

Abstract
AIMS
This open-label, two-period, randomized, crossover study was designed to determine the effect of CYP2C19 reduced function variants on exposure to active metabolites of, and platelet response to, prasugrel and clopidogrel.
METHODS
Ninety healthy Chinese subjects, stratified by CYP2C19 phenotype, were randomly assigned to treatment with prasugrel 10 mg or clopidogrel 75 mg for 10 days followed by 14 day washout and 10 day treatment with the other drug. Eighty-three subjects completed both treatment periods. Blood samples were collected at specified time points for measurement of each drug's active metabolite (Pras-AM and Clop-AM) concentrations and determination of inhibition of platelet aggregation (IPA) by light transmittance aggregometry. CYP2C19 genotypes were classified into three predicted phenotype groups: rapid metabolizers [RMs (*1/*1)], heterozygous or intermediate metabolizers [IMs (*1/*2, *1/*3)] and poor metabolizers [PMs (*2/*2, *2/*3)].
RESULTS
Pras-AM exposure was similar in IMs and RMs (90% CI 0.85, 1.03) and slightly lower in PMs than IMs (90% CI 0.74, 0.99), whereas Clop-AM exposure was significantly lower in IMs compared with RMs (90% CI 0.62, 0.83), and in PMs compared with IMs (90% CI 0.53, 0.82). IPA was more consistent among RMs, IMs and PMs in prasugrel treated subjects (80.2%, 84.2% and 80.2%, respectively) than in clopidogrel treated subjects (59.7%, 56.2% and 36.8%, respectively; P < 0.001).
CONCLUSIONS
Prasugrel demonstrated higher active metabolite exposure and more consistent pharmacodynamic response across all three predicted phenotype groups compared with clopidogrel, confirming observations from previous research that CYP2C19 phenotype plays an important role in variability of response to clopidogrel, but has no impact on response to prasugrel.
Keywords: clopidogrel, CYP2C19, genetic polymorphism, pharmacodynamic, pharmacokinetic, prasugrel
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Observations from clinical studies have demonstrated that:
CYP2C19 genotype does not have a clinically relevant effect on active metabolite concentrations or platelet inhibition in prasugrel-treated subjects.
Variability of response to clopidogrel is related to the presence of reduced function CYP2C19 alleles.
Lower concentrations of clopidogrel active metabolite and reduced platelet inhibition lead to increased adverse cardiovascular events in patients with acute coronary syndromes.
WHAT THIS STUDY ADDS
Observations from previous studies related to the effect of CYP2C19 genotype on plasma drug exposure and platelet response to prasugrel and clopidogrel are confirmed in this prospectively-stratified, randomized, crossover, genetic study in healthy Chinese subjects.
In this study, healthy subjects receiving prasugrel had higher exposure to its active metabolite and greater pharmacodynamic responses (inhibition of platelet aggregation) compared with clopidogrel treated subjects, regardless of CYP2C19-predicted phenotype group.
The exposure to the active metabolite of clopidogrel was lower in CYP2C19 IM compared with RM, and in CYP2C19 PM compared with IM. The inhibition of platelet aggregation was lowest in clopidogrel treated CYP2C19 PM subjects.
Introduction
Prasugrel, a thienopyridine prodrug, is metabolized in vivo to an adenosine diphosphate (ADP)-receptor antagonist that potently inhibits ADP-induced platelet aggregation. Prasugrel, co-administered with aspirin, has been approved in the European Union, United States and several other countries for the prevention of atherothrombotic events in patients with acute coronary syndromes (ACS) who undergo percutaneous coronary intervention (PCI). Clopidogrel is also a thienopyridine prodrug indicated for the reduction of atherosclerotic events in patients with recent stroke, recent myocardial infarction or established peripheral arterial disease. Although prasugrel and clopidogrel are both thienopyridine prodrugs that require in vivo conversion to their active metabolites, Pras-AM and Clop-AM, the pathways leading to active metabolite formation significantly differ between the two drugs (Figure 1) [1].
Figure 1.
