

Abstract
The synergistic combination of layer-by-layer (LbL) assembly and nanoporous membrane templating has greatly facilitated the creation of complex and functional nanotubular structures. The approach takes advantage of both the new properties conferred by assembling diverse LbL building blocks and the tight dimensional control offered by nanotemplating to enable new functionalities that arise from the highly anisotropic “one-dimensional” LbL-nanotube format. In this review, we aim to convey the key developments and provide a current snap-shot of such templated LbL nanoarchitectures. We survey recent developments that have enabled the assembly of polymers, biomolecules and inorganic nanoparticles “à la carte”, via electrostatic, covalent and specific (bio)recognition interactions. We also discuss the emerging mechanistic understanding of the LbL assembly process within the nanopore environment. Finally, we present a diverse range of LbL nanotube “devices” to illustrate the versatility of the nanotemplated LbL toolbox for generating functional soft nanotechnology.
1. Introduction
The co-assembly of functional polymeric, (bio)molecular and inorganic nanoparticulate building blocks poses new opportunities and alternatives to creating complex and tunable nanoarchitectures, especially as it is becoming possible to create genuinely new building block materials de novo by manipulating materials at the molecular level.1 It is also widely recognized that the functional properties of a nanomaterial can change dramatically when its structural dimensions coincide with the characteristic length-scale of a particular physical property.2,3 Highly anisotropic morphologies are therefore very interesting as they can address multiple functionalities over their various dimensions. As a result, the design and fabrication of “soft” nanowires and nanotubes—one-dimensional (1D) nanomaterials4—constitute a rapidly advancing branch of nanoscience that invokes the tools and concepts of (supra)molecular science and attempts to take advantage of the new properties and functionalities arising from the nanoscale.
Successful development of nanostructured entities requires suitable and convenient methods for their fabrication. It has been twenty years since Decher and Hong published their seminal works describing the concept of layer-by-layer (LbL) assembly.5 The method is based on the alternating electrostatic deposition of polycationic and polyanionic species onto a substrate. Deposition of a layer of material of one charge reverses the substrate surface charge (charge over-compensation) and enables the deposition of the next layer of the opposite charge. Its introduction represents a milestone in molecular design as it offers a simple and versatile bottom-up process for creating multilayered thin films. LbL assembly was originally demonstrated using polyelectrolytes.5 The fact that polymers are nanoscale objects translated into an unprecedented ability of the LbL technique to control film characteristics such as composition, thickness, and function on the nanoscale simply by varying the sequence, number and chemical nature of the polyelectrolyte layers.6 Further development led to the incorporation of other building blocks, including proteins, nanoparticles, and quantum dots, as long as they can participate in the charge overcompensation process.7 More recently, LbL assembly has been further extended beyond the domain of electrostatic interactions to multilayered systems stabilized by covalent bonding, hydrogen bonding or biospecific interactions.
Martin and collaborators pioneered the fabrication of 1D nanomaterials using the “template method”.8 The filling of pores within a nanoporous membrane, or “template”, generated nanorods, and the conformal deposition on the pore walls created nanotubes. Nanoporous anodic aluminum oxide (AAO) was adopted early as a robust nanotemplate for monodisperse 1D nanostructures, since AAO possesses uniform, cylindrical nanopores self-organized into a close-packed arrangement.9 Also, the nanoporous membranes are conveniently prepared by a “bottom-up” anodization process, and a large range of pore diameters (5-400 nm) and pore lengths (from nanometers to tens of microns) can be obtained by controlling a small set of anodization parameters. AAO nanotemplating therefore results in a convenient method for preparing monodisperse 1D nanomaterials which have dimensions replicating those of the AAO nanopores and which can be sensitively tuned with high precision. Nanotemplating by other nanoporous membranes soon followed. In particular, like AAO, track-etched polycarbonate (TEPC) membranes also exhibit straight, cylindrical nanopores and became widely utilized.10 Release of the 1D nano-objects from the AAO or TEPC is simply accomplished by selective dissolution of the nanoporous matrix. Undoubtedly, the convenience and conceptual simplicity of the template method have given a decisive impetus to the development of a plethora of nanotubes and nanowires.11
In 2003, Li12 and Caruso13 reported the first attempts to combine the template method and LbL assembly to create polyelectrolyte and heterostructured multilayered nanotubes. The very possibility of merging the versatility of both templating and LbL assembly marked a critical departure from traditional nanotemplating. This hybrid strategy enabled not only geometric control over the length and wall thickness of the resulting nanotubes, but also provided a versatile means of locally manipulating the wall components and properties by the sequential assembly of suitable building blocks that include polymers, nanoparticles, proteins, inorganic and organic functional molecules. Since its introduction, LbL assembly in nanoporous templates has led to a wide range of 1D soft functional nanoobjects displaying functionality that is controlled by the organizational arrangement14 and the properties of the building blocks, as well as the number of assembled layers making up the nanotubes.
This review presents the advances that have been made in the design, fabrication and application of LbL-mediated nanotubes. It is divided into four sections. The first and second encompass, respectively, a description of the current synthetic strategies towards LbL-mediated polymeric nanotubes and a discussion of the protocols important for preparing hybrid 1D soft nanoarchitectures. In the third, a discussion of the main mechanistic aspects of LbL assembly in the confined environment of nanopores is provided. Lastly, a diverse range of LbL nanotube “devices” are described, which serve to illustrate the potential of soft 1D nanostructures fabricated by the LbL nanotemplating strategy in various technological areas. It is hoped that the present contribution will illustrate the multidisciplinary breadth of LbL-mediated 1D nanostructure research and hence stimulate further advances in this emerging area of “soft nanotechnology”.
2. Template synthesis of one-dimensional soft nanostructures via layer-by-layer (LbL) assembly: Molecular interactions and building blocks à la carte
2.1. Hard nanoporous templates
Nanoporous anodic aluminum oxide (AAO) is formed through the electrolytic oxidation and etching (i.e. anodization) of pure Al.9 Track etched polycarbonate (TEPC) is prepared by the bombardment of a precursor polycarbonate film with heavy energetic ions produced by, e.g., a cyclotron.15 Both AAO and TEPC membranes are characterized by flat external surfaces and cylindrical pores that run parallel to each other and straight through the thickness of the membranes. The AAO pores are also characterized by a high-degree of ordering. However, this feature is not necessarily important for nanotemplated LbL assembly since the membranes are commonly sacrificed (dissolved) to release the deposited nanotubes. AAO can be etched in pH < 4.5 and pH > 8.516,17 and TEPC can be dissolved in common organic solvents such as dichloromethane.
Several lines of commercial TEPC, such as Nuclepore™, Poretics™ and ipPoreTM,18, are available from laboratory suppliers. Membranes with a wide range of pore diameters, from 15 nm to many microns, and membrane thickness 10 μm and up, can be sourced. Polyethylene terephthalate and polyimide membranes are also available. A more limited range of commercial AAO membranes can be obtained under the brand name AnodiscTM,19 or from specialized companies such as Synkera.20 Since anodization only requires a bench-top high voltage supply and a relatively simple two electrode electrochemical setup, many laboratories prepare their own samples for more precise and on-demand control over the pore diameter and pore length. Anodization protocols have been documented elsewhere.9,21,22,23,24
2.2. Electrostatic assembly of polyelectrolytes and dendrimers
Pioneering work from Li and his collaborators introduced the concept of layer-by-layer (LbL) assembly of polyelectrolyte multilayers within nanoporous templates in order to create polymeric tubular nanostructures displaying complex but well-controlled wall morphologies and adjustable wall thickness (Figure 1).25 The authors deposited polyallylamine hydrochloride (PAH)/sodium poly(styrene sulfonate) (PSS) from aqueous 0.5 M NaCl solutions within the ~300 nm pores of an anodic aluminum oxide (AAO) membrane by injecting polyelectrolyte solutions through the membrane under an applied pressure. Subsequent etching of the alumina matrix with an aqueous NaOH solution led to the release of flexible (PAH/PSS)3 nanotubes displaying physical length and outer diameter in agreement with the AAO template used (Figure 2). Interestingly, the nanotube walls were found to be much thicker than that of corresponding multilayer structures prepared on flat substrates (see section 4).
Figure 1.

Simplified representation of the formation of polyelectrolyte nanotubes through sequential assembly of polycations and polyanions inside the nanoporous template and the subsequent removal f the nanotemplate.
FIGURE 2.

Scanning electron micrographs at different magnifications (A-D) of polyelectrolyte nanotubes obtained via nanotemplated LbL assembly of poly(allylamine) and poly(styrene sulfonate). Reproduced with permission from Ai et al. J. Am. Chem. Soc., 2003, 125, 11140. Copyright 2003 American Chemical Society.
The same strategy has also been extended to functional polymers whose characteristics are compatible with the LbL technique. For example,26 the negatively charged conducting polymer polypyrrole (PPy) was used as a functional building block to fabricate conductive polymer nanotubes through the alternating deposition with positively charged PAH27 onto the inner pores of track-etched polycarbonate (TEPC) nanotemplates. Unlike the aforementioned work of Li et al., LbL assembly was accomplished simply by diffusion of PPY and PAH into the TEPC immersed in the polyelectrolyte solutions (60 min). Under these conditions, a six bilayer structure, (PPy/PAH)6, was reported as a critical condition for attaining mechanically stable nanotubes with an outer diameter of ~400 nm and length ~10 μm. In addition, their electroactive characteristics were measured by cyclic voltammetry (CV), and showed that PPy has a stable oxidation state in organic acid. The conductivity of the nanotubes was determined to be 8 mS/cm.
In a similar vein, soft nanotubes constituting exclusively of dendrimer polyelectrolytes were obtained using two versions of the same fourth generation phosphorus dendrimers, one with dendritic branches terminated by ammonium groups and another with carboxylate terminations.28,29 Analogous to (PPy/PAH)n, dendrimer nanotubes were also obtained by direct immersion of the AAO into dendrimer solution. Each dendrimer deposition step was followed by rinsing with pure water. The same process was repeated up to 20 bilayers. The dendrimer nanotubes were released from the template by immersing the AAO into a solution of chromium (III) in phosphoric acid, which facilitates the rapid dissolution of the alumina membrane and facile release of the nanotubes. These nanotubes, templated from AAO membranes with pores opened at both ends, also have a much higher aspect ratio (diameter ~400 nm, length ~80 μm, wall thickness ~40 nm) than the PPy/PAH assemblies described above.26
2.3. LbL assembly of metal organodiphosphonates
Strong ionic interactions using amphiphiles as modular building blocks have also been exploited to create metal-containing, nanoconfined supramolecular assemblies and one-dimensional soft nanostructures. The LbL deposition of organodiphosphonates as zirconium salts was originally introduced by Mallouk and co-workers as a strategy to achieve thin films with architectural control.30 The resulting films are markedly stable because the interlayers consist of Zr4+ ions strongly and multiply coordinated to phosphonate groups (analogous to high-melting, chemically robust zirconium phosphate and phosphonate bulk layered materials).31 Since the structure of the layered solid is determined by strong ionic interactions between the metal ions and the phosphate groups, the organic groups that are “along for the ride” in the structure can be arranged in predictable ways, even in confined geometries.
