

Abstract
The synthetic antimicrobial peptide CM15, a hybrid of N-terminal sequences from cecropin and melittin peptides, has been shown to be extremely potent. Its mechanism of action has been speculated to involve pore formation based on prior site-directed spin labeling studies. This study examines four single-site β-thiocyanatoalanine variants of CM15 in which the artificial amino acid side chain acts as a vibrational reporter of its local environment through the frequency and lineshape of the unique CN stretching band in the infrared spectrum. Circular dichroism experiments indicate that the placements of the artificial side chain have only small perturbative effects on the membrane-bound secondary structure of the CM15 peptide. All variant peptides were placed in buffer solution, in contact with dodecylphosphatidylcholine micelles, and in contact with vesicles formed from E. coli polar lipid extract. At each site, the CN stretching band reports a different behavior. Time-dependent attenuated total reflectance infrared spectra were also collected for each variant as it was allowed to remodel the E. coli lipid vesicles. These experiments agree with the previously proposed formation of toroidal pores, in which each peptide finds itself in an increasingly homogeneous and curved local environment without apparent peptide-peptide interactions. This work also demonstrates the excellent sensitivity of the SCN stretching vibration to small changes in peptide-lipid interfacial structure.
Keywords: antimicrobial peptides, cyanylated cysteine, nitrile vibrations, membrane-active peptides, site-directed labeling, toroidal pores
Characterizing the structure, structural distribution, and membrane binding geometry of peripheral membrane proteins (PMPs) is a challenge that requires new experimental approaches. Recent advances in crystallography using surfactants(1) and solid state NMR of oriented samples(2) have led to new insights into the structure and function of a number of such proteins and peptides. However, a hallmark of these studies is that the experimental approach used depends to a large extent on the lipid and protein system of interest without a single, unified approach that is applicable to proteins of any size in arbitrary lipid systems. EPR spectroscopy of site-directed spin labels has this sought-after flexibility when applied to PMPs,(3) with documented limitations due to the size and chemical nature of the most typical spin label(4) and the need for external solution- or lipid-phase paramagnetic species to address directly the extent of membrane burial.(3) Antimicrobial peptides (AMPs) are a subset of membrane-active species generating much recent interest due to their possible use as antibiotic agents in an era of global overuse of antibiotic drugs.(5) The relative simplicity of their sequences belies the complexity involved in their function, and with a few notable exceptions, the mechanism of action of many of these peptides has still not been clearly determined.(6) This challenge is largely due to a paucity of direct experimental techniques that can reveal the membrane-bound structure of such peptides in contact with their target native lipid systems (or other cellular structures) during periods relevant to their antimicrobial activity.
AMPs that disrupt target membranes have been proposed to do so by first binding to the surface of the bilayer.(7) This initial binding event is postulated to result in expansion of the outer leaflet of the lipid bilayer, after the peptide has reached a critical concentration, followed by membrane thinning. While the events that follow initial binding depend greatly on the particular AMP, several models of membrane disruption, leading to cell death from membrane destruction or permeabilization, have been proposed.(8) One general mechanism for the action of antimicrobial peptides is the formation of pores. The formation of tight pores mediated by peptide aggregation is referred to as the barrel-stave model, while the formation of wide pores, in which peptides are separated by intervening phospholipids and a strong local curvature is induced in the vicinity of the pore with the head groups knitting together the two leaflets, is called the toroidal pore model.(8) An alternative proposed mechanism of AMP membrane disruption is the detergent-like disintegration mechanism, in which the peptides permeate the lipid bilayer after reaching a critical concentration and remove pieces of the bilayer without translocating into the hydrophobic core.(8, 9) Analytical techniques with many different native length- and time scales have been used to show evidence (which is often indirect) of each of these mechanisms in varying AMP/lipid systems.
CM15 is a 15-residue long antimicrobial peptide that is a hybrid of the first seven residues of cecropin A and residues 2–9 of mellitin.(10, 11) Cecropin A is a naturally occurring, 37-residue, helical antimicrobial peptide found in the silk moth Hyalophora cecropia and mellitin is a 26-residue peptide found in the venom of the honey bee Apis mellifera. While mellitin has potent, broad-spectrum antimicrobial activity, it is extremely hemolytic, causing damage to host red blood cells. In contrast, cecropin A shows low cytotoxicity, but has lower antimicrobial activity.(8) The CM15 hybrid was first created by Andreu (11) to combine the favorable characteristics of each AMP. Although a number of cecropin-mellitin hybrids have been synthesized, CM15 is the shortest sequence displaying both broad-spectrum antimicrobial activity and low cytotoxicity.(11) Its short length facilitates its solid-phase synthesis, making CM15 an ideal AMP for detailed studies of its action on model membranes.
The interaction between CM15 and lipids has been studied by several groups since CM15’s introduction. Feix and co-workers used EPR-SDSL and LUVs formed from E. coli polar lipid extract to model CM15’s action on microbes.(8, 12) They generated six different single cysteine mutants of CM15 to attach the spin label at different sites and used the spin labels to map the peptide’s membrane immersion depth as a function of residue site. They concluded that the labels furthest towards hydrophobic face of the peptide are positioned at approximately 12.5 Å below the membrane surface, while the labels on the hydrophilic face reside are ~ 2.5 Å above the surface. These results suggest that CM15 initially binds with the helical axis ~5 Å below the membrane surface, in agreement with the initial docking step in the two-state peptide-bilayer interaction model proposed by Huang.(13) The Feix group also explored CM15’s ability to form pores in E. coli and Pseudomonas aeruginosas membranes via osmoprotection, which determines the size of transmembrane channels. These studies suggested that CM15 forms pores of diameter 2.2–3.8 nm.(14) Further EPR-SDSL studies suggested that the pores formed are not consistent with the tight “barrel-stave” pore model, since strong spin-spin coupling, which would be evidence of peptide-peptide contacts, was not observed.(15)
Bastos et al. (16) studied the interactions between CM15 and model LUVs formed from DMPC using FTIR centered on the lipid head groups. By following the lipid carbonyl stretch as a function of peptide concentration, they reported that the lipid remained ordered in the presence of increasing peptide. The group also concluded that the peptide becomes sequestered in localized pores, leaving the bulk lipid unperturbed. This report of pore formation by CM15 agrees with the conclusions made by the Feix group about the peptide’s mode of membrane perturbation.
Due to its relatively well-characterized initial orientation when bound to model membranes, CM15 has recently been used as a model system to develop new analytical techniques designed for general use in membrane-bound peptides and proteins. Zangger, et al. (17) employed NMR paramagnetic relaxation techniques to explore CM15’s orientation and immersion depth in DPC micelles. They found that the hydrophobic side of the helix interacts with the hydrophobic core of the micelle, while the hydrophilic side of the helix is closer to the micelle surface but remains immersed in micellar samples. Additionally, the length of the charged lysine residues on CM15 allows them to “snorkel” into DPC’s polar head group region in an analogous fashion to the “snorkeling” observed for charged or hydrophobic residues at lipid surfaces.(18) Most recently, we used two central residues of CM15 to demonstrate the ability of cyanylated cysteine (C*) to act as a site-specific infrared reporter of membrane binding.(19) The current work aims to characterize further CM15’s interactions with lipid-water surfaces in model and physiological lipid systems using this novel vibrational probe group, with ramifications for refining both the membrane-bound structure and remodeling activity of CM15 and understanding the particular sensitivity of cyanylated cysteine in a membrane-bound context.
