

Abstract
The structural factors responsible for the extraordinary rate enhancement (~1017) of the reaction catalyzed by orotidine 5′-monophosphate decarboxylase (OMPDC) have not been defined. Catalysis requires a conformational change that closes an active site loop and “clamps” the orotate base proximal to hydrogen-bonded networks that destabilize the substrate and stabilize the intermediate. In the OMPDC from Methanobacter thermoautotrophicus, a “remote” structurally conserved cluster of hydrophobic residues that includes Val 182 in the active site loop is assembled in the closed, catalytically active conformation. Substitution of these residues with Ala decreases kcat/Km with a minimal effect on kcat, providing evidence that the cluster stabilizes the closed conformation. The intrinsic binding energies of the 5′-phosphate group of OMP for the mutant enzymes are similar to that for the wild type, supporting this conclusion.
Orotidine 5′-monophosphate decarboxylase (OMPDC1) is one of Nature’s best catalysts: the reaction occurs with a rate enhancement of ~1017 and a proficiency of ~1023 M (2). The reaction coordinate includes a vinyl carbanion intermediate (Scheme 1) (3, 4).
Scheme 1.
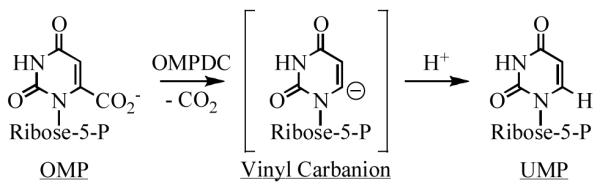
Structures of OMPDCs complexed with UMP or 6-hydroxyUMP, a n intermediate analog (5-9), reveal hydrogen-bonded networks proximal to 1) C6 of the pyrimidine (Asp 70-Lys 72-Asp 75 in the OMPDC from Methanobacter thermoautotrophicus; MtOMPDC); and 2) O2, N3, and O4 of the base (Ser 127-Gln 185). These destabilize the substrate (9) and stabilize the intermediate, although the structural strategy for the latter is uncertain.
The 5′-phosphate group binds in a conserved motif at the ends of the seventh and eighth β-strands of the (β/α)8-barrel structure, with interactions to backbone NH groups as well as the guanidinium group of a conserved Arg (Arg 203 in MtOMPDC). These interactions are important for catalysis: 1) kcat/Km for OMP exceeds that for orotidine by a factor of ~1011 for the OMPDC from Saccharomyces cerevisiae (ScOMPDC) (10); 2) kcat/Km for OMP exceeds that for 1-(β-D-erythrofuranosyl)orotic acid (EO; 5′-truncated OMP analog) by factors of 5.2 × 108 and 3.6 × 108 for ScOMPDC and MtOMPDC, respectively (11, 12); and 3 ) phosphite dianion ( H Pi) activates decarboxylation of EO by factors of 5.6 × 105 M−1 and 2.9 × 105 M−1 for ScOMPDC and MtOMPDC, respectively (11, 12). The “intrinsic binding energy” [IBE; (13)] of the 5′-phosphate/HPi 1) increases the affinity for the substrate (e.g., OMP vs. orotidine/EO); and 2) enables decarboxylation by juxtaposition of the substrate with the active site hydrogen-bonded networks (substrate destabilization and intermediate stabilization). How the IBE promotes catalysis is unknown but required to understand the structural basis for the rate enhancement.
A loop located at the end of the seventh β-strand closes over the active site when OMP binds (Figure 1). Although the active site loops differ in both length and sequence in divergent OMPDCs (12), each includes a spatially conserved Gln (Gln 185 in MtOMPDC) hydrogen-bonded to both the 5′-phosphate and O2 of the pyrimidine as well as a conserved Ser at the end of the fifth β-strand that also is hydrogen-bonded to N3 of the pyrimidine (Ser 127). We characterized the importance of these “clamp” residues in ScOMPDC (Gln 215-Ser 154) using EO/HPi and confirmed that the 5′-phosphate/HPi “switch” is required to activate the enzyme (14).