Martin and co-workers described the synthesis of α,ω diorganophosphonate/Zr layered nanotubes within the nanopores of alumina template membranes.32 The experimental protocol only required the alternate immersion of the nanotemplate into a solution of diorganophosphonate (1,10-decanediylbis(phosphonic acid), DBPA) and a solution of ZrO2+. After the desired number of immersion cycles, the alumina template was dissolved in 27 % H3PO4, and the layered DBPA/Zr nanotubes were collected by filtration. Kohli et al. also reported the synthesis of polydiacetylene nanotubes using the chemistry of 10,12-docosadiyndioic acid (DCDA) amphiphilic monomers inside nanoporous AAO templates. In combination with zirconium-carboxylate interlayer chemistry, UV irradiation of the DCDA-modified samples led to the formation of polymerized-DCDA multilayer nanotubes.33
2.4. Hydrogen bond mediated nanotube assembly
The ability of certain polymers to form strong hydrogen bonds along its polymer backbone permitted these relatively strong secondary forces to be exploited for the LbL assembly of multilayered thin films, and was introduced in 1997 on planar surfaces by the Rubner34 and Zhang groups.35 To date, the majority of work in non-electrostatic LbL assembly is still based on hydrogen bonding interactions.36 A typical example is the use of the carboxylic acid groups of poly(acrylic acid) (PAA) in combination with poly(4-vinyl-pyridine) (PVP). The oxygen of the carboxylic acid groups in PAA act as H-bond donors and the nitrogen atoms of the pyridine rings in PVP act as acceptors. To suppress electrostatic forces, H-bond assembly of PAA layers can be performed from methanolic solutions, while PVP assembly can be performed from either methanolic or ethanolic solutions.37 (PAA/PVP)5 nanotubes fabricated in this fashion exhibited good stability and flexibility, and as expected, the wall thickness of the nanotubes was strongly dependent on the number of multilayers assembled on the pore walls. The possibility of using organic solvents also fostered the application of hydrogen-bonded multilayers nanotubes in TEPC nanotemplates.38
The H-bonding nature of PVP/PAA nanotubes also introduced the possibility of pH-triggered (partial) disassembly of the nanotubes.39 Immersion of the nanotubes in a basic solution causes the release of PAA building blocks, thus promoting the formation of nanotubes with porous walls for which the pore size can be controlled by the immersion time. The porous nanotubes are stable at room temperature and this pH-control of nanotube “leakiness” (i.e. transport) could enable catalyst or drug carrier applications.
2.5. Covalent bonding and cross-linking of multilayers
Although non-covalent interactions have been the main driving forces for LbL assembly, covalent bonding has gained increased relevance in recent years as an alternative strategy for multilayer assembly.40 This methodology is particularly important when high stability of the multilayer assembly is required. Another interesting benefit is the increased compatibility of the multilayered assemblies with organic solvents, which in turn facilitates the use of polymeric building blocks that are only soluble/stable in non-aqueous solvents. For example, covalently-assembled nanotubes were obtained by amide bond formation between alternating layers of Polyethyleneimine (PEI) and poly(styrene-altmaleic anhydride) (PSMA) layers.41 (PEI/PSMA)5 nanotubes display uniform structure and good mechanical stability. In another interesting example, Li and co-workers reported the use of a dye molecule, 3,4,9,10-perylenetetracarboxylicdianhydride (PTCDA),42 to create templated polymer nanotubes featuring light-emitting properties.43 PEI was chosen as the macromolecular mediating unit displaying amine groups that are able to react with the PTCDA. Formation of covalent bonds was confirmed by IR spectroscopy whereas UV–visible spectroscopy indicated that the characteristic absorption of the PEI/PTCDA nanotubes increases linearly with the number of assembled layers. Linear growth was also corroborated by the linear increase of nanotube wall thickness upon increasing the number of deposition cycles. (PEI/PTCDA)6 nanotubes were flexible and the wall thickness was ~100 nm (each bilayer was ~16 nm). Finally, these light-emitting nanotubes were coated with a lipid bilayer membrane, thus creating nanotubular lipid nanostructures that may find potential applications as probes of intracellular environments, bioanalysis, or drug carriers.44
Another strategy toward covalent LbL deposition is the use of conventional polyelectrolyte assembly followed by chemical crosslinking of the pre-assembled multilayers. Detailed work by the Bruening group demonstrated that polyacrylic acid (PAA) and polyallylamine (PAH) display the ability to cross-link by heating, which results in a significant improvement of the structural stability of PAA/PAH multilayer assemblies.45 Subsequently, Caruso and co-workers described the fabrication of heat-stabilized PAA/PAH nanotubes within TEPC membranes.46 Cu2+ was also used as a coordinating agent in the sequential deposition of the PAA/PAH layers by addition of CuCl2 in the polyelectrolyte solutions. Interlayer cross-linking was accomplished by heating the membrane-supported nanotubes at 160°C for 4 h in air. It is worth mentioning that recent results from Lutkenhaus et al. revealed that the rate of amidation is strongly influenced by the film thickness and surface chemistry of the pore walls.47
2.6. Biorecognition-driven assembly of multilayers in nanopores
Bio-recognition-driven LbL assembly provides a versatile tool to generate bio-functional interfaces with well-defined architecture, topology, and biochemical functionality. To this end, a broad variety of biological components including DNA, enzymes and antibodies, have been utilized for planar film assembly.48 Recently, such LbL bioassemblies have been extended to porous templates to generate one-dimensional bionanostructures.
Martin and co-workers reported the formation of DNA nanotubes by the sequential deposition of complementary oligonucleotides with specific sequences in an AAO membrane.49 The nanotemplate was first immersed into a solution of 1,10-decanediylbis(phosphonic acid) (DOP) followed by a solution of ZrOCl2. The role of the DOP/Zr(IV) assembly was to act as a nanotube skin providing structural integrity to the oligonucleotide assembly located at the core of the templated one-dimensional structure. The inner environment of the nanotubes is constituted of multiple double-stranded DNA layers held together by the hybridization between complementary DNA. The DNA molecules comprising the nanotubes can be varied at will and the DNA can be released by changing the environmental conditions, such as temperature, which promote the melting of the duplexes.
The use of biotinylated-PAH as bifunctinal macromolecular ligands has also been demonstrated by Azzaroni et al.50 LbL assembly of biotinylated-PAH enables the facile modification of nanopore walls with biorecognition sites which can then be used for constructing a nanobiosensor. Streptavidin conjugation with biotin does not remove the assembled biotinylated-PAH from the channel surface. In fact, the bio-supramolecular multilayered structures were stabilized by the strong ligand-receptor interactions. Experiments also indicated that the assembled nanopore walls display good biospecificity and nonfouling properties.
3. Bottom up fabrication of hybrid nanotubes. Self-assembly of composite 1D nanostructures
3.1. Formation of heterostructured nanotubes via LbL assembly of inorganic nanomaterials and polyelectrolytes
One of the most attractive features of electrostatic LbL assembly is its intrinsic ability to incorporate a wide variety of components into the multilayered architecture. Several nanomaterials like quantum dots, metal colloids or clay can be easily processed as charged nanoparticles, thus enabling their incorporation as building blocks into polyelectrolyte multilayers.51 Thus the assembly of organic–inorganic hybrid nanotubes, with a controllable and regular thickness in the nanometer range, is a straightforward process as long as the charge reversal mechanism can be sustained with the chosen material. Within this framework, much of the information gathered during early studies on the growth of hybrid self-assembled multilayers on planar surfaces was used to design heterostructured polymer nanotubes.
In an early example, Caruso and co-workers reported the synthesis of hybrid nanotubes by incorporating CdTe quantum dots and gold nanoparticles, respectively, in polyelectrolyte multilayers (Figure 3). The authors also showed that the incorporation of these particular nanomaterials improves the structural stability of the nanotubes, thus requiring fewer layers to form mechanically stable hybrid nanotubes.
FIGURE 3.

SEM (a) and TEM(b) images (polyelectrolyte/gold nanoparticles)2 assemblies. SEM (c) and TEM(d) images (polyelectrolyte/CdTe nanoparticles)6 assemblies. Reproduced and adapted with permission from Liang et al. Adv. Mater. 2003, 15, 1849. Copyright 2003 Wiley-VCH Verlag GmbH & Co. KGaA.
Hybrid nanotubes can be also prepared by in-situ synthesis of nanoparticles inside the multilayer assembly. Möhwald et al. demonstrated that calcium carbonate can be biomimetically synthesized inside the cavities of the polyelectrolyte nanotubes by the catalysis of urease, with the size of the calcium carbonate precipitates controlled by the nanotube cavity dimensions. To form calcium carbonate-filled polyelectrolyte nanotubes, urease-loaded (PSS/PAH)6(Fe3O4/PAH)2 nanotubes prepared in TEPC were incubated in a solution 0.5 M urea + 1 M CaCl2 for 20 min at room temperature. Furthermore, the calcium carbonate-filled hybrid nanotubes also contained Fe3O4 nanoparticles and could be magnetically collected by applying an external magnetic field.52
Heterostructured titania-polyelectrolyte nanotubes were also successfully prepared by sequential LbL deposition of titanium(IV) bis(ammonium lactato) dihydroxide (TALH) and poly(ethylenimine) (PEI) inside the cylindrical pores of a TEPC templates, followed by calcination at various temperatures.53 TALH is stable in neutral solutions54 and represents an excellent candidate for the preparation of titania-based thin films using aqueous chemistry.55 Photocatalytically active titania nanotubes could be formed with various compositions and crystal phases, i.e.: pure anatase or rutile titania tubular architectures with well-defined diameters and wall thicknesses. Mallouk and collaborators have also shown that it is possible to combine the template synthesis with LbL assembly of polymer/colloid films constituted of TiO2/PSS, ZnO/PSS and ZnO/polyaniline to prepare rod-shaped devices that act as rectifiers.56
In a similar vein, negatively-charged polyoxometalates (POMs) and positively-charged polyelectrolytes were alternately coated onto the inner walls of TEPC in order to create a nanotubular catalytic reactor composed of Pt-loaded POM/polyelectrolyte nanotubes. The TEPC was first pretreated with PEI, PSS, and poly(allylamine hydrochloride) or poly(diallyldimethylammonium chloride) (PDMA) solutions to form a LbL multilayer nanotubular “foundation”.57 Sequential assembly of POM and the cationic polyelectrolyte was then performed on the PEI/PSS/PAH or PEI/PSS/PDMA, followed by final loading of Pt particles. The wall thickness as well as the inside diameter of the hybrid nanotubes were precisely controlled by repeating the desired number of bilayers of POM/polyelectrolyte (2.2 nm/step). These Pt-embedded POM/polyelectrolyte hybrid nanostructures were shown to exhibit catalytic activities for cyclohexene hydrogenation, indicating promising potential in designing reactors with specific sizes for specific reactions.
3.2. LbL assembly of bio-nanotubes using proteins, lipids and polypeptides
Among the advantages of the LbL technique is its use of (typically) mild conditions for film construction, which both industry and academia find valuable for creating interfacial architectures with protein organizations. Most biomacromolecules have charged residues on their peripheral surfaces and consequently their use as building blocks in electrostatic LbL assembly is directly compatible. For instance, a large variety of proteins have been assembled in combination with oppositely charged polyelectrolytes leading to different types interfacial architectures in which not only the number of layers but also the layering sequence can be controlled.58,59
Recent experiments revealed that one-dimensional nanostructures may prove successful for creating drug and gene delivery systems.60 Along these lines, cytochrome C,61 collagen62 and peroxidase63 nanotubes were prepared by LBL deposition using PSS as the complementary polyanionic building block. In a similar fashion, negatively charged L-α-dimyristoylphosphatidic acid (DMPA) was used as a counterpart to HSA to assemble phospholipid/protein nanotubes.64
Komatsu et al.65 demonstrated the versatility of the synthetic cationic polypeptide poly-L-arginine (PLA) as a mediating polyelectrolyte for assembling protein nanotubes incorporating HSA, ferritin, or myoglobin. In the case of HSA, it was also demonstrated that the formation of stable nanotubes is feasible using PEI as a polycation. As for the PLA/ferritin nanotubes, these nanostructures were used as precursors of solid nanotubes comprising α-Fe2O3 nanoparticles.66 The iron-storage protein ferritin and PLA were assembled in a LbL fashion into TEPC membranes (pore diameter, 400 nm) with subsequent dissolution of the template. Thereafter the (PLA/ferritin)3 nanotubes were calcinated at 500°C in air to yield iron oxide nanotubes that displayed superparamagnetic properties as well as efficient photocatalytic activity for the degradation of 4-chlorophenol.