The β-thiocyano group of C* leads to a CN stretching band that appears in the infrared absorption spectrum between 2153–2164 cm−1 in biologically relevant environments.(19–26) This band, which is moderately strong and can be observed clearly in aqueous samples of 25 μm thickness at approximately 1 mM concentration, is sensitive to its local environment in two ways. The frequency depends on water exposure (via a blue shift due to weak hydrogen bonds donated by water) and the local electrostatic environment (via a red shift with increasing solvent polarity).(22, 26) The lineshape in homogeneous environments is most typically a Voigt profile that varies from narrow and more Lorentzian in character to broad and Gaussian. This lineshape trend has been interpreted as evidence that C* can report on solvent dynamics through a motional narrowing mechanism in solvents with faster fs-ps dipolar reorientation dynamics.(26) The CN band of C* has been used to map electrostatic potential in enzyme active sites(21, 22, 24), at a binding interface between two structured proteins,(25) and also to document site-specific order/disorder transitions in both model peptides(27) and intrinsically disordered proteins.(20)
A similar nitrile-derivatized amino acid, p-cyano-phenylalanine(28), was used as a probe of membrane binding in peptides bound to membranes(29) and to the interior surface of reverse micelles.(30) The CN stretching band of this side chain exhibits a red shift qualitatively similar to that of C* on burial in the membrane and the resulting local exclusion of water. Two peptides derived from natural sequences, of which CM15 is one, were used recently in contact with DPC micelles to demonstrate that the CN stretching band of C* is also a sensitive probe of the membrane exposure of labeled side chains.(19) C* is projected to be a widely useful probe of membrane-protein interactions, since it can be incorporated into sequences of arbitrary size via site-directed mutagenesis to cysteine followed by chemical ligation of the CN group at the free thiol. Here C* is used to map the membrane-bound structure of CM15, a short peptide, but its usefulness is not limited to such short sequences.
Two centrally located residues in CM15’s sequence were chosen previously as sites for introduction of the C* vibrational probe group. One of these residues (Ala10) is known to be at least partially solvent-exposed and the other (Ile8) is deeply buried in contact with multiple lipid interfaces.(8, 12) In addition, for this study, one position at each end of the peptide sequence was chosen for attachment of the probe to examine peptide-lipid interactions and local structure and dynamics near the sequence termini. The last residue on either end of the sequence was avoided since the absolute termini help to cap the helix-forming sequence and have major effects on the overall helical propensity when binding to membranes.(14) To avoid the absolute C-terminus and possible capping effects, the second-to-last residue, Val14, was replaced with C*. When Lee, et al. compared the sequences of ten different cecropins, they found that all ten contained Trp2 and seven contained Lys3.(31) Because these two residues are highly conserved among cecropins, the fourth N-terminal position, Leu4, was selected as the site closest to the N-terminus to introduce the vibrational probe with minimal perturbation of the bound structure. Table 1 shows the chosen variant sequences. Figure 1 is a helical wheel diagram of membrane-bound CM15 based on previous EPR studies by the Feix group(8, 12, 15), showing the sites where C* was introduced as well as the residue positions in relation to the lipid-solvent interface.(12, 15)
Table 1.
Amino acid sequences of synthesized CM15 variants, where C*= cyanylated cysteine.
| Peptide | Sequence |
|---|---|
| Unmodified CM15 | Ac-KWKLFKKIGAVLKVL-NH2 |
| L4C* | Ac-KWKC*FKKIGAVLKVL-NH2 |
| I8C* | Ac-KWKLFKKC*GAVLKVL-NH2 |
| A10C* | Ac-KWKLFKKIGC*VLKVL-NH2 |
| V14C* | Ac-KWKLFKKIGAVLKC*L-NH2 |
Figure 1.
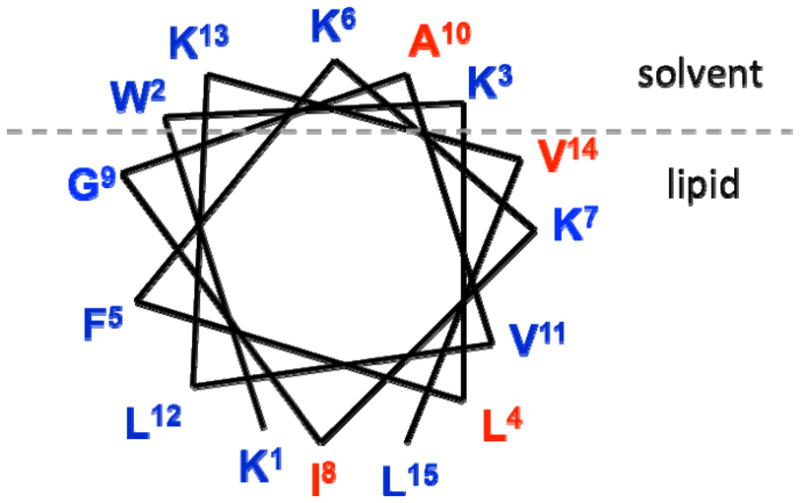
Helical wheel diagram for CM15 at the lipid/solvent interface based on previous studies (8, 12), with sites chosen for substitution with cyanylated cysteine in red.
The four chosen single-C* variants of CM15 are used here to examine its structure in three different environments: aqueous buffer solution, DPC micelles, and bacterial polar lipid extract from E. coli cells (BPL). The aqueous samples are used to show the sensitivity of the probe group to the peptide environment without the presence of lipid. The micelle-bound samples are used to produce CD spectra free of any spectral warping from scattered light and to indicate the basic propensities of side chains for burial at a generic phospholipid interface. The E. coli lipids present an environment in which CM15 adopts an active structure which porates the lipid vesicles. The spectrum of each side chain label is examined over time during this process to provide a site-specific probe of each residue’s micro-environment in the vesicle-bound structure while membrane remodeling occurs.
Experimental Procedures
Materials
Fmoc-labeled amino acids and all peptide synthesis reagents were purchased from Advanced Chem Tech, except N,N-diisopropylethylamine (DIEA; Pharmco-AAPER) and acetic anhydride (Aldrich). Peptide cleavage reagents were purchased from Aldrich and high-performance liquid chromatography (HPLC) solvents were purchased from Pharmco-AAPER. Sodium phosphate monobasic monohydrate (NaH2PO4 · H2O), sodium phosphate dibasic (Na2HPO4), MOPS (3-(N-morpholino)propanesulfonic acid), and KCl were purchased from J.T. Baker Chemical Co., Fisher Chemical, and Aldrich, respectively. 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) was purchased from Acros and NaCN and D,L-dithiothreitol (DTT) was purchased from Aldrich. All previously mentioned purchased items were used as received. Dodecylphosphocholine (DPC) and E. coli Bacterial Polar Lipid Extract (BPL) were purchased from Avanti Polar Lipids dissolved in chloroform. The composition of BPL is 67:23.2:9.8 (weight percent) phosphatidylethanolamine (PE): phosphatidylglycerol (PG): cardiolipin (CL). All aqueous solutions were prepared with doubly deionized, degassed, Milli-Q quality water.
Peptide synthesis and purification
All peptides were synthesized on a 0.1 mmol scale on an Applied Biosystems ABI 443A synthesizer using standard 9-fluorenylmethoxycarbonyl (Fmoc) chemistry. For each peptide, the C-terminal amino acid, all β-branched amino acids, and each single residue preceding a β-branched amino acid were double-coupled. A PAL resin solid-phase support was used to furnish a C-terminal carboxamide upon cleavage and acetic anhydride was used for N-acetylation. Purity of peptides was verified by reverse-phase HPLC using an analytical-scale Microsorb 100-5 C18 250 × 4.6 mm column (Varian, Inc.) with a flow rate of 1 mL/minute and a 0–100% or 20–70% gradient of acetonitrile in water with 0.1% trifluoroacetic acid as the modifier. Identity of peptides was confirmed via MALDI-MS performed at the Wistar Institute, Philadelphia.
