

Abstract
The presence of misfolded proteins elicits cellular responses including an endoplasmic reticulum (ER) stress response that may protect cells against the toxic buildup of misfolded proteins. Accumulation of these proteins in excessive amounts, however, overwhelms the “cellular quality control” system and impairs the protective mechanisms designed to promote correct folding and degrade misfolded proteins, ultimately leading to organelle dysfunction and cell death. Studies from multiple laboratories have identified the roles of several ER stress-induced cell death modulators and effectors. Earlier, we reported the role of the small co-chaperone protein p23 in preventing ER stress-induced cell death. p23 undergoes caspase-dependent cleavage to yield a 19-kD product (p19), and mutation of this caspase cleavage site not only blocks the formation of the 19-kD product but also attenuates the ER stress-induced cell death process triggered by various stressors. Thus, a critical question is whether p23 and/or p19 could serve as an in vivo marker for neurodegenerative diseases featuring misfolded proteins and cellular stress. In the present study, we used an antibody that recognizes both p23 and p19 as well as a specific neo-epitope antibody that detects only the p19 fragment. These antibodies were used to detect the presence of both these proteins in cells, primary neurons, brain samples from a mouse model of Alzheimer's disease (AD), and fixed human AD brain samples. While patients with severe AD did display a consistent reduction in p23 levels, our inability to observe p19 in mouse or human AD brain samples suggests that the usefulness of the p23 neo-epitope antibody is restricted to cells and primary neurons undergoing cellular stress.
Keywords: Endoplasmic reticulum, p23, HSP90, Alzheimer's disease, ER stress, Caspase, Programmed cell death
Introduction
Cell death pathways triggered by protein misfolding and associated endoplasmic reticulum (ER) stress have been implicated in all of the major neurodegenerative diseases (Kopito 2000; Forman et al. 2003; Hashimoto et al. 2003; Bredesen et al. 2006). Misfolded proteins elicit cellular stress responses including an ER stress response that serves to protect cells against the accumulation of these toxic proteins. Accumulation of these faulty proteins in excessive amounts, however, overwhelms the cellular protective “quality control” systems ultimately triggering programmed cell death (pcd) (Harding et al. 2002; Kaufman 2002; Selkoe 2003; Sitia and Braakman 2003; Rao and Bredesen 2004; Rao et al. 2004a; Bredesen et al. 2006). Cell death pathways triggered by misfolded proteins and other activators of ER stress display both cytochrome c/Apaf-1 dependent and independent activation of pcd (Morishima et al. 2002; Rao et al. 2002; Di Sano et al. 2006; Zhang and Armstrong 2006). Studies from multiple laboratories have identified the roles of several ER stress-induced cell death modulators and effectors through the use of biochemical, pharmacological, and genetic tools (Breckenridge et al. 2003; Rao and Bredesen 2004; Xu et al. 2005; Bredesen et al. 2006).
We recently described the role of the small co-chaperone protein p23 (a 23-kD HSP90-associated chaperone protein) in ER stress-induced cell death. Using several complementary approaches, we identified its role as an antiapoptotic protein (Rao et al. 2006). p23 is cleaved at D142 by caspases-3 and -7 to yield p19 (a 19-kD fragment) during ER stress-induced cell death (Rao et al. 2006). Mutating this caspase cleavage site not only blocked the appearance of p19 but also was associated with decreased cell death triggered by various ER stressors (Rao et al. 2006). These findings suggested that the presence of p19 may be an indication of cells undergoing some form of cellular stress leading to cell death and neurodegeneration. In order to determine whether this is the case, we examined the localization and levels of p23 and p19 in cells before and after undergoing ER stress, in brain samples from a mouse model of Alzheimer's disease (AD) and in human AD brain tissues using two antibodies: one that recognizes both the parent p23 and the cleaved p19 fragment as well as a custom neo-epitope antibody that specifically recognizes only p19. Our results indicate different localization and expression patterns of p23 and p19 in cells and human AD brains tissues. Our results also suggest the usefulness of the p23 neo-epitope antibody in detecting the p19 protein in stress-induced cells and primary neurons as well as its preliminary detection in human AD tissue.
Experimental Procedures
Cells, Culture Conditions, Plasmids, and Transfections
Human embryonic kidney 293T (HEK293T) and Apaf-1−/− immortalized mouse embryonic fibroblasts (Apaf-1−/− MEF) were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. We used Apaf-1−/− MEF as they are relatively resistant to cell death initiated by specific pro-apoptotic agents that utilize the intrinsic mitochondrial-dependent pcd pathway, including Fas and ceramide-mediated pathways (Rao et al. 2002, 2004b), but are susceptible to ER stress-induced cell death (Rao et al. 2001, 2002, 2004b, 2006). Thus, the MEF provided us with a system to assess whether cellular stress-mediated cell death is triggered by the activation of caspases without the involvement of the apoptosome.
Cortical neuronal cultures were prepared from 16-day Charles River CD1 mouse embryos and maintained as described earlier (Rao et al. 2006). To induce hypoxia, cultures were placed in modular incubator chambers (Billups-Rothenberg, Del Mar, CA) for 0–24 h at 37°C, under humidified 95% air/5% CO2 (control) or humidified 95% N2/5% CO2 (hypoxia) conditions. Cultures were then returned to normoxic conditions for the remainder (if any) of the 24 h.
N-terminal Flag-p23 (human) was subcloned into a pcDNA3 expression vector (Invitrogen) by PCR amplification of p23cDNA using 5′ p23 FLAG BamH1 and 3′ p23 Not1 primer pairs. The amplified N-terminal Flag-p23cDNAwas excised using BamH1 and Not 1 restriction enzymes and ligated into the pcDNA3 expression vector. Using the QuikChange Site-Directed Mutagenesis Kit (Stratagene), the uncleavable Flag-p23D142N mutant construct was generated (Rao et al. 2006). The p19 cDNA (p23 deletion construct missing the last 18 amino acid residues at the C-terminus) was produced from the full-length Flag-p23 after introducing a stop codon downstream of the D142 site. The sequences of all constructs were confirmed by DNA sequencing, and protein expression was verified by immunoblotting. Transient transfections of 293T cells with Lipofectamine 2000 (Invitrogen) were performed as described earlier (Rao et al. 2001, 2002, 2004b, 2006) and according to the manufacturer's instructions. Typically, 2×106 cells were seeded into 10-cm dishes and transfected 1 day later with 6 μg of the relevant construct using a ratio of 1 μg DNA/5 μl of transfection reagent. The transfection efficiency using these conditions was approximately 65–70%. Transient transfections of the Apaf-1−/− fibroblasts were performed using TransIT-Neural transfection reagent (Mirus Bio Corporation). Typically, 2×106 cells were seeded into 10-cm dishes and transfected 1 day later with 6 μg of the relevant construct using a ratio of 1 μg DNA/3 μl of the transfection reagent. The transfection efficiency using these conditions was approximately 40–50%.
