

Abstract
Bile acids have been shown to be important regulatory molecules for cells in the liver and gastrointestinal tract. They can activate various cell signaling pathways including the extracellular regulated kinase (ERK)1/2 and AKT as well as the G-protein coupled receptor (GPCR), TGR5/M-BAR. Activation of the ERK1/2 and AKT signaling pathways by conjugated bile acids has been reported to be pertussis toxin (PTX) and dominant negative Gαi sensitive in primary rodent hepatocytes. However, the GPCRs responsible for activation of these pathways have not been identified. Screening GPCRs in the lipid activated phylogenetic family, expressed in HEK293 cells, identified sphingosine 1-phosphate receptor 2 (S1P2) as being activated by taurocholate (TCA). TCA, taurodeoxycholic acid (TDCA), tauroursodeoxycholic acid (TUDCA), glycocholic acid (GCA), glycodeoxycholic acid (GDCA), and S1P-induced activation of ERK1/2 and AKT were significantly inhibited by JTE-013, a S1P2 antagonist, in primary rat hepatocytes. JTE-013 significantly inhibited hepatic ERK1/2 and AKT activation as well as short heterodimeric partner (SHP) mRNA induction by TCA in the chronic bile fistula rat. Knock down of the expression of S1P2 by a recombinant lentivirus encoding S1P2 shRNA, markedly inhibited the activation of ERK1/2 and AKT by TCA and S1P in rat primary hepatocytes. Primary hepatocytes prepared from S1P2 knock out (S1P2−/−) mice were significantly blunted in the activation of the ERK1/2 and AKT pathways by TCA. Structural modeling of the S1P receptors indicated that only S1P2 can accommodate TCA binding. In summary, all these data support the hypothesis that conjugated bile acids activate the ERK1/2 and AKT signaling pathways primarily via S1P2 in primary rodent hepatocytes.
Keywords: G protein coupled receptor, homology modeling, S1P receptor 2 knockout mice, cell signaling
Introduction
Over the past decade it has become clear that bile acids are important regulatory molecules in the liver and gastrointestinal tract and function much like hormones. Bile acids have been shown to activate specific nuclear receptors [farnesoid × receptor (FXR), pregnane × receptor (PXR), and Vitamin D receptor] and cell signaling pathways [i.e., c-jun N-terminal kinase1/2 (JNK1/2), ERK1/2, AKT] at physiological concentrations (1–6). In 2002, a Gαs protein coupled receptor, TGR5/M-BAR, was reported to be activated by both conjugated and unconjugated bile acids (7, 8). Subsequently, TGR5/M-BAR was shown to be involved in regulating energy expenditure by inducing the c-AMP-dependent thyroid hormone activating enzyme type 2 iodothyroxine deiodinase (D2). This enzyme converts metabolically inactive thyroxine (T4) into T3, a key hormone regulating energy metabolism in brown adipose tissue and muscle (9). TGR5/M-BAR also appears to play an important role in immune cells as bile acids are known to have immunoregulatory properties (10). TGR5/M-BAR is expressed in many cell types throughout the body including: neurons, astrocytes, cholangiocytes, macrophages, myocytes, and gallbladder epithelium (8, 11). This receptor may play a protective role in hepatic sinusoidal endothelial cells in the liver (12). However, the expression of TGR5/M-BAR in primary hepatocytes is very low.
Our previous studies indicate that conjugated bile acids activated the ERK1/2 and AKT signaling pathways via unidentified Gαi protein coupled receptor(s) in primary rodent hepatocytes and in vivo (13, 14). Unconjugated bile acids can also activate the ERK1/2 and AKT pathways by at least two different mechanisms. We have previously reported evidence that deoxycholic acid (DCA) can activate the ERK1/2 and AKT pathways by stimulating the synthesis of superoxide ions, which was shown to inactivate phosphotyrosine phosphatase(s) resulting in the activation of the epidermal growth factor receptor (EGFR) (15). In addition, other laboratories have reported that DCA, chenodeoxycholic acid (CDCA) and taurochenodeoxycholic acid (TCDCA) can activate matrix metalloproteinase(s) that generate transforming growth factor β (TGFβ), an EGFR ligand in cholangiocytes (16). Moreover, Raufman and co-workers reported that taurolithocholic acid (TLCA) and TDCA can activate the Gαs-coupled M3 muscarinic receptor in gastric chief cells as well as human colon cancer cells (17–19). The activation of the EGFR in colon cancer cells was by stimulation of matrix metalloproteinase gene expression resulting in the formation of heparin-binding EGF-like growth factor, also an EGFR ligand (20).
Sphingosine 1-phosphate (S1P) is a membrane-derived lipid mediator involved in the regulation of fundamental cellular responses. S1P is synthesized from sphingosine by either sphingosine kinase 1 (SphK1) or sphingosine kinase 2 (SphK2). SphK1 is located in the cytoplasm of mammalian cells and, following an external signal, translocates to the plasma membrane and converts sphingosine to S1P. S1P, a water soluble regulatory metabolite, is then actively transported by ATP-binding cassette transporter (ABC) C1 (ABCC1), and possibly ABCG2, in a regulated manner (21, 22). Exogenous S1P has been shown to activate, in an autocrine/paracrine manner, at least 5 different GPCRs located on the surface of mammalian cells. The GPCRs activated by S1P have been linked to the activation of various cell signaling pathways, including ERK1/2 and AKT. SphK2 is primarily located in the nucleus and is activated by phosphorylation by pERK1/2 to produce S1P, a powerful inhibitor of histone deacetylase 1 and 2 (23).
S1P2-mediated activation of ERK1/2 and AKT signaling cascades is linked to the regulation of gene expression, growth and differentiation in many cell types (21). In addition, the ERK1/2 signaling cascade has also been reported to be involved in the phosphorylation and stabilization of the small heterodimeric partner (SHP) (24). SHP is a nuclear receptor without a DNA binding domain that is induced by bile acids via a FXR element in the SHP promoter. SHP has been reported to play an important role in regulating metabolic pathways in the liver (25). Moreover, activation of the AKT pathway by TCA is linked to the regulation of glycogen synthase activation and up-regulation of FXR functional activity (14, 26).
In the current study, we report that conjugated, but not unconjugated, bile acids, can specifically activate the ERK1/2 and AKT signaling cascades through S1P2 in primary rodent hepatocytes.