Cyanylation of peptides
Each cleaved, lyophilized peptide was treated for 20 min with 0.01 M HCl and relyophilized to remove remaining trifluoroacetate ions. The HCl-treated peptides were redissolved in 50 mM sodium phosphate buffer, pH ~7.5, and treated with 100x DTT to yield the free thiol at the single cysteine side chain. DTT was separated from the peptides using size exclusion chromatography (28 cm × 10 mm column of Sephadex G-10 equilibrated in 50 mM sodium phosphate buffer, pH ~ 7) and the reduced peptides with buffer salts were lyophilized. The reduced peptides were redissolved in 50 mM sodium phosphate buffer, pH ~6.5, and treated with 5 molar equivalents of DTNB dissolved in 200 mM sodium phosphate buffer, pH 7.0, for 20 minutes to form a mixed disulfide at the cysteine, turning the solution yellow. The solution was then treated with 50 equivalents of NaCN dissolved in 50 mM sodium phosphate buffer, pH 6.5, to yield a thiocyanate at the cysteine, turning the solution orange. The cyanylated peptide was isolated using the 28 cm × 10 mm column of Sephadex G-10 equilibrated in either 20 mM sodium phosphate buffer, pH ~ 7 (for DPC experiments) or 20 mM MOPS and 100 mM KCl buffer solution, pH 6.5 (for BPL experiments). Peptide-containing fractions were concentrated ~5x using a Savant Speed Vac SCV100 centrifugal vacuum device, and the presence of the nitrile moiety on the peptide was verified using infrared spectroscopy. The final sample concentration was 2 mM in peptide.
Preparation of lipid-containg samples
DPC and BPL were dried down from chloroform stock solutions under a stream of nitrogen gas. For DPC samples, the resulting lipid film was hydrated with the speed vacuumed peptide solution, resulting in samples that were approximately 2 mM peptide in 100 mM DPC. For BPL samples, BPL was further dried for ~ 1 hour under vacuum and the resulting film was hydrated with 20 mM MOPS/100 mM KCl buffer, pH 6.5. Large unilamellar vesicles (LUVs) were prepared by freeze-thawing the hydrated BPL solution 5x, followed by extrusion 13x through 100 nm polycarbonate membrane filters, using a Mini-Extruder (Avanti Polar Lipids). The speed vacuumed peptide solution (30 μL) was added to the extruded BPL solution, resulting in samples that were approximately 1–2 mM peptide in 50 mM BPL.
Far-UV circular dichroism
CD spectra were collected between 190 and 250 nm on an Aviv model 410 spectropolarimeter. Speed vacuumed, cyanylated peptide samples were diluted 100x to yield a solution of 20–30 μM peptide in 1–2 mM sodium phosphate buffer. For the native sequence, lyophilized peptide was dissolved in 5 mM sodium phosphate buffer, pH 6.5–7.0, yielding a peptide concentration of ~ 30 μM. These samples were analyzed in a 1 mm quartz cell. For spectra in DPC, dried DPC was hydrated with the 2–3 mM peptide solution, resulting in a 100 mM DPC concentration, and samples were analyzed without dilution in a 0.1 mm quartz demountable cell (Starna Cells).
FTIR spectroscopy
For spectra in buffer and 100 mM DPC, cyanylated peptide samples were placed between two windows of a 22 μm CaF2 BioCell (BioTools, Jupiter, FL) placed inside a Biojack temperature-circulating jacket held at 25°C. Spectra were collected at 2 cm−1 resolution (1024 scans) using a Bruker Optics Vertex 70 FTIR spectrometer with a photovoltaic HgCdTe detector. A spectrum of buffer solution or 100 mM DPC in buffer solution was subtracted from the raw spectrum, and further baseline correction was accomplished by fitting the baseline outside the region from 2145 to 2180 cm−1 to a polynomial and subtracting the fit.
ATR-IR was used for peptides in 50 mM BPL due to the opacity of the samples and the additional advantage of selectively viewing species that cease to be well-suspended in solution. After adding peptide to extruded BPL solution and shaking slightly to homogenize, samples were immediately deposited manually onto a zinc selenide crystal in an ATR trough cell (Pike Technologies, Madison, WI) that was then sealed to maintain hydration. Spectra were collected at 2 cm−1 resolution (1024 scans) on the same FTIR spectrometer as above. A spectrum of 100 mM BPL in 20 mM MOPS/100 mM KCl was subtracted from the raw spectrum and baseline correction was accomplished as described above. Spectra of cysteine analogs in BPL were acquired approximately every half hour for twelve hours and changes in the spectra over time were documented.
Infrared Data analysis
Lineshapes for the CN stretching bands were analyzed with no assumption about the symmetry of the lineshape, in constrast to prior analyses of the same band in more homogeneous samples. The mode (most probable, or maximum) frequency was directly drawn from the data. Mean CN stretching frequencies were calculated using equation (1) for the first central moment of the distribution:
| (1) |
Where ω is the frequency in wavenumbers and I(ω) is the absorbance as a function of frequency. The variance for each CN stretching band was calculated as the second central moment of the distribution:
| (2) |
Results and discussion
Peptide synthesis and modification
All cleaved peptides were nearly analytically pure according to analytical HPLC, so repeated size exclusion chromatography associated with chemical ligation at cysteine was the only purification used. Cyanylation of cysteine residues followed published procedures with nearly quantitative yield according to infrared results showing a clear SCN stretching band. It is worth noting again that although this labeling methodology is applied here in the context of short, synthetic peptides, the general technique is expected to be of use in proteins of arbitrary size with an expression system that allows placement of a single cysteine residue at sites of interest. Indeed, the preparation of samples in this study is in some ways slightly more difficult than isolating single C* variants of larger proteins reported elsewhere(20, 22, 24) due to the lack of commercially available size-exclusion membranes effective for small peptides (<3000 Da) and the resulting requirement for more selective size-exclusion chromatography.
Far-UV circular dichroism
Figure 2 presents CD spectra of all CM15 variants, including the non-cysteine-containing original sequence, in aqueous buffer solution (diluted for solubility reasons) and in contact with DPC micelles (at the same high peptide concentrations as infrared measurements). Due to excessive light scattering and resulting wavelength-dependent spectral warping, CD spectra were not acquired for the cyanylated mutants in E. coli lipids. In the micellar CD experiments, the global secondary structure of CM15 in the lipid-bound state is not greatly perturbed by the placement of the artificial side chain at the four chosen sites. The DPC-bound CD spectra are nearly sumperimposable for all peptides except V14C*. Each spectrum exhibits significant α-helical character with a strong minimum at 208 nm and evidence of a weaker negative feature at 222 nm (yielding R values(32) of .67(V14C*)-.72(WT) for all DPC-bound peptides). For such a short peptide, determination of % helicity via fitting to standard helical and random coil spectra would not be particularly useful since the number of chromophores participating in the amide excitons is small, and in the presence of lipid, it is likely that the bound structural distribution of CM15 does not follow a simple two-state helix/coil structural distribution. In the case of V14C*, the shape of the spectrum remains similar to that of the other peptides but there is a decrease in the molar ellipticity measured for the peptide in contact with DPC micelles. This is likely due to partial unfolding of the peptide at the C-terminus, which is apparently increased by the presence of the less-hydrophobic C* in place of the more hydrophobic valine. This possibility is further discussed below in the context of IR results at the same site. A lack of complete quantitative agreement between all DPC-bound CD spectra indicates that there is a small perturbative influence of each C* placement on CM15’s DPC-bound structure; this is not particularly surprising in the context of recent measurements of C*’s effect on the helical stability of alanine-repeat peptides.(27) Whether these secondary structural changes affect the orientation of the peptide with respect to the micelle cannot be determined from CD spectra, which report only on the relationships between backbone amide groups of the peptide.
Figure 2.

Far-UV circular dichroism for all CM15 peptides in aqueous buffer and in DPC micelle solution. C* locations are marked on each plot.
Although the CD spectra for the cysteine mutants in DPC do not show large changes compared to the CD spectrum of the native peptide in DPC, the spectra in aqueous buffer are all significantly altered as compared to the native sequence. All buffer samples were highly visually transparent and displayed no macroscopic evidence of aggregation, so the assumption is that no inter-peptide aggregation occurred in any of these samples. The native sequence displays an aqueous CD spectrum with a minimum at 201 nm and a very weak higher-wavelength feature near 222 nm, which could be interpreted as a mainly random coil spectrum with some small residual signal from helical structures. The aqueous CD spectra for each mutant exhibit two minima at varying wavelengths and MREs, indicating that each peptide has a different latent helical propensity in an aqueous environment. The spectra with lower-wavelength minima generally display weaker negative high-wavelength features, suggesting that this group of mutants might be described as having different helical propensities lying along a general helix-random coil reaction coordinate. Interestingly, the L4C* and A10C* peptides appear to be more helical in buffer solution than the native sequence. The CD results in buffer indicate that the introduction of the vibrational probe at different sites causes steric changes that lead to a different average global secondary structure in aqueous buffer for the C*-containing variants, with some residual helical propensity remaining for all modified sequences as evidenced by the weak, high-wavelength negative feature in each of the aqueous buffer spectra. C* is not expected to be a conservative mutation at any of the chosen sites, but the basic helical propensity and mostly helical DPC-bound structure of the peptide are maintained in all C*-variant peptides.