Antibodies
The following antibodies were used for Western blotting: p23 monoclonal antibody (1:1,000, BD Biosciences), KDEL monoclonal antibody (1:500), GRP78 polyclonal antibody (1:500), anti-Hsp90 monoclonal antibody (1:500) (all from Stressgen) and glyceraldehyde-3-phosphate dehydrogenase monoclonal antibody (1:50,000, Research Diagnostics Inc).
Generation of the p23 neo-epitope antibody was based on the protocol that we used earlier to successfully generate an amyloid precursor protein (APP) neo-epitope antibody that recognizes APP that is cleaved at the caspase site Asp664 (reflecting the generation of the carboxy-terminal C31 fragment) (Banwait et al. 2008; Bredesen et al. 2010). Conventionally, neo-epitope antibodies recognize the newly created N- or C-terminus of protein degradation products but fail to recognize the same sequence of amino acids present in intact or undigested protein (Fosang et al. 2010). It is for this reason that we have designated the antibody as p23 neo-epitope antibody since it is expected to recognize the new C-terminus of cleaved p23.
Two rabbits were immunized with keyhole limpet hemocyanin (KLH)-CEDVDLPEVD that comprises the nine amino acids in p23 that precede the caspase cleavage site at aspartic acid D142 as well as an amino terminal cysteine residue to allow for coupling to KLH. After boosting the rabbits three times with the immunizing peptide over a 10-week period, the antisera were collected, pooled, and affinity purified by three successive chromato-graphic steps: (1) the antisera were first depleted of immunoglobulins recognizing intact p23 by adsorption to a bridging peptide (spanning the caspase cleavage site GDEDVDLPEVDGADDDSQ) immobilized onto Sepharose 4B by cyanogen bromide activation; (2) the flow-through from step (1) was applied to another Sepharose 4B column containing the immobilized immunizing peptide, and the column was washed and the specific antibody eluted via a pH gradient into 0.125 M borate buffer; and (3) the eluant from step (2) was again absorbed to the immobilized bridging peptide as described and the flow-through collected.
APP Transgenic Mouse
Mice overexpressing the human APP695 minigene carrying the Swedish (K670N, M671L) and Indiana (V717F) familial AD mutations downstream of the platelet-derived growth factor B-chain promoter have been previously described (PDAPP mice) (Mucke et al. 2000; Galvan et al. 2006; Nguyen et al. 2008). The use of animals in this study was in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and under a protocol approved by the Buck Institute's Animal Care and Use Program Committee, which is accredited by AAALAC.
Human Autopsy Material
Tissues from the Harvard Brain Tissue Resource Center (McLean Hospital, Belmont, MA) were used for the present study. The procedures were conducted in compliance with policies and principles contained in the Federal Policy for the Protection of Human Subjects. Eighteen postmortem human brains were used in this study. Twelve came from individuals with a clinical diagnosis of early (Braak stage I and II), moderate (Braak stage III and IV), or severe (Braak stage Vand VI) Alzheimer's disease (AD) by histological analyses, and six came from age-matched, normal controls. Ages at death ranged from 68 and 92 years, with a mean age of 78. Postmortem intervals ranged between 4.98 and 30.65 h, with a mean delay of 16.37 h. The gender, age at death, and postmortem interval were comparable in all groups. Brain tissues frozen in liquid nitrogen vapor or at −80°C were shipped along with formalin-fixed blocks of tissues. Subsequently, the formalin-fixed blocks of tissues were paraffin-embedded and sectioned for IHC, while frozen (non formalin-fixed) tissues were homogenized to make lysates for Western blotting.
Cell and Tissue Extracts and Western Blotting
Total cell extracts were prepared as previously described (Rao et al. 2001, 2002, 2004b, 2006). Briefly, untreated or treated cells were resuspended in RIPA buffer (50 mM Tris, pH 7.5, 0.5% deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 150 mM NaCl) with protease inhibitors (Complete Mini, Roche), sonicated for 10 s on ice, and centrifuged at 10,000×g for 10 min at 4°C. The supernatant was collected and protein concentration determined using Coomassie Plus protein assay reagent (Pierce). Protein (100–200 μg) was used for SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot analyses as described earlier (Rao et al. 2001, 2002, 2004b, 2006).
Dissected and frozen human brains or mouse hemi-brains were homogenized in ice-cold phosphate-buffered saline lysis buffer containing 0.05% Nonidet P-40, 0.25% sodium deoxycholate, 50 mmol/L Tris–HCl (pH 8.5), 100 nmol/L NaCl, 1 mmol/L EDTA (pH 8.0), (Roche) complete mini cocktail protease inhibitors, and 2 mg/mL b-glycerol phosphate. Samples were then centrifuged at 16,000×g for 10 min at 4°C and the resulting supernatant assayed for total protein concentration. Protein (100–200 μg) from total extracts was used for SDS–PAGE and Western blot analyses as described earlier (Rao et al. 2001, 2002, 2004b, 2006).
Immunocytochemistry
HEK293T cells or Apaf-1−/− MEF were plated at 60–75% confluency onto multiwell dishes or glass coverslips. Transfection of the relevant construct was carried out as mentioned above, and cells were treated with or without 0.5 μM thapsigargin. Cells were fixed in buffered 4% paraformaldehyde and permeabilized in 0.2% Triton in PBS. For the detection of p23, cells were incubated with anti-p23 monoclonal antibody (10 μg/mL, BD Transduction Labs) or anti p23 neo-epitope antibody (10 μg/mL) as primary antibody. Secondary antibodies included Alexa Fluor 488 donkey anti-mouse IgG or Alexa Fluor 488 donkey anti-rabbit IgG (both antibodies at 4 μg/mL, Invitrogen).
