

Background: Oxidative stress-induced DNA damage is repaired by proteins in the base excision pathway.
Results: We identified a novel interaction between two DNA repair proteins, OGG1 and PARP-1.
Conclusion: OGG1-PARP-1 binding has both a functional and biological consequence.
Significance: These results provide insight into the factors that regulate DNA repair under normal and oxidative stress conditions.
Keywords: DNA Damage, DNA Repair, Nucleic Acid Enzymology, Oxidative Stress, Protein-Protein Interactions, 8-Hydroxyguanine, Base Excision Repair, Poly(ADP-ribosyl)ation
Abstract
Human 8-oxoguanine-DNA glycosylase (OGG1) plays a major role in the base excision repair pathway by removing 8-oxoguanine base lesions generated by reactive oxygen species. Here we report a novel interaction between OGG1 and Poly(ADP-ribose) polymerase 1 (PARP-1), a DNA-damage sensor protein involved in DNA repair and many other cellular processes. We found that OGG1 binds directly to PARP-1 through the N-terminal region of OGG1, and this interaction is enhanced by oxidative stress. Furthermore, OGG1 binds to PARP-1 through its BRCA1 C-terminal (BRCT) domain. OGG1 stimulated the poly(ADP-ribosyl)ation activity of PARP-1, whereas decreased poly(ADP-ribose) levels were observed in OGG1−/− cells compared with wild-type cells in response to DNA damage. Importantly, activated PARP-1 inhibits OGG1. Although the OGG1 polymorphic variant proteins R229Q and S326C bind to PARP-1, these proteins were defective in activating PARP-1. Furthermore, OGG1−/− cells were more sensitive to PARP inhibitors alone or in combination with a DNA-damaging agent. These findings indicate that OGG1 binding to PARP-1 plays a functional role in the repair of oxidative DNA damage.
Introduction
Exposure of DNA to reactive oxygen species results in lesions that can have genotoxic or mutagenic consequences. Reactive oxygen species are generated either as a byproduct of normal cellular metabolism or through exposure to ultraviolet (UV) and ionizing radiation and environmental carcinogens (1–3). Different DNA repair mechanisms have evolved to combat the genotoxic effects of reactive oxygen species and to protect the integrity of the genome because the resulting lesions if unrepaired may lead to genomic instability and ultimately cellular transformation (4, 5).
7,8-Dihydro-8-oxoguanine (8-oxoG)2 is one of the major lesions produced by oxidative damage. This adduct is highly mutagenic because of its propensity to mispair with A residues thereby generating a G:C to T:A transversion mutation (6). These spontaneous mutations if unrepaired have been shown to increase tumorigenesis in selected cell types. 8-oxoG and other oxidatively modified bases is mainly repaired by the base excision repair (BER) pathway. BER is a multistep process that is initiated by the recognition and excision of the damaged base by a DNA glycosylase (4). The main enzyme for repairing 8-oxoG is 8-oxoguanine-DNA glycosylase (OGG1) (7–11). Although OGG1 is a bifunctional enzyme with both excision activity and the ability to cleave the abasic/apyrimidinic (AP) site, its AP lyase activity is considered weak but can be further stimulated by the AP endonuclease 1 (7–12). The resulting steps for repair are coordinated by several different proteins in the BER pathway (13).
It is important to determine the factors that influence the repair ability of OGG1, as defective activity may lead to increased mutations in genes that cause disease. In support of this idea, there is a positive correlation between high levels of 8-oxoG and several human cancers and aging (14, 15). Additionally, chromosomal loss of OGG1 has been reported in human lung, esophageal, and renal cancers, and OGG1−/− mice have a predisposition toward the development of lung adenoma/carcinomas (16–21). Recent findings suggest that OGG1 plays a role in preventing Ras mutations (22, 23), which have profound implications as nearly one-third of all human cancers harbor a Ras mutation (24). This accumulating evidence points to an essential role for OGG1 in maintaining genomic integrity of cells and that disruption of OGG1 function may be a critical step during carcinogenesis. Further evidence for this idea is the fact that several OGG1 polymorphisms have been found to be associated with various cancers (25, 26).
Multiple protein-protein interactions occur during the BER pathway to coordinate the highly intricate process of this pathway. In particular, recent evidence suggests that OGG1 can bind to different proteins in the BER and nucleotide excision repair pathways. Some of these binding partners have been reported to affect the incision activity of OGG1 in vitro including XRCC1, AP endonuclease 1, XPC, p300/CBP, Rad52, and the Rad9-Rad1-Hus1 complex (12, 27–31). However, many of these interactions have not been fully explored. In addition, OGG1 has been found to bind to other proteins, but little is known about how these interactions affect the BER pathway or whether complex formation has any biological or functional consequences.
We used an unbiased biochemical approach to determine functional binding partners for OGG1. Using this approach, we determined that PARP-1 specifically interacts with OGG1. PARP-1 is a molecular sensor of DNA breaks, and it plays a key role in repair of these breaks by either physically associating with or also by poly(ADP-ribosyl)ation of partner proteins including various nuclear proteins, histones, single-strand break repair proteins, BER proteins, and on PARP-1 itself (32, 33). Furthermore, PARP-1 is activated in response to DNA damage, and studies using knock-out cells and PARP-1 inhibitors show that PARP-1 is important for maintaining genomic integrity (34–36).
Here we have investigated the interaction of OGG1 and PARP-1 and its biological significance. We report that OGG1 and PARP-1 bind directly, and this complex is enhanced by oxidative stress. In support of a biological interaction, OGG1 stimulates PARP-1 activity, and cells deficient in OGG1 have reduced levels of poly(ADP-ribosyl)ation after DNA damage. In addition, inhibition of PARP-1 activity sensitizes OGG1−/− cells to DNA damage. Interestingly, activated PARP-1 negatively regulates OGG1 activity. Altogether, our results suggest that binding of OGG1 and PARP-1 plays a key role in the cellular response to oxidative stress and DNA damage.
EXPERIMENTAL PROCEDURES
Cell Lines and Transfections
HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). The pCMV-2B vector was from Stratagene, and the N-terminal FLAG-tagged wild-type OGG1 (pCMV2B-WT OGG1) was previously described (37). Plasmids were transfected into HeLa cells using FuGENE® 6 transfection reagent from Roche Applied Science according to manufacturer's directions. The cells were used 24 h after transfection.
Wild-type mouse embryo fibroblasts (MEF) and OGG1−/− MEFs were a gift from Dr. Yie Liu (NIA, NIH) and were repeatedly passaged to establish immortalized cell lines using standard procedures (38). Cells were maintained in DMEM containing 10% FBS.
Plasmids containing pGEX4T2-WT OGG1, OGG1 polymorphic variants, and OGG1 fragments were generated by PCR using pET-28a-OGG1 plasmids as templates (37). The PCR products were digested with EcoR1 and Xho1 and ligated into the pGEX4T2 vector. Plasmids were verified by sequencing.
Nuclear Extracts
Mouse brains and livers were harvested fresh, washed in PBS, and incubated in Buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl with protease, and phosphatase inhibitors) for 10 min. Samples were then Dounce-homogenized and centrifuged for 5 min at 400 × g. The supernatant was collected for the cytoplasmic extract, and the nuclear pellet was resuspended in 10 volumes of Buffer A and subsequently washed 2 times. The pellet was then resuspended in 0.5 ml of buffer A and centrifuged at 15,000 × g for 15 min. Nuclei were resuspended in 0.5 ml Buffer C (20 mm HEPES, pH 7.0, 25% glycerol, 0.42 m NaCl, 1.5 mm MgCl2, 0.2 mm EDTA with protease and phosphatase inhibitors), incubated for 30 min with rotation, and centrifuged at 15,000 × g for 15 min. The supernatant containing the nuclear extracts was collected, and the nuclear pellet was saved. The extraction with Buffer C was repeated, and the supernatants were pooled. Nuclear extracts were frozen in liquid nitrogen and stored at −80 °C. Thawed nuclear extracts were ultracentrifuged 2 times at 100,000 × g for 10 min. The procedure was performed at 4 °C.