A complementary observation of secondary structure would be the infrared amide I absorption band for each peptide. One particular advantage of nitriles as vibrational probes is that CN stretching vibrations are clearly visible in H2O solutions, which were used for all samples here. Collection of amide I signals from these samples would necessitate the use of D2O as the solvent, thus the current reliance on CD spectra to estimate the peptides’ secondary structure.
Infrared spectroscopy
Figure 3 shows the infrared absorption spectra in the nitrile stretching region for L4C*, I8C*, A10C*, and V14C* in aqueous buffer, DPC micelles, and E. coli lipid vesicles. The mean and mode frequencies and the calculated variances for each of the CN stretching bands in Figure 3 are presented in Table 2. In Figure 3, there is little variation in the frequency or linewidth of the aqueous samples, suggesting that the artificial side chain in each sample senses a similar aqueous environment without the water exclusion and resulting red shift that might be expected if the peptides were aggregated. A possible exception is A10C*: this is discussed below.
Figure 3.

Infrared CN stretching absorption bands for C*-labeled CM15 variants in aqueous buffer solution, DPC micelles, and BPL solution after 20 minutes of exposure. The non-scaled maximum absorbance of all samples was between 200 and 800 μO.D.
Table 2.
Analysis of CN stretching bands from infrared data in Figure 3: all units are cm−1.
| Buffer | DPC | E.coli lipids | |||||||
|---|---|---|---|---|---|---|---|---|---|
| (ω) | mode | σ | (ω) | mode | σ | (ω) | mode | σ | |
| L4C* | 2161.6 | 2161.5 | 4.7 | 2159.0 | 2159.6 | 5.0 | 2157.6 | 2157.7 | 5.2 |
| I8C* | 2162.8 | 2163.5 | 4.2 | 2158.2 | 2159.6 | 6.0 | 2160.2 | 2157.7 | 5.4 |
| A10C* | 2161.2 | 2160.6 | 7.3 | 2162.1 | 2160.1 | 8.4 | 2160.9 | 2161.5 | 8.6 |
| V14C* | 2162.8 | 2163.5 | 5.3 | 2159.8 | 2158.7 | 4.3 | 2160.5 | 2159.0 | 5.4 |
Time-dependent infrared spectra in the same spectral region for each labeled peptide in contact with E. coli lipids over a period of seven hours after initial mixing are presented in Figure 4A–D. The mean frequencies and variances of all CN bands as a function of time are shown in Figure 4E–F. The most obvious trend in all panels A–D of Figure 4 is an increase in the band’s signal intensity with time. Because the absorbance of the band is proportional to the sample concentration near the surface of the ATR crystal, the increase in the CN band’s absorbance with time indicates that the surface-localized peptide concentrations increased continuously over the seven hour time period. An increase in signal intensity is most likely due to separation of the peptide-lipid complex from the solution, which would lead to a higher concentration of peptide near the horizontal ATR crystal. This possible cause of the increase in signal intensity is supported by the observation of visible precipitates upon removing the sample from the IR spectrometer. These precipitates are likely due to the penetration and remodeling of lipid vesicles by CM15 during the waiting period. It is important to note that over several hours, the ATR signal from the peptide only increases by a factor of about 2 with the formation of these insoluble species, which are certainly not peptide aggregrates but are instead the end product of vesicles destroyed by CM15 and thus mainly composed of lipids (as evidenced by much greater IR absorbance in the C-H stretching region, data not shown).
Figure 4.

(A)–(D) Infrared CN stretching absorption bands for C*-labeled CM15 variants (with label location marked on each plot) as a function of time in BPL solution. (E) Average frequencies and (F) peak variances for all CN bands as a function of time.
A detailed inspection of the infrared results from each probe site begins in the center of the peptide and moves outwards.
A10C*
Although alanine is a hydrophobic residue, it has been reported that Ala10 is positioned on the solution-exposed side of the amphipathic α-helix formed when CM15 is in the membrane-bound state. EPR-SDSL studies showed that a long spin labeled side chain positioned at the tenth residue was solvent exposed when the peptide was bound to E. coli lipids.(8, 12) In contrast, recent NMR study concluded that residues on the hydrophilic side of CM15, including the short native Ala10, remain buried in DPC micelles, but they are closer to the micelle-solution surface than residues on the hydrophobic face of the helix.(17) Because Ala10 was characterized as a solvent-exposed residue by EPR, A10C* was previously chosen as a control for the examination of cyanylated cysteine’s sensitivity to membrane burial versus solvent-exposure.(19) If C* is sensitive to membrane burial and the tenth residue position is solvent-exposed, then there should be no change in the CN stretching band of A10C* in a lipid versus aqueous environment.
Solvent dependent studies of methyl thiocyanate (MeSCN) and other studies of C* in water-solvated peptides showed that the maximum infrared absorption of the aliphatic thiocyanate nitrile stretching band occurs at 2163 cm−1 in aqueous environments.(23, 26, 27) Thus, it was expected that A10C*’s CN stretch in buffer, DPC, and E. coli lipids would be centered at 2163 cm−1. In Figure 3A and Table 2, the mean CN stretching frequencies for A10C* are 2161.2 (buffer), 2162.1 (DPC), and 2160.9 cm−1 (E. coli lipids). The frequency modes are 2160.6 (buffer), 2160.1 (DPC), and 2161.5 cm−1 (E. coli lipids). The mean and mode frequencies of A10C*’s CN stretch do not differ to a large extent between the three environments. This result indicates that A10C* is in a similar solvent environment in all three samples, an observation that agrees qualitatively with the EPR observation of solvent exposure of the spin label in place of A10.(8, 12, 15)
However, the CN frequency is significantly lower than 2163 cm−1 in all three spectra, suggesting that the environment of C* is not fully aqueous regardless of membrane exposure. Bischak (20) examined MeSCN’s CN stretch in varying ratios of water:THF and showed that the CN stretch occurs near 2161 cm−1 in 80% water/20% THF; if this result is used as a benchmark, then the A10C* side chain is still in a predominantly solvent-exposed environment under all conditions, consistent with the wheel diagram in Figure 1.
CD data (Figure 2D) indicate that A10C* is at least partly helical (R=1.02, with lower MRE than in the bound form) in aqueous buffer. Due to the high occurrence of hydrophobic residues in the sequence, the peptide is likely at least partially collapsed, leading to interactions between the artificial side chain and hydrophobic or partly water-excluded regions of the peptide. When compared to the mean CN frequencies of the other cyanylated peptides in buffer, A10C*’s mean frequency is the lowest. While all of the other label sites are adjacent to a lysine, the tenth position is surrounded by two hydrophobic residues on either side. Thus C* is located in a more hydrophobic part of the peptide in A10C* than in the other single-C* variants. The CN frequency lower than 2163 cm−1 could be explained through interactions with hydrophobic nearest-neighbor side chains in the collapsed and/or folded aqueous peptide.