Figure 1.
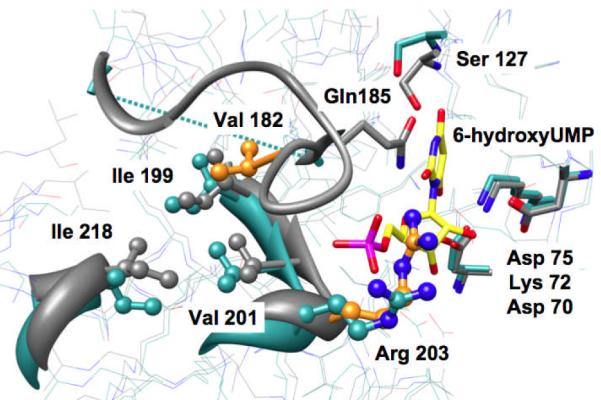
Superposition of the active site of wild type MtOMPDC in the absence (cyan; disordered loop depicted with the dotted line ) and presence ( grey ) of 6-hydroxyUMP. The carbons of 6-hydroxyUMP are highlighted in yellow; the carbons of Val 182 and Arg 203 in the liganded structure are highlighted in orange.
The structures of divergent OMPDCs reveal that OMP binding is always accompanied by a conformational change (Figure 1). The most obvious component is closure of the active site loop. However, the (β/α)8-barrel structure can be divided into two domains, one formed from the second, third, fourth, and fifth β-strands (where the hydrogen-bonded Asp 70-Lys 72-Asp 75 motif and Ser 127 are located) and the second from the sixth, seventh, eighth, and first β-strands (where the phosphate binding motif and the active site loop, including Gln 185, are located) (15). OMP binding reorients the domains, with the latter domain moving toward the former, enforcing the orotate carboxylate group to be juxtaposed vis a vis Asp 70- Lys 72-Asp 75 and, also, allowing formation of the Ser 127-Gln 185 “clamp”. Thus, the transition between the open and closed conformations is more complicated than “simple” hinge motion of the loop on the rigid framework of the (β/α)8-barrel structure. In this Report we identify “remote” residues involved in this conformational change and quantitate their importance in promoting and stabilizing the catalytically competent form of the enzyme.
The active site loop of MtOMPDC, P180-G181-V182-G183-A184-Q185-G186-G187-D188, is disordered in the absence of substrate but ordered and closed in its presence (Figure 1). In the liganded structure two residues in the loop make contacts with the (β/α)8-barrel scaffold: 1) Gln 185 is hydrogen-bonded to Ser 127 (vide infra); and 2) Val 182 is embedded in a hydrophobic cluster also formed by Ile 199, Val 201, and Ile 218. The conservation of this hydrophobic cluster in all OMPDCs suggests its importance in a common catalytic strategy. We probed this strategy by mutagenesis of these hydrophobic residues.
Ala substitutions for residues in the hydrophobic cluster cause substantial decreases in kcat/Km for decarboxylation of OMP but have little effect on kcat (Table 1). We determined high resolution X-ray structures (≤ 1.4 Å) for each mutant in the presence of 6-hydroxyUMP (Figure 2). The liganded structures superimpose well with that of wild type, with only small differences observed at the sites of the substitutions (panel A). The active sites are identical to that of wild type (panel B), explaining the minimal impact on kcat.
Table 1.