Biodegradable nanotubes can also be fabricated through the LbL assembly of anionic alginate (ALG) and cationic chitosan (CHI) onto the pore walls of polycarbonate membrane with the subsequent template removal by dissolution in CH2Cl2. The as-obtained CHI/ALG nanotubes were readily internalized into cells, presented low levels of cytotoxicity and were biodegraded in the presence of pancreatin.67 “Homoprotein assemblies” are also necessarily biodegradable, and Li and co-workers described the LbL construction of nanotubes using human serum albumin (HSA) alone. The isoelectric point (pI) of HSA is 4.8. The effective charge of HSA can therefore be reversed by adjusting the solution pH below and above the pI of the protein. Using this methodology, HSA was sequentially assembled on the inner walls of a nanoporous alumina matrix as a polycation (at pH 3.8) and as a polyanion (at pH 7.0). After the removal of the nanotemplate in NaOH solution, an array of HSA nanotubes was obtained without altering the secondary structure of the protein. Also, protein nanotubes based on heme-modified HSA were shown to reversibly bind O2 at room temperature. 68
Covalent LbL growth has been also explored as a route to create all-protein nanotubes. Martin and co-workers69 introduced the first report on protein nanotubes by alternately exposing the AAO membrane to a solution of the glucose oxidase (GOx) or hemoglobin (Hb) and then to a solution of glutaraldehyde, which serves as a cross-linking agent to hold the proteins together. After assembling the desired number of layers, the AAO was dissolved by immersion into a 5% phosphoric acid solution (24 h at 0 °C) to release the bio-nanostructures. Further characterization evidenced that GOx nanotubes catalyzed glucose oxidation whereas Hb nanotubes retained their heme electroactivity. The same strategy was used by Li and co-workers to build up electroactive LbL-grown cytochrome C nanotubes.61
4. LbL deposition of polyelectrolytes in nanopores: A physical picture of electrostatic assembly in nanoconfinement
Common characterization techniques typically used on flat substrates are not directly compatible with studies in nanopores and studying supramolecular assembly in nanogeometries is not a trivial task. Perhaps as a result, only a limited number of fundamental studies have been devoted to the assembly of polyelectrolyte multilayers in nanoporous templates. However, LbL deposition within nanopores is likely to be even more sensitive to the nature of the polyelectrolyte species and the ionic parameters than deposition on planar surfaces. This is because, as a polyelectrolyte enters a nanopore, the electrostatic potential around the polyelectrolyte may be altered due to charge regulation, and the local ion concentration, hence the local screening length, may deviate from bulk values. Although this effect may be small in the initial stages of assembly using “large” pores (e.g. 400 nm), the effect could quickly become appreciable as multilayers build-up (the pore cross-section decreases non-linearly) and if larger building blocks or smaller diameter nanotubes are desired.
Rubner and co-workers suggested that the presence of surface charge on the nanochannel walls provides a level of electrostatic repulsion over the width of the pore/channel sufficient to deplete the transport of the building blocks required for LbL assembly. This would also lead to a decreased level of charge overcompensation after the deposition of each layer.70 Recent work by Thayumanavan and coworkers71 indicates that the typical electrostatic assembly on planar surfaces cannot be straightforwardly extrapolated to small nanopores even when working with globular, compact charged dendrimers. Upon assembling cationic polypropyleneimine (PPI) dendrimers (second generation, G2, containing eight amino surface groups) on poly(acrylic acid) (PAA) modified TEPC nanopores, the pore diameter was reduced from 28 to 23 nm.72 However, the pore diameter was indirectly measured by the time-dependent transport of dye molecules through the membranes and introduces a degree of uncertainty in pore size determination. Notwithstanding, since the positively charged PPI dendrimers decorated the pore walls, it was expected that the anionic fluorescent probe calcein would diffuse through the pores more rapidly than the cationic probe rhodamine 6G. In contrast, experiments revealed no difference in the diffusion rates of the two probes and, more importantly, no difference between the diffusion of the dyes through PPI-G2/PAA-modified and unfunctionalized pores. At the same time, diffusion of rhodamine 6G was faster than calcein in PAA-modified nanopores. This suggests that within this range of pore diameter, PPI-G2 dendrimers have essentially neutralized the negative charge of PAA without charge-overcompensation and charge reversal.
The cross-over from “normal” assembly to a nanoconfined behavior based on pore diameter was studied by Lau et al.73 Structurally well-defined N,N-disubstituted hydrazine phosphorus-containing dendrimers of the fourth generation with diameters of approximately 7 nm (NN-G4) were used as a polyelectrolyte to study macromolecular assembly inside nanoporous AAO. Significantly, the authors were able to monitor the assembly of each layer in situ using the technique of nanoporous optical waveguide spectroscopy (OWS).74
On a planar surface, multilayers prepared from these NN-G4 polyelectrolyte dendrimers show a monotonic increase in layer thickness with the number of LbL deposition steps.75 NN-G4 multilayers also exhibit a lower degree of interlayer penetration than multilayers formed from linear polyelectrolytes because of their rigid internal hydrophobic structure and well-defined peripherally charged surfaces.76 Polyelectrolyte deposition within the cylindrical nanopores followed an initial linear deposition regime which, however, became completely inhibited when the pores were still significantly larger than the dendrimer diameter (Figure 4). At any particular ionic strength, the actual pore diameter at which deposition became hindered (defined by the pore constriction associated with the number of already deposited polyelectrolyte layers) converged to a common value regardless of the initial, native pore diameter. For example, the 7 nm dendrimers were unable to enter pores less than 30 nm in diameter at 0.1 M NaCl. Furthermore, significantly higher solution ionic strengths than needed on flat surfaces were required to deposit the same density of polyelectrolytes within the 30-116 nm nanopores studied. In fact, OWS revealed that pore deposition could be inhibited at all pore diameters investigated (30-116 nm) by adjusting the ionic strength of the deposition solution. These observations are consistent with the original suggestion of Rubner et al., and are corroborated by a recent molecular dynamics theoretical study.77 The initial charge inversion in the vicinity of the pore entrances likely created a repulsive potential that inhibited partitioning of polyelectrolytes in the pores. This “enhanced” ionic strength effect was subsequently utilized by Lau et al. to selectively deposit a polyelectrolyte multilayer atop the nanoporous membrane.
FIGURE 4.
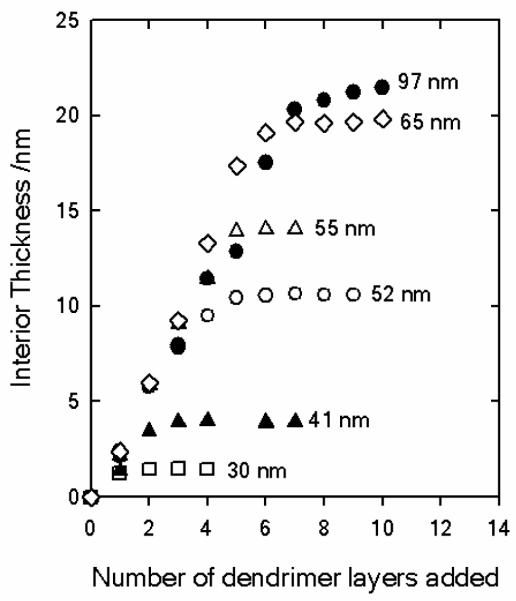
Thicknesses of the dendrimer polyelectrolyte layers deposited in the interior of the AAO nanomebranes from 100 mM NaCl aqueous solutions, as determined by optical waveguide spectroscopy (OWS). The data labels indicate the initial membrane pore diameters. In comparison, the dendrimer diameter is 7 nm. Reproduced and adapted with permission from Lazzara et al. ACS Nano, 2010, 4, 3909. Copyright 2010 American Chemical Society.
Other polyelectrolyte pairs behave in fundamentally different ways when deposited within nanopores. Jonas and co-workers described the multilayer assembly of cationic poly(vinylbenzylammonium chloride) (PVBAC) and anionic poly(styrene sodium sulfonate) (PSS) within TEPC membranes.78 Transmission electron microscopy was used to characterize the final wall thickness of LbL nanotubes deposited within pores 50-850 nm in diameter, which corresponded to 4 to 80 times the end-to-end distance of the polyelectrolyte chains in solution. Studies performed with the PVBAC/PSS assembly pair on flat model surfaces showed a linear growth, with the growth increment independent of the molar mass and substrate. However, upon assembling the same polyelectrolytes within the nanopores, a very different picture of multilayer growth emerged: the thickness of the nanotubes showed a marked dependence on the pore diameter, being proportional to pore diameter for small pores, but progressively deviating from this relationship for diameters above 250 nm. However, little dependence on the molar mass was found. These observations are consistent with the formation of a dense gel that filled the smaller sized nanopores, which, upon drying for TEM characterization, collapsed onto the pore walls to form nanotubes with wall thickness directly proportional to the pore diameter. Such a scenario was likely promoted by enhanced polyelectrolyte complexation and chain entanglement in the nanopores and the fact that polyelectrolyte chains are in a concentrated regime when passing in these confined channels. In such a regime, the end-to-end distance no longer describes the characteristic size of the system, and the growth of PVBAC/PSS multilayers in the nanopores is fundamentally different from what occurs on flat surfaces.
Jonas et al. also studied the pore-confined assembly of the quintessential assembly pair polyallylamine hydrochloride (PAH) and sodium poly(styrene sulfonate) (PSS).79 These investigations were performed using polyelectrolytes of different molar masses, ionic strengths and different templates with pore diameters ranging from 100~500 nm. The experiments showed the existence of two regimes in the PAH/PSS multilayer growth. The first was comparable to conventional LbL deposition as observed on flat surfaces, in which an increase in ionic strength leads to the formation of thicker multilayers (analogous to the dendrimer system used by Lau et al. above). The second regime was represented by slower kinetics and involved the formation of an entangled structure inside the pores (Figure 5). In close resemblance to aforementioned PVBAC/PSS assembly, sequential polyelectrolyte assembly in a confined geometry triggered the interconnection between polyelectrolyte chains, leading to the formation of a dense gel. The diffusion of polyelectrolytes in nanopores therefore becomes the controlling factor in this second regime, and dictates whether polymer nanorods (formed from a dense gel) or nanotubes (from multilayer assembly on pore walls) are generated.
FIGURE 5.

Schematic representation of the structural organization of polyelectrolyte multilayers in nanopores according to the scenario proposed of Jonas and co-workers. The cartoons describe both regimes prior to and after drying. Reproduced with permission from Roy et al. Langmuir, 2010, 26, 3350. Copyright 2010 American Chemical Society.
In this context, upon formation of the dense gel, nanoconfinement effects dominate both the degree of polymer interpenetration and the local structure of the multilayer. For instance, the dependence of multilayer thickness on the pore diameter is a strong indication that nanoconfinement effects govern the local topological features of the polyelectrolyte assembly. This effect could be even more pronounced in very small pores. Recently, Azzaroni et al.80 reported the LbL assembly of PAH/PSS in conical nanopores with pore tip dimensions close to 18 nm. The multilayer growth was monitored by measuring the pore conductance and the experimental values were described within a theoretical framework based on the Nernst-Planck-Poisson formalism to represent the ion transport across the nanopores. Their results suggest that increasing the number of PAH/PSS layers inside the nanopore imposes increasing topological restrictions to the incoming polyelectrolyte layers. In agreement with the scenario described by Jonas et al., the sequential confinement of polyelectrolyte layers within nanopores leads to film reorganization. Specifically, charge regulation within the nanopores lead to strong confinement, extensive polyelectrolyte intermixing at a molecular level, and complete ion pairing during sequential assembly. As a result, the net surface charge decreases after increasing the number of layers in the LbL assembly (Figure 6), which means that the overcompensation vanishes during multilayer growth within very small pores. The preceding discussion indicates that entry of polyelectrolyte species into small pores is an inherent complexity of the nanotemplated LbL strategy.
FIGURE 6.