For co-localization experiments, Apaf-1−/− MEF were plated into eight-well poly-D-lysine-coated culture slides. One day later, cells were treated with or without 0.5 μM thapsigargin for 12 or 24 h. Cells were incubated with 500 nM MitoTracker deep red 633 (Millipore) for 45 min at 37°C according to manufacturer's instructions, fixed in 4% PFA for 15 min, permeabilized in 0.2% Triton X-100 for 5 min, and blocked in 5% normal donkey serum for 1 h. For the detection of p23, cells were co-incubated with anti-p23 monoclonal antibody (10 μg/mL) or anti p23 neo-epitope antibody (10 μg/mL) and anti-calregulin (calreticulin) polyclonal antibody (4 μg/mL) or anti-β III Tubulin monoclonal antibody (8 μg/mL) and kept overnight at 4°C in 2.5% normal donkey serum. Calregulin was used as an ER marker protein (Xiao et al. 1999), while β-III Tubulin served as a marker for the cytosolic compartment. Secondary antibodies included Alexa Fluor 488 donkey anti-mouse IgG (green), Alexa Fluor 488 donkey anti-rabbit IgG (green), Alexa Fluor 555 donkey anti-goat IgG (red), or Alexa Fluor 555 donkey anti-rabbit IgG (red) (all at 4 μg/mL, Invitrogen). Slides were mounted in Vectashield with 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs H-1200). Z-stacks of cells were acquired on a Zeiss LSM or a Nikon PCM 2000 confocal mounted on Eclipse-E800 microscope equipped with Argon 488 and 543 HeNe laser filters. Z-stacks of the cells were acquired with voxel size of 0.048×0.048×0.25 um3 using PCI Imaging software. In Bitplane Imaris 4.1.3 Suite, individual cells were cropped out of stacks. Subsequently, each cell's data set was run through Huygens deconvolution software applying Classic MLE and using constant parameters for like primary/secondary combinations.
Evaluation of Cell Death
Assessment of cell death was carried out as previously described (Ellerby et al. 1999; Rao et al. 2001, 2002, 2004b, 2006). Cell death was determined as the percentage of dead cells over the total number of cells. Statistical significance was determined by two-way analysis of variance (ANOVA).
Results
Generation of the p23 Neo-epitope Antibody and Determining Its Specificity
A critical question regarding p19 and its relevance as a marker for cellular stress is whether it can be detected in vivo. Generating a neo-epitope-specific antibody that recognizes only p19 and not the parent p23 would not only specifically reveal the generation of p19 but also enable us to screen pathological samples of mouse or human brains for the presence of p19. We designed and generated an antibody directed against the neo-epitope of p23 after caspase cleavage at D142 (Fig. 1a). This antibody was screened for its specificity to recognize p19. As shown in Fig. 1b, treatment of Apaf1−/− MEFs with 0.5 μM thapsigargin resulted in the cleavage of p23 to p19. While the p23 monoclonal antibody recognized both the pro and cleaved forms of p23, the custom-made polyclonal antibody designed to recognize the neo-epitope specifically recognized only p19, confirming its specificity (Fig. 1b, middle panel). Incubation of the p23 neo-epitope antibody with an excess of the synthetic peptide immunogen resulted in the failure of the antibody to detect p19 (Fig. 1c), further indicating that this particular antibody can be designated as the p23 neo-epitope antibody.
Fig. 1.
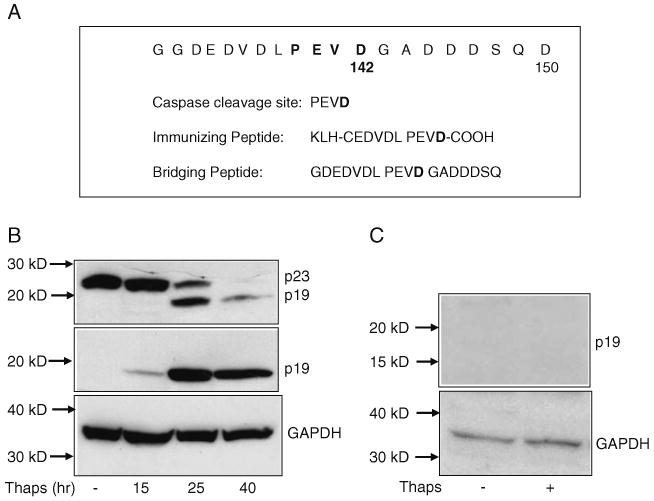
Generation of the anti-p23 neo-epitope antibody. a Partial amino acid sequence of p23 that includes the nine amino acids preceding the caspase cleavage site at aspartic acid D142 as well as an amino terminal cysteine residue, to allow coupling to KLH. Both the immunizing peptide and the bridging peptide were immobilized to Sepharose 4B for purification of the p23 neo-epitope antibody as described in “Experimental Procedures” Section. b Western blotting of p23 and p19 in cell extracts isolated from untreated or 0.5 μM thapsigargin-treated Apaf-1−/− MEF for various time periods. Following SDS–PAGE and Western blotting, membranes were probed with either the p23 antibody (top panel) or the p23 neo-epitope antibody (middle panel). GAPDH was used as a loading control. c Western blotting of p19 in cell extracts isolated from untreated or 0.5 μM thapsigargin-treated Apaf-1−/− MEF (24 h). Immunoblotting with the p23 neo-epitope antibody was performed after preadsorbing the antisera with the peptide immunogen. GAPDH was used as a loading control
To test the antibody specificity in cells, Apaf1−/− MEF were treated with 0.5 μM thapsigargin to induce ER stress. Immunocytochemistry was performed on untreated and thapsigargin-treated cells using the p23 neo-epitope antibody. As shown in Fig. 2a, immunoreactivity with the p23 neo-epitope antibody was observed only in thapsigargin-treated cells indicating the presence of p19 in the thapsigargin-treated MEF.
Fig. 2.