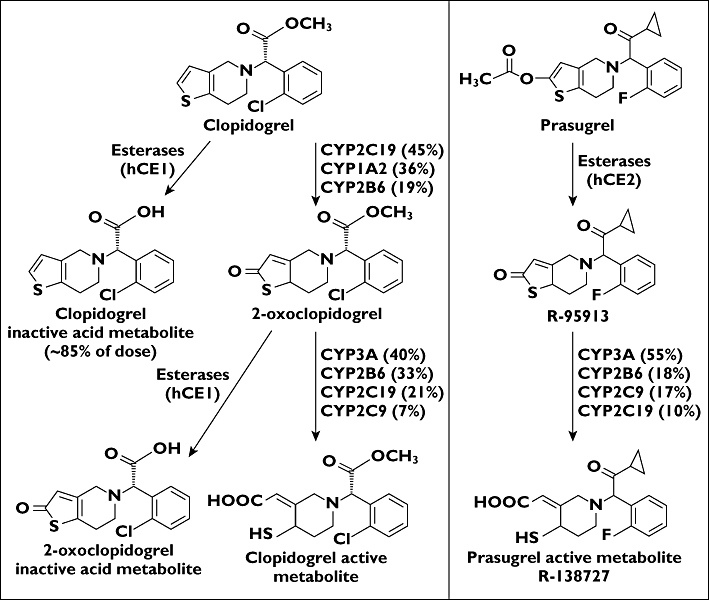
Prasugrel and clopidogrel metabolic pathways to their active metabolites
Multiple enzymes appear to be involved in the absorption and metabolism of prasugrel and clopidogrel [2–4]. The conversion of prasugrel and clopidogrel to Pras-AM and Clop-AM depends in part on the activity of cytochrome P450 (CYP) 2C19, which plays a greater role in the metabolism of clopidogrel than in that of prasugrel (Figure 1) [2, 3]. Prasugrel is not detected in plasma since it is rapidly hydrolyzed during absorption, forming the inactive thiolactone metabolite R-95913. R-95913 is metabolized to Pras-AM primarily by CYP3A4 and CYP2B6, and to a lesser extent by CYP2C9 and CYP2C19 [2, 5, 6]. Pras-AM is subsequently metabolized to inactive compounds by S-methylation or conjugation with cysteine [5]. Clopidogrel is converted to its active metabolite in the liver through two sequential CYP-mediated steps, being first converted to 2-oxoclopidogrel (via CYP2C19, CYP1A2 and CYP2B6) and then to Clop-AM (via CYP3A4, CYP2C19, CYP2C9 and CYP2B6) [3]. CYP2C19 contributes significantly to both oxidative steps in clopidogrel metabolism [3].
Pras-AM and Clop-AM produce their antiplatelet effects by irreversibly binding to the P2Y12 ADP receptor. Once exposed, platelets are inactivated for the remainder of their life span. In vitro studies have shown that, on a molar basis, Pras-AM and Clop-AM are equipotent in terms of inhibition of the platelet receptor P2Y12; however, compared with clopidogrel, prasugrel is metabolized to its active metabolite more efficiently, resulting in higher inhibition of platelet aggregation (IPA) and a more rapid onset of action [1, 7, 8].
Clinical studies have shown that the pharmacodynamic (PD) response to clopidogrel varies, with 20% to 30% of patients classified as poor responders, nonresponders or resistant to clopidogrel [9, 10]. Studies in healthy subjects have shown that IPA is decreased in subjects receiving clopidogrel who have at least one CYP2C19 reduced-function allele [11–15]; however, the PD response to prasugrel is not affected by polymorphisms associated with carrying CYP2C19 variants with reduced function [12, 16].
The presence of reduced function CYP2C19 alleles has also been associated with reduced responsiveness to, and thus reduced therapeutic benefit of, clopidogrel in cardiovascular patients [1, 16–19]. The major CYP2C19 alleles include three single-nucleotide polymorphisms (*2, *3, *17) [20]. The presence of CYP2C19 loss of function alleles *2 and *3 may result in lower exposure to Clop-AM and thus decreased platelet inhibition in clopidogrel treated patients [12], whereas carriers of *17 may have increased platelet response with an increased risk of bleeding, but not greater efficacy [21]. However, in a study designed to find variant CYP2C19 alleles associated with ethnic background, only 4% of Chinese subjects, compared with 18% of Swedes and Ethiopians, were CYP2C19*17 carriers [22].