For the lipid-bound samples, where A10C* is folded akin to the native sequence, cyanylated cysteine’s rotational freedom about the β-carbon and S atom may also cause the solvent-exposed nitrile group to come into transient contact with the membrane-solvent surface, possibly leading to membrane-bound A10C*’s slightly red shifted mean frequency compared to 2163 cm−1. Similar frequencies (near 2159–2161 cm−1) for this probe group have been recently observed by Webb et al. along a well-structured protein binding interface: the interpretation from that system suggested that partially water-exposed sites can exhibit such frequencies if their local electrostatic environments are suitably perturbed by directional electric fields(25), which is a distinct possibility near the polar head group region and the lipid-water interface. At this particular site in CM15, a clear explanation for the slightly low frequency of this mostly solvent-exposed probe in the membrane-bound form of the peptide is currently elusive. However, the environment of A10C* is nearly the same in the absence of lipid, in contrast to all of the other label sites. Our diagnosis of predominant solvent exposure of the A10C* side chain agrees with prior EPR results and disagrees with the NMR assignment of A10 as buried in DPC micelles. The disagreement with NMR could be due simply to the greater length of the artificial C* side chain (approximately 3–5 Å from the N atom to the backbone) or the spin-labeled MTSL side chain in EPR (~6–7 Å in length(33)) in place of the much shorter A10 (approximately 2.1 Å from the methyl H atoms to the backbone).
Edelstein et al. reported that the CN stretching band of cyanylated cysteine in alanine-repeat helical peptides is weakly sensitive to changes in the local secondary structure of the peptides: a more helical local secondary structure leads to a broader CN linewidth.(27) This linewidth change with secondary structure formation was also observed in experiments in which C* was placed in a domain of the measles virus nucleoprotein known to form a helix when bound to a physiological partner.(20) Here, the lineshape analysis in Table 2 indicates that A10C*’s CN stretching band broadens in E. coli lipids as compared to aqueous buffer. The variance of the CN stretching band is 8.6 cm−1 in lipids versus 7.3 cm−1 in buffer. Because A10C* adopts a helical structure in lipids, but is less structured in aqueous environments, the linewidth broadening upon membrane binding may be a result of the peptides’ forming a helical secondary structure while the probe remains mainly solvent-exposed.
When A10C* was exposed to E. coli lipids over time (Figure 4A), the frequency and linewidth did not change appreciably (Figure 4E–F). The probe at the tenth site retains the same degree of solvent exposure even after several hours of contact with bacterial lipids and noticeable changes in the lipid morphology. Thus the parallel orientation of CM15 vs. the membrane surface does not vary as CM15 acts on its lipid environment.
I8C*
Feix et al. determined that the eighth residue in CM15’s sequence is deeply membrane-buried in the presence of E. coli lipids.(8, 12, 15) NMR experiments in DPC micelles agreed, with Ile8 immersed in the hydrophobic core of the micelle.(17) Solvent dependent studies showed that the thiocyanate CN infrared absorption exhibits a red shift of up to 10 cm−1 in non-hydrogen-bonding, polar solvents such as THF and DMSO.(19, 23, 26, 34) If THF is a good model for the environment below the membrane interface, largely water-excluded but with substantial dipolar character, then I8C*’s CN stretch is expected to red shift by a significant amount in DPC and E. coli lipids versus in aqueous solution.
As expected, the I8C* CN stretching band in the DPC and lipid environments (Fig. 3B) is red shifted compared to the aqueous spectrum. Table 2 shows that the mean frequency exhibits a red shift of 4.6 cm−1 in DPC and a red shift of 2.6 cm−1 in E. coli lipids, while the mode shifts by 3.9 cm−1 in DPC and 5.8 cm−1 in E. coli lipids versus buffer. These results indicate that I8C* is buried when CM15 is membrane-bound and agree qualitatively with prior EPR-SDSL and NMR studies. The changes in mean and mode are a quantitative indication that the lineshape in E. coli lipids is skewed with a substantial shoulder at higher frequencies. The CN stretching band in E. coli lipids is also noticeably narrower than either the aqueous or DPC-bound bands.
The linewidth of methyl thiocyanate’s CN stretch narrows significantly in THF and alkane solvents compared to more dipolar solvents, such as water.(19, 26) This phenomenon may result from a smaller inhomogeneous frequency distribution imposed by the near-zero dipoles of the non-aqueous solvent molecules. I8C*’s spectra in buffer, DPC, and E. coli lipids indicate that the peptide’s CN stretching band narrows in E. coli lipids only. This result could be explained by peptide-lipid head group electrostatics. Feix measured CM15’s membrane-binding affinity in PE/PG versus E. coli lipids, which are composed of the anionic phospholipids PG and CL, as well as the zwitterionic phospholipid PE. They determined that CM15 had a greater binding affinity for the more anionic E. coli lipids.(35) CM15 should also have a greater binding affinity for E. coli lipids versus DPC, which is zwitterionic. The electrostatic attraction between the cationic, lysine-rich peptide and the anionic lipid head groups would cause the peptide to bury more uniformly in the vesicles, with the eighth residue further on average from the lipid-solvent interface than in the DPC samples. If the I8C* probe is buried more uniformly in the lipids and more excluded from the solution and the head groups (which interact strongly with lysine), then it would be exposed to a more homogeneous, less dipolar environment, resulting in a more narrow distribution of CN stretching frequencies.
This observation of homogeneity of burial in I8C* highlights a central difference of C* vs EPR spin labels near the membrane interface: the frequency variation (or the lineshape) of C* should be able to report directly on the distribution of environments sampled by a specific side chain, rather than just reporting the average exposure to lipid- or solution-phase paramagnetic species that yields a single depth number in EPR experiments.
Figure 4B exhibits two trends in the I8C* peptide’s CN stretch over the course of seven hours of exposure to BPL. The first trend is a decrease in the band’s mean frequency, shown quantitatively in the plot of mean frequency versus time (Figure 4E), from 2160.2 cm−1 after 20 minutes of incubation in the E. coli lipid solution to 2158.3 cm−1 after seven hours, a shift of −1.9 cm−1. The gradual red shifting of the mean frequency suggests that the probe is exposed to a less H-bonding environment over time. The second trend in Figure 4F is a decrease in variance over time, from 5.4 cm−1 after 20 minutes to 2.5 cm−1 after seven hours, as a small high-frequency shoulder vanishes from the spectrum. The I8C* probe’s local environment becomes increasingly homogeneous and less water-exposed with time.
L4C*
This site is membrane-buried when CM15 is bound to E. coli lipids, with a burial depth similar to that of the eighth residue according to EPR.(12) As expected, the CN stretching band in the DPC and lipid environments for L4C* (Figure 3C) is red shifted from the aqueous spectrum. The mean frequency exhibits a red shift of 2.6 cm−1 in DPC and a red shift of 4.0 cm−1 in E. coli lipids, while the mode shifts by 1.9 cm−1 in DPC and 3.8 cm−1 in E. coli lipids versus buffer. These results agree with the prior conclusion that L4C* is buried when CM15 is membrane-bound and also suggest a greater burial depth in anionic BPL than in DPC. The variance of L4C*’s CN stretching band is nearly the same between the three L4C* samples.
ATR-IR spectra of L4C* following exposure to BPL (Figure 4C) exhibit very little change over time. Both the mean frequency and variance of L4C*’s CN stretch remain relatively constant over time (Figure 4E–F), with a higher degree of scatter in the calculated variance than for other variants. The mean frequency was 2157.6 cm−1 after 20 minutes of incubation in the E. coli lipid solution and 2157.5 cm−1 after seven hours. The local environment of the L4C* probe does not change over time, in contrast to that of I8C* which also exhibits a buried probe group. A model explaining how pore formation might influence C*’s local environment, and thus the probe signals at the ends vs. the middle of the peptide, is discussed below.
V14C*
According to the conclusions of previous EPR-SDSL studies, CM15’s fourteenth residue is only slightly buried on average.(8, 12) NMR experiments indicated that Val14 is positioned on the hydrophilic side of CM15’s amphipathic α-helix, buried slightly inside, but close to the polar surface of the DPC micelle.(17) The V14C* CN stretching band (Figure 3D) clearly red shifts when the peptide is in DPC and E. coli lipids, compared to the aqueous spectrum. The mean frequency moves by −3.0 cm−1 in DPC − 2.3 cm−1 in BPL, while the mode shifts by −4.8 cm−1 in DPC and −4.5 cm−1 in E. coli lipids versus buffer. This probe is at least partly membrane-buried when bound to both DPC and BPL, agreeing qualitatively with the EPR conclusion of a near-interfacial depth at this site.(12) In contrast to magnetic experiments, however, the V14C* IR probe group reports the label’s distribution of environments rather than just the average.
