

Abstract
Hox proteins are transcription factors and key regulators of segmental identity along the anterior posterior axis across all bilaterian animals. Despite decades of research, the mechanisms by which Hox proteins select and regulate their targets remain elusive. We have carried out whole-genome ChIP-chip experiments to identify direct targets of Hox protein Ultrabithorax (Ubx) during haltere development in Drosophila. Direct targets identified include upstream regulators or cofactors of Ubx. Homothorax, a cofactor of Ubx during embryonic development, is one such target and is required for normal specification of haltere. Although Ubx bound sequences are conserved amongst various insect genomes, no consensus Ubx-specific motif was detected. Surprisingly, binding motifs for certain transcription factors that function either upstream or downstream to Ubx are enriched in these sequences suggesting complex regulatory loops governing Ubx function. Our data supports the hypothesis that specificity during Hox target selection is achieved by associating with other transcription factors.
Diversity in the regulation and function of Hox genes appear to be a major factor in the evolution of body plan in the animal kingdom1. Hox genes encode for homeodomain-containing transcription factors and regulate cell-fate specification to govern identity of body segments along the anterior posterior body axis in all bilateral animals1,2. Importance of Hox genes to determine organ identity is reflected in the observations that when Hox gene functions are altered, animals display dramatic homeotic transformations. For example, loss of function mutation in the Hox gene Ultrabithorax (Ubx) in Drosophila results in the transformation of balancing organs halteres to wings, resulting in famous four-winged fly, whereas ectopic expression of Ubx in developing wing discs leads to wing-to-haltere transformations3,4,5. Thus, alteration in organ identity by Hox gene mutations has served as a key paradigm to understand molecular mechanism of morphological development and evolution.
Considerable efforts have been made in the past two decades to understand events downstream of Hox proteins. Expression studies such as enhancer-trap and microarray analyses done in Drosophila and vertebrates have revealed that Hox proteins regulate a vast number of downstream targets1,6. Typical of such experiments, many of these genes are secondary targets of Hox proteins. Nevertheless, such studies have been immensely useful to understand the mechanism by which a given Hox protein specifies a developmental pathway. For example, Ubx specifies haltere development by fine tuning key signaling pathways such as Wingless7, Decapentaplegic8,9,10 and EGFR/Ras pathways11.
To understand mechanism of target selection it is essential to identify all direct targets of a given Hox protein and also analyze their cis-regulatory sequences to comprehend which sequences the Hox protein binds to in vivo. Candidate gene approaches have yielded a small number of direct targets of Ubx during embryonic1,6 or haltere development10,11,12,13,14, which are not sufficient for such analysis. However, in vitro analyses suggest that all Hox proteins display poor DNA-binding specificity and recognize very similar degenerate sequences containing -ATTA- core15,16,17,18. Such degenerate core sequences cannot explain how Hox proteins regulate their targets so specifically in vivo, a problem often referred to as ‘Hox-paradox'6.
We carried out whole genome Chromatin immuno precipitation coupled with-microarray (ChIP-chip) experiments to identify direct targets of Ubx during haltere development in Drosophila. We envisaged that such a study would help (i) better understand key early molecular mechanisms regulating haltere development and (ii) the mechanism by which Ubx selects its targets. We used polyclonal antibodies generated against N-terminal region of Drosophila Ubx protein to pull-down DNA fragments bound specifically by Ubx. Several of these fragments were further validated by ChIP-qPCR. Although large number of pulled-down sequences showed the presence of previously reported Ubx binding sites15,16, there was no significant enrichment for these motifs over the background sequences. Further sequence analysis, however, revealed enrichment for several motifs that are binding sites for other transcription factors such as GAGA factor (GAF) and MAD. ChIP-qPCR suggested that Ubx and GAF share several targets during haltere development and ChIP-Western suggested that they share binding elements in same space and time. Thus, our experimental results suggest that association with other transcription factors is key for achieving specificity in Hox target selection. Absence of a specific recognition motif may be one of the main reasons for versatility of Hox proteins in target selection. We also show that Homothorax (Hth), a cofactor of Ubx in the embryo, is a direct target of Ubx in the haltere and genetic analysis suggests a positive feedback loop between Homothorax and Ubx.
Results
Identification of direct targets of Ubx
Monoclonal antibodies have been used earlier for chromatin immuno-precipitation of Ubx10. However, polyclonal antibodies are better reagents for ChIP as recognition is not dependent on a single epitope. As homeodomain is common amongst several transcription factors, polyclonal antibodies against full-length Ubx are expected to recognize all Hox proteins and many other homeodomain-containing transcription factors. We therefore, generated rabbit polyclonal antibodies against N-terminal region of Drosophila Ubx protein lacking the homeodomain. Serum was tested and shown to be specific in immuno-staining and Western blot analysis and did not cross-react to any other protein in tissues lacking Ubx (Fig. 1A, Supplementary Fig. 1). We used this serum and performed ChIP using chromatin from wing discs of CbxHm/+ third instar larvae. CbxHm/+ over-express Ubx in the wing pouch and thereby show wing-to-haltere transforamtions5. These wing discs were used to focus on suppression of wing fate by Ubx, without mixing with targets involved in notum development. CbxHm/+ wing discs also provide more chromatin than wild type haltere discs and a previous study has reported that Cbx genetic background is more sensitive than wildtype in identifying certain true targets of Ubx function13.
Figure 1. Generation of polyclonal antibodies against Ubx.
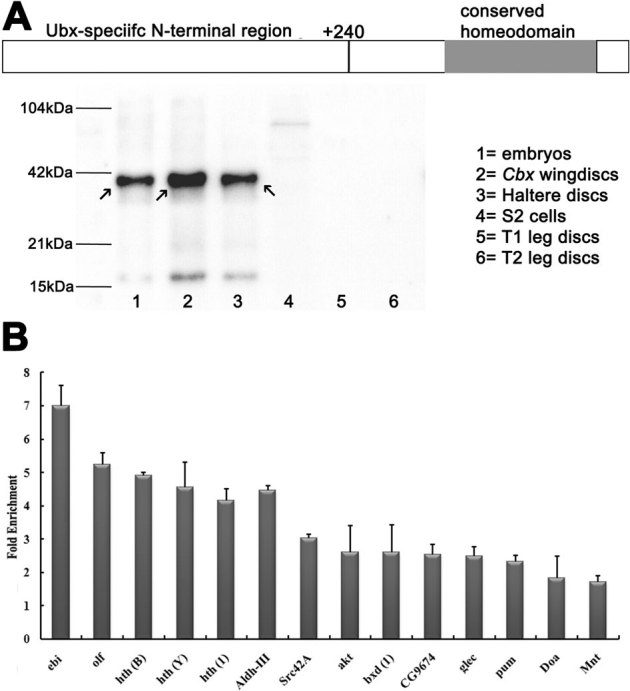
(A) Top: schematic of Ubx protein. N-terminal 240-aminoacid region was expressed for raising antibodies. Below: Western blot analysis to show specificity of the polyclonal antibodies raised against Ubx. See a single prominent band of full-length protein (arrows) detected in embryonal, haltere disc and CbxHm/+ wing disc lysates. S2 cells, T1 and T2 leg discs (which does not express Ubx) are used as negative control. Equal amount of protein was loaded in all the lanes. These antibodies were used to identify potential direct targets of Ubx. Validation of potential targets of Ubx. (B) Candidate regions were selected randomly from high, medium and low enrichment levels of ChIP-chip data and validated by two independent biological replicates of ChIP-qPCR for chromatin isolated from CbxHm/+ discs. Primers for qPCR were designed around the probes enriched in ChIP-chip. Bar height represent mean enrichment of test IP over mock IP and error bars represent deviation from the mean enrichment. Note, all 14 targets regions are enriched in both the replicates. Brackets next to gene name represent arbitrarily assigend names for multiple probes enriched for the same gene in ChIP-chip data. See Supplementary Material for details.