GST Purification and Precipitations
GST proteins were purified using standard procedures with some exceptions. For the OGG1 fragments in Fig. 2F, the samples were incubated in bacterial lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, 10% glycerol) containing an additional 225 mm NaCl and 50 μg/ml lysozyme for 30 min before sonication and subsequently 1% (w/v) sarcosine was added to each sample. For Fig. 6D, the various GST-OGG1 fusion proteins were eluted from the glutathione-Sepharose beads by thrombin cleavage.
FIGURE 2.

OGG1 binds directly to PARP-1. A, mouse liver nuclear extracts were incubated with immobilized GST or GST-OGG1 and were mock-treated (−) or treated (+) with DNase1. The proteins remaining associated with the GST fusion protein were probed with anti-PARP-1 antibodies. The amount of GST-OGG1 in the precipitations was determined using anti-OGG1 antibodies. NE, nuclear extracts. B and C, binding to GST-OGG1 or GST was assessed using an in vitro binding assay. GST-OGG1 or GST control (1 μg) were incubated with purified PARP-1 (0.25 μg), and samples were immunoblotted with anti-PARP-1 antibodies and reprobed with anti-OGG1 antibodies (C) or stained with Ponceau S (B) as loading controls. The arrow indicates GST-OGG1. C, GST-OGG1 incubated with PARP-1 in vitro was either mock-treated (−) or treated with DNase I (+). OGG1 retains binding to PARP-1 despite DNase treatment. D, HeLa cells transfected with FLAG vector control or FLAG-OGG1 were untreated (−) or treated for 30 min with 5 mm H2O2 (+). PARP-1 immunoprecipitates or lysates were probed with anti-FLAG antibodies or anti-PARP-1 antibodies. Increased binding of OGG1 to PARP-1 was also observed after treatment with lower concentrations of H2O2 (data not shown). E, schematic of GST-OGG1 fusion proteins is shown. HhH, Helix-hairpin-Helix; NLS, nuclear localization sequence; MLS, mitochondrial localization sequence. F, an in vitro binding assay was used to assess the binding of GST control, WT OGG1, and various fragments of OGG1 (1 μg) to purified PARP-1 (0.25 μg). GST precipitations were immunoblotted with anti-PARP-1 antibodies to identify PARP-1 binding and reprobed with anti-GST antibodies to visualize fusion proteins. G, a schematic of PARP-1 proteins is shown. H, purified His-tagged PARP-1 domains (1 μg) were incubated with GST-OGG1 (1 μg), and the precipitations were immunoblotted with anti-His antibodies and reprobed with anti-OGG1 antibodies to determine the amount of GST-OGG1 in the precipitations. The arrows indicate the bands corresponding to the DNA binding domain and the BRCT domain. Incubation with ethidium bromide (EthBr; +) abrogates the interaction between GST-OGG1 and the DNA binding domain.
FIGURE 6.

Binding of OGG1 polymorphic variants to PARP-1. A, shown is Coomassie colloidal blue staining of GST, WT OGG1, or OGG1 with the indicated amino acid mutations. Precipitations with the indicated GST fusion proteins were performed from liver nuclear extracts (NE; B) or with purified PARP-1 alone (C). Samples were probed with anti-PARP-1 antibodies. Anti-OGG1 antibodies were used to visualize the amount of GST-OGG1 fusion proteins. In C, the lane indicated WT alone indicates the WT fusion protein without the addition of PARP-1. D, an ELISA assay was used to measure the amount of PAR deposited by PARP-1 on immobilized histones. WT, R229Q, or S326C (0.5 μg) recombinant proteins were incubated with PARP-1 (2 ng) for 6 min before the addition of activated DNA and PARP-1 cocktail. The histograms show the averages ± S.E. from triplicate experiments. *, p < 0.05 and **, p < 0.01 for the indicated comparisons using one-way analysis of variance and Tukey's post-hoc test.
HeLa cells were washed twice with PBS and lysed in IP buffer (50 mm Tris-HCL pH 7.5, 150 mm NaCl, 1% Triton X-100, 2 mm EDTA, 1 mm dithiothreitol, protease and phosphatase inhibitors). Nuclear extracts were diluted 1:10 into IP buffer. Nuclear extracts (300 μg) or lysates were incubated with 20 μg of the appropriate GST fusion protein for 1 h at 4 °C. The samples were washed at least four times with IP buffer, and bound proteins were released from the beads by boiling in sample buffer. Samples were separated by SDS-PAGE and probed by immunoblotting with anti-OGG1 antibodies (Novus Biologicals), anti-PARP-1 polyclonal antibodies (Cell Signaling; clone 46D11), anti-Lamin B antibodies (Oncogene), or anti-HDAC2 antibodies (Abcam). In some cases, immunoblots were stained with Ponceau S to visualize loading of the GST fusion proteins. Alternatively, polyacrylamide gels were either stained with colloidal Coomassie Blue or with the Silver Stain Plus kit (Bio-Rad). For mass spectrometry, bands were excised from colloidal Coomassie Blue-stained gels and analyzed by nanoLC-MS/MS peptide sequencing technology (ProtTech, Inc.).
For in vitro binding experiments, GST-OGG1, OGG1 mutants or fragments, or GST control (1 μg) was incubated with recombinant high purity PARP-1 (250 ng; Alexis Biochemicals) in binding buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10% glycerol, 0.5% Triton X-100, 2 mm MgC1, 1 mm DTT) for 1 h at 4 °C. The samples were washed five times and separated by SDS-PAGE followed by immunoblotting with anti-PARP-1 (Clone 46D11 (Cell Signaling) or Clone C-2–10 (Biomol)) and reprobed with anti-OGG1 antibodies (Novus Biologicals) or anti-GST antibodies (Z-5:Santa Cruz Biotechnology) as a loading control. Alternatively, membranes were stained with Ponceau S to reveal both the GST and GST-OGG1 fusion proteins.
For experiments in Fig. 2, A and C, after washing the samples 2 times with IP or binding buffer, samples were washed an additional 2 times with NT2 buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm MgCl2, 0.05% Nonidet P-40) and treated with 10 units of DNase I (RNase-free, Ambion) in 0.1 ml of NT2 buffer at 37 °C for 15 min. Samples were then processed as above. In addition, DNase I-treated samples and controls were run on a DNA-agarose gel to verify DNA degradation.
His-tagged Protein Purification
His-tagged proteins corresponding to the BRCT domain (385–524 amino acids) and the catalytic domain (656–1014 amino acids) were purchased from Alexa Biochemicals. We used PCR to clone the DNA binding domain (1–373 amino acids) of PARP-1 into the pET28a His vector using PARP-1 cDNA as a template (39). We used a standard purification procedure from Qiagen that included a thrombin cleavage step to purify the protein to homogeneity. The binding assay with the PARP-1 domains and GST-OGG1 was performed as described in the previous section.