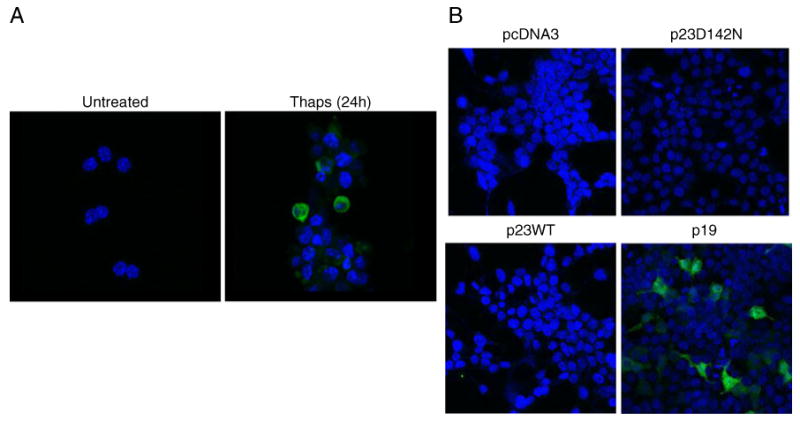
Specificity of the p23 neo-epitope antibody. a Immunofluo-rescent staining for p19 was performed in untreated or 0.5 μM thapsigargin-treated Apaf-1−/− MEF (24 h). Cells were stained with the p23 neo-epitope antibody as primary antibody and Alexa Fluor 488 donkey anti-rabbit IgG as secondary antibody. Nuclei were counterstained using VectaShield Mounting Medium with DAPI (blue). b Specificity of the p23 neo-epitope antibody in cells transfected with p23 constructs. HEK293T cells were transfected with various p23 constructs including WTp23, p19, or p23D142. Immunofluorescent staining for p19 was performed as described in “Experimental Procedures” Section. Cells were stained with the p23 neo-epitope antibody as primary antibody and Alexa Fluor 488 donkey anti-rabbit IgG as secondary antibody. Nuclei were counterstained using Vecta-Shield Mounting Medium with DAPI (blue)
The specificity of the antibodies was further confirmed by transfecting various p23 constructs including WTp23, p19, or p23D142N into HEK293T cells and staining the cells with the p23 neo-epitope antibody. Immunoreactivity with the p23 neo-epitope antibody was seen only in those cells that were transfected with p19 cDNA (Fig. 2b), further confirming its specificity for the p19 protein.
Hypoxia, ER Stress, and p23 Cleavage
Hypoxia-induced neuronal cell death proceeds through the ER stress pathway and involves several ER-associated proteins (Bando et al. 2003; Hori et al. 2004; Tajiri et al. 2004). To determine whether hypoxia-induced cellular stress triggers caspase-mediated p23 cleavage, we assessed the levels of p23 and p19 in cell extracts isolated from primary mouse cortical cells subjected to hypoxia. As shown in Fig. 3a, hypoxia triggered p23 cleavage resulting in the formation of the 19-kD fragment. While the p23 antibody recognized full-length p23, p19 was weakly visible in the 12- and 24-h hypoxic samples. In contrast, the presence of p19 was clearly observed using the p23 neo-epitope antibody (second panel). p19 levels increased with time of hypoxia, suggesting that longer exposure times to low oxygen levels triggered more p23 cleavage. Since p23 is an HSP90-associated chaperone protein and GRP78 is an indicator of ER stress (Rao et al. 2004b, 2006), we assessed the levels of these two proteins that were also found to increase with time of hypoxia.
Fig. 3.
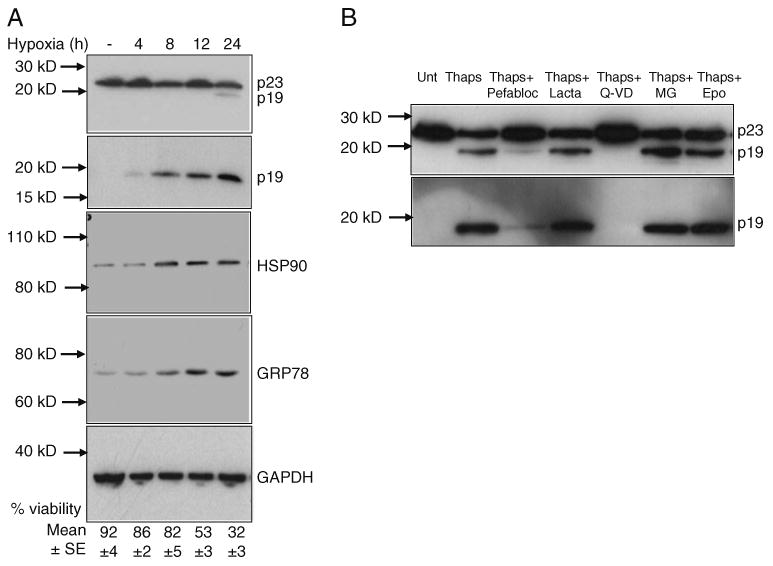
ER stress triggers p23 cleavage. a Western blot analysis of p23 and p19 before and after exposing cortical neurons to hypoxia. Cortical neuronal extracts isolated from normal and hypoxic cells were subjected to SDS–PAGE and Western blotting. Membranes were probed with the p23 monoclonal antibody to detect both p23 and p19 (top panel) or p23 neo-epitope antibody to detect p19 specifically (second panel). Cell extracts were also analyzed for HSP90 and GRP78 levels. GAPDH served as a loading control. Surviving versus apoptotic cells were quantified as described in the “Experimental Procedures” Section. b p23 cleavage is blocked by a caspase inhibitor Western blotting of p23 and p19 in cell extracts isolated from untreated or 0.5 μM thapsigargin-treated Apaf-1−/− MEF in the presence of various inhibitors including pefabloc (300 μM), lactacystin (lacta, 5 μM), Q-VD-OPh (Q-VD, 20 μM), MG262 (MG, 100 nM), or epoxomicin (Epo, 10 nM). Following SDS-PAGE and Western blotting, membranes were probed with the p23 monoclonal or p23 neo-epitope antibody
We next determined whether or not ER stress-mediated p23 cleavage and the appearance of p19 are blocked by specific inhibitors. As shown in Fig. 3b, addition of the pan caspase inhibitor Q-VD-OPh or the serine protease inhibitor Pefabloc completely blocked 0.5 μM thapsigargin-mediated p23 cleavage and appearance of p19 in Apaf1−/− MEF. The proteasome inhibitors including lactacystin, MG262, and epoxomycin were all ineffective at inhibiting thapsigargin-mediated p23 cleavage.
Cellular Localization of p23 and p19
To determine the intracellular localization of p23 and p19 before and after ER stress, we performed immunocyto-chemistry on untreated and 0.5 μM thapsigargin-treated Apaf1−/− MEF using either the p23 antibody or the p23 neo-antibody. Figure 4 is a representative figure from three independent experiments, and as shown in the figure, p23 is predominantly associated with the ER compartment in untreated cells, based on its co-localization with calregulin (calreticulin), an ER marker protein. Following thapsigargin treatment, a subpopulation of p23 gets associated with the cytosolic compartment as shown by its co-localization with cytosolic β-III Tubulin. While immunostaining with the p23 neo-epitope antibody did not show any staining in untreated cells, staining was predominantly observed in the cytosolic compartment following ER stress. While there may be some p19 staining in the ER after thapsigargin treatment, taking together all the three experiments, the localization of p19 seems to be predominantly cytosolic, suggesting that cellular stress triggers the localization of p19 and a subpopulation of p23 to the cytosolic compartment.