From three independent biological replicates of ChIP-chip, we identified 519 probes as enriched in test over mock (Supplementary Table S1). As chromatin hybridized to genomic array was ∼500 bp in size, we extended our probes (average 58 bp-long) to 500 bp in either direction and used these sequences (average 1058 bp-long), referred as ‘pulled down sequences' hereafter, for further analysis. While all probes were within non-coding parts of the genome, some of the pulled down sequences had coding regions included.
We first estimated phylogenetic conservation of pulled-down sequences by estimating PhastCons score, a measure of evolutionary conservation in twelve Drosophila species, mosquito, honeybee and red flour beetle, obtained from the UCSC genome browser. The average “per base PhastCons score” of the entire D. melanogaster genome is 0.363 whereas the same for the non-coding component of the genome is 0.334. In contrast, the PhastCons score for the pulled down sequences is 0.455 (0.441 after removing all the coding regions in these pulled-down sequences). These observations suggest that the pulled down portions are more conserved than the rest of the genome. This is reflective of possible functional role these sequences may play in regulating nearby genes.
Probes identified by ChIP-chip were assigned to their nearest genes within 2 kb in either direction. With this criterion, we found 493 unique genes as direct targets of Ubx (Supplementary Table S2). While several genes had more than one probe assigned to them, few probes were in gene rich or nested gene regions and were assigned to more than one gene. We extended those probes, which were not close to any gene within 2 kb, and looked for nearest genes up to 10 kb away (Supplementary Table S2). 50 probes, however, remained un-assigned. It is possible that certain Ubx binding sites are functional even if they are over 10 kb distances. For example, wg is the nearest gene for a probe at a distance of more than 10 kb. Wg is down-regulated in developing haltere19,20 and thus could be a good candidate for a direct target of Ubx. However, to avoid possible false-positives, we left the probes un-assigned if they were more than 10 kb away from any gene (Supplementary Table S2).
After assignment of probes to their targets, we examined if any of the known direct targets of Ubx are represented in our dataset. Several previously reported targets of Ubx6 in embryo and haltere discs were picked up by our ChIP-chip experiment e.g. antennapedia, thickveins, spalt major, scabrous, serpentine, connectin etc. (Supplementary Table S2). We further validated our data by ChIP-qPCR for few probes representing high, medium and low enrichment levels in ChIP-chip analyses. We performed qPCR with primers designed around these probes using independently prepared ChIP samples from two biological replicates. All the 14 probes tested showed enrichment in ChIP-qPCR assays (Fig. 1B), thus confirming the validity of ChIP-chip data. Furthermore, the relative degree of enrichment remained the same between ChIP-chip and ChIP-qPCR assays.
Functional classification of direct targets of Ubx
We classified all the potential targets of Ubx according to their function using experimental data from Flybase. Components of key signaling pathways (such as Wg, EGFR, Dpp etc.) and transcription factors are major categories amongst potential targets of Ubx based on their molecular function (Fig 2A, Supplementary Table S3). Genes that belong to realizator categories21 (such as cytoskeletal, chitin-binding, cuticular and other differentiation-specific proteins etc.) constituted relatively smaller component of Ubx targets (Fig. 2B). At the developmental level, as many as 70 genes known to be involved in wing development were identified as direct targets of Ubx (Fig. 2C). We also performed Gene Ontology (GO) analysis on the genes identified as direct targets of Ubx to identify over-represented categories using the open source platform BiNGO22. The analysis gave us a network with morphogenesis and cell signaling as the major nodes of the network (data not shown).
Figure 2. Classification of genes identified as potential targets of Ubx by ChIP-chip analysis.

Gene function is based on the summary information provided in Flybase, which in turn is based on consolidated experimental data. The summary for each gene was downloaded and the curation and tabulation was done manually. Out of total 589 genes experimental data for different categorized function was available for 271 genes and remaining 318 genes were of unknown/uncategorized function. Those 271 genes were classified based on their molecular function (A), cellular function (B) or developmental function (C). Please note a given gene may be represented in more than one category (for example, Wg pathway, wing development and cell proliferation). See Suppl. Table 2 for detailed functional information for each gene.
Amongst the Hox genes, antennapedia, sex combs reduced and deformed were identified as potential direct targets of Ubx. This is in confirmation with the general rule of posterior prevalence23, wherein posteriorly expressing Hox proteins negatively regulate the anteriorly expressing ones. Interestingly, Ubx binds to various chromatin modulators and members of Polycomb group (PcG) and Trithorax group (TrxG) genes hitherto known to function upstream of Hox genes e.g. trithorax, pipsqueak, polyhomeotic proximal, GAGA factor, osa, kismet, corto etc. (Supplementary Table S3). We also identified homothorax (hth), a cofactor of Ubx in the embryo24, as a direct target of Ubx and validated Ubx binding on three potential CREs of hth by independent ChIP-qPCR analysis (Fig. 1B). These observations suggest that Ubx regulates various upstream genes or cofactors, to maintain its own level/function via a feedback mechanism. We explore this possibility in later sections of this study.
Most of the genes differentially expressed between wing and haltere are secondary targets of Ubx
We also compared ChIP-chip data with the list of genes identified in microarray analyses as differentially expressed between developing wing and haltere. Out of total 542 genes reported by Mohit et al. (2006)13, only 26 were found to be direct targets of Ubx in our ChIP-chip data (Supplementary Table S4). Pavlapoulos and Akam (2011)25 used ectopically expressed Ubx in wing tissue and examined for global gene expression changes using microarray analyses at larval, prepupal and pupal stages of Drosophila development. Only 26 genes at larval, 45 genes in prepupal and 35 genes in pupal stages of this study are represented in our ChIP-chip data set (Supplementary Table S4).