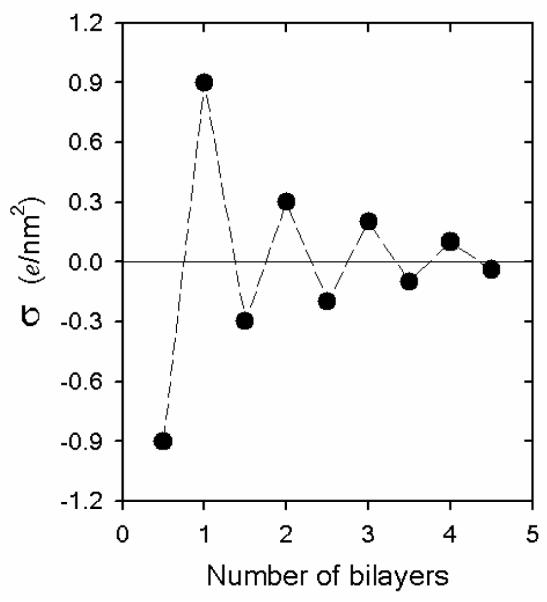
Representation of the nanopore surface charge density (σ) (obtained from the theoretical fittings) as a function of the number of PAH/PSS bilayers assembled on the conical pore walls (tip diameter = 18 nm). Reproduced and adapted with permission from Azzaroni et al. J. Am. Chem. Soc, 2010, 132, 8338. Copyright 2010 American Chemical Society.
5. Functional LbL-assembled nanotubes
5.1. Thermally-induced structural transformation of LbL-assembled nanotubes
Möhwald, Li and collaborators explored how Rayleigh instability can drive the transformation of LbL (PSS/PAH)n nanotubes into nano-capsules.81 PSS/PAH nanotubes were first assembled within nanoporous TEPC membranes. In the case of nanotubes with 8 bilayers, thermal annealing of the nanotubes dispersed in water above 120°C for 20 min led to the rupture of ion pairs between the polyelectrolyte layers and enabled the surface tension-driven structural transformation of the nanotubes into capsules. The original, multilayered nanotubes are stable at room temperature because the polyelectrolytes constituting the LbL-layers are electrostatically cross-linked and the mobility of the polyelectrolyte chains is rather slow. However, in the case of (PAH/PSS)8 nanotubes, increasing the temperature to 120°C provided sufficient thermal energy to overcome the stabilizing electrostatic interactions between ion pairs and increased the polyelectrolyte chain mobility. Consequently, Rayleigh instability82 broke up the nanotubular fluid-like PAH/PSS multilayers into smaller spherical droplets with the same enclosed volume but a reduced total surface area (Figure 7). The extent of this process depended on the number of polyelectrolyte multilayers constituting the nanotube walls. (PAH/PSS)12 nanotubes also transformed into capsules after typical hydrothermal treatment. However, no transformation in either size or shape was observed for (PAH/PSS)16 or (PAH/PSS)19 nanotubes when annealed under the same conditions. Transformation of these more robust, thicker nanotube LbL layers required an increase of the annealing temperature. For example, (PAH/PSS)19 nanotubes were transformed into capsule-like nanostructures when annealed at 138°C. Such a temperature-driven structural transformation83 of soft tubular architectures into vesicle-like nano-objects could represent an interesting approach to manipulating the topological features of hollow nanostructures for drug delivery.84
FIGURE 7.

Confocal laser scanning microscopy images of LbL-grown nanotubes (a) and the resulting nanocapsules (b) constituted of (PSS/PAH)8PAH assemblies after hydrothermal treatment at 121 °C. Images were taken in water and PAH was labeled with fluorescein isothiocyanate to facilitate the visualization of the nanostructures. Reproduced and adapted with permission from He et al. Langmuir, 2008, 24, 5508. Copyright 2008 American Chemical Society.
5.2. Soft nanoactuators based on swellable nanotube arrays
Macroscopic structural variations arising from nanoscale conformational changes provide promising new directions for exploiting soft materials as nanoactuators. In this context, Cohen et al. described an interesting technique for building up oriented arrays of substrate-bound LbL nanotubes that can undergo pH controllable mechanical actuation.85 The strategy takes advantage of the ability of PAH/PAA multilayers to undergo pronounced swelling-deswelling in the presence of pH variations. Supported nanotube arrays with specific orientations and spatial configurations were prepared by binding the PAH/PAA functionalized TEPC nanomembrane on an amine-treated glass substrate. This procedure relies on the fact that after LbL assembly, both the interior pore walls and the external faces of the TEPC membrane were functionalized with the PAH/PAA multilayer. Therefore, the glass substrate in contact with the TEPC membrane could interact with the LbL multilayer. Through heating at 60°C, the electrostatic adhesion between the PAH/PAA and the aminated glass substrate was enhanced, and the PAH/PAA nanotubes were secured on the substrate. The PAH/PAA assembled atop the other, unattached side of the TEPC membrane was then plasma etched away, and the TEPC dissolved in dichloromethane, to generate a supported array of nanotubes aligned normal to the substrate (Figure 8). Experiments performed under different pH conditions revealed that the reversible swelling-deswelling transitions of the PAH/PAA nanotube arrays led to significant changes in the nanotube dimensions. Interestingly, even in the highly swollen state, nanotubes remained intact as individual tubes without dissociating completely or merging irreversibly with neighbouring nanotubes (the heat treatment also enhanced the cohesion between the nanotube polyelectrolyte layers). Molecular rearrangements triggered by pH changes in the PAH/PAA multilayers further led to the actuation of movement of colloidal nanoparticles adsorbed on the nanotube array.
Figure 8.

(a) SEM images of substrate-attached LbL-grown (PAH/PAA) nanotube arrays. (b) Schematic illustration describing the dimensional changes of the tube arrays immersed in solutions with different pH conditions. Reproduced and adapted with permission from Chia et al. Langmuir 2009, 25, 14044. Copyright 2009 American Chemical Society.
5.3. Ion separation via pore assembled multilayers of charged polypeptides
Molecularly engineered nanomembranes have great potential for a wide variety of separation applications, including selective separation of ionic species from industrial waste solutions.86 Whilst traditional methods for separation of ionic species involve batch processing using ion exchange columns or liquid–liquid extraction, a membrane-based system offers the potential advantages of a continuous process, significant reduction in energy costs, and reduction of waste. Hollman and Bhattacharyya87 described the construction of highly permeable ion-selective membranes through the formation of polyelectrolyte multilayer assemblies within the inner pores of TEPC track-etched membranes with 200 nm pore diameters. Electrostatic LbL assembly was accomplished through alternate adsorption of cationic poly(L-lysine) (PLL) and anionic (poly(L-glutamic acid) (PLGA) polypeptides under convective flow conditions. Prior to initiating pore assembly, PLGA or PLL layers were covalently bound to the TEPC to create a robust charged platform for subsequent adsorption. Nonstoichiometric immobilization of charged multilayers within a confined pore geometry led to an enhanced volume density of ionizable groups in the membrane phase. The increase in the effective charge density of the nanopore allows for Donnan exclusion of ionic species.88 The manifestation of this exclusion phenomenon is even more pronounced in the case of divalent ions. Since polyelectrolyte-modified nanopores can create conditions of a “unipolar” solution,89,90 such nanopores allow very high degrees of permselectivity that yield ion selectivities that cannot be achieved with monolayer-modified nanopores.
The transport properties of the nanomebranes modified with the assembled peptides are highly dependent on the ionic strength of the solvent used for LbL adsorption. In the study of Hollman and Bhattacharyya, assemblies formed in high salt concentration showed lower pure water permeability and enhanced polypeptide adsorption as compared to assemblies formed in pure water. This observation was attributed to a reduction in the number of ionic interactions per adsorbed chain during the deposition process in pure water, which resulted in larger segments of the deposited polymers extending into the pore cross section and greater solvent resistance. This effect was even more dramatic upon increasing the number of bilayers.
Additionally, the use of weak polyelectrolyte assemblies introduces the possibility of controlling the ion permselectivity of different ionic species by changing the environmental pH (Figure 9). One interesting example is the pH-tunable separation of Na2SO4 and NaHAsO4 using nanomembranes modified with PLGA/PLL/PLGA assemblies. The pKa values associated with the SO42− and As(V) ions are 1.99 and 6.94, respectively. Hence, in the 3 < pH < 9 range SO42− remains divalent, while As(V) changes from a monovalent form in the low pH regime to a divalent form in the high pH regime. Consequently, in slightly acidic solutions separation of SO42− ions was substantially greater than As(V) species and its rejection was nearly constant above pH 6 (Figure 9). At the same time, lowering the solution pH also resulted in an enhanced protonation of the peptide side chains and conformational transition of the peptide backbone, which is reflected in a reduced level of SO42− rejection, and indicated the key role of the weak polyelectrolyte character of the PLGA assembly in the overall ion retention. Hence, the ion separation efficiency of the pore assembled polypeptide multilayers is governed by the interplay between the effective charges of the ions to be separated and the electrostatic state of the polypeptide assembly.
FIGURE 9.
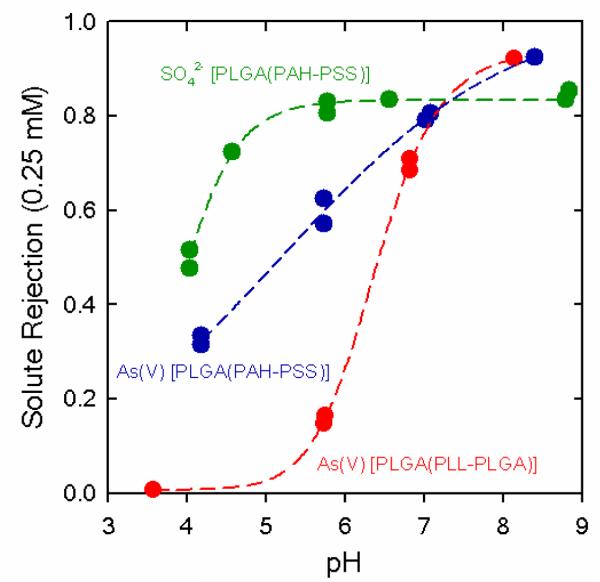
Effect of solution pH on the ion separation of 0.25 mM As(V) (Na2HAsO4) or Na2SO4 solutions using polycarbonate track-etched nanomembranes modified with PLGA-based assemblies. Reproduced and adapted with permission from Hollman et al. Langmuir 2004, 20, 5418. Copyright 2004 American Chemical Society.
5.4. Formation of catalytic nanomembranes
It has been shown that the LbL technique is fully compatible with the incorporation of metal colloids and nanoparticles in thin film architectures.91 Furthermore, in the case of catalytic materials, the LbL strategy permits addressing the nanomaterials into the film without inhibiting their catalytic properties. Bruening and colleagues described an interesting approach based on the use of porous membranes (AAO and TEPC) as a support for growing hybrid assemblies constituted of citrate-modified gold nanoparticles and polyelectrolytes.92 The membrane-supported one-dimensional assembly may prove very attractive for catalytic purposes in flow-through reactions, as it avoids not only the colloidal stabilization of the catalyst in the solvent media but also its loss from the reaction mixture. The AAO-confined hybrid assembly was prepared through the sequential flow of poly(acrylic acid) (PAA) aqueous solution, water, protonated poly(allylamine) (PAH) aqueous solution, water, and citrate-stabilized gold colloids through the membranes. The PAA/PAH layer provided a highly charged surface that enhanced colloid deposition. In the case of TEPC membranes, polystyrene sulfonate (PSS) was used for the formation of the precursor polyanionic layer. AAO membranes were also modified through direct adsorption of polyethyleneimine (PEI)/Au colloid bilayers by tuning the pH of the PEI solution to 8.5, which allowed direct polymer deposition on the alumina surface without a precursor PAA layer. The catalytic activity of the hybrid nanomembranes was characterized by reducing nitroaromatic compounds to their corresponding amino derivatives in the presence of NaBH4. Control experiments revealed that reduction does not occur in the absence of nanoparticles. In contrast, in the presence of the colloid-containing hybrid assemblies, more that 99 % of the 4-NP in a solution of 0.4 mM 4-NP + 20 mM NaBH4 can be reduced to 4-aminophenol, at a solution flux of 0.03 mL cm−2 s−1. Hence, colloid-modified membranes show remarkable catalytic conversions even at high flow rates. In addition, experimental results showed that the conversion increased as a function of the number of layers deposited for PEI/Au nanoparticle films when using a 25-fold excess of NaBH4. Interestingly, subsequent stability tests indicated that dry hybrid nanomembranes could be stored for several months without negative effects on its catalytic activity. These results illustrate the enormous potential of colloid-containing hybrid nanomembranes as a versatile catalytic platform which provides exquisite control over the arrangement and reaction environment of catalytic nanomaterials.