Fig. 4.
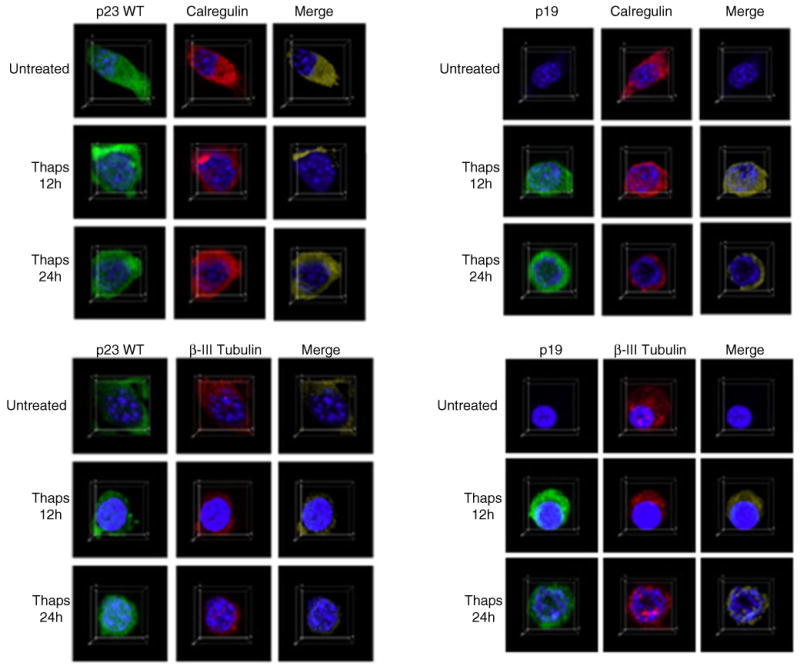
Co-localization of p23 and p19 before and after ER stress. Immunocytochemistry on untreated or 0.5 μM thapsigargin-treated Apaf1−/− MEF was performed using either the p23 monoclonal antibody or the p23 neo-epitope antibody as described in “Experimental Procedures” Section. Confocal images were acquired as described in “Experimental Procedures” Section. For the detection of p23 or p19, cells were co-incubated with the p23 monoclonal antibody or the p23 neo-epitope antibody and the anti-calregulin polyclonal antibody or the anti-β-III Tubulin monoclonal antibody as described in “Experimental Procedures” Section. Secondary antibodies included Alexa Fluor 488 donkey anti-mouse IgG (green), Alexa Fluor 488 donkey anti-rabbit IgG (green), Alexa Fluor 555 donkey anti-goat IgG (red), or Alexa Fluor 555 donkey anti-rabbit IgG (red). Calregulin served as an ER marker, while β-III Tubulin was used as a marker for the cytosolic compartment
Presence of p23 and p19 in a Mouse Model of AD and Human AD Brains
Since cleavage of p23 and appearance of p19 may be indications of cells undergoing some form of cellular stress leading to cell death, it prompted us to determine whether p23 cleavage is implicated in neurodegenerative diseases that feature misfolded proteins and ER stress. We used both p23 antibodies to determine the levels of p23 and p19 in extracts isolated from Tg-APP mouse brains (Galvan et al. 2006; Nguyen et al. 2008) as well as brains from normal human subjects and subjects at different stages of AD. Since both antibodies failed to detect p23 or p19 in mouse or human tissues by immunohistochemistry (data not shown), we examined the presence of these proteins by Western blot.
As shown in Fig. 5a, p23 levels did not vary between 8-month-old Tg and nonTg animals, and there was also no evidence of p23 cleavage. The p23 neo-epitope antibody also failed to detect the presence of p19 in all samples (data not shown). However, an increase in GRP78 levels in Tg-APP mouse brains was observed when compared with non-transgenic littermates, indicative of ER stress in these transgenic animals. Interestingly, a similar alteration in GRP78 levels was observed in humans with amnestic mild cognitive impairment, arguably the earliest form of AD (Owen et al. 2009). In order to assess the levels of p23 in older mice, we screened nonTg and Tg brains obtained from 13-month-old mice and compared them with samples obtained from 3-month-old mice. Again, there was no evidence of p23 cleavage in the older mice (Fig. 5b).
Fig. 5.
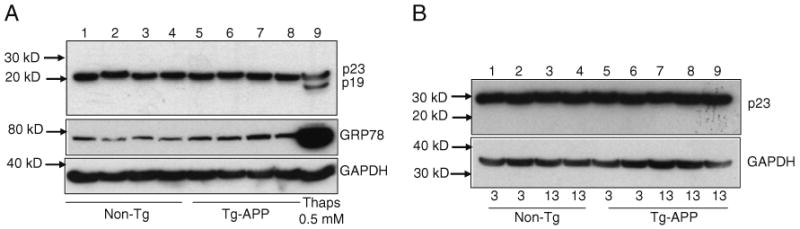
p23 levels in nonTg and Tg-APP mouse brains. a One hundred fifty micrograms each of mouse brain (hippocampal) extracts from approximately 8-month-old nonTg (lanes 1–4) and Tg-APP animals (lanes 5–8) were examined by Western blot using antibodies for p23 and GRP78. Cell lysate isolated from 0.5 μM thapsigargin-treated Apaf-1−/− MEF (lane 9) was used as a positive control. GAPDH served as a loading control. b One hundred micrograms each of mouse brain (hippocampal) extracts from 3- or 13-month-old nonTg (lanes 1–4) and Tg-APP animals (lanes 5–9) were examined by Western blot using the p23 antibody. GAPDH served as a loading control
Next, we examined p23 levels in postmortem samples of age-matched human subjects satisfying one of the following neuropathological diagnoses (Table 1): (1) neuropathologically normal (N; Braak stage 0–I, without evidence of other degenerative changes and lacking a clinical history of cognitive impairment), (2) E; early AD changes (Braak stage I–II, in the absence of discrete neuropathology), (3) M; moderate AD neuropathology (Braak stage III–IV), and (4) S; severe AD (Braak stage V–VI with neuropathological diagnosis of AD, in the absence of other neuropathology). p23 levels varied depending upon the severity of the AD and the age of the normal individuals. Generally, levels of p23 were higher in older normal (N) individuals and in individuals with early (E) or moderate (M) AD as opposed to those with severe (S) AD (Fig. 6a, b). All five individuals with severe AD had very low levels of p23. Furthermore, brain extracts from younger normal individuals (N4–6) had no detectable p23 (Fig. 6b). While the p23 neo-epitope antibody failed to detect the presence of p19 in all samples (data not shown), a low level of p19 expression was observed with the p23 antibody in one sample from a subject with moderate AD (Fig. 6c, M2, longer exposure of the film). The failure to detect p19 in these samples suggests that p19 is likely a transient species since it represents a caspase-cleavage product (Semple et al. 2007; Werner et al. 2007). Moreover, all of the severe AD patients (S1–S5) did display a consistent reduction in p23 levels, suggesting that the reduction may involve cleavage of p23 to the p19 fragment.
