-hydroxysterol Delta-reductase (Tm7sf2) in mice does not impair cholesterol biosynthesis. FEBS J. 2008;275:5034–5047. doi: 10.1111/j.1742-4658.2008.06637.x. [DOI] [PubMed] [Google Scholar]
- 33.Silve S, Dupuy PH, Ferrara P, Loison G. Human lamin B receptor exhibits sterol C14-reductase activity in Saccharomyces cerevisiae. Biochim. Biophys. Acta. 1998;1392:233–244. doi: 10.1016/s0005-2760(98)00041-1. [DOI] [PubMed] [Google Scholar]
- 34.Prakash A, Sengupta S, Aparna K, Kasbekar DP. The erg-3 (sterol delta14,15-reductase) gene of Neurospora crassa: generation of null mutants by repeat-induced point mutation and complementation by proteins chimeric for human lamin B receptor sequences. Microbiology. 1999;145(Pt 6):1443–1451. doi: 10.1099/13500872-145-6-1443. [DOI] [PubMed] [Google Scholar]
- 35.Wassif CA, Brownson KE, Sterner AL, Forlino A, Zerfas PM, Wilson WK, Starost MF, Porter FD. HEM dysplasia and ichthyosis are likely laminopathies and not due to 3beta-hydroxysterol Delta14-reductase deficiency. Hum. Mol. Genet. 2007;16:1176–1187. doi: 10.1093/hmg/ddm065. [DOI] [PubMed] [Google Scholar]
- 36.Shultz LD, Lyons BL, Burzenski LM, Gott B, Samuels R, Schweitzer PA, Dreger C, Herrmann H, Kalscheuer V, Olins AL, Olins DE, Sperling K, Hoffmann K. Mutations at the mouse ichthyosis locus are within the lamin B receptor gene: a single gene model for human Pelger-Huet anomaly. Hum. Mol. Genet. 2003;12:61–69. doi: 10.1093/hmg/ddg003. [DOI] [PubMed] [Google Scholar]
- 37.Waterham HR, Koster J, Mooyer P, Noort Gv G, Kelley RI, Wilcox WR, Wanders RJ, Hennekam RC, Oosterwijk JC. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am. J. Hum. Genet. 2003;72:1013–1017. doi: 10.1086/373938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clayton P, Fisher B, Mann A, Mansour S, Rossier E, Veen M, Lang C, Baasanjav S, Kieslich M, Brossuleit K, Gravemann S, Schnipper N, Karbasyian M, Demuth I, Zwerger M, Vaya A, Utermann G, Mundlos S, Stricker S, Sperling K, Hoffmann K. Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus. 2010;1:354–366. doi: 10.4161/nucl.1.4.12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsai S, Bartelmez S, Sitnicka E, Collins S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 1994;8:2831–2841. doi: 10.1101/gad.8.23.2831. [DOI] [PubMed] [Google Scholar]
- 40.Gaines P, Tien CW, Olins AL, Olins DE, Shultz LD, Carney L, Berliner N. Mouse neutrophils lacking lamin B-receptor expression exhibit aberrant development and lack critical functional responses. Exp. Hematol. 2008;36:965–976. doi: 10.1016/j.exphem.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaines P, Chi J, Berliner N. Heterogeneity of functional responses in differentiated myeloid cell lines reveals EPRO cells as a valid model of murine neutrophil functional activation. J. Leukoc. Biol. 2005;77:669–679. doi: 10.1189/jlb.1004567. [DOI] [PubMed] [Google Scholar]
- 42.Krakowiak PA, Wassif CA, Kratz L, Cozma D, Kovarova M, Harris G, Grinberg A, Yang Y, Hunter AG, Tsokos M, Kelley RI, Porter FD. Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum. Mol. Genet. 2003;12:1631–1641. doi: 10.1093/hmg/ddg172. [DOI] [PubMed] [Google Scholar]