Materials and Methods
Materials
Anti-actin antibody, JTE-013 and S1P were purchased from Cayman Chemicals (Ann Arbor, MI). All other antibodies were from Santa Cruz (Santa Cruz, CA). Pertussis toxin was from Calbiochem (San Diego, CA). Taurocholate (TCA) taurodeoxycholic acid (TDCA), glycodeoxycholic acid (GDCA), glycocholic acid (GCA), and tauroursodeoxycholic acid (TUDCA) and other chemicals were purchased from Sigma (St. Louis, MO). The 3×HA-tagged cDNAs of LPA1–3, CB1–2, and S1P3–5 were obtained from Guthrie cDNA Resource Center. GFP-tagged S1P1 and S1P2 were generated in Dr. Spiegel’s lab.
Animals
Male Sprague Dawley rats, 100 to 150 gms, were purchased from Harlan Laboratories (Frederick, MD). S1P2 knockout mice (S1P2−/−) were a gift from Dr. R. Proia (NIDDK). They were housed in a reverse light cycle (12:12 hours) for two weeks before use. Mice were bred in pathogen-free conditions, normal lighting and wild type and knockout mice were from the same litters. All animals were fed normal rodent chow and water ad libitum. All procedures were approved by the VCU IACUC committee that is accredited by AAALAC.
Bile fistula rat
Biliary fistulas and intraduodenal cannulas were placed in male Sprague-Dawley rats under brief anesthesia as previously described (14, 26). After surgery, they were placed in individual metabolic cages with water and normal chow ad libitum. All animals received continuous infusion of glucose-electrolyte replacement solution. After 48 h of chronic biliary diversion, TCA was infused at a rate of 1.05 ml per 100 g rat per h and at a concentration of 36 µmoles per 100 g rat per h for 3h. JTE-013 was intraperitoneally injected 2 h before TCA infusion at a dose of 2 mg/kg (27). At the end of the experiment, 0.1g of liver was harvested to isolate RNA as described previously (14), and the rest of the liver was flash-frozen in liquid nitrogen in several pieces. One piece was used to make total cell lysates (see Western blot analysis below). Animal research was conducted in conformity with PHS policy and with approval of the Institutional Animal Care and Use Committee of McGuire Veterans Affairs Medical Center of Richmond.
Primary rat and mouse hepatocyte cultures
Primary rat and mouse hepatocyte monolayer cultures were prepared from male Sprague-Dawley rats or wild type and S1P2−/− mice by the collagenase-perfusion technique of Bissell and Guzelian as described previously (28). Cells were plated at 2 × 106 cells per collagen-coated 60-mm dish in serum-free Williams E medium containing penicillin, dexamethasone (0.1 µM), and thyroxine (1 µM). In some experiments, PTX (300 ng/ml) was added 4 hours after plating and allowed to incubate 16 hours before starting experiments. Most experiments were conducted after 24 hours of culture, but shRNA experiments incubated 40 hours to allow for lentivirus-mediated gene expression before treatment.
Western blot analysis
Total cell lysates were prepared as described previously (14). Fifty µg of protein were resolved on 10% Bis-Tris NuPage gels and transferred to Nitrocellulose membranes. Immunoblots were blocked 1 hour at RT with 5% non-fat milk in TBS buffer and then incubated with antibodies to p-AKT, p-ERK, total-AKT, total-ERK, or actin in 1% BSA or non-fat milk for 24 hours at 4°C. Immunoreactive bands were detected using horseradish peroxidase–conjugated secondary antibodies and the Western Lightning Chemiluminescence Reagent Plus (PerkinElmer). The densities of immunoblot bands were analyzed using Image J computer software (NIH).
Transfection of HEK293 cells
Two methods were used. An Amaxa Nucleofector (Lonza Group, Ltd., Switzerland) was used according to the instructions to produce transient transfections of human embryonic kidney cells (ATCC HEK 293 cells). Five µg of DNA were used per 1 × 106 cells in 100 µl of Nucleofector solution. Program Q-001 was used for the 293 cells. The stable cell line was produced using FuGENE reagent (Roche). Cells were plated on 6-well plates and transfected with 1 µg DNA (pcDNA3 with appropriate insert) using 4 µl of FuGENE reagent in 2 ml of DMEM + 10% FCS. After 48 h, G418 was added to select the stable clones. The expression of each GPCR was confirmed by immunofluorescence staining followed by confocal microscopy. For GFP-S1P1 and GFP-S1P2, cells were inspected for GFP fluorescence and sorted by flow cytometry. The stable clones of each GPCR and vector only were maintained in culture medium containing G418 (100 µg/ml).
Lentiviral shRNA for down-regulating S1P2
The lentiviral vectors containing the stem loop sequences of shRNA specifically targeting the rat S1P2 and scrambled control sequence were a gift from Dr. Murthy (Department of Physiology and Biophysics, Virginia Commonwealth University, Richmond, VA). The silencing efficiency of each shRNA was confirmed by Western blot analysis in HEK293 cell transfected myc-tagged rat S1P2 using myc antibody as previously reported (29). The sequences for control shRNA and S1P2-shRNA used in this study are the following: Control shRNA: TCCTAAGGTTAAGTCGCCCTCG; S1P2-shRNA: AGGAACAGCAAG TTCTACTCA. The recombinant lentiviruses were produced by transient transfection of HEK293FT cells (from Invitrogen) using FuGene (Roche). Briefly, HEK293FT cells were cultured in high glucose DMEM, supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin (100 U/ml), 0.1mM non-essential amino acids, and 500 µg/ml of G418. The sub-confluent cells in a 10-cm culture dish were co-transfected with lentiviral vector (5 µg), and the lentiviral packaging vectors pRSV-REV (1.25 µg), pMDLg/pRRE (2.5 µg), and the vesicular stomatitis virus G glycoprotein (VSVG) expression vector pMD2G (1.5 µg). The viruses were collected from the culture supernatants on day 3 post-transfection, passed through a 22 ga needle and centrifuged at 2,000 × rpm for 7 minutes. The supernatants were then centrifuged at 26,000 rpm for 90 minutes at 4°C and the pellet was resuspended in 50 µl PBS. Titers were determined by infecting 293FT cells in a 24-well plate with serial dilutions of concentrated lentivirus, and counting EGFP-expressing cells after 48 h under fluorescent microscopy. (Transfecting units per ml = number of cells × dilution × 100) Primary hepatocytes were then infected (m.o.i.=10) with the appropriate amount of lentivirus for 40 hours and then treated with TCA (100 µM) or S1P (100 nM) for 20 minutes. Whole cell lysates were used to determine phosphorylation of AKT and ERK1/2 by Western blots. RNA was isolated to determine the knockdown efficiency of S1P2 shRNA by real time RT-PCR.