Presence of such small number of direct targets in microarray data suggests that i) Hox binding may induce subtle changes in gene expression (too subtle to be identified by microarray analysis) or regulatory landscape including chromatin conformation, ii) Such small changes in expression or chromatin structure can amplify and/or converge on downstream secondary targets leading to detectable changes in the expression levels of those genes, iii) expression changes in imaginal discs might be due to cumulative effect of changes over more than one developmental stage and (iv) not all data generated by ChIP and expression profiling experiments could be relevant to the function (but they are not the same as false positives) and hence may not overlap between the two kinds of experiments.
Functional analysis of Hth, a direct target of Ubx
Our ChIP-chip data identified three Ubx-binding regions on hth gene, all of which were validated by ChIP-qPCR (Fig. 1B). Hth has been shown to be required for embryonic, adult thorax and wing hinge development in Drosophila26,27,28. Its role in haltere development, if any, is hitherto not known. As in wing discs, in haltere discs too Hth is expressed in the peripodial membrane and in presumptive hinge and the notum regions. It is not expressed in the pouch region (Fig. 3). At the cellular level, Hth is required for the nuclear localization of (Extradenticle) Exd29,30 and therefore, down-regulation of hth and exd cause similar mutant phenotypes. However, no Ubx-binding regions were observed on exd in our ChIP-chip data. Hth and Exd are two cofactors known to associate with many Hox proteins including Ubx24,31 and increase their DNA binding affinity and specificity32. A previous study has shown that Ubx require Exd activity for maintaining their own levels and somatic clones of exd in halteres are associated with down-regulation of Ubx33. It is therefore interesting that Hth is also a direct target of Ubx, which was further investigated here.
Figure 3. Wildtype expression pattern of Hth in haltere imaginal discs is identical to its expression pattern in wing imaginal discs.
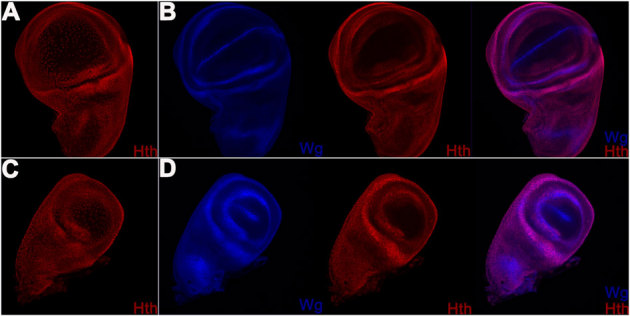
Wing (A, B) and haltere (C, D) imaginal discs stained with anti-Wg (blue) and guinea pig anti-Hth (red) antibodies. Please note strong expression of Hth in the peripodial membrane (A, C) and in the presumptive hinge and the notum (B, D). As in wing discs, in haltere discs too Hth is not expressed in the pouch region. In this and in subsequent images, all discs are shown with anterior to the left.
First, we examined if removal of Ubx affect Hth levels in haltere discs. We observed down-regulation of Hth levels in somatic clones of Ubx1 (a null allele of Ubx) in haltere discs (Fig. 4; Suppl Fig. 2). This data suggests that Ubx positively regulates hth expression, probably by binding to its regulatory regions identified by ChIP-chip experiments reported here. Reverse, however, was not true. We did not observe any change in Ubx levels in mitotic clones of hth allele hthP2 (data not shown), however, this could possibly due to the reason that hthP2 is not a complete null allele of hth34. We then attempted to down regulate Hth using transgenic UAS-RNAi transgene. We used three different RNAi lines against hth (VDRC id, 12763, 12764 and NIG id 17117R) and crossed to various GAL4 drivers expressed in thoracic imaginal discs. With MS1096-GAL4 driver, we observed significant down regulation of Hth in dorsal hinge regions (Fig. 5B). Similar to hthP2 clones, this reduction in Hth levels was not associated with any reduction in Ubx levels (Fig. 5B).
Figure 4. hth is down-regulated in somatic clones of Ubx in haltere discs.
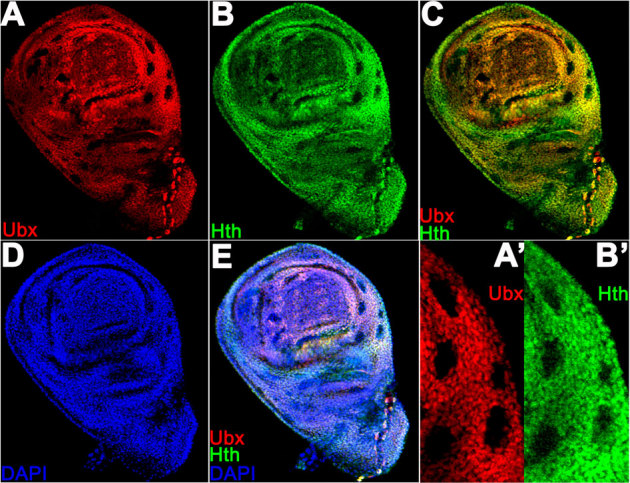
Mitotic clones of Ubx1 were generated as described in the Methods and the discs were stained for monoclonal anti-Ubx (red) and rabbit anti-Hth (green). Note, down-regulation of Hth expression in hinge and notum regions of disc proper cells. Staining seen in the pouch region with anti-Hth antibodies is probably the staining in the peripodial memrbane. A-E shows the same haltere in different channels. A' and B' shows a small region at higher magnificaiton. All the discs observed at 72–96 hours after AEL showed down-regulation of Hth in mitotic clones of Ubx, n = 12. Another representative haltere disc showing Ubx1 clones is shown in Suppl. Fig. S2.
Figure 5. Down regulation of Hth has no effect on Ubx expression.
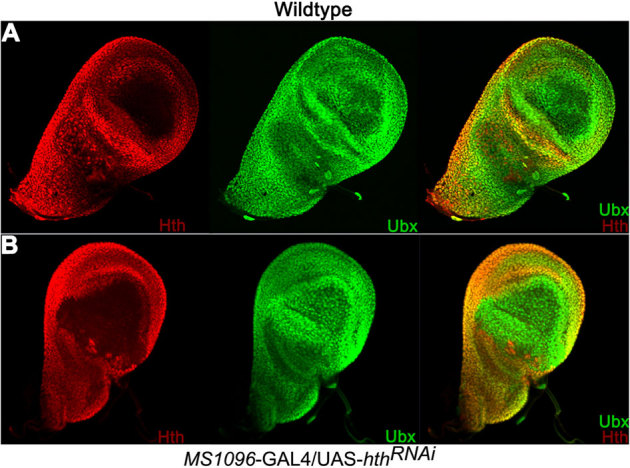
Wildtype (A) and MS1096-GAL4/UAS-hthRNAi (B) Haltere discs stained with guinea pig anti-Hth (red) and anti-Ubx (green) antibodies. Note severe reduction on Hth levels in dorsal hinge region due to the expression of hthRNAi. There is no change in the levels of Ubx expression in that region.