5.5. Biodegradable magnetic polypeptide nanotubes as DNA carriers
During the last decade, the biomaterials community showed a profound interest in developing magnetically-assisted DNA delivery systems assembled from iron oxide nanoparticles and funcational polymer building blocks,93 which could enable the manipulation of the local concentration of therapeutics in target tissues and lead to efficient treatment strategies. Along these lines, Möhwald, Li and collaborators fabricated plasmid-DNA carriers composed of biodegradable magnetic polypeptide nanotubes assembled from poly-L-lysine (PLL), poly-L-glutamic acid (PLGA) and magnetic nanoparticles.94 The nanotubes were assembled within the nanopores of TEPC templates and enabled the formation of tubular LbL nanostructures in which the mechanical stability could be improved by increasing the number of assembled PLL/PGA bilayers. Typically, 13.5 bilayers of PLL and PLGA were assembled before the deposition of two bilayers of PLL and magnetic Fe3O4 nanoparticles, which enabled the magnetic manipulation of the polypeptide tubes in solution. Positively charged PLL was always absorbed first on the pore walls of the TEPC membranes such that, after release of the nanotubes by dissolution of the supporting TEPC membrane in dichloromethane, negatively charged plasmid-DNA could be assembled onto the positively-charged, magnetic polypeptide nanotubes.
Experiments revealed that the plasmid-DNA/(PLL/PGA)13.5(Fe3O4/PLL)2 nanotubes, when dispersed in solution, could be rapidly aligned with a nearby magnetic field. The plasmid DNA was also fluorescently labeled with FITC, and the magnetic manipulation was corroborated by confocal laser scanning microscopy. In addition, α-chymotrypsin assays demonstrated the biodegradability of the polypeptide nanotubes; overnight incubation in an α-chymotrypsin solution led to the enzymatic hydrolysis of PLL and degradation of the nanotube walls. All these features herald the use of heterostructured polypeptide nanotubes as engineered nanomaterials acting as carriers for DNA delivery and release.
5.6. pH-induced hysteretic gating in nanoconfined LbL assemblies – Nanomembranes with adaptable transport properties
The rational design and construction of stimuli-responsive nanomembranes discriminating and/or propelling molecular transport represent an important factor in a wide variety of technological applications relying on “gated” transport processes, such as ultrafiltration or controlled delivery.95 Rubner et al. reported the construction of pH-responsive nanomembranes that show discontinuous changes in the permeation properties, i.e.: hysteretic gating, through the LbL assembly of PAH and PSS at a high pH conditions (pH > 9.0) into TEPC pores (Figure 10).96
FIGURE 10.

Changes in flux (a) and pore diameter (b) as a function of pH. The filled and open circles represent data generated after a pH 10.5 pretreatment and after a pH 2.5 pretreatment, respectively. Reproduced and adapted with permission from Lee et al. J. Am. Chem. Soc. 2006, 128, 8521. Copyright 2006 American Chemical Society.
It is well known that (PSS/PAH)n multilayers assembled at pH > 9 display discontinuous swelling/deswelling transitions as a function of environmental pH.97 These transitions are reversible, and arise from discontinuous changes in the degree of ionization of free amine groups within the PAH and a dramatic shift in the pKa of the polycation within the local environment of the multilayer assembly.98 Therefore, when PSS/PAH are assembled into nanomembrane pores, large discontinuous changes in the transmembrane flux were observed. Rubner et al. showed that PSS/PAH-modified membranes exhibited reversible gating properties upon alternating the pH conditions between pH 2.5 and 10.5.96 After the pH 10.5 pretreatment, the multilayer-modified pores remained “open” down to pH ~5. However, as the pH of permeated water was further lowered, the multilayers underwent a swelling transition that “closed” the pores. On the other hand, multilayer-modified pores pretreated at pH 2.5, retain their swollen “closed” structure up to pH 9.0. The hysteretic gating properties of the PSS/PAH-modified nanomembranes therefore introduces a new concept in stimuli-responsive nanomembranes in achieving “open” or “closed” states at a single pH condition depending on pretreatment conditions.
5.7. Uptake and delivery of ionic species with heterostructured magnetic polymer nanotubes
As described above PAH-containing multilayers assembled under high pH conditions undergo pronounced discontinuous swelling/deswelling transition as a function of solution pH. This interesting physical feature could be exploited to uptake low molecular weight anionic molecules within PAH-containing nanotubes after they have been “activated” with acidic solution (pH < 2.5). In the work of Rubner et al.,99 the inner LbL bilayers also contained Fe3O4 nanoparticles to allow the magnetic manipulation of the heterostrucutred nano-containers in solution. After the deposition of the Fe3O4 nanoparticles and PAH, the heterostructured nanotubes were released from the TEPC nanotemplate by dissolution in dichloromethane and methanol (9:1). The magnetic heterostructured nanotubes were utilized to take in a large amount of different anionic probe molecules, such as rose Bengal, acid red 8 or ibuprofen, after acid “activation”. Release studies demonstrated that these nanostructures were effective as “trapping and release” vehicles of anionic guest molecules, for which the diffusion dynamics is greatly influenced by the guest size, i.e.: larger molecules were released more slowly. Owing to their well defined magnetic properties these hetero-nanotubes can be easily manipulated and directed using magnetic fields.
5.8. Molecular recognition, discrimination and capture in protein nanotubes
Komatsu and co-workers described the construction of protein nanotubes displaying molecular capture properties as well as controllable ligand binding affinity and size selectivity.100 The nanostructures were prepared by conventional electrostatic LbL deposition of poly-L-arginine (PLA, Mw ~70 KDa) and human serum albumin (HSA) [(PLA/HSA)3] within nanoporous TEPC (pore diameter ~400 nm), with subsequent dissolution of the template in N,N-dimethylformamide (DMF) (Figure 11). HSA is anionic at pH > 4.8 and binds to the cationic PLA via electrostatic attraction. Molecular capture/recognition capabilities were demonstrated by complexing/binding of uranyl ion (UO22+), 3,3′-diethylthiacarbocyanine iodide (DTC) and zinc(II) protoporphyrin IX (ZnPP) to HSA. The molecular species were also able to diffuse through the multilayered walls of the nanotubes. It was demonstrated that HSA modified by site-specific mutations could enhance the binding properties of ZnPP. Furthermore, myristic acid also binds to the same domain of HSA as ZnPP, and ZnPP binding can be reversed in the presence of myristic acid by competitive binding. Protein nanotubes bearing a single avidin layer as an internal surface (assembled as the last layer within the nanotube) were also prepared and captured biotin efficiently. Subsequently biotin-labeled fluorescent latex beads sufficiently small to enter the pores were incorporated into the nanotubes.
FIGURE 11.

Scanning electron microscopy images at different magnifications (A-D) of (PLA/HSA)3 nanotubes prepared using polycarbonate track-etched nanomembranes. The figure also includes a descriptive cartoon showing the constituting building blocks of the multilayer assembly. Reproduced and adapted with permission from Qu et al. ACS Nano 2010, 4, 563. Copyright 2010 American Chemical Society.
In further studies, PLA/HSA protein nanotubes with an α-glucosidase (αGluD) interior surface were also prepared and displayed enzymatic activity.101 Experiments revealed that in aqueous media the protein nanotubes captured 4-methyl-umbelliferyl-α-D-glucopyranoside (MUGlc, a fluorogenic glucopyranoside) into their pore space which hydrolyzed to form α-D-glucose. Protein nanotubes displaying specific biorecognition elements in the interior of the one-dimensional nanostructure represent a potential strategy to create “on-demand” delivery (load & release) systems. The biocompatible exterior can also be modified to target specific tissues or to respond to biological stimuli.
5.9. Cascaded fluorescence resonance energy transfer (FRET) nanotubes for high-sensitivity biosensing
FRET occurs through the non-radiative Förster energy transfer from an excited donor to an acceptor fluorophore in close proximity (within 10 nm), and is observed through the quenching of the donor fluorescence and the subsequent increase in acceptor fluorescence intensity.102 The energy transferred can be further enhanced if multiple FRET pairs with overlapping energy bands are sequentially arranged in proximity to each other, from higher to lower emission frequencies, in a directional energy transfer cascade.103 Feng et al. exploited the cylindrical pore geometry of nanoporous AAO and the nanoscale spatial control of the LbL process to prepare an attractive implementation of cascaded FRET for high sensitivity DNA sensing (Figure12).104 The authors chose a series of water-soluble ZnxCd1-xSe alloy quantum dots (QDs) with emission wavelengths at 561 nm, 594 nm and 614 nm as the light-harvesting FRET cascade (i.e. QD561, QD594, QD 614). The QDs were made anionic by functionalization with mercaptoundecanoic acid (MUA) ligands to enable LbL assembly together with dendrimer polyelectrolytes having either terminal cationic (NH+Et2) or anionic (CH–COO−) moieties. To minimize fluorescence quenching by the alumina surface, three dendrimer bilayers were first deposited within the AAO (400 nm pores) before QD multilayers with the shortest emission wavelength, QD561, were deposited. This was followed by the LbL deposition of QD594 and then QD614 multilayers. Illumination at 460 nm excited all the QDs. However, the QD561 emission energy was transferred to the adjacent QD594, and the QD594 emission energy was similarly transferred to the QD 614 such that the only photoluminescence (PL) observed was at 614 nm. LbL preparation of nanotubular QD-FRET assemblies by AAO nanotemplating has several advantages. First, the AAO substrate provides for convenient handling during LbL deposition and rinsing, as opposed to the centrifugation and re-dispersion necessary for nanoparticle or nanowire-QD LbL assemblies.105 Second, the cylindrical pore geometry and the consequent concentric QD layer structure with acceptors deposited towards the pore centers ensure a slight excess of donor QDs—the LbL QD-FRET assemblies exhibited a 14-times enhancement in PL intensity as compared to equivalent assemblies with only QD614. Finally, the LbL QD-dendrimer assemblies were functionalized with single-stranded probe DNA. Complementary target DNA sequences, labeled with the Cy5 fluorescent dye (absorption from 580~680 nm), were subsequently detected as they hybridized with the probe DNA and were excited by the QD FRET cascade. A detection limit of 100 fM was demonstrated, which surpasses another highly sensitive DNA sensing technique, surface plasmon fluorescence spectroscopy.106
FIGURE 12.

Schematic of the cascaded FRET QD-dendrimer nanotubes. A) Self-assembled porous alumina membrane is used as a template. B) The pore walls are coated with 3-aminopropyl-dimethylethoxysilane to provide a positive surface charge. C) 3-bilayers of dendrimer polyelectrolytes are then deposited, starting with the negatively charged species. D) Negatively charged QDs are then deposited inside the template starting with QD561 (with luminescence maximum at k = 561 nm, i.e green), then QD594 (orange) and finally QD614 (red). Five QD/positively charged dendrimer bilayers were deposited for each QD species. E) After activation by N-hydroxysuccinimide (NHS) / 1-ethyl-3-(dimethylamino)propylcarbodiimide (EDC), probe DNA (pDNA) immobilization and hybridization with Cy5-labeled complementary DNA (tDNA) can be achieved inside the NTs. Reproduced and adapted with permission from Feng et al. Adv. Mater. 2007, 19, 1933. Copyright 2007 Wiley-VCH Verlag GmbH & Co. KGaA.