Homology modeling of S1P2 structure
A model of S1P2 was developed by homology to a rhodopsin model (1u19 in Protein Data Bank), using the Schrodinger Suite 2009 program. The rhodopsin model was based on low resolution experimental data (2.20Å), which was more accurate than previous studies (30, 31). S1P2 sequences were aligned with rhodopsin using the Structure Prediction of Schrodinger Suite 2009 program. Minor manual realignments were made to remove gaps in the seven transmembrane regions. Then a three-dimensional model for S1P2 was generated using a model of rhodopsin as a template. S1P2 was optimized to a root mean square (RMS) gradient of 0.5 kcal/(mol Å), by using a conjugate gradient algorithm under an OPLS2005 force field.
Docking calculation
The Glide docking method in the Schrodinger Suite 2009 program was used to predict the binding mode and abilities between ligands and receptor. The ligand molecules including S1P and TCA were prepared under an OPLS2005 force field. The binding pocket of S1P2 was defined according to the auto-searched results of the software and the results compared with other co-crystalline GPCR structures with ligands in the PDB database. All other parameters for docking calculations were set up as the default of the software.
Statistical Analysis
All of the experiments were repeated at least three times and the results were expressed as mean ± SD. One-way ANOVA was employed to analyze the differences between sets of data using GraphPad Prism (GraphPad, San Diego, CA). A value of p <0.05 was considered statistically significant.
Results
Screening G protein coupled receptors for activation by conjugated bile acids
The only currently known and characterized bile acid-activated GPCRs are TGR5/M-BAR (7, 8) and some muscarinic receptors (17–19). TGR5/M-BAR is phylogenetically related to members of the lipid activated family of GPCRs that include: S1P receptors (S1P1–5), lysophosphatidic acid receptors (LPA1–3), cannabinoid receptors (CB1–2), and orphan receptors GPR 3/6/12 (32). Our initial approach for identifying GPCRs activated by conjugated bile acids was to screen members of the lipid activated family by overexpressing the specific GPCR in HEK293 cells. The expression of functional GPCR was confirmed by immunofluorescence histochemisty followed by confocal microscopy. TCA was then added to the culture medium of cells expressing the individual receptor and the effects on the activation of the ERK1/2 and AKT signaling pathways determined by Western blot analysis. If activation occurred, sensitivity to PTX was next determined. The data presented in Fig. 1 shows that S1P1 and S1P2 were successfully expressed in the HEK293 cells. It also shows that TCA significantly activated S1P2, but not S1P1 expressed in HEK293 cells. In addition, there was no significant activation by TCA of other S1P receptors (S1P3–5), LPA1–3, or CB1, 2 when expressed in HEK293 cells (data not shown).
Fig. 1. Activation by TCA of S1P2 expressed in HEK293 cells.