Next we examined if down-regulation of hth has any phenotypic consequences on haltere morphology. All the three RNAi lines induced wing-type bristles on haltere when crossed with vg-GAL4 (penetrance 80%, n = 48 for VDRC ID 12764) and MS1096-GAL4 (penetrance 100%, n = 50 for VDRC ID 12763; penetrance 45%, n = 73 for VDRC ID 12764; penetrance 100%, n = 53 for NIG ID 17117R2) drivers (Fig. 6C, D), a phenotype much stronger than that in flies heterozygous for Ubx null alleles. To over-rule the possibility of ‘off-target effects', we carried out rescue experiment in which we combined RNAi against hth (VDRC ID 12763) with over-expression line for Hth and observed partial (12%, n = 11/91) to complete (42%, n = 39/91) suppression of the RNAi phenotype (Fig. 6F, G). We observed similar partial (11%, n = 8/70) to complete (45%, n = 32/70) rescue when human homologue of Hth, Meis1, along with RNAi against hth (NIG ID 17117R2) was used (data not shown). These results suggest that Hth is required for haltere specification.
Figure 6. Down regulation of Hth and Exd function causes haltere-to-wing transformations.

Genotypes of all halteres are as shown in the figure. Note appearance of wing-like bristles, characteristic of flies heterozygous for Ubx (B) when hth or exd is knocked down (C–E, H). The phenotype is far more severe than flies heterozygous for Ubx. Partial (F) to full (G) rescue of hthRNAi-induced phenotype is observed when Hth is over-expressed using a UAS-Hth construct suggesting the specificity of the phenotype. MS1096-GAL4 driver (which is on X-chromosome) was used as female parent, but both males and females showed comparable phenotypes.
To investigate the spatial specificity of these phenotypes, we crossed UAS-hthRNAitransgenes to hth-, tsh- and pnr-GAL4 drivers. We did not observe any down regulation of Hth with hth- and tsh-GAL4 drivers, perhaps the GAL4 drivers are weaker than MS1096-GAL4 driver. Alternatively, hth- and tsh-GAL4 drivers may express at later developmental stages compared to MS1096-GAL4 driver. A small number of tsh-GAL4/UAS-hthRNAianimals were pupal lethal (10%; n = 80) and they did not show any detectable haltere phenotype. With pnr-GAL4 driver, UAS-hthRNAitransgene caused significant reduction in Hth levels (Suppl. Fig. S3C) and the animals were lethal at pharate adult stage. As expected they all had severe notum as well as T3 dorsal cuticular phenotypes, but halters were normal as pnr-GAL4 driver down-regulated Hth expression only in the notum (Suppl. Fig. S3D, E). These results indicate that haltere phenotypes observed in vg- and MS1096-GAL4/UAS-hthRNAiflies reflect a role for Hth in haltere development.
As mutation in exd and hth cause many similar phenotypes, we also used a RNAi line against exd (VDRC ID 7803) which also caused wing-type bristles on the haltere (Fig. 6H; penetrance 97%, n = 74). However, simultaneous knock-down of exd and hth did not enhance bristle phenotype in haltere (data not shown). Further investigation is required to understand the mechanism of Hth function, either as a cofactor of Ubx and/or as a downstream effector of Ubx. If it indeed functions as a cofactor, our observations suggest positive feedback regulation, wherein Ubx positively regulates the levels of its cofactor Hth.
Search for in-vivo regulatory code for Ubx
To understand the mechanism by which Ubx selects its target genes in vivo, we analyzed ChIP data (519 fragments, each 1058 bp long) for motifs, if any, that may serve as a signature for Ubx to bind. Previously reported motif TTAATKR was investigated using both CLOVER35 and MATCH36 programs. CLOVER detected the presence of consensus Ubx motif (referred hereafter as ‘Ubx heptamers'; see Supplementary text for position weight matrix) in 319 out of 519 sequences, while MATCH detected Ubx heptamers in only 174 sequences under ‘minimize false positive' condition (stringent) and 517 sequences under ‘minimize false negative' condition (relaxed) after coding sequences had been filtered. This analysis suggested that under stringent statistical conditions, less than half the pulled-down sequences contained Ubx heptamers.
MATCH program was also used to investigate presence of binding sites listed in TRANSFAC database37 for various known transcription factor in pulled-down sequences versus the entire non-coding genome of Drosophila (∼89 MB) and randomly selected 519 sequence contigs of 1058 bp each from the fly genome (Table 1). Under ‘minimize false positive' conditions, we find that Ubx heptamers occur with a frequency of 0.417 per kilobase in the pulled-down sequences, while they occur with a frequency of 0.489 per kilobase in the entire non-coding genome and 0.453 per kilobase in randomly generated sequence contigs. We also searched for the frequency of occurrence for the two other degenerate sequences reported to be consensus-binding motifs for Ubx18,38. They too occur at marginally lower frequency in pulled-down sequences compared to randomly selected sequence contigs (Table 1).
Table 1. Frequency distribution (per kilobase) of binding motifs for various transcription factors 519 sequences, each 1058 bp long, pulled down by ChIP using anti-Ubx antibodies were searched for the presence of binding motifs for various transcription factor binding sites with MATCH using the motifs listed in TRANSFAC database (for Ubx, we also used motifs reported by Noyes et al., 2008 and Mann et al., 2009) under minimize false positive criterion. Similar analysis was done for randomly selected 519 contigs, each 1058 bp long from the fly genome and for the entire non-coding region of the fly genome. It should be noted that while binding motif for Ubx is not significantly enriched in the pulled down sequences, binding motifs for certain transcription factors such as GAF and MAD are significantly enriched. Others, although are statistically enriched, are represented at very low frequencies in both experimental and background sequences. The p-value was calculated from the z-score by taking the difference of the frequency of a motif in the ChIP-identified regions and its mean frequency in a sufficiently large set of random regions from the Drosophila genome (each consisting of 519 sequences 1058 bps long with coding regions masked), divided by the standard deviation of the frequency values for the random regions.
| Transcription factor | Sequences pulled down using anti-Ubx antibodies | Randomly selected sequences | Entire non-coding genome | Fold change in frequency | p-value |
|---|---|---|---|---|---|
| GAGA factor | 0.7361 | 0.1989 | 0.2098 | 3.7020 | <10−10 |
| MAD | 0.4281 | 0.2176 | 0.2163 | 1.9674 | <10−10 |
| Ubx (TRANSFAC) | 0.4165 | 0.4533 | 0.4886 | 0.9188 | 0.1383 |
| Ubx (Mann et al., 2009) | 8.170 | 10.377 | 10.635 | 0.7873 | <10−10 |
| Ubx (Noyes et al., 2008) | 1.147 | 1.479 | 1.557 | 0.7756 | 3.4×10−6 |
| Adf-1 | 0.0910 | 0.0313 | 0.0320 | 2.9110 | 7.1×10−7 |
| Grainyhead/Elf-1/NTF-1 | 0.0871 | 0.0385 | 0.0394 | 2.2648 | 2.6×10−8 |
| DREF | 0.0464 | 0.0235 | 0.0223 | 1.9787 | 0.0036 |
| OVO | 0.0387 | 0.0231 | 0.0228 | 1.6784 | 0.0207 |
| Snail | 0.0154 | 0.0088 | 0.0099 | 1.7563 | 0.0727 |
| Su(H) | 0.0038 | 0.0004 | 0.0005 | 8.6216 | 1.8×10−4 |
| dTCF | 0.0038 | 0.0006 | 0.0007 | 6.7526 | 0.0014 |
In contrast, binding sites for transcription factors GAF and MAD are significantly enriched in pulled-down sequences. For example, we find that GAF-binding motif occurs with a frequency of 0.736 per kilobase in pulled-down sequences, while it occurs with a frequency of 0.209 per kilobase in the non-coding genome and 0.198 per kilobase in randomly selected sequence contigs. TRANSFAC analyses also showed that pulled down sequences harbor several clusters of potential binding sites for various transcription factors within 200 bp region (Supplementary Table S5). Many of these clusters were of developmentally important transcription factors, suggesting enrichment for sequences that are already under developmental control. This suggests that Ubx may use the binding of other transcription factors as cofactors or collaborators to reach its targets.