6. Summary and Outlook
The introduction of layer-by-layer assembly and nanotemplating by nanoporous templates over two decades ago has enabled fundamental developments in nanotechnology. Their synergistic combination for the generation of LbL-nanotubes in recent years is an extremely versatile approach to designing and generating complex but well-defined, one-dimensional materials with innovative properties. The well-controlled cylindrical geometry and convenient availability of nanoporous AAO and TEPC templates have also given a decisive impetus to the development LbL nanotubes. Recent advances have enabled the assembly of polymers, biomolecules and inorganic nanoparticles “à la carte”. The control over composition, size, shape, topology, and thus function of one-dimensional nanosystems is an essential contribution of nanotemplated LbL “soft nanotechnology”.
The general procedure for LbL-assembly within nanoporous templates is analogous to deposition on planar substrates. Both linear and dendrimer polyelectrolytes and charged inorganic nanoparticles have been successfully incorporated into LbL deposited nanotubes. After deposition, the nanotemplates can be removed to release nanotubes by a variety of selective etchants as surveyed throughout this review. A plasma treatment to preferentially remove material deposited on the external template surface, before conventional solution template etching, may enhance etchant access and aid in complete template removal. With respect especially to electrostatic LbL nanotemplate deposition, attention should be paid in the selection of solution ionic strength, pH and polymer concentration since these factors control the physicochemical processes within the constrained spaces of the nanopores. In particular, charge regulation within nanopores as large as ~10 times the diameter of the deposition species can lead to inhibited pore partitioning of charged species (i.e. pore clogging). Initial reports of LbL nanotemplating have demonstrated a pressure-filter-template method to hydraulically assist polyelectrolyte transport through nanopores. Electrophoretic control could potentially also overcome pore clogging effects. Notwithstanding, slightly extended immersion times per deposition step (10~60 min), a proper selection of polyelectrolyte pairs, and the use of templates with pores open at both ends, have generally been sufficient in ensuring proper LbL assembly within nanopores of high aspect ratios.
In certain cases nanoconfinement can also induce strong charge complexation and lead to gel phase instead of LbL deposition for some polyelectrolyte species, or the inhibition of charge overcompensation and polyelectrolyte assembly, especially within relatively small pores. In situ characterization of the deposition process could help verify proper LbL assembly and identify undesirable structure generation within the nanopores. Recent demonstrations of in situ nanoporous optical waveguide spectroscopy and pore conductance measurements have proven their usefulness in characterizing the transition between linear and charge-dominated polyelectrolyte deposition regimes within nanoporous templates. Further development of such techniques that can monitor the film thickness and surface charge, and the development of new tools that can characterize the spatial arrangement and surface chemistry of the LbL species during the deposition process, could also enable the fabrication of more sophisticated LbL designs and catalyze the overall development of the nanotemplated LbL toolbox.
As with other supramolecular nanostructures, the preparation method, the chemical and physicochemical natures, geometry and the function of LbL-assembled nanotubes are intimately linked. The nanotemplate pore surface chemistry can significantly influence even the rate of covalent reactions within the nanopores, such as amidation for cross-linking LbL-deposited multilayers for enhanced nanotubes stability. Other non-electrostatic assembly methods that directly take advantage of covalent coupling, H-bonding and the specific interactions of biomolecules and proteins for LbL assembly have augmented the repertoire of nanotube designs. Device functionality may simply come through the ability to assemble together diverse components, such as with magnetic nanotube carriers of biomolecules. However, the LbL multilayer structure and the nanotube format have also been integral to enhanced functionality. Such is the case for a cascaded-FRET LbL nanotube biosensor, which depended on the nanoscale proximity and the concentric arrangement of the assembled quantum dot layers for proper device function. Functionality has also come through the manipulation of the multilayer structure of stimuli-responsive nanotubes. Temperature has been used to transform PSS/PAH nanotubes into nanocapsules. In addition, pH induced swelling has been utilized for nanotube mechanical actuators, for controlling the molecular transport through nanotubes/nanopores, as well as for loading nanotubes with (bio)molecules. The generation of biopolymer nanotubes also offers the promise of innovative developments in biodegradable and biocompatible drug delivery systems. In the case of the nanotube actuators, the nanotemplate pore alignment also played a crucial role in orienting the nanotubes during device preparation for its proper function. Nanoporous templates functionalized with nanotubular assemblies also represent an obvious design approach for membrane separation applications. This integration of device function and nanotemplate properties may be a promising direction for practical applications. The increasing mastery in the modular design for nanotemplated LbL assembly will also continue to expand the toolbox of “soft nanotechnology”.
Acknowledgements
The authors gratefully acknowledge support from the Max-Planck-Gesellschaft (MPG–Germany). O.A. is staff member of CONICET and gratefully acknowledges support from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT - Argentina) (PICT-PRH 163/08), Centro Interdisciplinario de Nanociencia y Nanotecnología (CINN, PAE 2006 37063, projects: PRH 2007-74 and PIDRI No. 74), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET - Argentina) (PIP 2009-0362), Alexander von Humbolt Stiftung (Germany), Fundación Petruzza and Laboratório Nacional de Luz Síncrotron (LNLS – Brazil). K.H.A.L. gratefully acknowledges support by grants HL104966 and EB005772 from the National Institutes of Health, USA.
References
- 1 (a).Bréchignac C, Houdy P, Lahmani M, editors. Nanomaterials and Nanochemistry. Springer-Verlag; Heidelberg: 2007. [Google Scholar]; (b) Ozin GA, Arsenault AC, Cademartiri L. Nanochemistry: A Chemical Approach to Nanomaterials. Royal Society of Chemistry; London: 2009. L. [Google Scholar]; (c) Vollath D. Nanomaterials: An Introduction to Synthesis, Properties and Applications. VCH-Wiley; Weinheim: 2008. [Google Scholar]
- 2.Champion Y, Fecht H-J, editors. Nano-Architectured and Nanostructured Materials. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 3 (a).Fendler JH, editor. Nanoparticles and Nanostructured Films: Preparation, Characterization and Applications. Wiley-VCH; Weinheim: 1998. [Google Scholar]; (b) Cassagneau TP, Fendler JH. Ch. 9. In: Hodes G, editor. Electrochemistry of Nanomaterials. Wiley-VCH; Weinheim: 2001. pp. 247–286. [Google Scholar]
- 4.Wang ZM, editor. One-Dimensional Nanostructures. Springer-Verlag; Heidelberg: 2008. [Google Scholar]
- 5 (a).Decher G, Hong JD. Ber. Bunsen-Ges. 1991;95:1430–1434. [Google Scholar]; (b) Decher G, Hong JD. Makromol. Chem., Macromol. Symp. 1991;46:321–327. [Google Scholar]
- 6 (a).Decher G. Ch. 1. In: Decher G, Schlenoff JB, editors. Multilayer Thin Films. Wiley-VCH; Weinheim: 2002. pp. 1–46. [Google Scholar]; (b) Shia X, Shen M, Möhwald H. Prog. Polym. Sci. 2004;29:987–1019. [Google Scholar]; (c) Ariga A, Ji Q, Hill JP, Vinu A. Soft Matter. 2009;5:3562–3571. [Google Scholar]
- 7 (a).Decher G. Science. 1997;277:1232. [Google Scholar]; (b) Caruso F. Adv. Mater. 2001;13:11. [Google Scholar]; (c) Peyratout CS, Dähne L. Angew. Chem., Int. Ed. 2004;43:3762. doi: 10.1002/anie.200300568. [DOI] [PubMed] [Google Scholar]; (d) Quinn JF, Johnston APR, Such GK, Zelikin AN, Caruso F. Chem. Soc. Rev. 2007;36:707. doi: 10.1039/b610778h. F. [DOI] [PubMed] [Google Scholar]; (e) Ariga K, Hill JP, Ji Q. Phys. Chem. Chem. Phys. 2007;9:2319. doi: 10.1039/b700410a. [DOI] [PubMed] [Google Scholar]; (f) Caruso F. Chem.Eur. J. 2000;6:413. doi: 10.1002/(sici)1521-3765(20000204)6:3<413::aid-chem413>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; (g) Zhang X, Chen H, Zhang HY. Chem. Commun. 2007:1395. doi: 10.1039/b615590a. [DOI] [PubMed] [Google Scholar]; (h) Bertrand P, Jonas AM, Laschewsky A, Legras R. Macromol. Rapid Commun. 2000;21:319–348. [Google Scholar]
- 8 (a).Cai Z, Lei J, Liang W, Menon V, Martin CR. Chem. Mater. 1991;3:960–967. [Google Scholar]; (b) Martin CR. Science. 1994;266:1961–1966. doi: 10.1126/science.266.5193.1961. [DOI] [PubMed] [Google Scholar]; (c) Parthasarathy R, Martin CR. Nature. 1994;369:298–301. doi: 10.1038/369298a0. [DOI] [PubMed] [Google Scholar]; (d) Martin CR. Acc. Chem. Res. 1995;28:61–68. [Google Scholar]