The CN stretching band of V14C* narrows slightly in DPC as compared to in buffer. The variance of the DPC spectrum (4.3 cm−1) is smaller than that of the buffer spectrum (5.3 cm−1). On the other hand, the broadening observed for V14C*’s CN stretch in E. coli lipids suggests a greater heterogeneity in the probe’s environment when bound to BPL. The frequency range covered by the BPL spectrum for V14C* indicates that the probe is able to sample both aqueous and substantially non-aqueous environments, and thus the line-broadening mechanism previously observed due to structural formation around a fully aqueous C* probe group (20, 27) is not expected to be relevant at this particular site. The V14C* peptide’s CD spectrum exhibits a lower MRE in contact with DPC micelles: this might be due to local fraying at the C terminus of the helix caused by the presence of the probe. Some fraying of the C terminus of this short peptide is also anticipated regardless of the artificial C* residue, since helical peptides tend to unfold more easily at the C terminus.(36) Either fraying at the C-terminal end of the CM15 helix or the inherent rotational flexibility of the C* side chain allows the V14C* probe group to sample a greater diversity of local environments than any of the other probe groups located at different sites in the bound CM15 peptides. Figure 5 shows the CN stretch after 20 minutes of lipid exposure fit to two Gaussians of similar width, one centered at 2164.1 cm−1 and one at 2157.2 cm−1. This fit provides evidence of two possible frequency subpopulations resulting from C* in solvent-exposed and lipid-buried environments.
Figure 5.
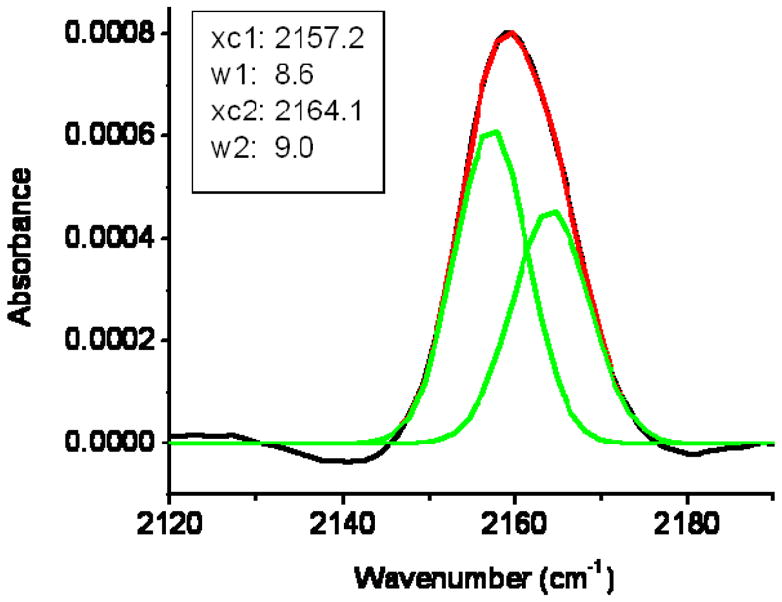
CN stretching band for V14C* in BPL solution after 20 minutes of exposure, fit to two Gaussians with variable widths (w) and frequencies (xc).
When exposed to BPL over time, the mean CN frequency for V14C* (Figures 4D and 4E) jumps from 2161.5 cm−1 after 20 minutes of lipid exposure to 2159.4 cm−1 after one hour, then remains relatively constant over the remainder of the seven hour period. Although the variance data are scattered, the CN stretching band’s linewidth does not change systematically with time. These results suggest that with a possible change during the first hour, the fourteenth position does not becoming increasingly buried or solvent-exposed with time and remains accessible to both the solvent and the membrane.
Pore formation by CM15
Figure 4 largely agrees with a previous proposal for CM15’s mechanism of membrane disruption. The trends in the C* nitrile stretching bands’ mean frequencies and variances (Figures 4E–F) suggest only small, if any, changes over time in the orientation of CM15 with respect to the water-lipid interface that are consistent with the toroidal pore model of AMP membrane perturbation.(7)
Figure 4A shows that the A10C* probe group remains in a similar, solvent-exposed environment during exposure to E. coli lipids throughout a seven hour time period. If CM15 were disrupting the membrane via a detergent-like carpet mechanism, the interfacial orientation of individual peptides would be expected to be more random and disordered. The detergent mechanism would likely cause the A10C* probe to be solvent-exposed in some peptides and lipid-buried in others, resulting in broad and time-dependent CN band IR spectra as peptide-coated, micelle-like particles were created and released. This is clearly not the case in Figure 4A.
Feix, et al. reported that CM15 initially binds parallel to the membrane surface with a spin label at the tenth residue position solvent-exposed.(8, 12) If CM15 formed pores in the bilayer, it would insert itself perpendicular to the surface as the outer lipid leaflet expanded. If CM15 were simply to reorient itself with respect to the bilayer axis, then the tenth residue should remain solvent exposed, as our experiments indicate it does. A lack of evidence for vibrational coupling in the IR spectra, which agrees with the previously observed lack of label-label coupling in EPR experiments,(15) agrees with the EPR-based suggestion that the pores formed are toroidal with lipid head groups intervening between peptides. In such a scenario, even if the peptide were inserted across the bilayer, the A10C* probe would still be partially exposed to the polar head groups of the lipid and partly to the solvent, just as it is when the peptide is surface-bound. Figure 6 depicts the proposed orientations of all labeling sites with respect to the lipid bilayer before and after pore formation.
Figure 6.

Cartoon representation of the positioning of CM15 during pore formation in BPL bilayers, with IR label sites indicated. Graphical distances do not correspond to any physical scale. Light blue=aqueous solution, dark blue=polar head groups, yellow=hydrophobic lipid chains, red cylinder=CM15 helix.
Figure 4B suggests that CM15’s eighth residue becomes slightly more water-excluded after hours of incubation in the BPL vesicle solution. CM15, as a whole, is most likely not becoming immersed more deeply in the bilayer, according to A10C*’s CN stretch. The greater burial of the probe at the eighth label site while the probe in the tenth site remains in the same relative position with respect to the membrane interface could be due to changes in membrane curvature, consistent with pore formation. The walls of a pore should have a higher degree of local curvature than the vesicle surface, caused by the insertion of polar head groups along the pore’s surface. If the eighth residue position coincided with the portion of the pore surface with the highest degree of curvature, then L8C* would be buried more uniformly in the hydrophobic interior compared to its original environment at the unperturbed bilayer surface. Figure 6 suggests how the 8th residue’s local environment after pore formation could become more alkane as the result of membrane curvature, while the 10th residue’s solvent exposure remains unchanged.
Figures 4C and 4D indicate that the environments around the probe groups at the 4th and 14th positions do not change appreciably with time. This lack of change is also consistent with toroidal pore formation as schematically represented in Figure 7, in which the environment of the peptide’s ends remains nearly the same while the curvature near the center of the peptide changes slightly as a result of pore formation.
The results in Figure 4 say nothing about the relative orientation or registry between peptides or the orientation of individual peptides with respect to the bilayer plane or the axes of the pore. Presumably, multiple peptides are involved in the formation of each pore, but they do not interact closely enough for the relatively short-range C* probe groups to report any clear peptide-peptide interactions. Spin labels, which have a much larger length scale for observable label-label interactions, also failed to show any peptide-peptide interactions within 20 Å,(15) and this was used previously to rule out barrel-stave poration as CM15’s mechanism of action. Toroidal pore formation was proposed based on experiments using peptide-bound spin labels at primarily the fourth position:(15) this study adds a more comprehensive map of the lipid exposure of the rest of the peptide.