We also performed a de-novo motif search using MEME39 (see Supplementary Text for details). MEME analysis, however, did not identify any motif that is common to all the probes pulled down by ChIP. Interestingly, some of the frequently occurring motifs identified by MEME analysis were similar to the consensus binding sites of proteins known to be upstream to Ubx such as Polycomb and Trithorax group response elements (PREs and TREs)37,40,41 suggesting these factors and Ubx may regulate common set of targets (Fig. 7A; Supplementary table S5).
Figure 7. Ubx and GAF may regulate common downstream targets.

(A) Multiple motifs were enriched in MEME analysis of sequences pulled down as potential direct targets of Ubx. Many of these motifs resemble previously known motifs for polcomb (a) and Trithorax (b, e) proteins and general RNA polymerase II associated factors Aef1 (c) and Top2 (h). d, f, g and i are the new motifs not related to any known DNA-binding factors. The numbers below each motifs represents their respective E-value derived by MEME, lower E-value represents higher occurrence and significance for each of the motifs in given dataset. (B) GAF ChIP-qPCR showing enrichment for direct targets of Ubx, ebi and hth. Same set of primers were used to detect enrichment as was used for Ubx ChIP-qPCRs (Fig. 1A). bxd and trl regions were used as positive controls. Bar height represent mean enrichment of Test ChIP over mock ChIP and error bars represent deviation from mean enrichment. Three differnet probes within hth, and one probe near ebi all of which were enriched in pulled-down sequecnes, were validated by ChIP-qPCR. (C) Chromatin immunoprecipitation of Ubx from CbxHm/+ wing disc lysate followed by Westerb blot analysis for GAF. ChIP-Western suggests that GAF is associated with Ubx on chromatin. Densitometric analyses using ImageJ suggested a ∼12-fold specific enrichment for GAF in anti-Ubx lane compared to IgG lane.
As shown in Table 1 and Fig. 7A, GAGA motif and MAD binding sites are enriched in sequences pulled down using anti-Ubx antibodies. Collaboration of Ubx with MAD is already shown to be required for repression of salm in haltere discs42. GAGA like motif serves as recognition sequences for GAGA-binding factor (GAF, encoded by gene Trl), a member of TrxG proteins43. Subsequent work suggested that GAF has both PcG and TrxG like activity44,45,46.
To examine association between Ubx and GAF on specific common targets during haltere specification, we carried out ChIP on CbxHm/+ imaginal discs using anti-GAF antibodies46 followed by qPCR for different regions with predicted GAGA motifs in the vicinity. We used the same primer pairs that were earlier used for Ubx ChIP-qPCR. We observed that GAF binds to all the four chromatin-regions tested, suggesting that those regions are bound by both GAF and Ubx (Compare Fig. 1B and Fig. 7B).
As shown in Fig. 7B, GAF binds to as many as three regulatory regions of hth. We also examined if GAF regulates hth expression. Only one combination of Trl alleles are viable beyond embryonic stages and they did not show any haltere phenotypes. Down regulation of GAF using RNAi method did not result in considerable decrease in its protein levels nor was there any effect on Hth levels (data not shown). In the absence of appropriate genetic tools, functional role of GAF in regulating Hth and specifying haltere remains inconclusive.
ChIP-Western was carried out to further validate if Ubx and GAF together bind to chromatin in vivo. ChIP was carried out for chromatin obtained from CbxHm/+ discs using anti-Ubx antibodies followed by a Western blot analysis on pulled down chromatin using anti-GAF antibodies. We observed enrichment of GAF in this complex suggesting that Ubx can indeed associate with GAF on DNA in vivo (Fig. 7C).
To conclude, results presented here on our attempts to identify direct targets of Ubx by ChIP-chip analyses have provided new insights into how a Hox protein selects its targets and regulates their expression.
Discussion
How Hox proteins select downstream targets to specify cell fate along the anterior-posterior body axis across various animals has remained a long-standing question. This is an important question considering the fact that all Hox proteins are master control genes, only few layers down in the hierarchy of genes that control development since fertilization. Nevertheless, the developmental events that they control are complex and always involve multiple genes immediately downstream of a Hox gene. There are, however, a few exceptions to this rule, wherein mutations in a single downstream gene could cause homeotic phenotypes to the same degree as mutations in Hox genes. For example, homeotic transformations of certain neuroblast lineages caused by loss-of-function mutations in abd-A or Abd-B are indistinguishable from phenotypes by the over-expression of their target gene cycE47,48. In contrast, Hox-mediated specification of segmental/organ/tissue identities during development involves complex regulation of downstream events by Hox proteins leading to their canalization ensuring irreversibility of those identities. Such a mechanism, however, would be dependent on elaborate spatio-temporal regulation of Hox proteins themselves as change in their expression pattern would lead to major phenotypic consequences. Complex upstream regulatory circuitry involving Polycomb and Trithorax group of proteins in addition to maternal, gap, pair-rule and segment polarity genes in regulating expression of Hox genes in Drosophila reflects the importance of Hox gene regulation in development. This regulatory mechanism is largely conserved across the animal kingdom1. While our attempt to identify and analyze direct targets of Ubx is primarily to understand the mechanism of Hox control, it is also aimed at connecting this complex circuitry of Hox regulation to the events downstream of a Hox protein.
Hox proteins are thought to be at the end of a series of hierarchical regulatory steps. Further downstream, they are thought to regulate real building blocks of development such as differentiation factors, often referred to as realizator genes21. While comparative analyses of downstream genes of various Hox proteins using microarray approach in embryos suggested that realizator genes constitute major class of Hox responsive genes49, microarray analysis of developing discs identified significant number of signaling molecules and transcription factors as targets of Hox proteins13. In this study, we have identified more than 500 in vivo Ubx bound sequences in third instar larval discs of Drosophila (Supplementary Table S3); most of the sequences belonged to genes encoding for transcription factors and signaling molecules. Our analysis reveals that realizator genes, such as those coding for cytoskeletal, cuticular, chitin-binding proteins, constitute only a small subset of direct targets of Ubx (Fig. 2B; Supplementary Table S3). It is likely that many realizator genes are secondary rather than direct targets, whereas, transcription factors and cell signaling molecules constitute the major categories of genes directly regulated by Ubx. These in turn may regulate large number of realizator genes to specify haltere development.