Of interest, recent studies have presented evidence of poor clinical outcomes, including an increased risk of major adverse cardiovascular events (MACE) for clopidogrel treated patients with ACS who are carriers of reduced function CYP2C19 alleles, particularly for those who undergo PCI [16, 19, 23]. In contrast, an analysis of two studies evaluating the effects of CYP2C19 genotype on clinical outcomes found that the efficacy of clopidogrel compared with placebo was not affected by CYP2C19 genotype in some patients with ACS [20]. However, one explanation for this different finding may be that only 18% of the patients underwent PCI and 14.5% underwent PCI with stent placement. Because of the demonstrated negative effect of reduced function CYP2C19 on the generation of Clop-AM and the reduced PD response to clopidogrel, a black box warning was recently added to the clopidogrel label indicating the key role of CYP2C19 in clopidogrel metabolism and efficacy (http://products.sanofi-aventis.us/PLAVIX/PLAVIX.html) [24].
The genetic variants resulting in reduced CYP2C19 function are more frequent in East Asians than in Caucasians [25, 26]. In Chinese, the major CYP2C19 genotypes are *1/*1 and *1/*2, each of which comprise about 43% of the population [25]. In Caucasians these genotypes comprise approximately 40% and 19% of the population, respectively. Since the reduced-function *1/*2 allele is ‘enriched’ in Asians compared with Caucasians, studies that evaluate the effect of this allele can be sufficiently powered with fewer Asian subjects than Caucasian subjects. The current study was therefore conducted in a healthy Chinese population to evaluate the effects of this variation using a prospectively stratified design. Specifically, the objective of this study was to determine the effect of predicted CYP2C19 phenotype on plasma exposure to the active metabolites and platelet response to Pras-AM and Clop-AM.
METHODS
Study design
This single-centre, open-label, two-period, randomized, crossover study was conducted at the Lilly-NUS Centre for Clinical Pharmacology Pte Ltd, National University of Singapore, between 7 December 2006 and 2 April 2008. The study was conducted in accordance with the Declaration of Helsinki and consistent with applicable guidelines for good clinical practice. The study was approved by the local ethics committee (National Healthcare Group – Domain Specific Review Board, reference number C/06/361) and written informed consent was obtained from each subject.
Subjects
Subjects were healthy men and women of Chinese ancestry (having all four grandparents of Chinese origin) 21 to 60 years of age, and had a body mass index (BMI) of 18.5 to 29.9 kg m−2 with no clinically important physical findings or abnormalities in laboratory results, including platelet function tests at screening. Genetic testing for determination of CYP2C19 genotype was also performed, unless such data for a subject were already available. Exclusion criteria included a history of bleeding disorders or reasonable suspicion of vascular malformations at screening. Aspirin, nonsteroidal anti-inflammatory drugs and medications other than study drugs were not permitted within 14 days prior to or at any time during the study unless deemed necessary by the clinical investigator to treat adverse events.
Treatment administered
Study participants were randomly assigned to daily treatment with prasugrel 10 mg or clopidogrel 75 mg for 10 days during period 1, followed by a minimum 14 day washout before beginning period 2 daily treatment with the other drug for 10 days (Figure 2). Study drug was administered in the clinical research unit (CRU) on days 1, 2 and 10 of each period, with other dosing days conducted on an outpatient basis.
Figure 2.