The infrared results here could be used to validate recent molecular dynamics simulations of pore formation by antimicrobial peptides, which tend to exhibit a pronounced lack of registry in the peptides even as pores form and expand.(37, 38) Mixed quantum mechanical/molecular mechanics simulations could also be useful in further interpretation of the frequencies observed in Figure 3 and 4 for the probe CN vibration. One possible issue in this work pointed out by a reviewer is a disconnect in the time scale; according to stopped flow experiments, the initial time scale for formation of the first pores created by amphipathic peptides can be seconds, rather than minutes or hours (and according to some MD simulations it can be as fast as 10s–100s of ns), so it is possible that there is already some formation of pores at the earliest time points in Figure 4. This would mean that any time evolution observed in Fig. 4 is mostly due to evolution of the more macroscopic lipid structure and small resulting changes in the peptide’s bound environment. Even in the event that pore formation is not the process revealed in the Fig. 4 time dependence, all of the IR data are still consistent with toroidal pore formation and maintenance of a mostly helical secondary structure, and the data clearly do not agree with either barrel-stave pore formation or surfactant-like disintegration.
Methodological ramifications
From this study, a few general obervations can be made regarding the possible general use of this infrared probe strategy for mapping the structural relationship between a membrane protein and its heterogeneous environment. With just four sites in a small peptide, it is clear in this context that the C* probe moiety can be a very sensitive reporter of its local environment. Both the frequency and lineshape of the CN stretching band show significant variations at the four sites of interest in CM15/lipid samples, and new structural conclusions can be drawn from these variations. There is only small perturbation of the lipid-bound structure as a result of the artificial amino acid substitution; as long as this residue is not used to replaced charged residues, it can be placed in largely arbitrary positions along a membrane-active domain. Since C* does weakly perturb helical folding and the secondary structures of CM15 variant peptides here are not quanititatively identical, careful consideration should be applied in the placement of this probe group.
In order to facilitate the global use of C* to understand more complicated membrane-bound proteins from a site-specific point of view, there is a clear need for more systematic experimental work to clarify the exact reporting ability of the CN stretching band in the broad contexts of lipid bilayers and the lipid/solution interface, which each present extremely heterogeneous environments with respect to local polarity, charge, and dynamics. In this study, a few residues near the solution/lipid boundary were investigated. Before meaningful conclusions can be drawn in much more complicated systems, there is a need for a clear and systematic study of the dependence of the CN stretching band’s frequency and lineshape on both its depth dependence in the bilayer and its response through the head group and interfacial regions. This band’s response to its structural environment has been shown by a number of recent studies involving enzymatic active sites and binding interfaces to be quite complicated,(20–22, 25) with site-specific responses that do not appear to vary systematically based on solely either H-bonding or electrostatic factors according to either experiment or increasingly sophisticated theory.(23, 39–41) Therefore, a thorough study of the response of this particular probe group to lipid-bound and lipid-influenced environments must be an experimental priority going forward. Already we can see that its complicated and sensitive response may lead to its wide, and likely quite illuminating, use in many different possible systems.
Conclusions
C* is a generally useful probe group for evaluating protein-membrane contacts in PMPs. In the case of CM15’s interaction with BPL, C* is able to show in a site-specific way how the peptide interacts with its target membrane interface. The data from single-C* variants are consistent with toroidal pore formation and clearly rule out detergent-like disintegration as the mechanism of lipid disruption.
This study demonstrates several unique features of the C* probe for revealing protein-membrane interactions. C*’s frequency and lineshape are clearly sensitive to small changes in the local environment around the probe group, including varying levels of membrane binding and water exposure. Unlike most non-vibrational site-specific probes, the CN band of C* is able to report on the local structural distrbution, which is important in peptides and proteins that can interact with a variety of lipid, solvent, and peptide-based partners in the complex environment of a lipid-water interface. In the case of CM15, this ability to report on the heterogeneity of the probe’s local environment leads to functional conclusions, among which is the confirmation of the toroidal pore model for the mechanism of action.
Further interpretation of the current data might become possible in light of additional systematic study of the C* probe in contact with lipids. In particular, most of the interpretations here are predicated on the C* frequency’s being largely dependent on the presence or absence of water molecules around the CN moiety. A systematic study of the depth dependence of C*, as it passes out of the solvent, through the polar head groups, and into the alkane interior of the bilayer, will be crucial to the general ability of C* to report on orientation and surface structure in membrane proteins of arbitrary depth and orientation vs the bilayer, rather than relatively well-characterized PMPs or antimicrobials like CM15.
Acknowledgments
Dr. Jimmy Feix is thanked by KNA for important suggestions at a formative stage of this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding sources for this work.
Funding sources:
KNA: summer fellowship made possible by a grant from the Howard Hughes Medical Institute to Haverford College
ARV: scholarship from the Arnold and Mabel Beckman Foundation
CHL: Cottrell College Science Award from Research Corporation, a New Faculty Start-Up Award from the Dreyfus Foundation, and grant R15-GM088749 from the National Institute of General Medical Sciences.
Abbreviations
- AMPs
antimicrobial peptides
- ATR
attenuated total reflectance
- BPL
bacterial polar lipids, extracted from E. coli
- C*
β-thiocyanato-alanine, or cyanylated cysteine
- CD
far-UV circular dichroism
- CM15
sequence hybrid of residues 1-7 of cecropin and residues 2-9 of mellitin
- DPC
dodecylphosphatidylcholine
- DMPC
1,2-dimyristoyl-s,n-glycero-3-phosphocholine
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- EPR
electron paramagnetic resonance
- FTIR
Fourier transform infrared spectroscopy
- LUVs
GUVs, SUVs, large, giant, and small unilamellar vesicles
- NMR
nuclear magnetic resonance
- PMPs
peripheral membrane proteins
References
- 1.Caffrey M. Crystallizing Membrane Proteins for Structure Determination: Use of Lipidic Mesophases. Ann Rev Biophys. 2009;38:29–51. doi: 10.1146/annurev.biophys.050708.133655. [DOI] [PubMed] [Google Scholar]
- 2.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klug CS, Feix JB. Biophysical Tools for Biologists: Vol 1 in Vitro Techniques. Elsevier Academic Press Inc; San Diego: 2008. Methods and applications of site-directed spin Labeling EPR Spectroscopy; pp. 617–658. [DOI] [PubMed] [Google Scholar]
- 4.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: Implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 5.Melo M, Ferre R, Castanho M. Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nat Rev Microbiol. 2009;7:245–250. doi: 10.1038/nrmicro2095. [DOI] [PubMed] [Google Scholar]
- 6.Brogden KA. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 7.Huang H. Action of antimicrobial peptides: Two-state model. Biochemistry. 2000;39:8347–8352. doi: 10.1021/bi000946l. [DOI] [PubMed] [Google Scholar]