It has been proposed that Ubx sits at the top of gene regulatory networks (GRNs) acting like an input/output switch without receiving any feedback from the downstream targets50. Direct targets identified by ChIP-chip analysis reported here include Hth, a cofactor of Ubx during embryonic development. Genetic analysis suggested a positive feedback loop between Hth and Ubx. Our model suggests that Ubx (and probably GAF) may activate hth expression by binding to its CREs, which might help to localize Exd into the nucleus. This nuclear Exd protein may in turn activate Ubx. Although not validated here, identification of PcG proteins (negative regulators of Ubx levels) and GAF (positive regulator of Ubx) as direct targets of Ubx adds another dimension to auto-regulation of Ubx levels in the developing haltere. These results have implications on the hierarchy of Hox regulated networks since topology of the network would change considerably due to feedback from downstream targets.
Mechanisms regulating spatio-temporal expression of Hox genes are conserved across different animals. Nevertheless, there is sufficient diversity in developmental pathways, which are regulated by a Hox protein in different animals. Indeed, we did not observe one common motif or mechanism that explains the regulation of all direct targets of Ubx. Although Ubx heptamers15 are present in large number of its targets, they do not appear to help Ubx identify its targets as they are equally distributed in the entire non-coding genome of Drosophila. Absence of consensus mechanism in target selection and regulation may be the reason for a given Hox protein to generate enormous diversity in body plan from a common ground plan.
There is a possibility that other transcription factor pre-existing in the same milieu as Ubx may help Ubx select and regulate its targets. Preliminary data presented here raises the possibility that GAF might function as a co-factor of Ubx to help identify its targets and/or regulate their expression. Additional experiments such as Immuno-precipitation, Electro mobility shift assays (EMSA) and transgenic analysis are underway to understand precise mechanism by which they together regulate downstream genes. Currently, we are also analyzing genome wide association between Ubx and GAF by ChIP-sequencing using anti-Ubx and anti-GAF antibodies on wild type wing and haltere and CbxHm/+ wing discs. Preliminary ChIP-seq data suggest that GAF and Ubx may share more than 100 targets during haltere development (PA unpublished results).
Thus, it is probable that specificity of target selection in vivo by Ubx is achieved with the help of other factors, which might serve as a docking platform which is recognized by Hox proteins specifically in a tissue and context specific manner. This hypothesis is consistent with the thumb-rule that Hox proteins modify pre-existing ground plans e.g., Ubx modifies wing fate in meta-thoracic segment to haltere fate. Thus, Hox proteins can be thought of as versatile modifying factors which can associate with pre-existing platforms of transcription factors in different combinations and modify the fate of a cell.
Note: Two recent publications in PLoS One51,52 report similar approaches to identifying direct targets of Ubx during haltere development. In both the publications, ChIP-chip (test vs input DNA using Affymetrix platform as against test vs mock using Agilent platform reported here) method has been used to identify targets of Ubx. Slattery et al. (2011)51 have carried out ChIP-chip using antibodies against full-length Ubx (including conserved homeodomain) on wildtype haltere discs, while Choo et al. (2011)52 have used antibodies against YFP on haltere discs of Ubx::YFP transgenic flies. While no functional validation is included in those publications, they both indicate that (a) targets of Ubx include large number of wing-patterning genes, several signaling molecules and transcription factors (b) chromatin accessibility appears to be key for Ubx to bind to its targets. Our work used a modified approach wherein antibodies against N-terminus fragment of Ubx are used on CbxHm/+ wing discs. These antibodies are specific to endogenous Ubx and not cross-react with any other homeodomain-containing proteins. Nevertheless, all three reports have certain common observations particularly on the identification of large number of wing patterning genes as targets of Ubx. Functional validation and analysis presented here go a step further in reporting complex regulatory loop leading to canalization of molecular mechanisms help in specifying haltere fate.
Methods
Fly strains and Genetics
Canton-S was used as wild type strain. The flies were reared at 25°C unless mentioned otherwise. Clones were generated by FLP/FRT techniques53. Ubx clones were generated54 using P[FRT] 82BUbx1 as described20 and hth clones using FRT82BhthP2 as described28. The GAL4/UAS system55 was used to induce ectopic function of different gene products. The GAL4 drivers employed in this study were: vg-GAL456 and MS1096-GAL457. Following UAS stocks were used: UAS-Hth30; UAS-hthRNAi (VDRC#12763 and 12764 NIG# 17117R), UAS-exdRNAi (VDRC# 7802)58 and UAS-Meis129.
Immunohistochemistry
Immunohistochemical staining was performed as described59. Primary antibodies used were rabbit anti-Hth 1∶500 (kind gift from Adi Salzberg), guinea pig anti-Hth 1∶200 (kind gift from Natalia Azpiazu); rabbit anti-Ubx (in this study) 1∶1000; Monoclonal anti-Ubx54 1∶30. Secondary antibodies used were AlexaFluor488 (1∶1000); AlexaFluor568 (1∶1000); AlexaFluor594 (1∶1000); all from Molecular Probes, USA. Images were captured using either Leica confocal (TCS SP5 AOBS) or Zeiss confocal (LSM510); processed using manufacturer's software and adjusted for contrast and brightness using Adobe Photoshop.
Generation of polyclonal antibodies against truncated Ubx
To avoid cross reactivity, homeodomain was removed by amplification of truncated N-terminal region of Ubx by PCR using primer sequences given in Supplementary Text. Rabbit anti-Ubx serum was raised against the bacterially produced protein as described in the Supplementary Text. Specificity of this serum was confirmed by probing against lysates from different tissues with or without Ubx by Western blot analysis and on wild type wing, haltere and CbxHm/+ wing discs by immuno-histochemistry (Fig. 1 and Supplementary Fig. 1). We could detect specific Ubx signal and no cross reactivity was observed when probed on PVDF membrane spotted with pure abdominal-A (Abd-A) protein (data not shown).
Chromatin immuno precipitation (ChIP)
The ChIP protocol used is modified from Upstate Ez ChIP kit (Catalog # 17-371). We used CbxHm/+ wing discs, which ectopically express Ubx in the pouch region to focus on suppression of wing fate by Ubx. For each pull down ∼120 CbxHm/+ wing discs were used. Discs were dissected from wandering third instar larvae and fixed in 1% formaldehyde solution for 20 minutes at room temperature. After washing 2 times in cold PBS, the discs were incubated in protease inhibitors and nuclear lysis buffer for 20 minutes and subjected to sonication in Diagenode bioruptor (UCD-200), to obtain average 500 bp size chromatin. These chromatin fragments were immuno-precipitated with 1∶500 rabbit anti-Ubx (this study) or rabbit anti-GAF (described in46). An equal amount of chromatin without antibody was used as mock IP control. All the pull down reactions were carried out in 300 µl volume.