- 43.Matabosch X, Ying L, Serra M, Wassif CA, Porter FD, Shackleton C, Watson G. Increasing cholesterol synthesis in 7-dehydrosterol reductase (DHCR7) deficient mouse models through gene transfer. J. Steroid Biochem. Mol. Biol. 2010;122:303–309. doi: 10.1016/j.jsbmb.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zwerger M, Herrmann H, Gaines P, Olins AL, Olins DE. Granulocytic nuclear differentiation of lamin B receptor-deficient mouse EPRO cells. Exp. Hematol. 2008;36:977–987. doi: 10.1016/j.exphem.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geiger SK, Bar H, Ehlermann P, Walde S, Rutschow D, Zeller R, Ivandic BT, Zentgraf H, Katus HA, Herrmann H, Weichenhan D. Incomplete nonsense-mediated decay of mutant lamin A/C mRNA provokes dilated cardiomyopathy and ventricular tachycardia. J. Mol. Med. (Berl) 2008;86:281–289. doi: 10.1007/s00109-007-0275-1. [DOI] [PubMed] [Google Scholar]
- 46.Guthridge MA, Stomski FC, Thomas D, Woodcock JM, Bagley CJ, Berndt MC, Lopez AF. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells. 1998;16:301–313. doi: 10.1002/stem.160301. [DOI] [PubMed] [Google Scholar]
- 47.Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, Lopez AF. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114:1289–1298. doi: 10.1182/blood-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kent D, Copley M, Benz C, Dykstra B, Bowie M, Eaves C. Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin. Cancer Res. 2008;14:1926–1930. doi: 10.1158/1078-0432.CCR-07-5134. [DOI] [PubMed] [Google Scholar]
- 49.Rodrigues HG, Vinolo MA, Magdalon J, Fujiwara H, Cavalcanti DM, Farsky SH, Calder PC, Hatanaka E, Curi R. Dietary free oleic and linoleic acid enhances neutrophil function and modulates the inflammatory response in rats. Lipids. 2010;45:809–819. doi: 10.1007/s11745-010-3461-9. [DOI] [PubMed] [Google Scholar]
- 50.Padovese R, Curi R. Modulation of rat neutrophil function in vitro by cis- and trans-MUFA. Br. J. Nutr. 2009;101:1351–1359. doi: 10.1017/S0007114508076307. [DOI] [PubMed] [Google Scholar]
- 51.Mastrangelo AM, Jeitner TM, Eaton JW. Oleic acid increases cell surface expression and activity of CD11b on human neutrophils. J. Immunol. 1998;161:4268–4275. [PubMed] [Google Scholar]
- 52.Schiavoni G, Bennati AM, Castelli M, Fazia MA, Beccari T, Servillo G, Roberti R. Activation of TM7SF2 promoter by SREBP-2 depends on a new sterol regulatory element, a GC-box, and an inverted CCAAT-box. Biochim. Biophys. Acta. 2010;1801:587–592. doi: 10.1016/j.bbalip.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 53.Gaines P, Lamoureux J, Marisetty A, Chi J, Berliner N. A cascade of Ca(2+)/calmodulin-dependent protein kinases regulates the differentiation and functional activation of murine neutrophils. Exp. Hematol. 2008;36:832–844. doi: 10.1016/j.exphem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zwerger M, Kolb T, Richter K, Karakesisoglou I, Herrmann H. Induction of a massive endoplasmic reticulum and perinuclear space expansion by expression of lamin B receptor mutants and the related sterol reductases TM7SF2 and DHCR7. Mol. Biol. Cell. 2010;21:354–368. doi: 10.1091/mbc.E09-08-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olins AL, Rhodes G, Welch DB, Zwerger M, Olins DE. Lamin B receptor: Multi-tasking at the nuclear envelope. Nucleus. 2010;1:53–70. doi: 10.4161/nucl.1.1.10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruan B, Wilson WK, Schroepfer GJ., Jr. An alternative synthesis of 4,4-dimethyl-5 alpha-cholesta-8,14,24-trien-3 beta-ol, an intermediate in sterol biosynthesis and a reported activator of meiosis and of nuclear orphan receptor LXR alpha. Bioorg. Med. Chem. Lett. 1998;8:233–236. doi: 10.1016/s0960-894x(98)00016-x. [DOI] [PubMed] [Google Scholar]