Immunoprecipitations
Immunoprecipitations were performed essentially as previously described (40). In brief, HeLa cells were treated with 5 mm H2O2 in serum-free media for 30 min, washed with PBS, and lysed in IP buffer (see above). PARP-1 was immunoprecipitated with anti-PARP1 polyclonal antibodies (Alexis Biochemicals; ALX-210-302). The immunoprecipitates were separated by SDS-PAGE and probed by immunoblotting with anti-FLAG® monoclonal antibodies (Clone M2 (Sigma)) and reprobed with anti-PARP1 monoclonal antibodies (Biomol International; Clone C-2-10).
Poly(ADP-ribose) (PAR) Assays
To measure whether OGG1 affects PARP activity, we used the HT Universal Colorimetric PARP assay kit from Trevigen. PARP activity is determined by the amount of PAR deposited onto immobilized histone proteins. The procedure was performed according to the recommendations of the manufacturer with the exception that 0.5 μg of OGG1 (New England Biolabs) or Fpg (New England Biolabs) was incubated with 2 ng of the PARP-high specific activity-enzyme for 6 min before the addition of the PARP substrate cocktail and activated DNA. Similar results were also obtained with OGG1 from Trevigen. In Fig. 6D, we used recombinant wild-type (WT) OGG1, R229Q, and S326C proteins that were purified as described in the GST purification section. In Fig. 3B, the assays were performed without activated DNA. Absorbance at 630 nm was measured using an ELISA plate reader. Measurements were taken without PARP enzyme and were subtracted as the background. The data were normalized to PARP-1 incubated with activated DNA.
FIGURE 3.

OGG1 stimulates the poly(ADP-ribosyl)ation activity of PARP-1. A and B, PARP-1 activity was measured by determining the amount of PAR deposited on immobilized histones in an ELISA assay. The addition of OGG1 (0.5 μg) increased the amount of PAR synthesis by PARP-1 (2 ng). B, ELISA assays were performed with or without activated DNA. The histograms show the averages + S.E. from triplicate (A) and quadruplicate (B) experiments. **, p < 0.01 compared with untreated control by Student's t test for A. *, p < 0.05 and ***, p < 0.001 for the indicated comparisons in B using one-way analysis of variance and Tukey's post-hoc test.
Immunofluorescence Microscopy
For PAR immunofluorescence, WT or OGG1−/− MEFs were untreated or treated with 500 μm H2O2 for 10 min. Cells were then fixed and stained with anti-PAR antibodies (Clone 10HA, Trevigen) and 4′,6-diamidino-2-phenylindole (DAPI) essentially as previously described (39). Pictures were taken using a Zeiss Observer D1 microscope with a AxioCam1Cc1 camera at a set exposure time. Nuclei were counted as PAR-positive if the fluorescence intensity was at least 200% above background levels for untreated cells. The background fluorescence intensity was indistinguishable between untreated WT and OGG1−/− cells. Percentage of PAR positive nuclei was calculated by counting the number of PAR positive nuclei per DAPI-stained nuclei in the different cell lines. A total of ∼1000–1300 cells were counted from triplicate coverslips for each experiment and repeated in three independent experiments.
Staining for 8-oxoG was performed as previously described (30) with some modifications. In brief, cells were pretreated for 30 min with 5 μm ABT-888 and then treated with or without 25 μm menadione for 30 min in serum-free media. Cells were fixed for 20 min in acetone:methanol (1:1), washed with PBS, and then incubated with 1 pg/ml pepsin in 0.01 n HCl for 30 min. Cells were then incubated with 2 n HCl for 10 min and sodium borate for 5 min, washed with PBS, and then blocked with PBS containing 10% goat serum, 1% BSA for 30 min. After a quick wash with PBS, cells were stained with anti-8-oxoG monoclonal antibodies (Millipore, 1:250) for 30 min. After washing with PBST (PBS, 0.5% BSA, 0.1% Tween 20), cells were exposed to Alexa-488 conjugated secondary antibodies (Invitrogen) for 30 min, washed with PBST, and stained with DAPI. All incubations were performed at room temperature.
Single Cell Gel Electrophoresis (Comet) Assay
WT or OGG1−/− MEFs were untreated or treated for 30 min with 100 μm H2O2, which would induce various base lesions and also single-strand breaks at this concentration (41). Comet assays were performed under alkaline conditions as previously described (42, 43). The comets were visualized using an Eclipse E-400 fluorescence microscope (Nikon, Japan) attached to a Pulnix video camera (Kinetic Imaging, LTD, Liverpool, UK) and were analyzed using Komet 5.5 software (Kinetic Imaging LTD). Olive tail moment was used as a measure of DNA damage level (44, 45).
DNA Incision Activity Assay
A HPLC 30-mer oligonucleotide (GAAGAGAGAAAGAGAXAAGGAAAGAGAGAA) containing 8-oxoG at position X and the complementary oligonucleotides containing a C opposite X were obtained from Midland Certified Reagent Company (Midland, TX). The 5′- 32P-labeling of the duplex oligonucleotide was performed essentially as previously described (46). To measure OGG1 glycosylase/AP lyase activity, 320 nm OGG1 (New England Biolabs) was incubated with either 180 nm PARP-1, 1.2 μl of PARP-1 cocktail, or 0.01 nm PAR (all from Trevigen) in 12-μl reactions containing (20 mm Tris-HCl, pH 7.4, 100 mm NaCl, and 0.15 μg/μl BSA) for 10 min at 4 °C, then 8 μl of radiolabeled oligonucleotides (1.3 nm) were added. All molar values indicate final concentration in the reaction. In Fig. 5, B and C, various concentrations of both OGG1 and PARP-1 were used in the incision assays as indicated in the figure. The reactions were incubated at 37 °C for 30 min and incubated in stop solution (20 mm HEPES, pH 7.5, 0.5% SDS, 5 mm EDTA, 40 μg/ml Proteinase K) for 15 min at 37 °C. To cleave DNA at abasic sites, 10% piperidine (final concentration) was added to each sample, and the reactions were incubated at 90 °C for 15 min. Samples were then precipitated by the addition of 3 m sodium acetate, pH 5.3, 4 mg/ml glycogen in absolute ethanol and subsequently incubated on dry ice for 15 min. The precipitates were collected by centrifugation for 30 min at 20,000 × g. Samples were resuspended in loading buffer (1× Tris borate EDTA, 90% deionized formamide, 0.1% bromphenol blue, 0.1% xylene cyanol), heated at 95 °C for 5 min, and run on 20% acrylamide gels containing 7 m urea. Radioactivity was measured using a Storm Phosphorimager and quantified using ImageQuant software (GE Healthcare).
FIGURE 5.

PARP-1 inhibits OGG1 activity. A, recombinant OGG1 (320 nm) was incubated with buffer, PARP-1 cocktail (PARP-1 ct), PARP-1 (180 nm), PAR, or PARP-1 and PARP-1 cocktail and reacted with a 5′-end-labeled oligonucleotide duplex containing an 8-oxoG/C mismatch. After a 30-min incubation at 37 °C, the cleavage products were analyzed using 20% denaturing gels and phosphorimaging. A representative experiment is shown in the left panel, and the histogram represents the mean ± S.E. from four independent experiments. The percent incision was calculated by taking the amount of cleaved substrate (lower band) normalized to the amount of uncleaved substrate (top band). The data were normalized to the incision activity of OGG1 alone (100%). B, incision assays were performed as in A with the exception that different concentrations of PARP-1 with or without PARP cocktail were added to the OGG1 (320 nm) incision assay. The percent incision was calculated as above, and the histogram represents the mean ± S.E. from five independent experiments for PARP-1 alone and three independent experiments for activated PARP-1. C, different concentrations of OGG1 were incubated with or without PARP-1 (180 nm), and the incision assays were performed as in A. Incision was calculated by taking the amount of cleaved substrate (lower band of A) normalized to the amount of uncleaved substrate (top band of A). The histogram represents the mean ± S.E. from three independent experiments. For A–C, *, p < 0.05; **, p < 0.01; ***, p < 0.001 for the indicated comparisons using one-way analysis of variance and Tukey's post-hoc test. D, HeLa cells were untreated or treated with menadione (25 μm), ABT-888 (5 μm), or both menadione and ABT-888. Cells were stained with anti-8-oxoG antibodies and DAPI as described under “Experimental Procedures.”