- 9.Masuda Hideki, Fukuda K. Science. 1995;268:1466–1468. doi: 10.1126/science.268.5216.1466. [DOI] [PubMed] [Google Scholar]
- 10 (a).Zheng SP, Tao C, He Q, Zhu HF, Li JB. Chem. Mater. 2004;16:3677–3681. [Google Scholar]; (b) Tian Y, He Q, Tao C, Cui Y, Li JB. Chem. Eur. J. 2006;12:4808–4812. doi: 10.1002/chem.200600208. [DOI] [PubMed] [Google Scholar]
- 11 (a).Steinhart M, Wendorff JH, Greiner A, Wehrspohn RB, Nielsch K, Schilling J, Choi J, Gösele U. Science. 2002;296:1997. doi: 10.1126/science.1071210. [DOI] [PubMed] [Google Scholar]; (b) Steinhart M, Wendorff JH, Wehrspohn RB. ChemPhysChem. 2003;4:1171–1176. doi: 10.1002/cphc.200300733. [DOI] [PubMed] [Google Scholar]; (c) Steinhart M, Wehrspohn RB, Gösele U, Wendorff JH. Angew. Chem. Int. Ed. 2004;43:1334–1344. doi: 10.1002/anie.200300614. [DOI] [PubMed] [Google Scholar]; (d) Steinhart M. Adv. Polym. Sci. 2008;220:123–187. [Google Scholar]; (e) Zhao L, Steinhart M, Gösele U, Schlecht S. Adv. Mater. 2008;20:1218–1221. [Google Scholar]
- 12.Ai S, Lu G, He Q, Li J. J. Am. Chem. Soc. 2003;125:11140–11141. doi: 10.1021/ja0356378. [DOI] [PubMed] [Google Scholar]
- 13.Liang Z, Susha AS, Yu A, Caruso F. Adv. Mater. 2003;15:1849–1853. [Google Scholar]
- 14 (a).He Q, Cui Y, Ai S, Tian Y, Li J. Curr. Op. Colloid. Interf. Sci. 2009;14:115–125. [Google Scholar]; (b) Manne S, Warr GG, editors. Supramolecular Structure in Confined Geometries. American Chemical Society; Washington, DC: 1999. [Google Scholar]
- 15 (a).Price PB, Walker RM. J. Appl. Phys. 1962;33:3407–3412. [Google Scholar]; (b) Fleischer RL, Alter HW, Furman SC, Price PB, Walker RM. Science. 1972;178:255–263. doi: 10.1126/science.178.4058.255. [DOI] [PubMed] [Google Scholar]
- 16.Lau KHA, Duran H, Knoll W. J. Phys. Chem. B. 2009;113:3179–3189. doi: 10.1021/jp809593d. [DOI] [PubMed] [Google Scholar]
- 17.Lau KHA, Tan L-S, Tamada K, Sander MS, Knoll W. J. Phys. Chem. B. 2004;108:10812–10818. [Google Scholar]
- 18.it4ip s.a. Seneffe; Belgium: http://www.it4ip.be. [Google Scholar]
- 19.Whatman International Ltd . Kent, UK: http://www.whatman.com. [Google Scholar]
- 20.Synkera Technologies, Inc . Longmont, USA: http://www.synkera.com. [Google Scholar]
- 21.O’Sullivan JP, Wood GC. Proc. Royal Soc. London A. 1970;317:511. [Google Scholar]
- 22.Jessensky O, Muller F, Gosele U. Appl. Phys. Lett. 1998;72:1173. [Google Scholar]
- 23.Nielsch K, Choi J, Schwirn K, Wehrspohn RB, Gosele U. Nano Lett. 2002;2:677. [Google Scholar]
- 24 (a).Masuda H, Satoh M. Jpn.. J. Appl. Phys. 1996;35:L126–L129. [Google Scholar]; (b) Masuda H, Yamada H, Satoh M, Asoh H, Nakao M, Tamamura T. Appl. Phys. Lett. 1997;71:2770–2772. [Google Scholar]
- 25.Ai SF, Lu G, He Q, Li JB. J. Am. Chem. Soc. 2003;125:11140–11141. doi: 10.1021/ja0356378. [DOI] [PubMed] [Google Scholar]
- 26.Ai SF, He Q, Tao C, Zheng SP, Li JB. Macromol. Rapid. Comm. 2005;26:1965–1969. [Google Scholar]
- 27.Zheng SP, Tao C, He Q, Zhu HF, Li JB. Chem. Mater. 2004;16:3677–3681. [Google Scholar]
- 28.Kim DH, Karan P, Goering P, Leclaire J, Caminade A-M, Majoral J-P, Gösele U, Steinhart M, Knoll W. Small. 2005;1:99–102. doi: 10.1002/smll.200400024. [DOI] [PubMed] [Google Scholar]
- 29 (a).Caminade A-M, Majoral J-P. J. Mater. Chem. 2005;15:3643–3649. [Google Scholar]; (b) Caminade A-M, Majoral J-P. Chem. Soc. Rev. 2010;39:2034–2047. doi: 10.1039/b926408f. [DOI] [PubMed] [Google Scholar]
- 30.Cao G, Hong H-G, Mallouk TE. Acc.Chem.Res. 1992;25:420–427. [Google Scholar]
- 31.Katz HE, Schilling ML, Ungashe S, Putvinski TM, Chidsey CE. Ch. 3. In: Bein T, editor. Supramolecular Architecture: Synthetic Control in Thin Films and Solids. American Chemical Society; Washington: 1992. pp. 24–32. [Google Scholar]
- 32.Hou S, Harrell CC, Trofin L, Kohli P, Martin CR. J. Am. Chem. Soc. 2004;126:5674–5675. doi: 10.1021/ja049537t. [DOI] [PubMed] [Google Scholar]
- 33.Gatebe E, Herron H, Zakeri R, Rajasekaran PR, Aouadi S, Kohli P. Langmuir. 2008;24:11947–11954. doi: 10.1021/la801948z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stockton WB, Rubner MF. Macromolecules. 1997;30:2717–2725. [Google Scholar]
- 35.Wang L, Wang ZQ, Zhang X, Shen JC, Chi LF, Fuchs H. Macromol. Rapid. Commun. 1997;18:509–514. [Google Scholar]
- 36 (a).Sukhishvili SA, Granick S. J. Am. Chem. Soc. 2000;122:9550–9551. [Google Scholar]; (b) Sukhishvili SA, Granick S. Macromolecules. 2002;35:301–310. [Google Scholar]; (c) Quinn JF, Caruso F. Langmuir. 2004;20:20–22. doi: 10.1021/la0360310. [DOI] [PubMed] [Google Scholar]; (d) Yang SY, Rubner MF. J. Am. Chem. Soc. 2002;124:2100–2101. doi: 10.1021/ja017681y. [DOI] [PubMed] [Google Scholar]
- 37.Such GK, Johnston APR, Caruso F. Chem Soc. Rev. 2011;40:19–29. doi: 10.1039/c0cs00001a. [DOI] [PubMed] [Google Scholar]
- 38.Tian Y, He Q, Tao C, Cui Y, Li JB. Chem. Eur. J. 2006;12:4808–4812. doi: 10.1002/chem.200600208. [DOI] [PubMed] [Google Scholar]
- 39 (a).Fu Y, Bai SL, Cui SX, Qiu DL, Wang ZQ, Zhang X. Macromolecules. 2002;35:9451. [Google Scholar]; (b) Zhang HY, Fu Y, Wang D, Wang LY, Wang ZQ, Zhang X. Langmuir. 2003;19:8497. [Google Scholar]
- 40 (a).Liu YL, Bruening ML, Bergbreiter DE, Crooks RM. Angew. Chem. Int. Ed. 1997;36:2114–2116. [Google Scholar]; (b) Kohli P, Blanchard GJ. Langmuir. 2000;16:4655–4661. [Google Scholar]; (c) Serizawa T, Nanameki K, Yamamoto K, Akashi M. Macromolecules. 2002;35:2184–2189. [Google Scholar]; (d) Duan L, He Q, Yan XH, Cui Y, Wang KW, Li JB. Biochem. Biophys. Res. Comm. 2007;354:357–362. doi: 10.1016/j.bbrc.2006.12.223. [DOI] [PubMed] [Google Scholar]; (e) Such GK, Quinn JF, Quinn A, Tjipto E, Caruso F. J. Am. Chem. Soc. 2006;128:9318–9319. doi: 10.1021/ja063043+. [DOI] [PubMed] [Google Scholar]
- 41.Tian Y, He Q, Tao C, Cui Y, Ai SF, Li JB. J. Nanosci. Nanotechnol. 2006;6:2072–2076. doi: 10.1166/jnn.2006.369. [DOI] [PubMed] [Google Scholar]
- 42 (a).Zhi L, Wu J, Li J, Stepputat M, Kolb U, Müllen K. Adv. Mater. 2005;17:1492–1496. [Google Scholar]; (b) Djuristic AB, Fritz T, Leo K. Opt. Commun. 2000;183:123–132. [Google Scholar]
- 43.Tian Y, He Q, Li JB. Langmuir. 2006;22:360–362. doi: 10.1021/la0524768. [DOI] [PubMed] [Google Scholar]
- 44.He Q, Tian Y, Möhwald H, Li JB. Soft Matter. 2009;5:300–303. [Google Scholar]
- 45.Balachandra AM, Dai J, Bruening ML. Macromolecules. 2002;35:3171–3178. [Google Scholar]
- 46.Liang ZJ, Susha AS, Yu AM, Caruso F. Adv. Mater. 2003;15:1849–1853. F. [Google Scholar]
- 47.Jang W-S, Jensen AT, Lutkenhaus JL. Macromolecules. 2010;43:9473–9479. [Google Scholar]
- 48 (a).Azzaroni O, Álvarez M, Abou-Kandil AI, Yameen B, Knoll W. Adv. Funct. Mater. 2008;18:3487–3496. [Google Scholar]; (b) Anzai J.-i., Kobayashi Y, Nakamura N, Nishimura M, Hoshi T. Langmuir. 1999;15:221. [Google Scholar]; (c) Cassier T, Lowack K, Decher G. Supramol. Sci. 1998;5:309. [Google Scholar]; (d) Pallarola D, Queralto N, Knoll W, Ceolín M, Azzaroni O, Battaglini F. Langmuir. 2010;26:13684–13696. doi: 10.1021/la100486g. [DOI] [PubMed] [Google Scholar]; (e) Pallarola D, Queralto N, Knoll W, Azzaroni O, Battaglini F. Chem. Eur. J. 2010;16:13970–13975. doi: 10.1002/chem.201001407. [DOI] [PubMed] [Google Scholar]; (f) Pallarola D, Queralto N, Battaglini F, Azzaroni O. Phys. Chem. Chem. Phys. 2010;12:8071–8083. doi: 10.1039/c000797h. [DOI] [PubMed] [Google Scholar]
- 49.Hou S, Wang JH, Martin CR. J. Am. Chem. Soc. 2005;127:8586–8587. doi: 10.1021/ja042343t. [DOI] [PubMed] [Google Scholar]
- 50.Ali M, Yameen B, Neumann R, Ensinger W, Knoll W, Azzaroni O. J. Am. Chem. Soc. 2008;130:16351–16357. doi: 10.1021/ja8071258. [DOI] [PubMed] [Google Scholar]
- 51 (a).Fendler JH. Ch. 9. In: Decher G, Schlenoff JB, editors. Multilayer Thin Films. Wiley-VCH; Weinheim: 2002. pp. 245–269. [Google Scholar]; (b) Kotov NA. Ch. 8. In: Decher G, Schlenoff JB, editors. Multilayer Thin Films. Wiley-VCH; Weinheim: 2002. pp. 207–243. [Google Scholar]
- 52.He Q, Möhwald H, Li JB. Macromol. Rapid Commun. 2009;30:1538–1542. doi: 10.1002/marc.200900261. [DOI] [PubMed] [Google Scholar]
- 53.Yu A, Lu GQM, Drennan J, Gentle IR. Adv. Funt. Mater. 2007;17:2600–2605. [Google Scholar]
- 54 (a).Baskaran S, Song L, Liu J, Chen YL, Graff GL. J. Am. Ceram. Soc. 1998;81:401. [Google Scholar]; (b) Hanprasopwattana A, Rieker T, Sault AG, Datye AK. Catal. Lett. 1997;45:165. [Google Scholar]; (c) Mockel H, Giersig M, Willig F. J. Mater. Chem. 1999;9:3051. [Google Scholar]
- 55 (a).Mayya KS, Gittins DI, Caruso F. Chem. Mater. 2001;13:3833. [Google Scholar]; (b) Shi XY, Cassagneau T, Caruso F. Langmuir. 2002;18:904. [Google Scholar]; (c) Caruso F, Shi XY, Caruso RA, Susha A. Adv. Mater. 2001;13:740. [Google Scholar]
- 56 (a).Kovtyukhova NI, Martin BR, Mbindyo JKN, Mallouk TE, Cabassi M, Mayer TS. Mater. Sci. Eng. C. 2002;19:255–262. [Google Scholar]; (b) Kovtyukhova NI, Martin BR, Mbindyo JKN, Smith PA, Razavi B, Mayer TS, Mallouk TE. J. Phys. Chem. B. 2001;105:8762–8769. [Google Scholar]
- 57.Ma Z, Liu Q, Cui Zhi-Min, Shao-Wei Bian, Wei-Guo Song. J. Phys. Chem. C. 2008;112:8875–8880. [Google Scholar]