A. Representative images of the GFP-tagged S1P1 and S1P2 stably expressed in HEK293 cells. B. Effect of TCA on ERK1/2 activation. HEK293 cells expressing GFP-tagged S1P1 or S1P2 or GFP only were treated with TCA (5 or 50 µM) or vehicle control for 20 minutes at 37°C. The protein levels of phosphor-ERK1/2 and total-ERK1/2 were determined by Western blot analysis. *p<0.05 compared to vehicle control, n=3.
Saturation curves of S1P and TCA activation of ERK and AKT in primary rat hepatocytes
First, we tested the ability of the natural ligand of S1P2 (S1P) to activate ERK and AKT in primary rat hepatocytes. S1P showed a concentration-dependent increase of phosphor-AKT (p-AKT) and phosphor-ERK (p-ERK) at nM levels. The data presented in online supplemental Fig. 1 shows that both p-ERK and p-AKT levels were saturated at 50–100 nM S1P. We further determined whether TCA had a similar effect on ERK and AKT activation as S1P in primary rat hepatocytes. As shown in Fig. 2A, TCA also dose-dependently induced activation of ERK1/2 and AKT, but at µM levels. Both p-ERK1/2 and p-AKT levels were saturated at 50–100 µM TCA (Fig. 2B).
Fig. 2. Saturation curves of TCA-induced ERK1/2 and AKT activation in primary rat hepatocytes.

Primary rat hepatocytes were treated with different amounts of TCA (0, 5, 10, 25, 50 and 100 µM) for 30 minutes. The cells were harvested to prepare total cell lysates. The protein levels of phosphorylated ERK1/2 (p-ERK1/2) and AKT (p-AKT) were determined by Western blot analysis and normalized with total ERK1/2 (T-ERK1/2) and total AKT (T-AKT) as described in Materials and Methods. A. Representative images of Western blots of p-ERK1/2, T-ERK1/2, p-AKT, and T-AKT. B. Saturation curves of TCA-induced ERK1/2 and AKT activation. *p<0.05 compared to control, n=4.
JTE-013, an S1P2 antagonist, blocked the activation of the ERK1/2 pathway by TCA and S1P in primary rat hepatocytes
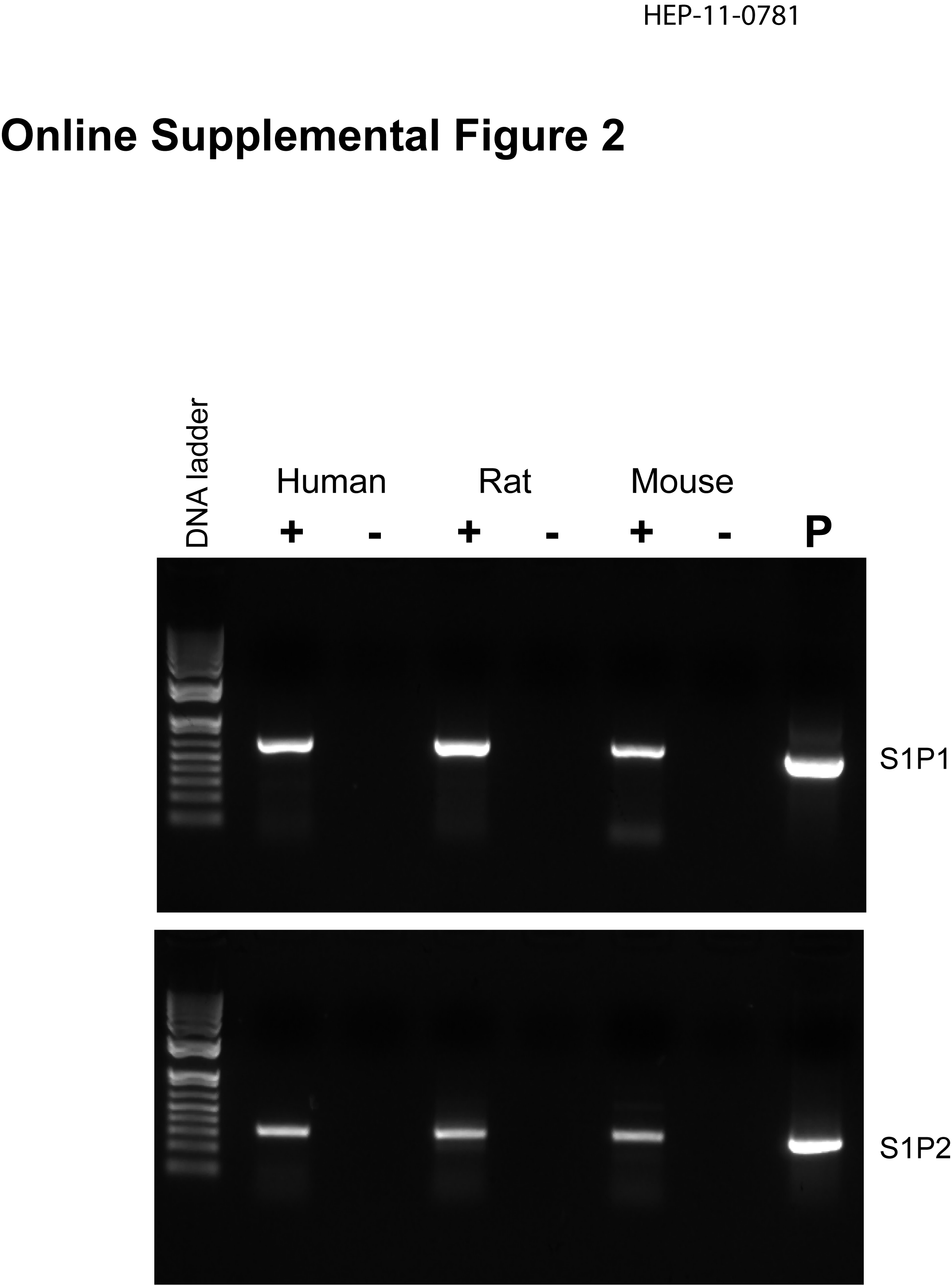
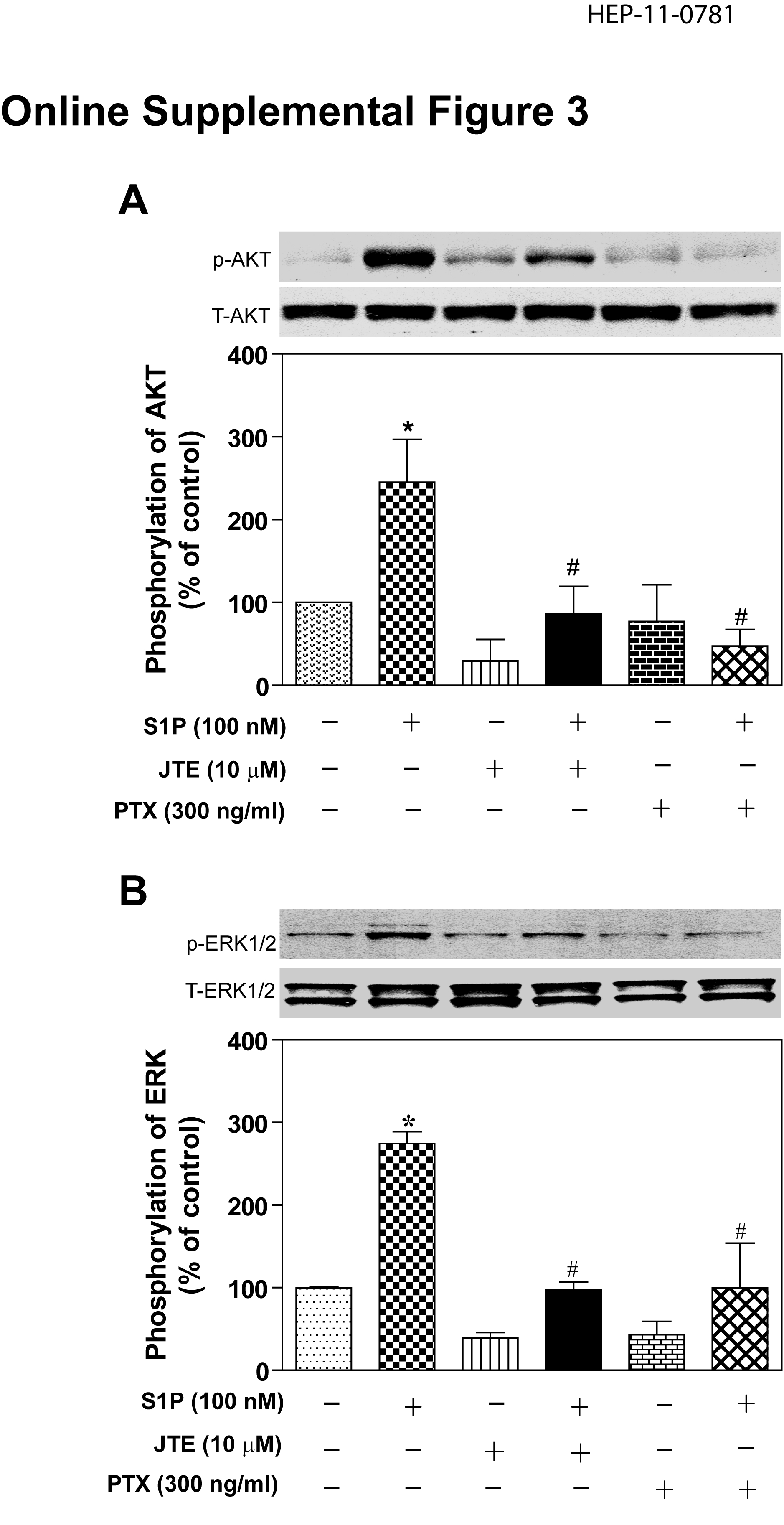
There have been several papers reporting that S1P receptors are expressed and functional in the liver (33, 34). The expression of S1P1 and S1P2 in mouse, rat and human primary hepatocytes was identified by RT-PCR (online supplemental Fig.2) and further confirmed by DNA sequencing. The addition of S1P (100 nM) to primary rat hepatocytes significantly activated both the ERK1/2 and AKT signaling pathways (online supplemental Fig.3). Moreover, JTE-013, a S1P2 chemical antagonist, as well as PTX blocked the activation of both ERK1/2 and AKT by S1P (online supplemental Fig.3). We have previously reported that TCA-induced ERK1/2 and AKT activation was PTX-sensitive in primary rat hepatocytes (13). In this study, we identified that TCA-induced ERK1/2 and AKT activation was also significantly blocked by JTE-013 (10 µM) in primary rat hepatocytes (Fig.3). We further examined whether other conjugated bile acids have similar effects on ERK1/2 and AKT activation. As shown in Fig. 4, in addition to TCA, TDCA, TUDCA, GCA, and GDCA also induced activation of ERK1/2 in primary rat hepatocytes and their effects were blocked by JTE-013 to different degrees. These results indicated that S1P2 may be a major GPCR in the activation of the ERK1/2 and AKT signaling pathways by conjugated bile acids and S1P in primary hepatocytes.