Colony Formation and Cell Survival Assays
For the colony formation assays, 400 WT or OGG1−/− MEF cells were plated in 60-mm dishes in triplicate and were untreated or treated with 2.5 μm ABT-888 (Enzo Life Sciences), 20 μm H2O2, or ABT-888 and H2O2 for 7 days. ABT-888 was added 30 min before the addition of H2O2. The media with or without the different treatments was changed once during the time of the assay. Colonies were stained with crystal violet, and only colonies with >50 cells were counted.
WT or OGG1−/− MEF cells (3000/well) were plated in a 96-well plate. The following day cells were untreated or treated for 24 h with 5 μm ABT-888, 70 μm H2O2, or ABT-888 and H2O2. Cells were pretreated with ABT-888 for 30 min before the addition of H2O2. Cell survival was measured using a MTT assay (Sigma).
RESULTS
PARP-1 Interacts with OGG1 in Vitro and in Vivo
To identify proteins that interact with OGG1, we first purified a GST fusion protein containing wild-type OGG1 (GST-OGG1; Fig. 1A). We used this GST-OGG1 fusion protein to precipitate proteins from mouse brain and liver nuclear extracts (Fig. 1) and confirmed that our nuclear extracts contained nuclear proteins including OGG1, Lamin B, and HDAC2 (Fig. 1B). A prominent band of ∼113 kDa was present in both GST-OGG1 precipitations from mouse liver and brain but not with the GST-OGG1 protein alone or with GST control (Fig. 1, C and D). Mass spectrometry analysis revealed that the prominent ∼113-kDa protein associating with GST-OGG1 in this band was PARP-1 (Fig. 1, C and D). This band also contained a smaller number of peptides corresponding to HNRP (Hnrpu, heterogeneous nuclear ribonucleoprotein U), ZBP-148 (zinc finger protein 148), SP3 (Sp3 transcription factor isoform 1), Lfc (Lymphoid blast crisis like 1), and Xrn2 (5′-3′ exoribonuclease 2). However, we chose to focus on PARP-1 because mass spectrometry identified this protein as the most prominent binder to OGG1. An additional lower molecular weight band was excised, and mass spectrometry analysis revealed that this band contained XRCC1 peptides, indicating that our GST system can be used to identify previously known binding partners of OGG1 (data not shown) (31).
FIGURE 1.

Identification of proteins binding to OGG1. A, Coomassie colloidal blue staining of purified GST and GST-OGG1 (20 μg) proteins is shown. B, nuclear extracts from mouse brain and liver were immunoblotted with the indicated antibodies (cyt) cytosolic fraction (nuc) nuclear fraction. C, GST-OGG1 and GST control were used to precipitate proteins from mouse brain and liver nuclear extracts (NE). GST precipitations were analyzed on a 12% polyacrylamide gel and silver-stained. −, GST fusion protein alone. pdown, pulldown. D, GST-OGG1 was used to precipitate proteins from mouse brain and liver. Samples were analyzed on an 8% polyacrylamide gel and stained with Coomassie colloidal blue. The arrow points to the band, corresponding to PARP-1, that was excised and analyzed by mass spectrometry. −, GST fusion protein alone.
To confirm that PARP-1 associates with OGG1, we immunoblotted GST-OGG1 precipitations from mouse nuclear extracts with PARP-1 antibodies. We observed that PARP-1 binds to OGG1 in nuclear extracts from both the mouse liver and brain and in whole cell lysates from HeLa cells (Fig. 2A and supplemental Fig. S 1A). Using an in vitro binding assay with purified proteins alone, we found that OGG1 binds directly to both unmodified and auto(ADP-ribosyl)ated PARP-1 (Fig. 2, B and C; supplemental Fig. S3). The interaction between OGG1 and PARP-1 was not disrupted by DNase or ethidium bromide treatment, thus confirming that the binding observed between OGG1 and PARP-1 is due to protein-protein interactions and not through DNA (Fig. 2, A and C, supplemental Fig. S1). In addition, we wanted to perform the reciprocal experiment to determine whether we could detect OGG1 binding to PARP-1 in PARP-1 immunoprecipitations. Co-immunoprecipitation experiments revealed that PARP-1 binds to OGG1 in vivo. Furthermore, this binding was enhanced by oxidative stress, suggesting that these proteins interact in response to oxidative stress-induced DNA damage (Fig. 2D). Additionally, the increased association of OGG1 and PARP-1 after oxidative stress was observed despite DNase treatment, suggesting that it is mediated through a protein-protein interaction (supplemental Fig. S1B).
To further characterize the interaction between PARP-1 and OGG1, we generated GST-tagged fragments of OGG1 corresponding to different and overlapping regions of OGG1 (Fig. 2E). Incubation of these fusion proteins with PARP-1 in an in vitro binding assay (Fig. 2, E and F) revealed that PARP-1 binds to the N-terminal region of OGG1, specifically within amino acids 79–180 of OGG1 (Fig. 2F). In addition, we mapped the region where OGG1 binds to PARP1 (Fig. 2, G and H). We found that OGG1 binds to the BRCT domain of PARP-1, a domain that is important in mediating protein-protein interactions. To a lesser extent, GST-OGG1 also bound to the DNA binding domain; however, this binding was abrogated by ethidium bromide, suggesting that it is mainly mediated through DNA (Fig. 2H).
OGG1 Stimulates PARP-1 Activity
We wanted to examine whether OGG1 can affect PARP activity. We initially used an in vitro assay to test this idea and found that OGG1 stimulates the poly(ADP-ribosyl)ation activity of PARP-1 on immobilized histones (Fig. 3A). This effect was more pronounced when the assay was performed in the presence of activated DNA, which stimulates PARP-1 activity (Fig. 3B). To make certain that PARP-1 is directly activated by OGG1 and to exclude the possibility that purified OGG1 proteins were contaminated with DNA, we used recombinant OGG1 purified from other laboratories. Consistent with the findings in Fig. 3 using commercial OGG1 from New England Biolabs, recombinant OGG1 from other sources also activated PARP-1 (supplemental Fig. S2A). In addition, we found that the bacterial glycosylase Fpg, which has cleavage activity similar to OGG1 but lacks the specific interaction with PARP-1, did not affect PARP activity (supplemental Fig. S2B). This suggests that under these experimental conditions cleavage of 8-oxoG in the chromatin DNA does not substantially contribute to the OGG1-mediated activation of PARP-1 in this assay.
To further address whether OGG1 affects PARP-1 function, we examined whether the lack of OGG1 alters the poly(ADP-ribosyl)ation activity of PARP-1 after oxidative stress. We treated cells with 500 μm H2O2 for 10 min, which is a time course, and concentration of this DNA-damaging agent that has been commonly used to study the activation of PARP-1 (47). To examine how reduction of OGG1 may affect PARP-1 activity, we used mouse embryo fibroblasts (MEFs) derived from OGG1−/− or WT mice (38). In both cell lines the basal level of PAR production was very low, as observed by immunofluorescence labeling of cells for PAR (Fig. 4A, left panels). Treatment with H2O2 for 10 min induced a dramatic increase in the amount of PAR synthesis in the WT MEFs (Fig. 4A, right panels). However, there was a significant reduction in the synthesis of PAR in OGG1−/− cells. We quantified this and found that there was a significant decrease in the number of PAR-positive nuclei in OGG1−/− cells compared with WT control in response to oxidative stress (Fig. 4B). Impaired PAR formation in response to H2O2 was also observed in HeLa cells that were generated to stably knockdown OGG1 (data not shown).