- 57.Pascual-Garcia M, Carbo JM, Leon T, Matalonga J, Out R, Van Berkel T, Sarrias MR, Lozano F, Celada A, Valledor AF. Liver X receptors inhibit macrophage proliferation through downregulation of cyclins D1 and B1 and cyclin-dependent kinases 2 and 4. J. Immunol. 2011;186:4656–4667. doi: 10.4049/jimmunol.1000585. [DOI] [PubMed] [Google Scholar]
- 58.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’connell RM, Cheng G, Saez E, Miller JF, Tontonoz P. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 59.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 60.Chen Q, Ross AC. Retinoic acid regulates cell cycle progression and cell differentiation in human monocytic THP-1 cells. Exp. Cell Res. 2004;297:68–81. doi: 10.1016/j.yexcr.2004.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huwait EA, Greenow KR, Singh NN, Ramji DP. A novel role for c-Jun N-terminal kinase and phosphoinositide 3-kinase in the liver X receptor-mediated induction of macrophage gene expression. Cell. Signal. 2011;23:542–549. doi: 10.1016/j.cellsig.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J. Biol. Chem. 2003;278:10443–10449. doi: 10.1074/jbc.M213071200. [DOI] [PubMed] [Google Scholar]
- 63.Kannan KB, Barlos D, Hauser CJ. Free cholesterol alters lipid raft structure and function regulating neutrophil Ca2+ entry and respiratory burst: correlations with calcium channel raft trafficking. J. Immunol. 2007;178:5253–5261. doi: 10.4049/jimmunol.178.8.5253. [DOI] [PubMed] [Google Scholar]
- 64.Blank N, Gabler C, Schiller M, Kriegel M, Kalden JR, Lorenz HM. A fast, simple and sensitive method for the detection and quantification of detergent-resistant membranes. J. Immunol. Methods. 2002;271:25–35. doi: 10.1016/s0022-1759(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 65.Fitzky BU, Witsch-Baumgartner M, Erdel M, Lee JN, Paik YK, Glossmann H, Utermann G, Moebius FF. Mutations in the Delta7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc. Natl. Acad. Sci. U. S. A. 1998;95:8181–8186. doi: 10.1073/pnas.95.14.8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wassif CA, Maslen C, Kachilele-Linjewile S, Lin D, Linck LM, Connor WE, Steiner RD, Porter FD. Mutations in the human sterol delta7-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome. Am. J. Hum. Genet. 1998;63:55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Waterham HR, Koster J, Romeijn GJ, Hennekam RC, Vreken P, Andersson HC, FitzPatrick DR, Kelley RI, Wanders RJ. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am. J. Hum. Genet. 2001;69:685–694. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oosterwijk JC, Mansour S, van Noort G, Waterham HR, Hall CM, Hennekam RC. Congenital abnormalities reported in Pelger-Huet homozygosity as compared to Greenberg/HEM dysplasia: highly variable expression of allelic phenotypes. J. Med. Genet. 2003;40:937–941. doi: 10.1136/jmg.40.12.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olins AL, Olins DE. Cytoskeletal influences on nuclear shape in granulocytic HL-60 cells. BMC Cell Biol. 2004;5:30. doi: 10.1186/1471-2121-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–247. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- 71.Kumaran RI, Spector DL. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 2008;180:51–65. doi: 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008;4:e1000039. doi: 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Filippov A, Oradd G, Lindblom G. Lipid lateral diffusion in ordered and disordered phases in raft mixtures. Biophys. J. 2004;86:891–896. doi: 10.1016/S0006-3495(04)74164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]