Fig. 3. Effect of JTE-013 on TCA-induced activation of ERK1/2 and AKT in primary rat hepatocytes.

Cells were pre-incubated with JTE-013 (10 µM) for 30 minutes and then treated with 100 µM TCA for 20 minutes at 37°C. The protein levels of phosphorylated ERK1/2 (p-ERK1/2) and AKT (p-AKT) were determined by Western blot analysis and normalized with total ERK1/2 (T-ERK1/2) and total AKT (T-AKT) as described in Materials and Methods. *p<0.05 compared to vehicle control, #p<0.05 compared to TCA group, n=3.
Fig. 4. Effect of JTE-013 on conjugated bile acid-induced activation of ERK1/2 and AKT in primary rat hepatocytes.

Cells were pre-incubated with JTE-013 (10 µM) for 30 minutes and then treated with individual conjugated bile acids (TCA, TDCA, TUDCA, GCA, GDCA, 50 µM) or S1P (100 nM) for 30 minutes at 37°C. The protein levels of phosphorylated ERK1/2 (p-ERK1/2) and AKT (p-AKT) were determined by Western blot analysis and normalized with total ERK1/2 (T-ERK1/2) and total AKT (T-AKT) as described in Materials and Methods. The representative Western blot images are shown. The relative density was determined by Image J software.
TCA/S1P-mediated ERK1/2 and AKT activation was markedly inhibited by S1P2 lentiviral shRNA in primary rat hepatocytes
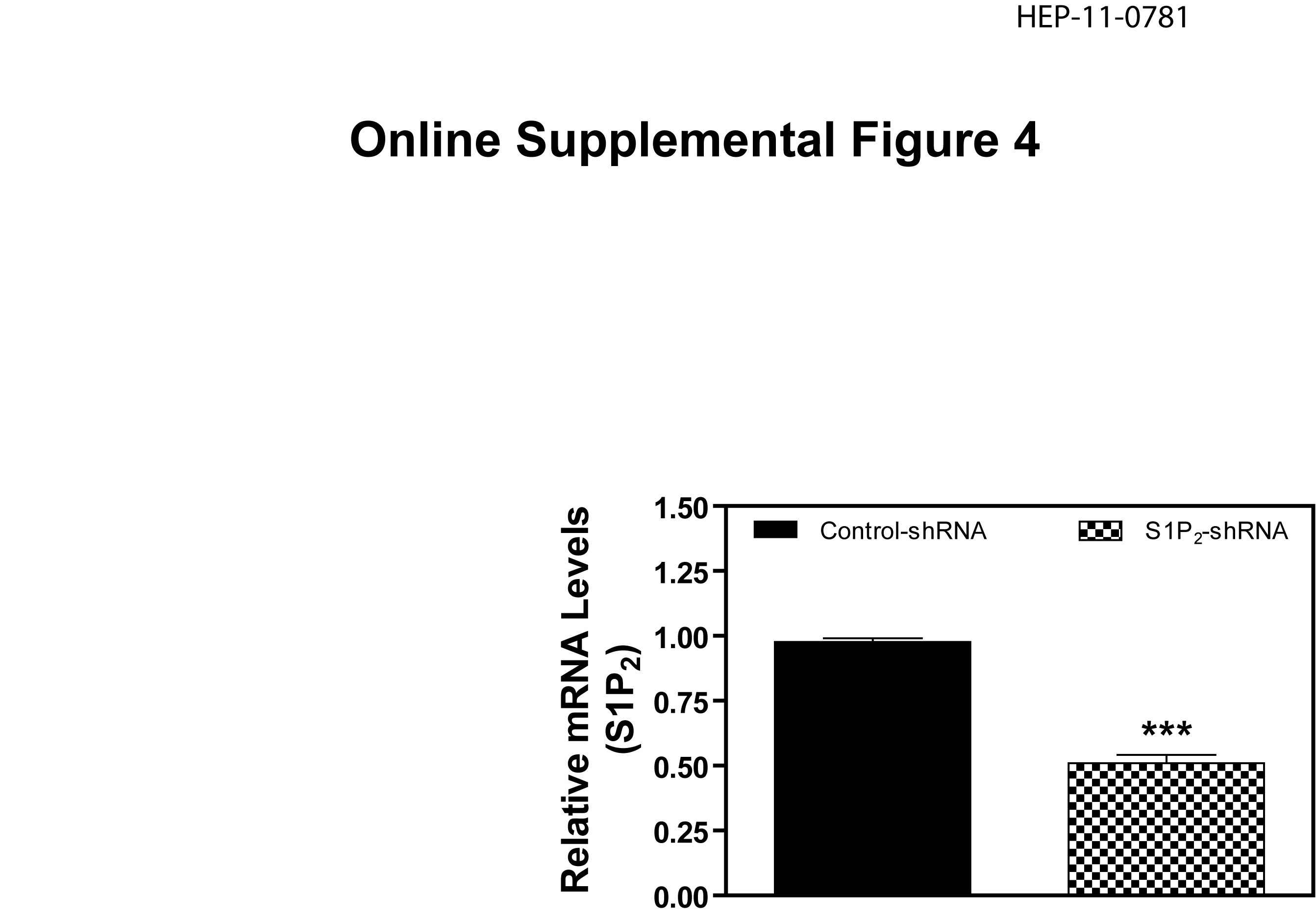
In order to further identify the role of S1P2 in TCA-mediated activation of ERK and AKT, lentiviral shRNA specifically targeting rat S1P2 was constructed. The knockdown efficiency was determined by real-time RT-PCR in primary rat hepatocytes. The mRNA level of S1P2 was reduced by 50% after 48-h transduction of lentiviral shRNA (online supplemental Fig.4). Transduction of primary rat hepatocytes with S1P2 shRNA lentivirus markedly inhibited the activation of ERK1/2 (60%) and AKT (70 %) by TCA as compared to a control lentivirus. Moreover, shRNA against S1P2 also inhibited S1P-mediated activation of ERK1/2 (51%) and AKT (72%) (Fig. 5)
Fig. 5. Effect of S1P2 shRNA on activation of pAKT and pERK by TCA and S1P.