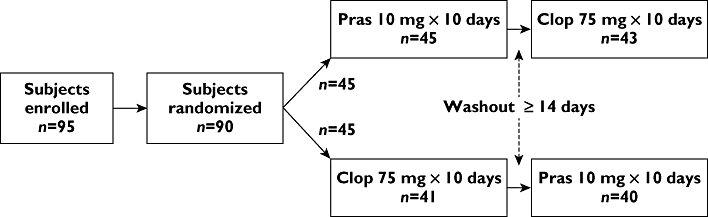
Study design and patient disposition. Ninety subjects received ≥1 dose of study drug. In period 1, 45 subjects completed 10 days of prasugrel and 41 subjects completed 10 days of clopidogrel. In period 2, 43 subjects completed 10 days of clopidogrel and 40 subjects completed 10 days of prasugrel. Clop, clopidogrel; n, number of subjects; Pras, prasugrel
Pharmacokinetic measurements
Plasma concentrations of Pras-AM and Clop-AM were measured to enable estimation of PK parameters for each metabolite. Subjects were admitted to the CRU the day before PK sampling days and remained in the CRU until discharge on the mornings of days 2 and 11. Blood samples (3 ml each) were collected via a venous cannula or direct venipuncture into ethylenediamine tetraacetic acid (EDTA) tubes at specified time points on days 1 and 10 in each of the two study periods. The blood samples were reacted with 25 µl of 500 µm 3′-methoxyphenacyl bromide in acetonitrile within 30 s of collection to derivatize and stabilize the active metabolites [27]. Plasma samples harvested from blood samples were stored at approximately −70°C in labelled polypropylene tubes until shipment on dry ice to the analytical laboratory.
Plasma concentrations were determined using validated liquid chromatography methods with tandem mass spectrometric detection as previously described [27, 28]. The lower limit of quantitation was 0.5 ng ml−1 for both Pras-AM and Clop-AM.
Pharmacodynamic measurements
Blood samples for PD measurements were collected at various time points on days 1, 2, 3, 5, 9 and 10 of each period; venous blood samples of approximately 9 ml (two tubes of 4.5 ml each) were obtained via direct venipuncture. Platelet aggregation using light transmittance aggregometry (LTA) was measured at the CRU on a platelet aggregation profiler-4 optical aggregometer, with temperature maintained at 37°C. Platelet aggregation in platelet-rich plasma was measured with 20 µm ADP as agonist. Platelet counts in platelet-rich plasma were adjusted to approximately 250 000 µl–1 using autologous platelet-poor plasma. In addition, citrated blood samples (2 ml each) were collected to perform an exploratory analysis assessing the correlation of LTA platelet aggregation results with those using the Accumetrics VerifyNow™ (VN) P2Y12 assay system (Accumetrics, San Diego, California, United States) as well as the flow cytometric assessment of vasodilator-stimulated phosphoprotein (VASP) phosphorylation using a commercially available kit (BioCytex, Marseille, France). In addition, subjects were characterized as PD nonresponders if they had ≤20% IPA to 20 µm ADP as assessed by LTA [29], >240 P2Y12 reactivity units (PRU) as assessed by the VN P2Y12 assay [30] or >50% platelet reactivity index (PRI) as assessed by VASP phosphorylation [31].
Genotype and phenotype groups
A 6 ml blood sample was collected at screening to determine CYP2C19 genotype by polymerase chain reaction. During the study, DNA was extracted from peripheral blood anticoagulated with EDTA using standard methods to genotype for CYP2C19. The Drug Metabolizing Enzyme and Transporter gene assay system [32] was used to generate genotype data. Using validated computer systems, the genetic variants measured were converted to the common consensus allele nomenclature [33] for CYP2C19. Genotype assignments were also reviewed manually.
Subjects were classified by CYP2C19 genotype into metabolic phenotypes based on functional predictions established by the literature [33, 34]. The three classifications consisted of rapid metabolizers [RMs; wild-type homozygotes having two alleles with normal enzyme activity (*1/*1)], heterozygous or intermediate metabolizers [IMs; heterozygotes with one allele having normal enzyme activity and one allele having little or no activity (1*/2* and 1*/3*)] and poor metabolizers [PMs; two alleles with little or no activity (2*/2* and 2*/3*)] (Table 1) [35, 36].
Table 1.