ChIP-Western
Pulled down immune complex from ChIP was washed twice with low salt binding buffer (Upstate, Catalog # 17-371) and once with TE; protein A beads with immune complex were boiled for 5 min with protein loading buffer with BME, reverse cross linked at 65°C for 5 hours, loaded on 12% SDS-PAGE gel and processed for Western Blot Hybridization using standard protocol as described in Supplementary Text. Quantitative estimation was carried out by densitometric analysis using Image J software to look for enrichment of GAF between anti-Ubx and anti-IgG lanes. We used antibody Heavy chain bands to normalize the enrichment between anti-Ubx and anti-IgG lanes.
Hybridization to array, data processing and analysis
Agilent Drosophila whole-genome array, 2004 build with ∼488,000 probes (each ∼58 bp long and with average 233 bp spacing) were used for three biological replicates of ChIP-chip. For the first replicate, we processed DNA pulled down from a single ChIP experiment, whereas for the remaining two replicates we pooled DNA obtained from 3–4 different ChIP carried out in parallel. This was done to account for experimental variability. LM-PCR and Hybridization to array was performed as per the manufacturer's protocol (Agilent Technologies Inc, CA, USA). Standard analysis algorithms were used for feature extraction and data processing. The updated Agilent design file (build April 2006 dm3) was used to extract the data. Raw data is available on http://www.ncbi.nlm.nih.gov/geo/ with accession number GSE28778. Further details for data processing and sequence analysis can be found in Supplementary Text.
Identification of Transcription Factor binding sites using MATCH and TRANSFAC
Transcription factor binding sites were identified using the MATCH36 program for the motifs listed in TRANSFAC database37 Release 2010.2 (C) Biobase GmbH. Scripts written for sequence analyses are available on request.
Author Contributions
Author contribution PA and LSS designed the experiments. RY raised anti-Ubx antibodies, PA performed the experiments. FH did the Bioinformatics analysis. PA, FH and LSS wrote the paper.
Supplementary Material
Supplementary text and figures
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5
Acknowledgments
The Authors thank R Mishra, R White, Y Graba, N Azpiazu, A Salzberg, M Sato, J Bernues, Bloomington, VDRC and NIG Stock Centers for antibodies and fly stocks. Members of LSS and R Mishra lab for help with experiments and discussion. N Rangarajan and A Mehla for help with confocal microscopy; R Palparthi for help with data analysis; E Sanchez-Herrero and R Mishra for helpful suggestions and critical reading of the manuscript. We thank Council of Scientific and Industrial Research, India for a fellowship to PA and Department of Atomic Energy and Department of Science and Technology (Government of India) for research grants to LSS.
References
- Pearson J. C., Lemons D. & McGinnis W. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 6, 893–904 (2005). [DOI] [PubMed] [Google Scholar]
- Mcginnis W., Krumiauft R. & Hill M. Homeobox genes and axial patterning. Cell 66, 283–302 (1992). [DOI] [PubMed] [Google Scholar]
- Lewis E. B. A gene complex controlling segmentation in Drosophila. Nature 276, 565–570 (1978). [DOI] [PubMed] [Google Scholar]
- Cabrera C. V., Botas J. & Garcia-Bellido A. Distribution of proteins in mutants of bithorax complex genes and its transregulatory genes. Nature 318, 569–571 (1985). [Google Scholar]
- White R. A. H. & Akam M. E. Contrabithorax mutations cause inappropriate expression of Ultrabithorax products in Drosophila. Nature 318, 567–569 (1985). [Google Scholar]
- Hueber S. D. & Lohmann I. Shaping segments: Hox gene function in the genomic age. BioEssays 965–979 (2008). [DOI] [PubMed] [Google Scholar]
- Mohit P., Bajpai R. & Shashidhara L. S. Regulation of Wingless and Vestigial expression in wing and haltere discs of Drosophila. Development 1537–1547 (2003). [DOI] [PubMed] [Google Scholar]
- Crickmore M. A. & Mann R. S. Hox control of organ size by regulation of morphogen production and mobility. Science 313, 63–68 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Navas L. F., Garaulet D. L. & Sánchez-Herrero E. The ultrabithorax Hox gene of Drosophila controls haltere size by regulating the Dpp pathway. Development 133, 4495–4506 (2006). [DOI] [PubMed] [Google Scholar]
- Makhijani K., Kalyani C., Srividya T. & Shashidhara L. S. Modulation of Decapentaplegic gradient during haltere specification in Drosophila. Dev. Biol. 302, 243–255 (2007). [DOI] [PubMed] [Google Scholar]
- Pallavi S. K., Kannan R. & Shashidhara L. S. Negative regulation of Egfr/Ras pathway by Ultrabithorax during haltere development in Drosophila. Dev. Biol. 296, 340–352 (2006). [DOI] [PubMed] [Google Scholar]
- Hersh B. M. & Carroll S. B. Direct regulation of knot gene expression by Ultrabithorax and the evolution of cis-regulatory elements in Drosophila. Development 132, 1567–1577 (2005). [DOI] [PubMed] [Google Scholar]
- Mohit P. et al. Modulation of AP and DV signaling pathways by the homeotic gene Ultrabithorax during haltere development in Drosophila. Dev. Biol. 291, 356–367 (2006). [DOI] [PubMed] [Google Scholar]
- Hersh B. M. et al. The UBX-regulated network in the haltere imaginal disc of D. melanogaster. Dev. Biol. 302, 717–727 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker S. C., Young K. E., Kessler D. P. V. & Beachy P. A. Optimal DNA sequence recognition by the Ultrabithorax homeodomain of Drosophila. EMBO J. 10, 1179–1186 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker S. C. et al. The degree of variation in DNA sequence recognition among four Drosophila homeotic proteins. EMBO J. 13, 3551–3560 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M. F. et al. Variation in Homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133, 1266–1276 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes M. B. et al. Analysis of Homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133, 1277–1289 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherbee S. D., Halder G., Kim J., Hudson A. & Carroll S. B. Ultrabithorax regulates genes at several levels of the wing-patterning hierarchy to shape the development of the Drosophila haltere. Genes Dev. 12, 1474–1482 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashidhara L. S., Agrawal N., Bajpai R., Bharathi V. & Sinha P. Negative regulation of dorsoventral signaling by the homeotic gene Ultrabithorax during haltere development in Drosophila. Dev. Biol. 212, 491–502 (1999). [DOI] [PubMed] [Google Scholar]
- García-Bellido A. Genetic control of wing disc development in Drosophila. Ciba Found Symp. 29, 161–182 (1975). [DOI] [PubMed] [Google Scholar]