Table 1. Information about the human sample number, gender, AD status, and age of the brain samples analyzed.
| Number/gender | AD status | Age |
|---|---|---|
| N1/F | Normal | 63 |
| N2/M | Normal | 76 |
| N3/F | Normal | 97 |
| E1/M | Early | 70 |
| E2/M | Early | 75 |
| E3/M | Early | 85 |
| M1/M | Moderate | 68 |
| M2/M | Moderate | 80 |
| M3/M | Moderate | 89 |
| S1/M | Severe | 69 |
| S2/F | Severe | 75 |
| S3/M | Severe | 88 |
| N4/F | Normal | 53 |
| N5/M | Normal | 20 |
| N6/M | Normal | 23 |
| M4/F | Moderate | 84 |
| S4/F | Severe | 94 |
| S5/M | Severe | 77 |
Numbers 1, 2, 3 etc. indicate individual samples
N normal, E early AD, M moderate AD, and S severe AD pathology
Fig. 6.
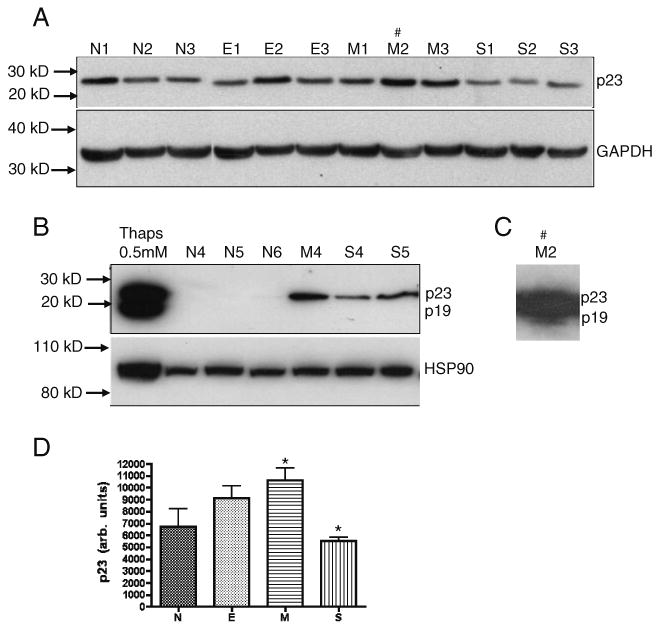
p23 levels in hippocampal extracts from individuals at different stages of Alzheimer's disease. a, b One hundred micrograms each of human brain extracts from normal (N), early AD (E), moderate AD (M), and severe AD (S) pathology were examined by Western blot using the p23 monoclonal antibody. Normal samples (N1–N3) were run in an independent blot and placed along side the AD samples. Numbers indicate individual samples. Cell lysate isolated from 0.5 μM thapsigargin-treated Apaf-1−/− MEF (b, lane 1) was used as a positive control. HSP90 was also examined in this subset of samples (b). GAPDH served as loading control. c A longer exposure of the membrane blot (a, lane M2) revealed the presence of p19 in the brain extract from subject M2 with moderate AD. d p23 band density (integrated density value) of samples represented in a expressed graphically as arbitrary units and with denotations of significance obtained by one-way ANOVA (*p<0.05)
Since p23 is an HSP90-associated chaperone protein, we also assessed the expression of HSP90 in a subset of samples. Levels of HSP90 remained unchanged in the controls as well as the AD samples (Fig. 6b). Shown in Fig. 6d is the p23 band density (arbitrary units) of samples represented in Fig. 6a expressed graphically.
Discussion
p23 is a widely expressed protein and is involved in the binding, folding, and processing of proteins and in the assembly of steroid hormone receptor and telomerase complexes, events that are closely associated with the endoplasmic reticulum (Johnson et al. 1994; Johnson and Toft 1995; Weaver et al. 2000; Felts and Toft 2003). Treatment of cells with ER stress-inducing agents resulted in the cleavage of p23 to yield p19 (Rao et al. 2006; Bakhshi et al. 2008; Chinta et al. 2008; Chinta et al. 2009). We were successful in generating an antibody designated the p23 neo-epitope antibody that specifically recognizes p23 cleaved at the caspase site D142, reflecting the generation of p19. The p23 neo-epitope antibody specifically recognized p19 in (1) whole cells or cell extracts isolated from cells transfected with p19cDNA, (2) whole cells or cell extracts isolated from cells treated with thapsigargin and, (3) cell extracts isolated from primary cortical cells exposed to hypoxia. Immunocytochemistry and/or Western blot analysis with a commercially available monoclonal antibody that recognizes both p23 and p19 or the p23 neo-epitope antibody that specifically recognizes p19 indicate that (1) p23 is cleaved to yield p19 during ER stress-induced cell death, (2) while p23 is predominantly associated with the ER compartment in untreated cells, a subpopulation of p23 gets distributed in the cytosolic compartment following ER stress, and (3) p19 staining was observed predominantly in the cytosolic compartment of stressed cells.
We had previously shown that downregulation of the expression of p23 by immunodepletion and RNAi resulted in enhanced ER stress-induced apoptosis, suggesting a role for p23 as an antiapoptotic protein. Moreover, uncleavable p23 (mutation of the caspase cleavage site p23D142N) not only blocked the cleavage by caspases and formation of the 19-kD product but also attenuated the cell death process (Rao et al. 2006). The above-mentioned results together with our earlier observations (Rao et al. 2006; Bakhshi et al. 2008; Chinta et al. 2008; Chinta et al. 2009) support the notion that prolonged ER stress resulting in p23 cleavage and generation of p19 renders cells more susceptible to pcd. It has been observed that the entire C-terminus of p23 is required for its chaperone activity and to enhance the assembly of protein complexes (Weikl et al. 2000; Oxelmark et al. 2003). Thus, it is possible that ER stress-induced cleavage of p23 at the D142 site, resulting in p19, abolishes the ability of p23 to act as an antiapoptotic protein.