Three hours after plating, rat primary hepatocytes were transduced with appropriate lentivirus encoding scramble control shRNA sequence or specific shRNA to S1P2 for 40 h and then treated with TCA (100 µM) or S1P (100 nM) for 20 minutes. The protein levels of phosphorylated ERK1/2 (p-ERK1/2) and AKT (p-AKT) were determined by Western blot analysis and normalized with actin as described in Materials and Methods. **p<0.01 compared to control shRNA group treated with TCA, n=3; #p<0.05 compared to control shRNA group treated with S1P, n=3.
Primary hepatocytes prepared from S1P2−/− mice are blunted in the activation of ERK1/2 and AKT by TCA
Primary hepatocytes were prepared from S1P2−/− mice and matched wild-type control mice. It was observed that male, but not female, S1P2−/− mice livers were difficult to infuse with collagenase solution and the hepatocyte cell yields from male mice livers were quite low. Therefore, all experiments were run with hepatocytes prepared from female S1P2−/− mice. The activation of the ERK1/2 and AKT pathways by TCA was significantly decreased by 30% and 44%, respectively, in hepatocytes prepared from S1P2−/− mice livers as compared to wild-type controls (Fig. 6). Similarly, S1P-induced ERK1/2 and AKT activation also was reduced by about 40% in the absence of S1P2 (Fig.6).
Fig. 6. Activation of the ERK1/2 and AKT pathways in primary mouse hepatocytes prepared from S1P2−/− and control mice.

Primary mouse hepatocytes were prepared from the S1P2−/− mouse and wild type littermate and treated with TCA (100 µM) for 20 minutes. The protein levels of phosphorylated ERK1/2 (p-ERK1/2) and AKT (p-AKT) were determined by Western blot analysis and normalized with actin as described in Materials and Methods. **p<0.01 compared to wild type treated with TCA, n=3; ##p<0.01 compared to wild type treated with S1P, n=3.
Effect of JTE-013 on TCA-induced SHP expression
Our recent study shows that TCA-meditated SHP induction was blocked by PTX in primary rat hepatocytes (26). In order to determine whether TCA-mediated activation of S1P2 is correlated to its effect on SHP-induction, we first examined the effect of JTE-013 on TCA-induced SHP expression in primary rat hepatocytes. As shown in Fig.7A, TCA rapidly induced SHP mRNA expression, which was significantly inhibited by JTE-013. We further examined the effect of JTE-013 on TCA-mediated ERK1/2 and AKT activation as well as SHP expression in the chronic bile fistula rat model. Rats were injected (ip, 2 mg/kg) with JTE-013 2 h before the perfusion of TCA. As shown in Fig.7B, TCA-mediated ERK1/2 and AKT activation was significantly inhibited by JTE-013. Furthermore, TCA-induced SHP mRNA expression was also markedly inhibited by JTE-013 (Fig. 7B).
Fig. 7. Effect of JTE-013 on TCA-induced SHP expression.

A. Primary rat hepatocytes were pre-incubated with JTE-013 (10 µM) for 30 minutes and then treated with TCA (50 µM) for 0.5, 1, 2, and 3 h at 37°C. The mRNA levels of SHP were determined by real-time RT-PCR as described in Materials and Methods. *p<0.05 compared to control group, n=3; #p<0.05 compared to TCA group, n=3. B. Bile fistula rats were i.p injected with JTE-013 (20 mg/kg) 2 h before TCA infusion. Rat livers were harvested after infusion with TCA for 3h. The protein levels of p-ERK1/2 and p-AKT were determined by Western blot analysis and normalized with actin and the mRNA levels of SHP were determined by real-time RT-PCR as described in Materials and Methods. Representative Western blot images are shown. ***p<0.001 compared to vehicle control with TCA, n=3; ###p<0.001 compared to TCA-treated group, n=3.
Homology modeling of the S1P2 docking to S1P or TCA
A model of the S1P2 was generated based on homology to rhodopsin as described in Materials and Methods. Docking calculations were used to predict binding sites and amino acid hydrogen bonding with S1P and taurocholate. The model shown in Fig. 8 predicts that S1P, a high affinity ligand, hydrogen bonds to three amino acid residues (Ser6, Leu173 and Glu177) of the S1P2. In contrast, TCA, a low affinity agonist, is predicted to hydrogen bond only to Leu 173. Efforts to model TCA into the putative binding pocket of other S1P receptors were unsuccessful.
Fig. 8. Modeling of S1P and TCA in the binding site of S1P2.

(A) Model S1P2 with bound S1P or TCA. (B) Specific amino acid residues interacting with bound S1P and TCA.
Discussion
We have previously reported that conjugated bile acids rapidly activate the ERK1/2 and AKT signaling pathways in a PTX sensitive manner in primary rat hepatocytes and in the chronic bile fistula rat (13, 14). Activation of the AKT pathway by TCA was shown to activate glycogen synthase activity in primary rat hepatocytes (14). Moreover, the addition of both insulin and TCA showed an additive effect on glycogen synthase activity in this system. Furthermore, TCA was shown to repress the gluconeogenic genes, phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6 phosphatase (G-6-Pase), in both primary hepatocytes and the chronic bile fistula rat (26). Repression of PEPCK and G-6-Pase mRNA by TCA was shown to be PTX sensitive in primary rat hepatocytes (26). In addition, both insulin and TCA had an additive effect on repressing glucose synthesis in primary rat hepatocytes (14). Finally, it was discovered that the activation of the AKT pathway was required for optimal induction of SHP mRNA, an FXR target gene, by TCA in primary rat hepatocytes (26). SHP has been reported to play an important role in the regulation of bile acid, glucose and lipid metabolism in the liver (25). It has been reported that the activation of the ERK1/2 pathway plays an important role in regulating the rate of turnover of SHP protein (24). These data all suggest that conjugated bile acids may be important regulators of hepatic glucose and lipid metabolism via activation of specific Gi protein coupled receptor(s) and FXR in a coordinated manner.
Bile acids are known to be involved in hepatocyte cell survival pathways. In this regard, hydrophobic bile acids have been reported to be cytotoxic and to induce apoptosis in hepatocytes (35). In contrast, hydrophilic bile acids such as ursodeoxycholic acid (UDCA) and conjugates have been reported to prevent apoptosis possibly via the activation of the AKT and ERK1/2 signaling pathways (36, 37). Conjugates of UDCA have been shown to activate the ERK1/2 and AKT signaling pathways in primary hepatocytes in culture (36, 37). The current data suggest that activation of these signaling pathways by TUDCA may be via the S1P2 in primary hepatocytes (Fig. 4). Other laboratories have reported that bile acids may increase the cellular cyclic-AMP which prevents apoptosis (38). The movement of ABC transporters in hepatocytes from intracellular locations to the canalicular membrane as well as their activity may also be partially controlled by the activation of PI3 kinase via the S1P2 (39–41). The data suggest that activation of S1P2 may have effects on survival pathways and movement of ABC transporters in primary hepatocytes.