FIGURE 4.

Reduced levels of OGG1 impair poly(ADP-ribosyl)ation of cellular proteins after oxidative stress. A, WT or OGG1−/− MEFs were either control-treated or treated with 500 μm H2O2 for 10 min and stained with anti-PAR antibodies and DAPI. B, percentage of PAR-positive nuclei were calculated by counting the number of PAR-positive nuclei per DAPI-stained nuclei in the indicated cell lines. A total of ∼1000–1300 cells were counted from triplicate coverslips for each experiment. The histogram shows the averages normalized to WT MEFs ± S.E. from three independent experiments. **, p < 0.01 compared with control using Student's t test. C, WT or OGG1−/− MEFs were untreated (−) or treated for 10 min with 500 μm H2O2 (+). Lysates were probed with anti-PARP-1 antibodies and anti-GAPDH antibodies as a protein loading control. The relative levels of PARP-1 were not significantly different between WT (1.0) and OGG1−/− MEFs (1.07). The numbers in parentheses show the average relative levels of PARP-1 normalized to GAPDH and to WT cells from three independent experiments. D, shown is accumulation of DNA damage in OGG1−/− cells in response to H2O2 treatment. WT or OGG1−/− MEFs were untreated or treated with 100 μm H2O2 for 30 min. Oxidative DNA damage was analyzed using the alkaline comet assay. The histogram represents the mean of five independent experiments ± S.E. *, p < 0.05 comparing OGG1−/− to WT using Student's t test.
It has previously been reported that PARP expression may regulate the levels of OGG1 (48). However, the effects we observed on PAR synthesis are likely not due to decreased expression of PARP-1, as PARP-1 protein levels were not changed significantly in the MEFs regardless of OGG1 expression (Fig. 4C). In addition, PARP-1 expression was not altered during this time course of treatment with H2O2 (Fig. 4C).
The lower amount of PAR in OGG1−/− cells could also be due to a decreased number of strand breaks as 8-oxoG and possibly other lesions are not properly incised in these cells. To test this possibility, we performed the single cell gel electrophoresis (comet) assay under alkaline conditions, which measures alkaline-sensitive sites that include single-strand breaks, alkaline labile sites, and transient repair sites. In the absence of DNA damage, there was very little difference in the amount of endogenous DNA damage between WT and OGG1−/− MEFs (Fig. 4D), consistent with the very low PAR levels in untreated cells (Fig. 4A). However, after treatment with H2O2 there was a significant increase in the amount of DNA damage in OGG1−/− cells compared with WT cells (Fig. 4D), indicating that the number of strand breaks in OGG1−/− cells after H2O2 treatment may not explain the lower levels of PAR in these cells. Nevertheless, we cannot exclude the possibility that we may be unable to detect an in vivo difference in strand breaks, as different alkaline-sensitive sites are measured under these experimental conditions.
PARP-1 Modulates OGG1 Activity
To determine whether PARP-1 alters the incision activity of OGG1, we incubated the purified proteins with a radiolabeled DNA duplex containing a single 8-oxoG lesion. OGG1 effectively cleaved the substrate at the 8-oxoG lesion (Fig. 5, A and C). Preincubation of PARP-1 alone with OGG1 has a slight concentration-dependent inhibitory effect on OGG1 activity, although this effect is non-significant (Fig. 5, A and B). However, activated PARP-1 significantly inhibits the incision activity of OGG1 in a concentration-dependent manner (Fig. 5, A and B). Furthermore, we found that OGG1 incision activity increased with its concentration and that activated PARP-1 inhibited OGG1 activity at all concentrations (Fig. 5C). To ensure that decreased 8-oxoG cleavage was due to PARP-1 activity and not factors such as NAD+ that are in the PARP-1 cocktail and necessary for PARP-1 activation, we incubated OGG1 in the absence of PARP-1 and in the presence of the PARP-1 cocktail. We did not observe any significant change in activity when OGG1 was incubated with the PARP-1 cocktail relative to OGG1 alone (Fig. 5A). In addition, similar to these results, PARP-1 impaired OGG1 activity when incubated with NAD+ alone (data not shown). Interestingly, under the conditions of our assay, preincubation of OGG1 with PAR did not significantly affect OGG1 incision activity (Fig. 5A), suggesting that PARP-1 is important for modulating OGG1 activity.
Given our in vitro data that PARP-1 inhibits OGG1 activity, we wanted to qualitatively examine whether PARP-1 influences the level of the OGG1-sensitive base lesion 8-oxoG in cells. Consistent with previous reports, untreated cells have an endogenous background level of 8-oxoG (Fig. 5D) (28, 30, 49). The levels of 8-oxoG were similar to untreated cells when cells were incubated with the PARP inhibitor ABT-888. However, treatment of cells with the DNA-damaging agent menadione increased the level of 8-oxoG. Interestingly, inhibiting PARP-1 in the presence of menadione reduced the levels of 8-oxoG to close to background levels (Fig. 5D). These data suggest that PARP-1 activity influences 8-oxoG levels in vivo.
Several OGG1 polymorphisms have been reported and correlate with diseases including cancer and Alzheimer disease (25, 26, 50, 51). Two frequently observed OGG1 polymorphisms include the R229Q and S326C mutations in the C terminus of OGG1 (17, 52). Previously, we and others have shown that these OGG1 variants have defective enzymatic activity and other unique properties distinct from the wild-type enzyme (37, 52–54). Therefore, we wanted to investigate whether these polymorphisms affect binding to PARP-1. We purified GST-tagged polymorphic variants of OGG1 and found that both the S326C and R229Q proteins were able to bind PARP-1 similar to OGG1 wild-type both in liver and brain nuclear extracts and in an in vitro binding assay (Fig. 6, A–C).
To test whether the polymorphic variants activate PARP-1, we initially performed ELISA assays that measure the amount of PAR deposited by PARP-1 on immobilized histones. We found that both the R229Q and S326C polymorphic proteins were defective in activating PARP-1 compared with wild-type OGG1 (Fig. 6D). In addition, we incubated the various GST fusion proteins with activated PARP-1, washed with buffer containing detergent to remove unbound PARP-1, and ran these samples on an SDS-polyacrylamide gel. As expected, immunoblotting with anti-PAR antibodies revealed that there was very little PAR in the GST lane, indicating that activated PARP does not bind to GST. We observed a decrease in poly(ADP-ribosyl)ation when the R229Q and S326C polymorphic variants were incubated with activated PARP-1 compared with wild-type OGG1 (supplemental Fig. S3). Similar effects were observed when the assays were performed without activated DNA (supplemental Fig. S3). However, analogous to Fig. 3B, more substantial PARP-1 activation was observed when the assays were performed in the presence of activated DNA. To confirm that PARP-1 activation was not due to DNA contamination in our OGG1 preparations, we treated wild-type OGG1 with DNase before adding it to the ribosylation reaction. We found that DNase treatment did not affect the ability of OGG1 to stimulate PARP-1 activity (data not shown), indicating that OGG1 activates PARP-1 through a protein-protein interaction.