- Maere S., Heymans K. and Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in biological networks. Bioinformatics 21, 3448–3449 (2005). [DOI] [PubMed] [Google Scholar]
- Maeda R. K. & Karch F. The ABC of the BX-C: the bithorax complex explained. Development 133, 1413–1422 (2006). [DOI] [PubMed] [Google Scholar]
- Mann R. S. & Markus A. Hox proteins meet more partners. Curr. Opin. Genetics Dev. 8, 423–429 (1998). [DOI] [PubMed] [Google Scholar]
- Pavlopoulos A. & Akam M. Hox gene Ultrabithorax regulates distinct sets of target genes at successive stages of Drosophila haltere morphogenesis. Proc. Natl. Acad. Sci. USA 108, 2855–2860 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casares F. & Mann R. S. A dual role for homothorax in inhibiting wing blade development and specifying proximal wing identities in Drosophila. Development 127, 1499–1508 (2000). [DOI] [PubMed] [Google Scholar]
- Azpiazu N. & Morata G. Function and regulation of homothorax in the wing imaginal disc of Drosophila. Development 127, 2685–2693 (2000). [DOI] [PubMed] [Google Scholar]
- Aldaz S., Morata G. & Azpiazu N. Patterning function of homothorax / extradenticle in the thorax of Drosophila. Development 132, 439–446 (2005). [DOI] [PubMed] [Google Scholar]
- Rieckhof G. E., Casares F., Ryoo H. D., Abu-shaar M. & Mann R. S. Nuclear translocation of extradenticle requires homothorax , which rncodes an extradenticle-related homeodomain protein. Cell 91, 171–183 (1997). [DOI] [PubMed] [Google Scholar]
- Pai C. Y., et al. C. The Homothorax homeoprotein activates the nuclear localization of another homeoprotein, Extradenticle and suppresses eye development in Drosophila. Genes Dev. 12, 435–446 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann R. S. & Chan S. K. Extra specificity from extradenticle: the partnership between HOX and PBX/EXD homeodomain proteins. Trends Genet. 12, 258–262 (1996). [DOI] [PubMed] [Google Scholar]
- Jaffe L., Capovilla M., Botas J. & Mann R. S. The DNA binding specificity of Ultrabithorax is modulated by cooperative interactions with extradenticle , another homeoprotein. Cell 76, 603–615 (1994). [DOI] [PubMed] [Google Scholar]
- Azpiazu N. & Morata G. Functional and regulatory interactions between Hox and extradenticle genes. Genes Dev. 12, 261–273 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M. et al. Engrailed cooperates with extradenticle and homothorax to repress target genes in Drosophila. Development 130, 741–751 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith M. C. et al. Detection of functional DNA motifs via statistical over-representation. Nucl. Acids Res. 32, 1372–81 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kel A. E. et al. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucl. Acids Res. 31, 3576–9 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucl. Acids Res. 24, D108–D110 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann R. S., Lelli K. M. & Joshi R. Hox Specificity: Unique Roles for Cofactors and Collaborators. Curr. Topics Dev. Biol. 88, 63–101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. L. et al. MEME Suite: tools for motif discovery and searching. Nucl. Acids Res. 37, W202–W208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringrose L., Rehmsmeier M., Dura & Paro R. Genome-wide prediction of Polycomb / Trithorax response elements in Drosophila melanogaster. Dev. Cell 5, 759–771 (2003). [DOI] [PubMed] [Google Scholar]
- Schwartz Y. B. et al. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat. Genet. 38, 700–705 (2006). [DOI] [PubMed] [Google Scholar]
- Walsh C. M. & Carroll S. B. Collaboration between Smads and a Hox protein in target gene repression. Development 134, 3585–3592 (2007). [DOI] [PubMed] [Google Scholar]
- Farkas et al. The Trithorax-like gene encodes the Drosophila GAGA factor. Nature 371, 806–808 (1994). [DOI] [PubMed] [Google Scholar]
- Bejarano F. & Busturia A. Function of the Trithorax-like gene during Drosophila development. Dev. Biol. 268, 327–341 (2004). [DOI] [PubMed] [Google Scholar]
- Bhat K. M. et al. The GAGA factor is required in the early Drosophila embryo not only for transcriptional regulation but also for nuclear division. Development 122, 1113–1124 (1996). [DOI] [PubMed] [Google Scholar]
- Mishra K., Chopra V. S., Srinivasan A. & Mishra R. K. Trl-GAGA directly interacts with lola like and both are part of the repressive complex of Polycomb group of genes. Mech. Dev. 120, 681–689 (2003). [DOI] [PubMed] [Google Scholar]
- Berger C., Pallavi S. K., Prasad M., Shashidhara L. S. & Technau G. M. A critical role for Cyclin E in cell fate determination in the central nervous system of Drosophila melanogaster. Nat. Cell Biol. 7, 56–62 (2005). [DOI] [PubMed] [Google Scholar]
- Kannan R., Berger C., Myneni S., Technau G. M. & Shashidhara L. S. Abdominal-A mediated repression of Cyclin E expression during cell-fate specification in the Drosophila central nervous system. Mech. Dev. 127, 137–145 (2010). [DOI] [PubMed] [Google Scholar]
- Hueber S. D. et al. Comparative analysis of Hox downstream genes in Drosophila. Development 134, 381–392 (2007). [DOI] [PubMed] [Google Scholar]
- Erwin D. H. & Davidson E. H. The evolution of hierarchical gene regulatory networks. Nat. Rev. Genet. 10, 141–148 (2009). [DOI] [PubMed] [Google Scholar]
- Slattery M., Ma L., Négre N., White K. P. & Mann R. S. Genome-wide tissue-specific occupancy of the Hox protein Ultrabithorax and Hox cofactor Homothorax in Drosophila. Plos One 6, e14686 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo S. W., White R. & Russell S. Genome-wide analysis of the binding of the Hox protein Ultrabithorax and the Hox cofactor Homothorax in Drosophila. Plos One 6, e14778 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T. & Rubin G. M. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223–1237 (1993). [DOI] [PubMed] [Google Scholar]
- White R. A. H. & Wilcox M. Protein products of the Bithorax Complex in Drosophila. Cell 39, 163–171 (1984). [DOI] [PubMed] [Google Scholar]
- Brand A. H., & Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415 (1993). [DOI] [PubMed] [Google Scholar]
- Simmonds A. J., Brook W. J., Cohen S. M. & Bell J. B. Distinguishable functions for engrailed and invected in anterior-posterior patterning in the Drosophila wing. Nature 376, 424–427 (1995). [DOI] [PubMed] [Google Scholar]
- Guillen I. et al. The function of engrailed and the specification of Drosophila wing pattern. Development 121, 3447–3456 (1995). [DOI] [PubMed] [Google Scholar]
- Dietzl G. et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156 (2007). [DOI] [PubMed] [Google Scholar]
- Patel N. H., et al. Expression of Engrailed proteins in arthropods, annelids and chordates. Cell 58, 955–968 (1989). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary text and figures
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5