Summary of demographic and CYP2C19-predicted phenotype results
| Demographic characteristics | |||
|---|---|---|---|
| n= 90 randomized subjects; 51 men, 39 women | |||
| Age (years) | Body weight (kg) | BMI (kg m−2) | |
| Mean (SD) | 34 (11.2) | 63.0 (9.7) | 22.6 (2.4) |
| Range | 21–60 | 43.4–101 | 21.8–29.6 |
| Genetic results | |||
| CYP2C19 predicted phenotype | RM | IM | PM |
| CYP2C19-predicted phenotype CYP2C19 genotype | *1/*1 | *1/*2*1/*3 | *2/*2*2/*3 |
| Number of completing subjects (n= 83) | 34 (41%) | 38 (46%) | 11 (13%) |
BMI, body mass index; RM, rapid metabolizers; IM, intermediate metabolizers; n, number of subjects; PM, poor metabolizers; SD, standard deviation.
Safety analyses
Safety data were collected in the form of vital signs, clinical examinations, laboratory tests and adverse events. A follow-up visit for each subject was conducted within 14 days of the subject's last dose of study drug.
Pharmacokinetic analysis
Pharmacokinetic parameter estimates of Pras-AM and Clop-AM were calculated by standard non-compartmental methods of analysis using WinNonlin® Enterprise Version 5.0.1 and were summarized by treatment and CYP2C19-predicted phenotype. Following the dose of prasugrel or clopidogrel on days 1 and 10, the estimated PK parameters included the area under the plasma concentration–time curve (AUC) from the time of dosing through the sampling time of the last measurable concentration [AUC(0,tlast)], the maximum observed plasma concentration (Cmax), and the time of observed Cmax (tmax). Log transformed Cmax and AUC estimates were evaluated to estimate ratios of least squares (LS) geometric means and the corresponding 90% confidence intervals (CIs). An analysis of covariance model was used to detect differences in PK parameter estimates between groups.
Statistical analyses accounted for differences in body weight among the three predicted phenotype groups. The inclusion of body weight in the statistical model as an intrinsic factor was based on an integrated analysis of 16 clinical pharmacology studies of prasugrel in which the effect of body weight on AUC(0,tlast) and Cmax was statistically significant; higher Pras-AM exposure was associated with lower body weight [37].
Pharmacodynamic analysis
The PD effects of prasugrel and clopidogrel were compared with each CYP2C19-predicted phenotype group. The PD parameter was IPA measured in response to 20 µm ADP using LTA. Estimates of IPA were calculated for all subjects at each time point using the following formula:
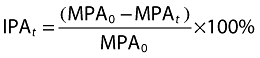 |
where MPA0 is the maximum platelet aggregation (MPA) at baseline (pre-dose), MPAt is the MPA at time t and IPAt is the IPA at time t. If baseline MPA in the two periods did not differ at a 0.05 significance level using a linear mixed model, then the average of these values was used; otherwise, the corresponding pre-dose MPA for each period was used in the calculation of IPA. A linear mixed effects model was used to test for prasugrel or clopidogrel effect on IPA between predicted phenotype groups with baseline (pre-dose) IPA as a continuous covariate. The fixed categorical effects included phenotype, time and phenotype by time. Subject and subject by time were fitted as random effects. For each phenotype, the LS geometric mean and two-sided 90% CI were estimated at each time point. The mean differences between phenotypes and the corresponding two-sided 90% CIs were also estimated at each time point.
Results
Subjects
Ninety-five healthy Chinese subjects were enrolled, five of whom were not dosed due to physician or subject decision (Figure 2). Ninety subjects received at least one dose of study drug. Seven were withdrawn before completing the study (six during period 1 and one during period 2) as a result of adverse events considered to be unrelated to study drug. Table 1 summarizes the demographic characteristics and CYP2C19-predicted phenotypes of the study population. Subjects were tested for the *17 allele; however, none were found to be carriers.