- 58 (a).Lvov Y, Ariga K, Ichinose I, Kunitake T. J. Am. Chem. Soc. 1995;117:6117. [Google Scholar]; (b) Ariga K, Ji Q, Hill JP. Adv. Polym. Sci. 2010;229:51–87. [Google Scholar]
- 59.Dougherty SA, Zhang D, Liang J. Langmuir. 2009;25:13232–13237. doi: 10.1021/la901909n. [DOI] [PubMed] [Google Scholar]
- 60 (a).Champion JA, Mitragotri S. Pharm. Res. 2008;26:244–249. doi: 10.1007/s11095-008-9626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Nat. Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tian Y, He Q, Cui Y, Li JB. Biomacromolecules. 2006;7:2539–2542. doi: 10.1021/bm060412l. [DOI] [PubMed] [Google Scholar]
- 62.Landoulsi J, Roy CJ, Dupont-Gillain C, Demoustier-Champagne S. Biomacromolecules. 2009;10:1021–1024. doi: 10.1021/bm900245h. [DOI] [PubMed] [Google Scholar]
- 63.Yu A, Liang Z, Caruso F. Chem. Mater. 2005;17:171–175. [Google Scholar]
- 64.Lu G, Ai AF, Li JB. Langmuir. 2005;21:1679–1682. doi: 10.1021/la047771r. [DOI] [PubMed] [Google Scholar]
- 65.Qu X, Lu G, Tsuchida E, Komatsu T. Chem. Eur. J. 2008;14:10303–10308. doi: 10.1002/chem.200800771. [DOI] [PubMed] [Google Scholar]
- 66.Qu X, Kobayashi N, Komatsu T. ACS Nano. 2010;4:1732–1738. doi: 10.1021/nn901879d. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y, He Q, Duan L, Cui Y, Li JB. Biomaterials. 2007;28:3083–3090. doi: 10.1016/j.biomaterials.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 68.Lu G, Komatsu T, Tsuchida E. Chem. Comm. 2007:2980–2982. doi: 10.1039/b704135g. [DOI] [PubMed] [Google Scholar]
- 69.Hou S, Wang J, Martin CR. Nano Lett. 2005;5:231–234. doi: 10.1021/nl048305p. [DOI] [PubMed] [Google Scholar]
- 70.Kim JY, DeRocher JP, Mao P, Han J, Cohen RE, Rubner MF. Chem. Mater. 2010;22:6409–6415. [Google Scholar]
- 71.Savariar EN, Sochat MM, Klaikherd A, Thayumanavan S. Angew. Chem., Int. Ed. 2009;48:110–114. doi: 10.1002/anie.200804136. [DOI] [PubMed] [Google Scholar]
- 72.Savariar EN, Krishnamoorthy K, Thayumanavan S. Nature Nanotechnology. 2008;3:112–117. doi: 10.1038/nnano.2008.6. [DOI] [PubMed] [Google Scholar]
- 73.Lazzara TD, Lau KHA, Abou-Kandil AI, Caminade A-M, Majoral J-P, Knoll W. ACS Nano. 2010;4:3909–3920. doi: 10.1021/nn1007594. [DOI] [PubMed] [Google Scholar]
- 74.Lau KHA, Tan LS, Tamada K, Sander MS, Knoll W. J. Phys. Chem. B. 2004;108:10812–10818. [Google Scholar]
- 75 (a).Yu Y, Feng C, Caminade A-M, Majoral J-P, Knoll W. Langmuir. 2009;25:13680–13684. doi: 10.1021/la901988r. [DOI] [PubMed] [Google Scholar]; (b) Kim BS, Lebedeva OV, Kim DH, Caminade AM, Majoral J-P, Knoll W, Vinogradova OI. Langmuir. 2005;21:7200–7206. doi: 10.1021/la0504208. [DOI] [PubMed] [Google Scholar]
- 76 (a).Launay N, Caminade AM, Lahana R, Majoral J-P. Angew. Chem., Int. Ed. 1994;33:1589–1592. [Google Scholar]; (b) Caminade AM, Majoral J-P. Acc. Chem. Res. 2004;37:341–348. doi: 10.1021/ar020077n. [DOI] [PubMed] [Google Scholar]
- 77.Carrillo J-MY, Dobrynin AV. ACS Nano. 2011;5:3010–3019. doi: 10.1021/nn200065q. [DOI] [PubMed] [Google Scholar]
- 78.Alem H, Blondeau F, Glinel K, Demoustier-Champagne S, Jonas AM. Macromolecules. 2007;40:3366–3372. [Google Scholar]
- 79.Roy CJ, Dupont-Guillan C, Demoustier-Champagne S, Jonas AM, Landoulsi J. Langmuir. 2010;26:3350–3355. doi: 10.1021/la903121e. [DOI] [PubMed] [Google Scholar]
- 80.Ali M, Yameen B, Cervera J, Ramírez P, Neumann R, Ensinger W, Knoll W, Azzaroni O. J. Am. Chem. Soc. 2010;132:8338–8348. doi: 10.1021/ja101014y. [DOI] [PubMed] [Google Scholar]
- 81.He Q, Song W, Möhwald H, Li J. Langmuir. 2008;24:5508–5513. doi: 10.1021/la703738m. [DOI] [PubMed] [Google Scholar]
- 82.Toimil-Molares ME, Balogh AG, Cornelius TW, Neumann R, Trautmann C. Appl. Phys. Lett. 2004;85:5337. [Google Scholar]
- 83 (a).Bar-Ziv R, Moses E, Nelson P. Biophys. J. 1998;75:294. doi: 10.1016/S0006-3495(98)77515-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reinecke AA, Dobereiner HG. Langmuir. 2003;19:605. [Google Scholar]; (c) Chen JT, Zhang MF, Russell TP, P. T. Nano Lett. 2007;7:183. doi: 10.1021/nl0621241. [DOI] [PubMed] [Google Scholar]; (d) Müller R, Dähne L, Fery AJ, J. A. Phys. Chem. B. 2007;111:8547. doi: 10.1021/jp068762p. [DOI] [PubMed] [Google Scholar]
- 84 (a).Martin CR, Kohli P. Nat. Rev. Drug Discovery. 2003;2:29. doi: 10.1038/nrd988. P. [DOI] [PubMed] [Google Scholar]; (b) Caruso F, Caruso RA, Möhwald H. Science. 1998;282:1111. doi: 10.1126/science.282.5391.1111. [DOI] [PubMed] [Google Scholar]
- 85.Chia K-K, Rubner MF, Cohen RE. Langmuir. 2009;25:14044–14052. doi: 10.1021/la9015959. [DOI] [PubMed] [Google Scholar]
- 86 (a).Gupta CK, Mukherjee TK. Hydrometallurgy in Extraction Processes. vol. 2. CRC Press; Boca Raton: 1987. [Google Scholar]; (b) Wiesner MR, Chellam S. Environ. Sci. Technol. 1999;33:360A. doi: 10.1021/es993008l. [DOI] [PubMed] [Google Scholar]
- 87.Hollman AA, Bhattacharyya D. Langmuir. 2004;20:5418–5424. doi: 10.1021/la049688+. [DOI] [PubMed] [Google Scholar]
- 88 (a).Calvo A, Fuertes MC, Yameen B, Williams FJ, Azzaroni O, Soler-Illia GJAA. Langmuir. 2010;26:5559–5567. doi: 10.1021/la9038304. [DOI] [PubMed] [Google Scholar]; (b) Calvo A, Yameen B, Williams FJ, Soler-Illia GJAA, Azzaroni O. J. Am. Chem. Soc. 2009;131:10866–10868. doi: 10.1021/ja9031067. [DOI] [PubMed] [Google Scholar]; (c) Calvo A, Yameen B, Williams FJ, Azzaroni O, Soler-Illia GJAA. Chem Comm. 2009:2553–2555. doi: 10.1039/b822489g. [DOI] [PubMed] [Google Scholar]
- 89.Tagliazucchi M, Azzaroni O, Szleifer I. J. Am. Chem. Soc. 2010;132:12404–12411. doi: 10.1021/ja104152g. [DOI] [PubMed] [Google Scholar]
- 90 (a).Yameen B, Ali M, Neumann R, Ensinger W, Knoll W, Azzaroni O. Chem. Comm. 2010;46:1908–1910. doi: 10.1039/b920870d. [DOI] [PubMed] [Google Scholar]; (b) Yameen B, Ali M, Alvarez M, Neumann R, Ensinger W, Knoll W, Azzaroni O. Polym. Chem. 2010;1:183–192. [Google Scholar]; (c) Yameen B, Ali M, Neumann R, Ensinger W, Knoll W, Azzaroni O. Nano Lett. 2009;9:2788–2793. doi: 10.1021/nl901403u. [DOI] [PubMed] [Google Scholar]; (d) Yameen B, Ali M, Neumann R, Ensinger W, Knoll W, Azzaroni O. J. Am. Chem. Soc. 2009;131:2070–2071. doi: 10.1021/ja8086104. [DOI] [PubMed] [Google Scholar]
- 91 (a).Schmitt J, Decher G, Dressick WJ, Brandow SL, Geer RE, Shashidhar R, Calvert JM. Adv. Mater. 1997;9:61–65. [Google Scholar]; (b) Malikova N, Pastoriza-Santos I, Schierhorn M, Kotov NA, Liz-Marzán LM. Langmuir. 2002;18:3694–3697. [Google Scholar]; (c) Huang H, Yang X. Colloids Surf., A. 2003;226:77–86. [Google Scholar]; (d) Chirea M, García-Morales V, Manzanares JA, Pereira C, Gulaboski R, Silva F. J. Phys. Chem. B. 2005;109:21808–21817. doi: 10.1021/jp0537815. [DOI] [PubMed] [Google Scholar]; (e) Ostrander JW, Mamedov AA, Kotov NA. J. Am. Chem. Soc. 2001;123:1101–1110. doi: 10.1021/ja0029578. [DOI] [PubMed] [Google Scholar]; (f) Jiang C, Markutsya S, Tsukruk VV. Langmuir. 2004;20:882–890. doi: 10.1021/la0355085. [DOI] [PubMed] [Google Scholar]
- 92.Dotzauer DM, Dai J, Sun L, Bruening ML. Nano Lett. 2006;6:2268–2272. doi: 10.1021/nl061700q. [DOI] [PubMed] [Google Scholar]
- 93.Scherer F, Anton M, Schillinger U, Henke J, Bergemann C, Krueger A, Gansbacher B, Plank C. Gene Ther. 2002;9:102. doi: 10.1038/sj.gt.3301624. [DOI] [PubMed] [Google Scholar]; (b) Chorny M, Polyak B, Alferiev IS, Walsh K, Friedman G, Levy RJ. FASEB J. 2007;21:2510. doi: 10.1096/fj.06-8070com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.He Q, Tian Y, Cui Y, Möhwald H, Li J. J. Mater. Chem. 2008;18:748–754. [Google Scholar]
- 95.Calvo A, Yameen B, Williams FJ, Azzaroni O, Soler-Illia GJAA. Chem. Comm. 2009:2553–2555. doi: 10.1039/b822489g. [DOI] [PubMed] [Google Scholar]
- 96.Lee D, Nolte AJ, Kunz AL, Rubner MF, Cohen RE. J. Am. Chem. Soc. 2006;128:8521–8529. doi: 10.1021/ja0608803. [DOI] [PubMed] [Google Scholar]
- 97 (a).Hiller J, Rubner MF. Macromolecules. 2003;36:4078–4083. [Google Scholar]; (b) Itano K, Choi JY, Rubner MF. Macromolecules. 2005;38:3450–3460. [Google Scholar]
- 98.Choi J, Rubner MF. Macromolecules. 2005;38:116–124. [Google Scholar]
- 99.Lee D, Cohen RE, Rubner MF. Langmuir. 2007;23:123–129. doi: 10.1021/la0612926. [DOI] [PubMed] [Google Scholar]
- 100.Qu X, Komatsu T. ACS Nano. 2010;4:563–573. doi: 10.1021/nn901474y. [DOI] [PubMed] [Google Scholar]
- 101.Komatsu T, Terada H, Kobayashi N. Chem. Eur. J. 2011;17:1849–1854. doi: 10.1002/chem.201001937. [DOI] [PubMed] [Google Scholar]
- 102.Wu P, Brand L. Anal. Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 103 (a).Berggren M, Dodabalapur A, Slusher RE, Bao Z. Nature. 1997;389:466–469. [Google Scholar]; (b) Serin JM, Brousmiche DW, Fréchet JMJ. Chem. Commun. 2002;38:2605–2607. doi: 10.1039/b207905d. [DOI] [PubMed] [Google Scholar]
- 104 (a).Feng CL, Zhong XH, Steinhart M, Caminade A-M, Majoral J-P, Knoll W. Adv. Mater. 2007;19:1933–1936. [Google Scholar]; (b) Feng CL, Zhong XH, Steinhart M, Caminade A-M, Majoral J-P, Knoll W. Small. 2008;4:566–571. doi: 10.1002/smll.200700453. [DOI] [PubMed] [Google Scholar]
- 105.Lee J, Govorov AO, Kotov NA. Nano Lett. 2005;5:2063–2069. doi: 10.1021/nl051042u. [DOI] [PubMed] [Google Scholar]
- 106.Sato A, Menges B, Knoll W. J. Appl. Phys. 2009;105:014701. [Google Scholar]