We had earlier shown that caspase-mediated cleavage of p23 abolishes the interaction of p23 with PUMA thereby allowing PUMA to associate with Bax and trigger pcd (Rao et al. 2006). These findings are significant since they indicate that ER stress-induced cell death may require cross-talk between the ER and cytosolic compartments with p23 and p19 coordinating the ER–cytosolic death pathway. Since cleavage of p23 is associated with increased cell death, it is possible that p19 displays pro-apoptotic activity analogous to cleaved protein fragments of Bcl-2 and Bid (Cheng et al. 1997; Li et al. 1998). Studies are ongoing to determine (a) if p19 is pro-apoptotic, (b) if p19 interacts with pro-death molecules, and (c) the mechanism by which these interacting partners elicit pcd.
Since both the p23 monoclonal antibody and the p23-neospecific antibody failed to detect p23 in human or mouse tissues by immunohistochemical staining, we examined p23 and p19 expression in mouse and human AD brains by immunoblotting. In the mouse model of AD, we did not find cleavage of p23 in Tg-APP mice. The regulation of p23 abundance and activity in the transgenic mouse model of AD and in humans may not be similar; moreover, staging of AD in mice may not necessarily correspond to staging of the disease in humans, as the appearance of plaques precedes symptoms in humans, but follows symptom onset in mice. Finally, some features of the human disease, such as overt cell loss and pathology are not represented in the mice (Selznick et al. 1999; Dewachter et al. 2000; Gotz et al. 2004; McGowan et al. 2006). Thus, the 8- or 13-month-old AD mice that we chose for the analysis may not be in a state of chronic stress leading to p23 cleavage.
The profile is slightly different in human brain samples—p23 expression varied depending upon the severity of the AD and the age of the normal individuals. Levels of p23 were consistently higher in brain extracts from subjects with moderate AD (with one of the samples exhibiting cleaved p23) as opposed to younger normal individuals or those with early cognitive changes or even severe AD. A somewhat similar pattern was also observed with cleaved APP (Banwait et al. 2008) and nuclear p21-activated kinase (Nguyen et al. 2008). Of further interest, three brain extracts from younger normal individuals (N4–6, ages 20–53, Fig. 5c) had no detectable p23. The failure to detect p23 expression in these normal individuals suggests that other factors may influence p23 expression (Di Domenico et al. 2010). All five severe AD individuals had very low levels of p23. We speculate that as the disease advances, the activity and expression of p23 may be increased or decreased at two different levels depending on its function (e.g., protein–protein interaction or chaperone activity). In addition, because housekeeping genes may change expression in postmortem tissues of neurodegenerative diseases (Banwait et al. 2008; Nguyen et al. 2008), the level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) may not accurately reflect equal protein concentration and neuron number across groups; therefore, the low levels of p23 in severe AD samples might be due to changes in p23 protein expression or secondary protein loss as a result of cell death.
Our inability to observe p19 in mouse or human brain samples suggests that the usefulness of the p23 neo-epitope antibody is restricted to cells and primary neurons undergoing cellular stress. However, it should be noted that given a limited sample size, it is not particularly surprising that we did not observe the p19 fragment which may represent a transient species since it represents a caspase-cleavage product (Semple et al. 2007; Werner et al. 2007). Of note, the severe AD patients did display a consistent reduction in p23 levels. While there may be several reasons for the steady-state levels of p23 to be lower in this phase of the disease, it is also quite possible that the reduction may be due to cleavage to the p19 fragment.
Taken together, these findings suggest that the expression of p23 is generally higher in older normal individuals and in those with moderate AD (and in one case also observed as cleaved protein) possibly in an effort to stave off ER stress and neurodegeneration. Additional studies will be needed to determine the significance, if any, of the presence of p23 and p19 in AD and other neurodegenerative diseases that feature misfolded proteins.
Acknowledgments
We thank members of the Bredesen laboratory for helpful comments and discussions, Dr. David Toft (Mayo Medical School, Rochester, MN) for the human p23 cDNA, and Molly Susag and Rowena Abulencia for administrative assistance. This work was supported by grants from the National Institutes of Health NS33376 to D.E.B. and R.V.R and AG034427-02 to D.E.B.
Abbreviations
- ER
Endoplasmic reticulum
- pcd
Programmed cell death
- AD
Alzheimer's disease
- GRP
Glucose-regulated protein
- MEF
Mouse embryonic fibroblasts
Contributor Information
Karen S. Poksay, The Buck Institute for Research on Aging, 8001 Redwood Blvd, Novato, CA 94945, USA
Surita Banwait, The Buck Institute for Research on Aging, 8001 Redwood Blvd, Novato, CA 94945, USA.
Danielle Crippen, The Buck Institute for Research on Aging, 8001 Redwood Blvd, Novato, CA 94945, USA.
Xiao Mao, The Buck Institute for Research on Aging, 8001 Redwood Blvd, Novato, CA 94945, USA.
Dale E. Bredesen, The Buck Institute for Research on Aging, 8001 Redwood Blvd, Novato, CA 94945, USA; University of California, San Francisco, CA 94143, USA
Rammohan V. Rao, Email: rrao@buckinstitute.org, The Buck Institute for Research on Aging, 8001 Redwood Blvd, Novato, CA 94945, USA.