Pharmacokinetic results
Figure 3 presents mean plasma concentration–time profiles on day 10 in subjects categorized by CYP2C19-predicted phenotype. Mean plasma concentration–time curves for Pras-AM and Clop-AM were similar for days 1 and 10. Following prasugrel dosing, IMs and RMs had similar Pras-AM concentration–time profiles, but PMs had lower Pras-AM concentrations than did IMs or RMs. Following clopidogrel dosing, both PMs and IMs had lower concentrations of active metabolite than did RMs and PMs had lower concentrations than did IMs. Of note, Pras-AM concentrations in PMs were higher than those of Clop-AM in RMs. A comparison of PK parameter estimates of Pras-AM and Clop-AM by predicted CYP2C19 phenotype on day 10 is summarized in Table 2. The data in Table 2 are adjusted for body weight. The molecular weights of Pras-AM (349.4 g mol−1) and Clop-AM (355.9 g mol−1) differ by less than 2%, and therefore comparison of the relative exposures shown in Table 2 is valid.
Figure 3.
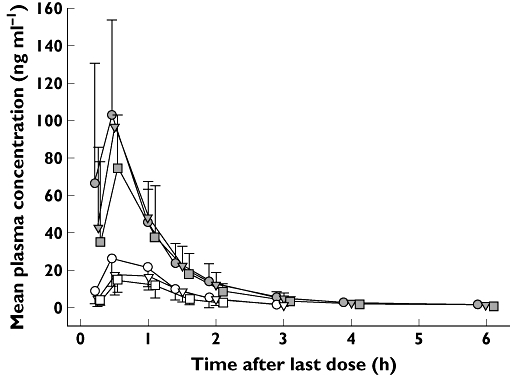
Mean plasma concentration of Pras-AM and Clop-AM (day 10) by CYP2C19-predicted phenotype. Clop-AM, clopidogrel active metabolite, CYP, cytochrome P450; RM, rapid metabolizer; IM, intermediate metabolizer; PM, poor metabolizer; Pras-AM, prasugrel active metabolit. Prasugrel RM ( ); Clopidogrel RM (
); Clopidogrel RM ( ); Prasugrel IM (
); Prasugrel IM ( ); Clopidogrel IM (
); Clopidogrel IM ( ); Prasugrel PM (
); Prasugrel PM ( ); Clopidogrel PM (
); Clopidogrel PM ( )
)
Table 2.
Comparison of PK parameter estimates of prasugrel and clopidogrel active metabolites by CYP2C19-predicted phenotype following daily doses of prasugrel 10 mg or clopidogrel 75 mg for 10 days
| All subjects | Subject 105 excluded | ||||||
|---|---|---|---|---|---|---|---|
| LS geometric means* | Ratio to RM (90% CI) | Ratio to IM (90% CI) | LS Geometric Means* | Ratio to RM (90% CI) | Ratio to IM (90% CI) | Subject 105 | |
| Phenotype | Prasugrel active metabolite | ||||||
| AUC(0,tlast) (ng ml−1 h) | |||||||
| RM | 101.4 | 101.1 | |||||
| IM | 95.0 | 0.94 (0.85, 1.03)† | 94.8 | 0.94 (0.86, 1.03)† | |||
| PM | 80.9 | 0.80 (0.69, 0.93) | 0.85 (0.74, 0.99) | 87.5 | 0.87 (0.75, 1.00) | 0.92 (0.80, 1.06) | 42.40 |
| Cmax (ng ml−1) | |||||||
| RM | 102.4 | 102.2 | |||||
| IM | 92.9 | 0.91 (0.76, 1.08)† | 92.8 | 0.91 (0.77, 1.08)† | |||
| PM | 85.9 | 0.84 (0.64, 1.09) | 0.92 (0.71, 1.20) | 94.5 | 0.93 (0.71, 1.21) | 1.02 (0.78, 1.33) | 38.29 |
| Clopidogrel active metabolite | |||||||
| AUC(0,tlast) (ng ml−1 h) | |||||||
| RM | 29.6 | 29.5 | |||||
| IM | 21.3 | 0.72 (0.62, 0.83) | 21.3 | 0.72 (0.62, 0.83) | |||
| PM | 13.9 | 0.47 (0.38, 0.59) | 0.66 (0.53, 0.82) | 15.2 | 0.51 (0.41, 0.65) | 0.71 (0.57, 0.89) | 6.51 |
| Cmax (ng ml−1) | |||||||
| RM | 27.6 | 27.5 | |||||
| IM | 19.9 | 0.72 (0.60, 0.87)† | 19.9 | 0.72 (0.61, 0.86)† | |||
| PM | 15.1 | 0.55 (0.41, 0.72) | 0.76 (0.58, 0.99) | 16.7 | 0.60 (0.46, 0.80) | 0.84 (0.63, 1.10) | 6.43 |
Adjusted for body weight as an intrinsic factor.