Our current study strongly suggests that the S1P2 is the major GPCR activated by TCA and other conjugated bile acids in hepatocytes. This conclusion is based on the following lines of experimentation: 1) screening of individual GPCRs in the lipid activated phylogenetic family showed that only S1P2 was significantly activated by TCA in HEK293 cells (Fig. 1). These cells lack a bile acid transporter; hence, TCA, a very hydrophilic bile acid, must activate cell signaling pathways by binding to cell surface receptors. In addition, S1P2 mRNA is highly expressed in mouse, rat and human hepatocytes (Online supplemental Fig. 2). In contrast, TGR5/M-BAR mRNA level is very low compared to S1P2 in primary hepatocytes; 2) a chemical antagonist (JTE-013) of S1P2 markedly inhibited the activation of ERK1/2 and AKT signaling pathways in primary rat hepatocytes by TCA and S1P (Fig. 3); 3) JTE-013 significantly inhibited the activation of ERK1/2 and AKT as well as SHP mRNA induction by TCA both in primary rat hepatocytes and in the chronic bile fistula rat (Fig. 7); 4) knock down of S1P2 expression using specific lentiviral shRNA markedly inhibited the activation of ERK1/2 and AKT signaling pathways by conjugated bile acids in primary rat hepatocytes (Fig. 5); 5) S1P2−/− mouse hepatocytes were blunted in the activation of the ERK1/2 and AKT pathway by TCA as compared to control wild-type mouse hepatocytes (Fig. 6); and 6) homology modeling of S1P2 showed that it was the only S1P receptor that would accommodate binding to TCA (Fig. 8). All these data point to S1P2 as another GPCR activated by bile acids.
TCA rapidly activates the ERK1/2 and AKT signaling pathways in primary rat hepatocytes and following intestinal infusion into a biliary diverted rat (14, 26). In addition, TCA also is an excellent activator of FXR in primary rat hepatocytes and in vivo models (26). Our current model suggests that both the ERK1/2 and the AKT pathways are activated by TCA via S1P2. These data indicate that S1P2 may play an important role in the regulation of hepatic glucose, bile acid and lipid metabolism via coordinate activation of ERK1/2, AKT and FXR. In this regard, we have observed that S1P2−/− mice have fatty livers as compared to wild type control mice (unpublished).
In summary, TCA activated the S1P2 in rodent hepatocytes and in vivo causing activation of both the ERK1/2 and AKT pathways in primary hepatocytes. Activation of the AKT pathway appears to be essential for the optimal activation of the nuclear receptor, FXR, by conjugated bile acids (26). The coordinate activation of cell signaling pathways and FXR via S1P2 may help conjugated bile acids control hepatic glucose, bile acid, and lipid metabolism during the feed/fast cycle.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Financial Support: The work was supported by National Institutes of Health Grant R01 DK-057543
List of Abbreviations
- ERK
extracellular signal-regulated kinase
- AKT
protein kinase B
- GPCR
G protein coupled receptor
- TGR5/M-BAR
membrane type bile acid receptor
- PTX
pertussis toxin
- S1P2
sphingosine 1-phosphate receptor 2
- TCA
taurocholate
- TDCA
taurodeoxycholic acid
- TUDCA
tauroursodeoxycholic acid
- GCA
glycocholic acid
- GDCA
glycodeoxycholic acid
- SHP
short heterodimeric partner
- S1P2−/−
S1P2knock out
- FXR
farnesoid × receptor
- PXR
pregnane × receptor
- JNK
c-jun N-terminal kinase
- D2
iodothyroxine deiodinase
- DCA
deoxycholic acid
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- CDCA
chenodeoxycholic acid
- TCDCA
taurochenodeoxycholic acid
- TGFβ
transforming growth factor β
- TLCA
taurolithocholic acid
- S1P
sphingosine 1-phosphate
- SphK
sphingosine kinase
- ABC
ATP-binding cassette transporter
- GFP
green fluorescence protein
- PEPCK
phosphoenolpyruvate carboxykinase
- G-6-Pase
glucose-6-phosphatase
- LPA
lysophosphatidic acid receptor
- CB
cannabinoid receptor
- UDCA
ursodeoxycholic acid
References
- 1.Scotti E, Gilardi F, Godio C, Gers E, Krneta J, Mitro N, De Fabiani E, et al. Bile acids and their signaling pathways: eclectic regulators of diverse cellular functions. Cell Mol Life Sci. 2007;64:2477–2491. doi: 10.1007/s00018-007-7280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cell Signal. 2008;20:2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Edwards PA. FXR signaling in metabolic disease. FEBS Lett. 2008;582:10–18. doi: 10.1016/j.febslet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 6.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Itadani H, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 8.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 10.Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-Protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology. 2011 doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas C, Auwerx J, Schoonjans K. Bile acids and the membrane bile acid receptor TGR5--connecting nutrition and metabolism. Thyroid. 2008;18:167–174. doi: 10.1089/thy.2007.0255. [DOI] [PubMed] [Google Scholar]
- 12.Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, Selbach O, Haussinger D, et al. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. doi: 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]
- 13.Dent P, Fang Y, Gupta S, Studer E, Mitchell C, Spiegel S, Hylemon PB. Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology. 2005;42:1291–1299. doi: 10.1002/hep.20942. [DOI] [PubMed] [Google Scholar]
- 14.Fang Y, Studer E, Mitchell C, Grant S, Pandak WM, Hylemon PB, Dent P. Conjugated bile acids regulate hepatocyte glycogen synthase activity in vitro and in vivo via Galphai signaling. Mol Pharmacol. 2007;71:1122–1128. doi: 10.1124/mol.106.032060. [DOI] [PubMed] [Google Scholar]
- 15.Fang Y, Han SI, Mitchell C, Gupta S, Studer E, Grant S, Hylemon PB, et al. Bile acids induce mitochondrial ROS, which promote activation of receptor tyrosine kinases and signaling pathways in rat hepatocytes. Hepatology. 2004;40:961–971. doi: 10.1002/hep.20385. [DOI] [PubMed] [Google Scholar]
- 16.Werneburg NW, Yoon JH, Higuchi H, Gores GJ. Bile acids activate EGF receptor via a TGF-alpha-dependent mechanism in human cholangiocyte cell lines. Am J Physiol Gastrointest Liver Physiol. 2003;285:G31–G36. doi: 10.1152/ajpgi.00536.2002. [DOI] [PubMed] [Google Scholar]