Loss of OGG1 Sensitizes Cells to PARP-1 Inhibitors
Currently, PARP inhibitors are being used clinically as single-agent anticancer drugs or in combination with different DNA-damaging agents to enhance chemosensitization (55, 56). We wanted to examine whether cells deficient in OGG1 display an altered sensitivity to PARP inhibitors alone or in combination with a DNA-damaging agent. To mimic the physiological conditions of treatment, we exposed cells to prolonged low doses of the PARP inhibitor ABT-888 alone or in combination with H2O2. Incubation with the PARP inhibitor alone had minimal effects on WT cells, but both colony formation and cell survival were significantly lower in OGG1−/− cells than WT cells when treated with ABT-888 alone (Fig. 7). Moreover, combining ABT-888 and H2O2 was also effective in reducing colony formation and cell survival in OGG1−/− cells compared with wild-type cells (Fig. 7). In addition, a dose-dependent effect was observed when we used varying concentrations of ABT-888 alone and in combination with a higher concentration of H2O2 over a shorter time course (supplemental Fig. S4). Similar to previous findings, there was no significant difference in either cell survival or colony formation between WT and OGG1−/− cells when exposed to low doses of H2O2 alone (57).
FIGURE 7.
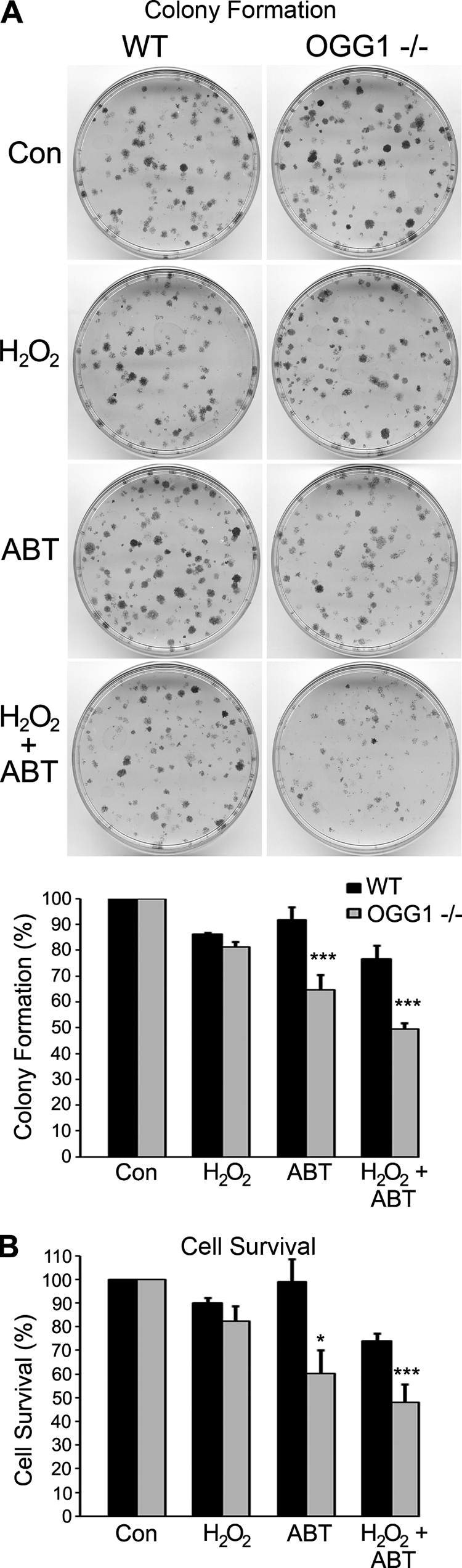
Loss of OGG1 and inhibition of PARP-1 impairs colony formation and cell survival in response to DNA damage. A, colony formation assay of MEFs either untreated (Con) or treated with H2O2, ABT-888 or both H2O2 and ABT-888 are shown. B, cell survival of MEFs was measured after incubation for 24 h with the indicated treatments. Both histograms show the normalized averages ± S.E. from five independent experiments. *, p < 0.05 and ***, p < 0.001 comparing OGG1−/− to WT using one-way analysis of variance and Tukey's post-hoc test.
To confirm that the effects of ABT-888 that we observed are through PARP inhibition, we used RNA interference to down-regulate PARP-1 expression in WT and OGG1−/− cells (supplemental Fig. S4, C and D). ABT-888 decreased cell viability of OGG1−/− cells transfected with control siRNA, similar to untransfected OGG1−/− cells (supplemental Fig. 4, B and D). However, OGG1−/− cells transfected with PARP-1 siRNA were insensitive to the effects of ABT-888 alone or in combination with H2O2 (supplemental Fig. 4D), indicating that the decreased cell survival caused by ABT-888 treatment is mainly through inhibiting PARP.
DISCUSSION
Normal cells are continuously faced with oxidative DNA damage. Various DNA repair mechanisms, including the BER pathway, are important to protect genomic integrity and to aid in the prevention of mutations that could cause disease or cell death. It is, therefore, important to unravel the intricate works of the different DNA repair pathways and to characterize the various interactions that occur for repair to proceed. Here, we have uncovered a novel protein-protein interaction between the DNA glycosylase OGG1 and the DNA sensor protein PARP-1. Our results indicate that PARP-1 binds to OGG1 through the N terminus of OGG1 and that this interaction is enhanced by oxidative stress. Cells with decreased OGG1 expression have a defect in the poly(ADP-ribosyl)ation of cellular proteins after treatment with a DNA-damaging agent. This deficiency could be caused by decreased PARP-1 expression and/or activity. We found that PARP-1 expression is not significantly altered in OGG1−/− MEFs compared with WT MEFs. Therefore, it is likely that decreased PARP-1 activity contributes to impaired PAR synthesis of nuclear proteins after DNA damage. In agreement with this idea, we found that OGG1 can stimulate PARP-1 activity in vitro. Alternatively, OGG1 expression may enhance only PARP-1 automodification rather than influencing PAR formation on target proteins. Nevertheless, our data demonstrate that OGG1 can stimulate PARP-1 activity, which may explain the lack of poly(ADP-ribosyl)ation in cells with decreased levels of OGG1. However, we cannot rule out that there may be differences in the protein expression and/or activity of other components of the oxidative stress pathway that may contribute to optimal PAR formation in control versus OGG1−/− cells. Nonetheless, these data indicate a potential functional importance of the OGG1-/PARP-1 complex in the early response to DNA damage.
It is interesting that under our experimental conditions PAR binding was not sufficient to affect OGG1 incision activity. Rather, incubation of OGG1 with PARP-1 activated by a PARP cocktail or NAD+ alone inhibited the ability of OGG1 to excise damaged DNA lesions, suggesting that PARP-1 activity is important for regulating OGG1. In the absence of cofactors, we did find that PARP-1 alone had a slight, although non-significant effect on OGG1 activity. This may be due to PARP-1 binding to DNA and occluding the target lesion. However, in response to DNA damage, the catalytic activity of PARP-1 is highly stimulated, which leads to automodification of PARP-1, suggesting that under conditions of oxidative stress PARP-1 activity may be important for regulating OGG1. In support of this idea, OGG1 binding to PARP-1 is enhanced by DNA damage and OGG1 binds to auto(ADP-ribosyl)ated PARP-1. Furthermore, inhibition of PARP-1 reduced the level of 8-oxoG in response to a DNA-damaging agent, indicating that PARP-1 plays a role in influencing the level of the OGG1 sensitive base lesion 8-oxoG. It will be important in the future to examine the effect of PARP-1 on 8-oxoG and potentially other oxidative base lesions through direct measurements in the cellular DNA.