Small differences between RMs and IMs in ‘All subjects’ and ‘Subject 105 excluded’ are due to the effect on fitted models of excluding this subject. AUC(0,tlast), area under the plasma concentration-time curve from the time of dosing through the sampling time of the last measurable concentration; CI, confidence interval; Cmax, maximum observed plasma concentration; CYP, cytochrome P450; RM, rapid metabolizer (phenotype *1/*1); IM, intermediate metabolizer (phenotype *1/*2 & *1/*3); LS, least squares; PM, poor metabolizer (phenotype *2/*2 and *2/*3).
Data are also presented from a sensitivity analysis, excluding subject 105. This particular subject from the PM phenotype group had atypically low active metabolite exposure for both prasugrel and clopidogrel on both days 1 and 10, despite having a relatively low body weight (59.9 kg) and BMI (19.0 kg m−2). The retrospective analysis was conducted to account for any disproportionate influence this anomalous subject may have had on comparisons in the multivariate analysis. For Pras-AM, the LS geometric means ratios (90% CI) of AUC(0,tlast) with and without subject 105 data were 0.85 (0.74, 0.99) and 0.92 (0.80, 1.06), respectively, for the comparison of PMs to IMs, and 0.80 (0.69, 0.93) and 0.87 (0.75, 1.00), respectively, for the comparison of PMs with RMs. Therefore, after excluding subject 105 data, the exposure to Pras-AM was not significantly different between PMs and RMs or IMs. For Clop-AM, excluding subject 105 data slightly changed the LS geometric means ratios and 90% CIs between phenotype groups, but did not change conclusions. Data presented in Table 2 also illustrate that, although Pras-AM and Clop-AM AUCs were not compared statistically, exposures to Pras-AM were numerically several fold higher than those of Clop-AM for all predicted phenotype groups.
Pharmacodynamic results
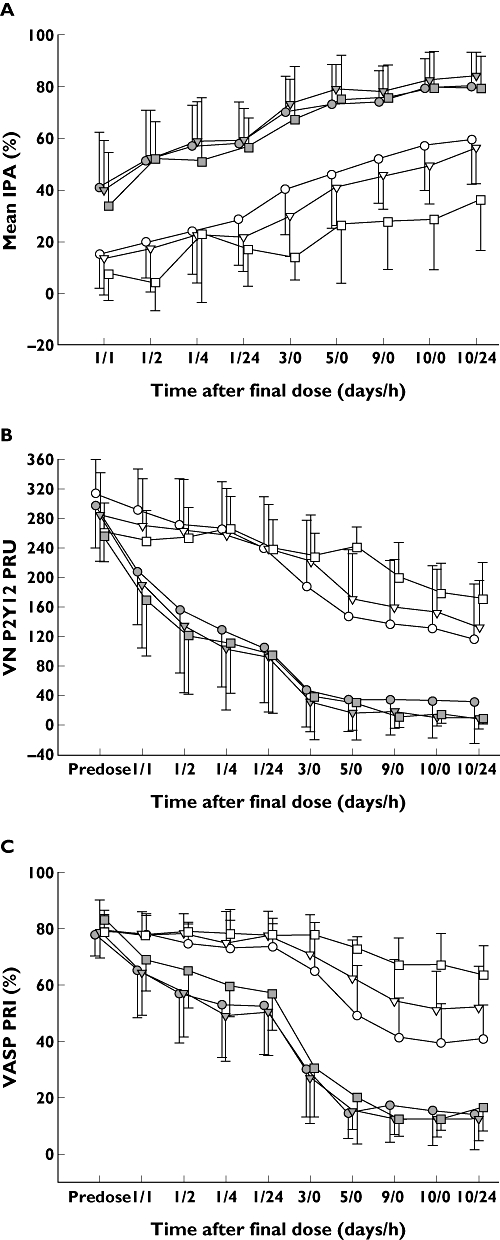
A summary of IPA to 20 µm ADP (LTA) for day 10 is presented in Table 3 and Figure 4 shows mean PD results for all sampling times for days 1 through 10. Mean IPA was higher (P < 0.05) for prasugrel than for clopidogrel for all three phenotype groups across all time points. Although statistical analyses were not performed, platelet aggregation results obtained for PRU using the VN P2Y12 device, and for PRI as assessed by VASP phosphorylation, showed similar trends to those calculated for IPA to 20 µm ADP using LTA following 10 days of prasugrel 10 mg and clopidogrel 75 mg.