- 8.Sato H, Felix J. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim Biophys Acta-Biomembranes. 2006;1758:1245–1256. doi: 10.1016/j.bbamem.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 9.Sharon M, Oren Z, Shai Y, Anglister J. 2D-NMR and ATR-FTIR study of the structure of a cell-selective diastereomer of melittin and its orientation in phospholipids. Biochemistry. 1999:15305–15316. doi: 10.1021/bi991225t. [DOI] [PubMed] [Google Scholar]
- 10.Andreu D, Merrifield RB, Steiner H, Boman HG. N-terminal analogs of cecropin A- synthesis, anibacterial activitity, and conformational properties. Biochemistry. 1985;24:1683–1688. doi: 10.1021/bi00328a017. [DOI] [PubMed] [Google Scholar]
- 11.Andreu D, Ubach J, Boman A, Wahlin B, Wade D, Merrifield R, Boman H. Shortened cecropin-A melittin hybrids-significant size reduction retains potent antibiotic activity. FEBS Lett. 1992;296:190–194. doi: 10.1016/0014-5793(92)80377-s. [DOI] [PubMed] [Google Scholar]
- 12.Bhargava K, Feix JB. Membrane binding, structure, and localization of cecropin-mellitin hybrid peptides: A site-directed spin-labeling study. Biophys J. 2004;86:329–336. doi: 10.1016/S0006-3495(04)74108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H. Molecular mechanism of antimicrobial peptides: The origin of cooperativity. Biochim Biophys Acta-Biomembranes. 2006;1758:1292–1302. doi: 10.1016/j.bbamem.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Sato H, Feix J. Osmoprotection of bacterial cells from toxicity caused by antimicrobial hybrid peptide CM15. Biochemistry. 2006;45:9997–10007. doi: 10.1021/bi060979m. [DOI] [PubMed] [Google Scholar]
- 15.Pistolesi S, Pogni R, Feix JB. Membrane insertion and bilayer perturbation by antimicrobial peptide CM15. Biophys J. 2007;93:1651–1660. doi: 10.1529/biophysj.107.104034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bastos M, Bai G, Gomes P, Andreu D, Goormaghtigh E, Prieo M. Energetics and partition of two cecropin-melittin hybrid peptides to model membranes of different composition. Biophys J. 2008:2128–2141. doi: 10.1529/biophysj.107.119032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zangger K, Respondek M, Goebl C, Hohlweg W, Rasmussen K, Grampp G, Madl T. Positioning of Micelle-Bound Peptides by Paramagnetic Relaxation Enhancements. J Phys Chem B. 2009;113:4400–4406. doi: 10.1021/jp808501x. [DOI] [PubMed] [Google Scholar]
- 18.Lew S, Caputo GA, London E. The effect of interactions involving ionizable residues flanking membrane-inserted hydrophobic helices upon helix-helix interaction. Biochemistry. 2003;42:10833–10842. doi: 10.1021/bi034929i. [DOI] [PubMed] [Google Scholar]
- 19.McMahon HA, Alfieri KN, Clark KAA, Londergan CH. Cyanylated Cysteine: A Covalently Attached Vibrational Probe of Protein Lipid Contacts. J Phys Chem Lett. 2010;1:850–855. doi: 10.1021/jz1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bischak CG, Longhi S, Snead DM, Costanzo S, Terrer E, Londergan CH. Probing Structural Transitions in the Intrinsically Disordered C-Terminal Domain of the Measles Virus Nucleoprotein by Vibrational Spectroscopy of Cyanylated Cysteines. Biophys J. 2010;99:1676–1683. doi: 10.1016/j.bpj.2010.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fafarman AT, Sigala PA, Herschlag D, Boxer SG. Decomposition of Vibrational Shifts of Nitriles into Electrostatic and Hydrogen-Bonding Effects. J Am Chem Soc. 2010;132:12811–12813. doi: 10.1021/ja104573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fafarman AT, Webb LJ, Chuang JI, Boxer SG. Site-specific conversion of cysteine thiols into thiocyanate creates an IR probe for electric fields in proteins. J Am Chem Soc. 2006;128:13356–13357. doi: 10.1021/ja0650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh KI, Choi JH, Lee JH, Han JB, Lee H, Cho M. Nitrile and thiocyanate IR probes: Molecular dynamics simulation studies. J Chem Phys. 2008;128:154504. doi: 10.1063/1.2904558. [DOI] [PubMed] [Google Scholar]
- 24.Sigala PA, Fafarman AT, Bogard PE, Boxer SG, Herschlag D. Do ligand binding and solvent exclusion alter the electrostatic character within the oxyanion hole of an enzymatic active site? J Am Chem Soc. 2007;129:12104–12105. doi: 10.1021/ja075605a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stafford AJ, Ensign DL, Webb LJ. Vibrational Stark Effect Spectroscopy at the Interface of Ras and Rap1A Bound to the Ras Binding Domain of RalGDS Reveals an Electrostatic Mechanism for Protein-Protein Interaction. J Phys Chem B. 2010;114:15331–15344. doi: 10.1021/jp106974e. [DOI] [PubMed] [Google Scholar]
- 26.Maienschein-Cline MG, Londergan CH. The CN stretching band of aliphatic thiocyanate is sensitive to solvent dynamics and specific solvation. J Phys Chem A. 2007;111:10020–10025. doi: 10.1021/jp0761158. [DOI] [PubMed] [Google Scholar]
- 27.Edelstein L, Stetz MA, McMahon HA, Londergan CH. The Effects of alpha-Helical Structure and Cyanylated Cysteine on Each Other. J Phys Chem B. 2010;114:4931–4936. doi: 10.1021/jp101447r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Getahun Z, Huang CY, Wang T, De Leon B, DeGrado WF, Gai F. Using nitrile-derivatized amino acids as infrared probes of local environment. J Am Chem Soc. 2003;125:405–411. doi: 10.1021/ja0285262. [DOI] [PubMed] [Google Scholar]
- 29.Tucker MJ, Getahun Z, Nanda V, DeGrado WF, Gai F. A new method for determining the local environment and orientation of individual side chains of membrane-binding peptides. J Am Chem Soc. 2004;126:5078–5079. doi: 10.1021/ja032015d. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee S, Chowdhury P, DeGrado WF, Gai F. Site-specific hydration status of an amphipathic peptide in AOT reverse micelles. Langmuir. 2007;23:11174–11179. doi: 10.1021/la701686g. [DOI] [PubMed] [Google Scholar]
- 31.Lee J, Boman A, Sun C, Andersson M, Jornvall H, Mutt V, Boman H. Antibacterial peptides from pig intestine-isolation of a mammalian cecropin. Proc Nat Acad Sci USA. 1989;86:9159–9162. doi: 10.1073/pnas.86.23.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudha TS, Vijayakumar EKS, Balaram P. Circular dichroism studies of helical oligopeptides: can 3–10 and a-helical conformations be chiroptically distinguished. Int J Pept Prot Res. 1983;22:464–468. doi: 10.1111/j.1399-3011.1983.tb02116.x. [DOI] [PubMed] [Google Scholar]
- 33.Rabenstein M, Shin Y. Determination of the distance between two spin labels attached to a macromolecule. Proc Nat Acad Sci USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh KI, Lee JH, Joo C, Han H, Cho M. beta-Azidoalanine as an IR probe: Application to amyloid Aβ (16–22) aggregation. J Phys Chem B. 2008;112:10352–10357. doi: 10.1021/jp801558k. [DOI] [PubMed] [Google Scholar]
- 35.Sato H, Feix J. Lysine-Enriched Cecropin-Mellitin Antimicrobial Peptides with Enhanced Selectivity. Antimicrob Agents Ch. 2008;52:4463–4465. doi: 10.1128/AAC.00810-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silva R, Kubelka J, Bour P, Decatur SM, Keiderling TA. Site-specific conformational determination in thermal unfolding studies of helical peptides using vibrational circular dichroism with isotopic substitution. Proc Natl Acad Sci USA. 2000;97:8318–8323. doi: 10.1073/pnas.140161997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bond PJ, Khalid S. Antimicrobial and Cell-Penetrating Peptides: Structure, Assembly and Mechanisms of Membrane Lysis via Atomistic and Coarse-Grained Molecular Dynamic Simulations. Prot Pept Lett. 2010;17:1313–1327. doi: 10.2174/0929866511009011313. [DOI] [PubMed] [Google Scholar]
- 38.Marrink SJ, de Vries AH, Tieleman DP. Lipids on the move: Simulations of membrane pores, domains, stalks and curves. Biochim Biophys Acta -Biomembranes. 2009;1788:149–168. doi: 10.1016/j.bbamem.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Cho M. Vibrational solvatochromism and electrochromism: Coarse-grained models and their relationships. J Chem Phys. 2009;130:094505. doi: 10.1063/1.3079609. [DOI] [PubMed] [Google Scholar]
- 40.Choi JH, Cho M. Vibrational solvatochromism and electrochromism of infrared probe molecules containing C≡O, C≡N, C=O, or C-F vibrational chromophore. J Chem Phys. 2011;134:154513. doi: 10.1063/1.3580776. [DOI] [PubMed] [Google Scholar]
- 41.Choi JH, Oh KI, Lee H, Lee C, Cho M. Nitrile and thiocyanate IR probes: Quantum chemistry calculation studies and multivariate least-square fitting analysis. J Chem Phys. 2008;128:134506. doi: 10.1063/1.2844787. [DOI] [PubMed] [Google Scholar]