Kinetic Constants for OMP, EO and EO/HPi and Intrinsic Binding Energies of the 5′-Phosphate Group of OMP at pH 7.1 and 25 °C.
| MtOMPDC |
kcat OMP s−1 |
kcat/Km OMP M−1 s−1 |
ΔΔG‡ kcal/mola |
kcat/Km EO M−1 s−1 |
ΔΔG‡ kcal/mola |
(kcat/Km)/K D EO•HPib M−2 s−1 |
ΔΔG‡ kcal/mola |
5′-Phosphate IBEc kcal/mol |
|---|---|---|---|---|---|---|---|---|
| Wild type | 4.6 | 2.9 × 106 | 8.7 × 10−3 | 2500 | 11.6 d | |||
| V182A | 3.4 | 1.4 × 105 | 1.8 | 1.3 × 10−3 | 1.1 | 190 | 1.5 | 10.9 |
| I199A | 3.9 | 9.1 × 105 | 0.7 | 1.9 × 10−3 | 0.9 | 980 | 0.6 | 11.8 |
| V201A | 4.0 | 9.5 × 105 | 0.7 | 3.1 × 10−3 | 0.6 | 690 | 0.8 | 11.5 |
| I218A | 3.3 | 2.8 × 105 | 1.4 | 2.3 × 10−3 | 0.8 | 340 | 1.2 | 11.0 |
| V182A/I199A | 3.1 | 4.9 × 104 | 2.4 | 3.9 × 10−4 | 1.8 | 81 | 2.0 | 11.0 |
| V182A/V201A | 2.5 | 4.9 × 104 | 2.4 | 5.0 × 10−4 | 1.7 | 30 | 2.6 | 10.9 |
Calculated from the ratio of the second-order or third-order rate constants for the wild type and mutant enzyme.
Third-order rate constant for reaction of EO/HPi.
Transition state stabilization by the 5′-phosphate group of OMP, calculated from the ratio of the values of kcat/Km for OMP and EO. IBEs for phosphite dianion can be calculated from the ratio of (kcat/Km)/KD for EOHPi and kcat/Km for EO.
Figure 2.
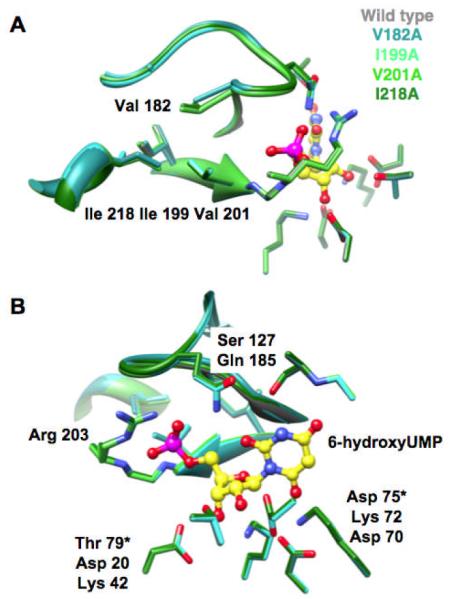
Superpositions of the 6-hydroxyUMP-liganded structures of wild type MtOMPDC and the single mutants in the hydrophobic cluster. Panel A, hydrophobic cluster; panel B, active sites.
The mutated residues are remote from the active site (Figures 1 and 2). Thus, the effects of the substitutions on kcat/Km cannot be explained by altered direct interactions with the substrate. Instead, the effects can be explained by decreased stabilities of the closed conformation in which the substrate is destabilized (9) and the anionic intermediate is stabilized. A consistent model (Scheme 2) is that an equilibrium of open (Eo) and closed (Ec) conformations exists in the absence or presence of substrate (KC’ or KC; KC’ << 1). The substitutions destabilize Ec (decrease KC and KC ’), so kcat/Km is decreased (the energy difference between Eo + S and [Ec•S]‡ is increased). However, the invariance of kcat establishes that the substitutions do not alter the reactivitiy of Ec•S (the energy difference between Ec•S and [Ec•S]‡). The energy to form Ec•S from Eo + S is achieved from 1) interactions of the 5′-phosphate group with its binding motif (IBE; vide infra), and 2) an increased concentration of OMP to form Ec•S (KC/KS or, equivalently, KC’/KS’, although the former is expected to be the relevant pathway).