Taking these results together, we hypothesize that OGG1 binding to PARP is important for the early steps of repair of oxidative DNA damage. At sites of DNA damage, OGG1 would excise the damaged lesion and activate PARP-1. In vivo, OGG1 may also stimulate PARP-1 activity by creating abasic sites that AP endonuclease 1 would convert to single-strand breaks (13). Activated PARP-1 would then poly(ADP-ribosyl)ate itself and other nuclear proteins. PAR synthesis at the damaged sites would then serve to recruit important DNA repair proteins such as XRCC1. The scaffolding protein XRCC1 binds preferentially to PARP-1 when it is poly(ADP-ribosyl)ated, and its recruitment affects the repair process by stimulating most of the repair enzymes (58). OGG1 and PARP-1 both bind to XRCC1 and may form a multiprotein complex in cells (31, 58), suggesting that OGG1/PARP1 binding may be important for recruiting proteins to damaged sites. PARP-1 may then inhibit the glycosylase activity of OGG1, and OGG1 would then be released from the damaged DNA, enabling other proteins to facilitate repair (4, 32, 33).
Our data indicate that in the absence of OGG1, the poly(ADP-ribosyl)ation activity of PARP-1 is impaired, and decreased PAR was observed on nuclear proteins. This suggests that the formation of PAR-dependent multiprotein complexes may be impaired in cells lacking OGG1. This impairment in the recruitment and binding of BER proteins may then delay the DNA repair process or potentially lead to the accumulation of damaged DNA. Importantly, several of the OGG1 polymorphisms have been shown to have decreased catalytic activity, which may hinder the activation of PARP-1 and the early steps in the BER pathway (37, 51–54). Indeed, both the R229Q and S326C have an impaired ability to activate PARP-1 compared with wild-type OGG1. This difference may be due to the fact that the S326C polymorphism exists as a dimer and has decreased enzymatic activity compared with wild-type and the R229Q polymorphism has decreased activity and is a thermolabile protein (37, 51–54). Future work lies in further understanding the interplay between the OGG1 polymorphisms and PARP-1. Nevertheless, it is interesting to speculate that inadequate DNA repair by these polymorphisms could help to explain the association of these proteins with increased cancer risk (59, 60). In addition, loss of heterozygosity of OGG1 has been observed in lung, renal, and esophageal cancers (17–21), which may also contribute to the disease process. In support of this idea, OGG1−/− mice have a predisposition for the development of lung tumors (16), and polymorphisms of OGG1, including the S326C and R229Q variants, have been positively associated with several diseases, such as various cancers and Alzheimer disease (59, 60).
Our results provide evidence that in the absence of OGG1, cells are more sensitive to PARP inhibitors as a single agent or in combination with a DNA-damaging agent, H2O2. This increased sensitivity could have clinical relevance because cancer cells that express lower levels of OGG1 may be more susceptible to PARP inhibitors such as ABT-888, olaparib, or iniparib as single agents or when used in combination with other chemotherapeutic agents. As many PARP inhibitors are currently being used to treat various cancers (55, 56), it will be interesting to determine whether OGG1 tumor expression correlates with efficacy of treatment with these anti-cancer drugs. In addition, it will be important to further characterize the relationship between OGG1 and PARP-1 in various cancers.
To our knowledge this represents the first example of PARP-1 binding to a DNA glycosylase. It will be interesting in the future to determine whether PARP-1 binds to and regulates the activity of other glycosylases. Nonetheless, the results reported here provide important insight into the factors that regulate the BER pathway and increase our understanding of the complex interactions that occur in response to oxidative stress.
Supplementary Material
Acknowledgments
We thank Robert Brosh, Brian Berquist, and Deborah Croteau for critical reading of the manuscript and Jeff Hill, David M. Wilson III, Daniel McNeill, and Hansen Du for helpful discussions and technical help. We also thank Yie Liu for the generous gift of the wild-type and OGG1−/− primary MEFs and Vilhelm Bohr (NIA, NIH) and Samuel Wilson (NIEHS, NIH) for the generous gifts of recombinant OGG1.
This work was supported by the National Institutes of Health National Institute on Aging, Intramural Research Program (NIA).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- 8-oxoG
- 7,8-dihydro-8-oxoguanine
- PAR
- poly(ADP-ribose)
- MEF
- mouse embryo fibroblasts
- BER
- base excision repair
- OGG1
- 8-oxoguanine DNA glycosylase
- AP
- abasic/apyrimidinic
- PARP-1
- poly(ADP-ribose) polymerase 1
- IP
- immunoprecipitation
- BRCT
- BRCA1 C-terminal
- WT
- wild-type.
REFERENCES
- 1. Barnes D. E., Lindahl T. (2004) Annu. Rev. Genet. 38, 445–476 [DOI] [PubMed] [Google Scholar]
- 2. Cadet J., Berger M., Douki T., Ravanat J. L. (1997) Rev. Physiol. Biochem. Pharmacol. 131, 1–87 [DOI] [PubMed] [Google Scholar]
- 3. Dizdaroglu M. (1991) Free Radic. Biol. Med. 10, 225–242 [DOI] [PubMed] [Google Scholar]
- 4. David S. S., O'Shea V. L., Kundu S. (2007) Nature 447, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hegde M. L., Hazra T. K., Mitra S. (2008) Cell Res. 18, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shibutani S., Takeshita M., Grollman A. P. (1991) Nature 349, 431–434 [DOI] [PubMed] [Google Scholar]
- 7. Aburatani H., Hippo Y., Ishida T., Takashima R., Matsuba C., Kodama T., Takao M., Yasui A., Yamamoto K., Asano M. (1997) Cancer Res. 57, 2151–2156 [PubMed] [Google Scholar]
- 8. Arai K., Morishita K., Shinmura K., Kohno T., Kim S. R., Nohmi T., Taniwaki M., Ohwada S., Yokota J. (1997) Oncogene 14, 2857–2861 [DOI] [PubMed] [Google Scholar]
- 9. Radicella J. P., Dherin C., Desmaze C., Fox M. S., Boiteux S. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 8010–8015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roldán-Arjona T., Wei Y. F., Carter K. C., Klungland A., Anselmino C., Wang R. P., Augustus M., Lindahl T. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 8016–8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenquist T. A., Zharkov D. O., Grollman A. P. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 7429–7434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hill J. W., Hazra T. K., Izumi T., Mitra S. (2001) Nucleic Acids Res. 29, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sung J. S., Demple B. (2006) FEBS J. 273, 1620–1629 [DOI] [PubMed] [Google Scholar]
- 14. Barja G. (2004) Trends Neurosci. 27, 595–600 [DOI] [PubMed] [Google Scholar]