- 17.Raufman JP, Zimniak P, Bartoszko-Malik A. Lithocholyltaurine interacts with cholinergic receptors on dispersed chief cells from guinea pig stomach. Am J Physiol. 1998;274:G997–G1004. doi: 10.1152/ajpgi.1998.274.6.G997. [DOI] [PubMed] [Google Scholar]
- 18.Raufman JP, Chen Y, Cheng K, Compadre C, Compadre L, Zimniak P. Selective interaction of bile acids with muscarinic receptors: a case of molecular mimicry. Eur J Pharmacol. 2002;457:77–84. doi: 10.1016/s0014-2999(02)02690-0. [DOI] [PubMed] [Google Scholar]
- 19.Raufman JP, Cheng K, Zimniak P. Activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig Dis Sci. 2003;48:1431–1444. doi: 10.1023/a:1024733500950. [DOI] [PubMed] [Google Scholar]
- 20.Cheng K, Xie G, Raufman JP. Matrix metalloproteinase-7-catalyzed release of HB-EGF mediates deoxycholyltaurine-induced proliferation of a human colon cancer cell line. Biochem Pharmacol. 2007;73:1001–1012. doi: 10.1016/j.bcp.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol. 2010;688:141–155. doi: 10.1007/978-1-4419-6741-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, Nagahashi M, Harikumar KB, et al. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem. 2010;285:10477–10486. doi: 10.1074/jbc.M109.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miao J, Xiao Z, Kanamaluru D, Min G, Yau PM, Veenstra TD, Ellis E, et al. Bile acid signaling pathways increase stability of Small Heterodimer Partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev. 2009;23:986–996. doi: 10.1101/gad.1773909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim MK, Chanda D, Lee IK, Choi HS, Park KG. Targeting orphan nuclear receptor SHP in the treatment of metabolic diseases. Expert Opin Ther Targets. 2010;14:453–466. doi: 10.1517/14728221003652463. [DOI] [PubMed] [Google Scholar]
- 26.Cao R, Cronk ZX, Zha W, Sun L, Wang X, Fang Y, Studer E, et al. Bile acids regulate hepatic gluconeogenic genes and farnesoid X receptor via G(alpha)i-protein-coupled receptors and the AKT pathway. J Lipid Res. 2010;51:2234–2244. doi: 10.1194/jlr.M004929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imasawa T, Koike K, Ishii I, Chun J, Yatomi Y. Blockade of sphingosine 1-phosphate receptor 2 signaling attenuates streptozotocin-induced apoptosis of pancreatic beta-cells. Biochem Biophys Res Commun. 2010;392:207–211. doi: 10.1016/j.bbrc.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bissell DM, Guzelian PS. Degradation of endogenous hepatic heme by pathways not yielding carbon monoxide. Studies in normal rat liver and in primary hepatocyte culture. J Clin Invest. 1980;65:1135–1140. doi: 10.1172/JCI109767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu W, Huang J, Mahavadi S, Li F, Murthy KS. Lentiviral siRNA silencing of sphingosine-1-phosphate receptors S1P1 and S1P2 in smooth muscle. Biochem Biophys Res Commun. 2006;343:1038–1044. doi: 10.1016/j.bbrc.2006.03.079. [DOI] [PubMed] [Google Scholar]
- 30.Parrill AL, Baker DL, Wang DA, Fischer DJ, Bautista DL, Van Brocklyn J, Spiegel S, et al. Structural features of EDG1 receptor-ligand complexes revealed by computational modeling and mutagenesis. Ann N Y Acad Sci. 2000;905:330–339. doi: 10.1111/j.1749-6632.2000.tb06573.x. [DOI] [PubMed] [Google Scholar]
- 31.Parrill AL, Wang D, Bautista DL, Van Brocklyn JR, Lorincz Z, Fischer DJ, Baker DL, et al. Identification of Edg1 receptor residues that recognize sphingosine 1-phosphate. J Biol Chem. 2000;275:39379–39384. doi: 10.1074/jbc.M007680200. [DOI] [PubMed] [Google Scholar]
- 32.Hama K, Aoki J. LPA(3), a unique G protein-coupled receptor for lysophosphatidic acid. Prog Lipid Res. 2010;49:335–342. doi: 10.1016/j.plipres.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Ikeda H, Watanabe N, Ishii I, Shimosawa T, Kume Y, Tomiya T, Inoue Y, et al. Sphingosine 1-phosphate regulates regeneration and fibrosis after liver injury via sphingosine 1-phosphate receptor 2. J Lipid Res. 2009;50:556–564. doi: 10.1194/jlr.M800496-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SW, Kim M, Chen SW, Brown KM, D'Agati VD, Lee HT. Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P1 receptor activation. Lab Invest. 2010;90:1209–1224. doi: 10.1038/labinvest.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amaral JD, Viana RJ, Ramalho RM, Steer CJ, Rodrigues CM. Bile acids: regulation of apoptosis by ursodeoxycholic acid. J Lipid Res. 2009;50:1721–1734. doi: 10.1194/jlr.R900011-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiao L, Yacoub A, Studer E, Gupta S, Pei XY, Grant S, Hylemon PB, et al. Inhibition of the MAPK and PI3K pathways enhances UDCA-induced apoptosis in primary rodent hepatocytes. Hepatology. 2002;35:779–789. doi: 10.1053/jhep.2002.32533. [DOI] [PubMed] [Google Scholar]
- 37.Schoemaker MH, Conde de la Rosa L, Buist-Homan M, Vrenken TE, Havinga R, Poelstra K, Haisma HJ, et al. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology. 2004;39:1563–1573. doi: 10.1002/hep.20246. [DOI] [PubMed] [Google Scholar]
- 38.Reinehr R, Haussinger D. Inhibition of bile salt-induced apoptosis by cyclic AMP involves serine/threonine phosphorylation of CD95. Gastroenterology. 2004;126:249–262. doi: 10.1053/j.gastro.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 39.Kipp H, Arias IM. Trafficking of canalicular ABC transporters in hepatocytes. Annu Rev Physiol. 2002;64:595–608. doi: 10.1146/annurev.physiol.64.081501.155793. [DOI] [PubMed] [Google Scholar]
- 40.Misra S, Ujházy P, Varticovski L, Arias IM. Phosphoinositide 3-kinase lipid products regulate ATP-dependent transport by sister of P-glycoprotein and multidrug resistance associated protein 2 in bile canalicular membrane vesicles. Proceedings of the National Academy of Sciences. 1999;96:5814–5819. doi: 10.1073/pnas.96.10.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu D, Wakabayashi Y, Lippincott-Schwartz J, Arias IM. Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proc Natl Acad Sci U S A. 2011;108:1403–1408. doi: 10.1073/pnas.1018376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.