References
- Bakhshi J, Weinstein L, Poksay KS, Nishinaga B, Bredesen DE, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in mouse melanoma cells: effect of curcumin. Apoptosis. 2008;13:904–914. doi: 10.1007/s10495-008-0221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando Y, Katayama T, Kasai K, Taniguchi M, Tamatani M, Tohyama M. GRP94 (94 kDa glucose-regulated protein) suppresses ischemic neuronal cell death against ischemia/reperfusion injury. Eur J Neurosci. 2003;18:829–840. doi: 10.1046/j.1460-9568.2003.02818.x. [DOI] [PubMed] [Google Scholar]
- Banwait S, Galvan V, Zhang J, Gorostiza OF, Ataie M, Huang W, et al. C-terminal cleavage of the amyloid-beta protein precursor at Asp664: a switch associated with Alzheimer's disease. J Alzheimers Dis. 2008;13:1–16. doi: 10.3233/jad-2008-13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE, John V, Galvan V. Importance of the caspase cleavage site in amyloid-beta protein precursor. J Alzheimers Dis. 2010;22:57–63. doi: 10.3233/JAD-2010-100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in dopaminergic cells: effect of paraquat. Neuromolecular Med. 2008;10:333–342. doi: 10.1007/s12017-008-8047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Poksay KS, Kaundinya G, Hart M, Bredesen DE, Andersen JK, et al. Endoplasmic reticulum stress-induced cell death in dopaminergic cells: effect of resveratrol. J Mol Neurosci. 2009;39(1–2):157–168. doi: 10.1007/s12031-008-9170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, van Dorpe J, Spittaels K, Tesseur I, Van Den Haute C, Moechars D, et al. Modeling Alzheimer's disease in transgenic mice: effect of age and of presenilin1 on amyloid biochemistry and pathology in APP/London mice. Exp Gerontol. 2000;35:831–841. doi: 10.1016/s0531-5565(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Di Domenico F, Sultana R, Tiu GF, Scheff NN, Perluigi M, Cini C, et al. Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: an investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res. 2010;1333:72–81. doi: 10.1016/j.brainres.2010.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M, Cecconi F. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J Biol Chem. 2006;281:2693–2700. doi: 10.1074/jbc.M509110200. [DOI] [PubMed] [Google Scholar]
- Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- Felts SJ, Toft DO. p23, a simple protein with complex activities. Cell Stress Chaperon. 2003;8:108–113. doi: 10.1379/1466-1268(2003)008<0108:paspwc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Lee VM, Trojanowski JQ. ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci. 2003;26:407–410. doi: 10.1016/S0166-2236(03)00197-8. [DOI] [PubMed] [Google Scholar]
- Fosang AJ, Last K, Stanton H, Golub SB, Little CB, Brown L, et al. Neoepitope antibodies against MMP-cleaved and aggrecanase-cleaved aggrecan. Methods Mol Biol. 2010;622:312–347. doi: 10.1007/978-1-60327-299-5_19. [DOI] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, et al. Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, et al. Transgenic animal models of Alzheimer's disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry. 2004;9:664–683. doi: 10.1038/sj.mp.4001508. [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Rockenstein E, Crews L, Masliah E. Role of protein aggregation in mitochondrial dysfunction and neuro-degeneration in Alzheimer's and Parkinson's diseases. Neuromolecular Med. 2003;4:21–36. doi: 10.1385/NMM:4:1-2:21. [DOI] [PubMed] [Google Scholar]
- Hori O, Ichinoda F, Yamaguchi A, Tamatani T, Taniguchi M, Koyama Y, et al. Role of Herp in the endoplasmic reticulum stress response. Genes Cells. 2004;9:457–469. doi: 10.1111/j.1356-9597.2004.00735.x. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Toft DO. Binding of p23 and hsp90 during assembly with the progesterone receptor. Mol Endocrinol. 1995;9:670–678. doi: 10.1210/mend.9.6.8592513. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Beito TG, Krco CJ, Toft DO. Characterization of a novel 23-kilodalton protein of unactive progesterone receptor complexes. Mol Cell Biol. 1994;14:1956–1963. doi: 10.1128/mcb.14.3.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Galvan V, Huang W, Banwait S, Tang H, Zhang J, et al. Signal transduction in Alzheimer disease: p21-activated kinase signaling requires C-terminal cleavage of APP at Asp664. J Neurochem. 2008;104:1065–1080. doi: 10.1111/j.1471-4159.2007.05031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, et al. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer's disease and mild cognitive impairment: implications for progression of AD. J Proteome Res. 2009;8:471–482. doi: 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxelmark E, Knoblauch R, Arnal S, Su LF, Schapira M, Garabedian MJ. Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J Biol Chem. 2003;278:36547–36555. doi: 10.1074/jbc.M305960200. [DOI] [PubMed] [Google Scholar]
- Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, et al. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, Del Rio G, et al. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004a;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- Rao RV, Poksay KS, Castro-Obregon S, Schilling B, Row RH, Del Rio G, et al. Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J Biol Chem. 2004b;279:177–187. doi: 10.1074/jbc.M304490200. [DOI] [PubMed] [Google Scholar]
- Rao RV, Niazi K, Mollahan P, Mao X, Crippen D, Poksay KS, et al. Coupling endoplasmic reticulum stress to the cell-death program: a novel HSP90-independent role for the small chaperone protein p23. Cell Death Differ. 2006;13:415–425. doi: 10.1038/sj.cdd.4401761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EM, Jr, et al. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1020–1026. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Semple JI, Smits VA, Fernaud JR, Mamely I, Freire R. Cleavage and degradation of Claspin during apoptosis by caspases and the proteasome. Cell Death Differ. 2007;14:1433–1442. doi: 10.1038/sj.cdd.4402134. [DOI] [PubMed] [Google Scholar]
- Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Weaver AJ, Sullivan WP, Felts SJ, Owen BA, Toft DO. Crystal structure and activity of human p23, a heat shock protein 90 co-chaperone. J Biol Chem. 2000;275:23045–23052. doi: 10.1074/jbc.M003410200. [DOI] [PubMed] [Google Scholar]
- Weikl T, Muschler P, Richter K, Veit T, Reinstein J, Buchner J. C-terminal regions of Hsp90 are important for trapping the nucleotide during the ATPase cycle. J Mol Biol. 2000;303:583–592. doi: 10.1006/jmbi.2000.4157. [DOI] [PubMed] [Google Scholar]
- Werner ME, Chen F, Moyano JV, Yehiely F, Jones JC, Cryns VL. Caspase proteolysis of the integrin beta4 subunit disrupts hemidesmosome assembly, promotes apoptosis, and inhibits cell migration. J Biol Chem. 2007;282:5560–5569. doi: 10.1074/jbc.M603669200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Chung TF, Pyun HY, Fine RE, Johnson RJ. KDEL proteins are found on the surface of NG108-15 cells. Brain Res Mol Brain Res. 1999;72:121–128. doi: 10.1016/s0169-328x(99)00188-6. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Armstrong JS. Bax and the mitochondrial permeability transition cooperate in the release of cytochrome c during endoplasmic reticulum-stress-induced apoptosis. Cell Death Differ. 2006;14:703–715. doi: 10.1038/sj.cdd.4402072. [DOI] [PubMed] [Google Scholar]