Scheme 2.

We also used the two part EO•HPi substrate. The values of kcat/Km for EO are decreased relative to that for wild type (Table 1); these can be explained by decreased populations of Ec (Scheme 2), assuming that EO, without a 5′-phosphate group, is unable to promote the transition from Eo to Ec. The values of kcat/Km for OMP and kcat/Km for EO allow calculation of the IBE for the 5′-phosphate group of OMP (Table 1).
HPi activates the mutants as judged by the values of the third-order rate constant, (kcat/Km)EO•HPi/Kd. The equivalent changes (ΔΔG‡) in kcat/Km for both OMP and EO and the third-order rate constant indicate that all three measure the effects of the substitutions on the values of KC.
The values of the IBEs for the 5′-phosphate group for the mutants are the same as that for the wild type, establishing that decarboxylation in the Ec•S complex occurs with equivalent amounts of ground state destabilization (9) and transition state stabilization (as also reflected by the invariant values of kcat). The IBEs provide further support for the role of the “remote” hydrophobic cluster in stabilizing Ec relative to Eo but do not directly participating in catalysis.
Our experiments implicate a structurally conserved hydrophobic cluster, Val 182, Ile 199, Val 201, and Ile 218 in MtOMPDC, in stabilizing the closed conformation required for catalysis. Its identification provides evidence that structural elements distal from the active site, in addition to the proximal active site loop that closes to “clamp” the substrate, are required for OMPDC’s extraordinary catalytic efficiency and proficiency.
Supplementary Material
Footnotes
SUPPORTING INFORMATION AVAILABLE
Descriptions of the experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by NIH Grants GM039754 to J.P.R. and GM065155 to J.A.G.
Abbreviations: OMP, orotidine 5′-monophosphate; OMPDC, OMP decarboxylase; MtOMPDC, OMPDC from Methanobacter thermoautotrophicus; ScOMPDC, OMPDC from Saccharomyces cerevisiae; EO, 1-(β-D-erythrofuranosyl)-orotic acid; HPi, phosphate dianion; IBE, intrinsic binding energy.
REFERENCES
- (1).Wood BM, Chan KK, Amyes TL, Richard JP, Gerlt JA. Biochemistry. 2009;48:5510–5517. doi: 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- (3).Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J. Am. Chem. Soc. 2007;129:12946–12947. doi: 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J. Am. Chem. Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc. Natl. Acad. Sci. U A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Appleby TC, Kinsland C, Begley TP, Ealick SE. Proc atl Acad Sci U S A. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Harris P, Navarro Poulsen JC, Jensen KF, Larsen S. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- (8).Wu N, Mo Y, Gao J, Pai EF. Proc. Natl. Acad. Sci. U S A. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chan KK, Wood BM, Fedorov AA, Fedorov EV, Imker HJ, Amyes TL, Richard JP, Almo SC, Gerlt JA. Biochemistry. 2009;48:5518–5531. doi: 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sievers A, Wolfenden R. Bioorg. Chem. 2005;33:45–52. doi: 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- (11).Amyes TL, Richard JP, Tait JJ. J. Am. Chem. Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- (12).Toth K, Amyes TL, Wood BM, Chan KK, Gerlt JA, Richard JP. Biochemistry. 2009;48:8006–8013. doi: 10.1021/bi901064k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jencks WP. Adv. Enzymol. Rel. Areas. Mol. Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- (14).Barnett SA, Amyes TL, Wood BM, Gerlt JA, Richard JP. Biochemistry. 2008;47:7785–4487. doi: 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Harris P, Poulsen JC, Jensen KF, Larsen S. J. Mol. Biol. 2002;318:1019–1029. doi: 10.1016/S0022-2836(02)00200-0. [DOI] [PubMed] [Google Scholar]
- (16).Go MK, Amyes TL, Richard JP. Biochemistry. 2009;48:5769–5778. doi: 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.