- 15. Hamilton M. L., Van Remmen H., Drake J. A., Yang H., Guo Z. M., Kewitt K., Walter C. A., Richardson A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 10469–10474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakumi K., Tominaga Y., Furuichi M., Xu P., Tsuzuki T., Sekiguchi M., Nakabeppu Y. (2003) Cancer Res. 63, 902–905 [PubMed] [Google Scholar]
- 17. Kohno T., Shinmura K., Tosaka M., Tani M., Kim S. R., Sugimura H., Nohmi T., Kasai H., Yokota J. (1998) Oncogene 16, 3219–3225 [DOI] [PubMed] [Google Scholar]
- 18. Audebert M., Chevillard S., Levalois C., Gyapay G., Vieillefond A., Klijanienko J., Vielh P., El Naggar A. K., Oudard S., Boiteux S., Radicella J. P. (2000) Cancer Res. 60, 4740–4744 [PubMed] [Google Scholar]
- 19. Wikman H., Risch A., Klimek F., Schmezer P., Spiegelhalder B., Dienemann H., Kayser K., Schulz V., Drings P., Bartsch H. (2000) Int. J. Cancer 88, 932–937 [DOI] [PubMed] [Google Scholar]
- 20. Lu R., Nash H. M., Verdine G. L. (1997) Curr. Biol. 7, 397–407 [DOI] [PubMed] [Google Scholar]
- 21. Hagiwara A., Kitajima Y., Sato S., Miyazaki K. (2005) Oncol. Rep. 13, 1009–1016 [PubMed] [Google Scholar]
- 22. Saxowsky T. T., Meadows K. L., Klungland A., Doetsch P. W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 18877–18882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie Y., Yang H., Cunanan C., Okamoto K., Shibata D., Pan J., Barnes D. E., Lindahl T., McIlhatton M., Fishel R., Miller J. H. (2004) Cancer Res. 64, 3096–3102 [DOI] [PubMed] [Google Scholar]
- 24. Bos J. L. (1989) Cancer Res. 49, 4682–4689 [PubMed] [Google Scholar]
- 25. Hung R. J., Hall J., Brennan P., Boffetta P. (2005) Am. J. Epidemiol. 162, 925–942 [DOI] [PubMed] [Google Scholar]
- 26. Weiss J. M., Goode E. L., Ladiges W. C., Ulrich C. M. (2005) Mol. Carcinog. 42, 127–141 [DOI] [PubMed] [Google Scholar]
- 27. D'Errico M., Parlanti E., Teson M., de Jesus B. M., Degan P., Calcagnile A., Jaruga P., Bjørås M., Crescenzi M., Pedrini A. M., Egly J. M., Zambruno G., Stefanini M., Dizdaroglu M., Dogliotti E. (2006) EMBO J. 25, 4305–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Souza-Pinto N. C., Maynard S., Hashiguchi K., Hu J., Muftuoglu M., Bohr V. A. (2009) Mol. Cell. Biol. 29, 4441–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park M. J., Park J. H., Hahm S. H., Ko S. I., Lee Y. R., Chung J. H., Sohn S. Y., Cho Y., Kang L. W., Han Y. S. (2009) DNA Repair 8, 1190–1200 [DOI] [PubMed] [Google Scholar]
- 30. Bhakat K. K., Mokkapati S. K., Boldogh I., Hazra T. K., Mitra S. (2006) Mol. Cell. Biol. 26, 1654–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marsin S., Vidal A. E., Sossou M., Ménissier-de Murcia J., Le Page F., Boiteux S., de Murcia G., Radicella J. P. (2003) J. Biol. Chem. 278, 44068–44074 [DOI] [PubMed] [Google Scholar]
- 32. Bürkle A. (2006) Free Radic. Res. 40, 1295–1302 [DOI] [PubMed] [Google Scholar]
- 33. Schreiber V., Dantzer F., Ame J. C., de Murcia G. (2006) Nat. Rev. Mol. Cell Biol. 7, 517–528 [DOI] [PubMed] [Google Scholar]
- 34. Masutani M., Suzuki H., Kamada N., Watanabe M., Ueda O., Nozaki T., Jishage K., Watanabe T., Sugimoto T., Nakagama H., Ochiya T., Sugimura T. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 2301–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Z. Q., Stingl L., Morrison C., Jantsch M., Los M., Schulze-Osthoff K., Wagner E. F. (1997) Genes Dev. 11, 2347–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Murcia J. M., Niedergang C., Trucco C., Ricoul M., Dutrillaux B., Mark M., Oliver F. J., Masson M., Dierich A., LeMeur M., Walztinger C., Chambon P., de Murcia G. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 7303–7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hill J. W., Evans M. K. (2006) Nucleic Acids Res. 34, 1620–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Z., Rhee D. B., Lu J., Bohr C. T., Zhou F., Vallabhaneni H., de Souza-Pinto N. C., Liu Y. (2010) PLoS Genet. 6, e1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. von Kobbe C., Harrigan J. A., May A., Opresko P. L., Dawut L., Cheng W. H., Bohr V. A. (2003) Mol. Cell. Biol. 23, 8601–8613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Noren N. K., Foos G., Hauser C. A., Pasquale E. B. (2006) Nat. Cell Biol. 8, 815–825 [DOI] [PubMed] [Google Scholar]
- 41. Ward J. F., Evans J. W., Limoli C. L., Calabro-Jones P. M. (1987) Br J. Cancer Suppl. 8, 105–112 [PMC free article] [PubMed] [Google Scholar]
- 42. Singh N. P., McCoy M. T., Tice R. R., Schneider E. L. (1988) Exp. Cell Res. 175, 184–191 [DOI] [PubMed] [Google Scholar]
- 43. Trzeciak A. R., Barnes J., Ejiogu N., Foster K., Brant L. J., Zonderman A. B., Evans M. K. (2008) Free Radic. Biol. Med. 45, 1631–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumaravel T. S., Jha A. N. (2006) Mutat. Res. 605, 7–16 [DOI] [PubMed] [Google Scholar]
- 45. Olive P. L., Banáth J. P., Durand R. E. (1990) Radiat. Res. 122, 86–94 [PubMed] [Google Scholar]
- 46. Nyaga S. G., Lohani A., Evans M. K. (2008) Biochem. Biophys. Res. Commun. 376, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amé J. C., Hakmé A., Quenet D., Fouquerel E., Dantzer F., Schreiber V. (2009) Methods Mol. Biol. 464, 267–283 [DOI] [PubMed] [Google Scholar]
- 48. Harris J. L., Jakob B., Taucher-Scholz G., Dianov G. L., Becherel O. J., Lavin M. F. (2009) Hum. Mol. Genet. 18, 4102–4117 [DOI] [PubMed] [Google Scholar]
- 49. Conlon K. A., Zharkov D. O., Berrios M. (2003) DNA Repair 2, 1337–1352 [DOI] [PubMed] [Google Scholar]
- 50. Mao G., Pan X., Zhu B. B., Zhang Y., Yuan F., Huang J., Lovell M. A., Lee M. P., Markesbery W. R., Li G. M., Gu L. (2007) Nucleic Acids Res. 35, 2759–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson D. M., 3rd, Kim D., Berquist B. R., Sigurdson A. J. (2011) Mutat. Res. 711, 100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hill J. W., Evans M. K. (2007) Cancer Detect. Prevent. 31, 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dherin C., Radicella J. P., Dizdaroglu M., Boiteux S. (1999) Nucleic Acids Res. 27, 4001–4007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yamane A., Kohno T., Ito K., Sunaga N., Aoki K., Yoshimura K., Murakami H., Nojima Y., Yokota J. (2004) Carcinogenesis 25, 1689–1694 [DOI] [PubMed] [Google Scholar]
- 55. Rouleau M., Patel A., Hendzel M. J., Kaufmann S. H., Poirier G. G. (2010) Nat. Rev. Cancer 10, 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zaremba T., Curtin N. J. (2007) Anticancer Agents Med. Chem. 7, 515–523 [DOI] [PubMed] [Google Scholar]
- 57. Xie Y., Yang H., Miller J. H., Shih D. M., Hicks G. G., Xie J., Shiu R. P. (2008) Carcinogenesis 29, 722–728 [DOI] [PubMed] [Google Scholar]
- 58. Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G. (1998) Mol. Cell. Biol. 18, 3563–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goode E. L., Ulrich C. M., Potter J. D. (2002) Cancer Epidemiol. Biomarkers Prev. 11, 1513–1530 [PubMed] [Google Scholar]
- 60. Sidorenko V. S., Zharkov D. O. (2008) Molekuliarnaia biologiia 42, 891–903 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.