Table 3.
Summary of day 10 IPA to 20 µm ADP (LTA) for 10 mg prasugrel and 75 mg clopidogrel by CYP2C19-predicted phenotype
| LS mean IPA (90% CI) | (Prasugrel – Clopidogrel) | |||
|---|---|---|---|---|
| Phenotype | Prasugrel | Clopidogrel | Difference (90% CI) | P value |
| RM | 80.2 (75.9, 84.5) | 59.7 (55.3, 64.2) | −20.4 (−25.5, −15.4) | <0.001 |
| IM | 84.2 (80.0, 88.4) | 56.2 (52.2, 60.3) | −28.0 (−32.7, −23.2) | <0.001 |
| PM | 80.2 (72.3, 88.2) | 36.8 (29.0, 44.5) | −43.5 (−52.4, −34.5) | <0.001 |
ADP, adenosine diphosphate; CI, confidence interval; CYP, cytochrome P450; RM, rapid metabolizer (phenotype *1/*1); IM, intermediate metabolizer (phenotype *1/*2 and *1/*3); IPA, inhibition of platelet aggregation; LS, least squares; LTA, light transmittance aggregometry; PM, poor metabolizer (phenotype *2/*2 and *2/*3).
Figure 4.
Mean platelet aggregation as assessed by LTA IPA to 20 µM ADP (A), VN P2Y12 assay (B) and VASP phosphorylation (C) following daily doses of 10 mg prasugrel and 75 mg clopidogrel according to CYP2C19-predicted phenotype. Differences in IPA (A) between prasugrel and clopidogrel were statistically significant (P < 0.05) at all time points. ADP, adenosine diphosphate; CYP, cytochrome P450; RM, rapid metabolizer; IM, intermediate metabolizer; IPA, inhibition of platelet aggregation; LTA, light transmittance aggregometry; PM, poor metabolizer; PRI, platelet reactivity index; PRU, P2Y12 reactivity units; VASP, vasodilator-stimulated phosphoprotein; VN, Accumetrics VerifyNow™. Prasugrel RM ( ); Clopidogrel RM (
); Clopidogrel RM ( ); Prasugrel IM (
); Prasugrel IM ( ); Clopidogrel IM (
); Clopidogrel IM ( ); Prasugrel PM (
); Prasugrel PM ( ); Clopidogrel PM (
); Clopidogrel PM ( )
)
For prasugrel treated subjects, CYP2C19-predicted phenotype group was not associated with platelet response. For clopidogrel treated subjects, CYP2C19-predicted phenotype group was consistently associated with a dampened PD effect for IMs and for PMs across all platelet aggregation measures. Figure 5 shows individual PK and PD responses to prasugrel and clopidogrel post dose on day 10 stratified by CYP2C19-predicted phenotype group and indicates the threshold for nonresponse for each PD assay. For clopidogrel, the number of nonresponders was similar for IPA and PRU but comparatively larger for PRI.
Figure 5.
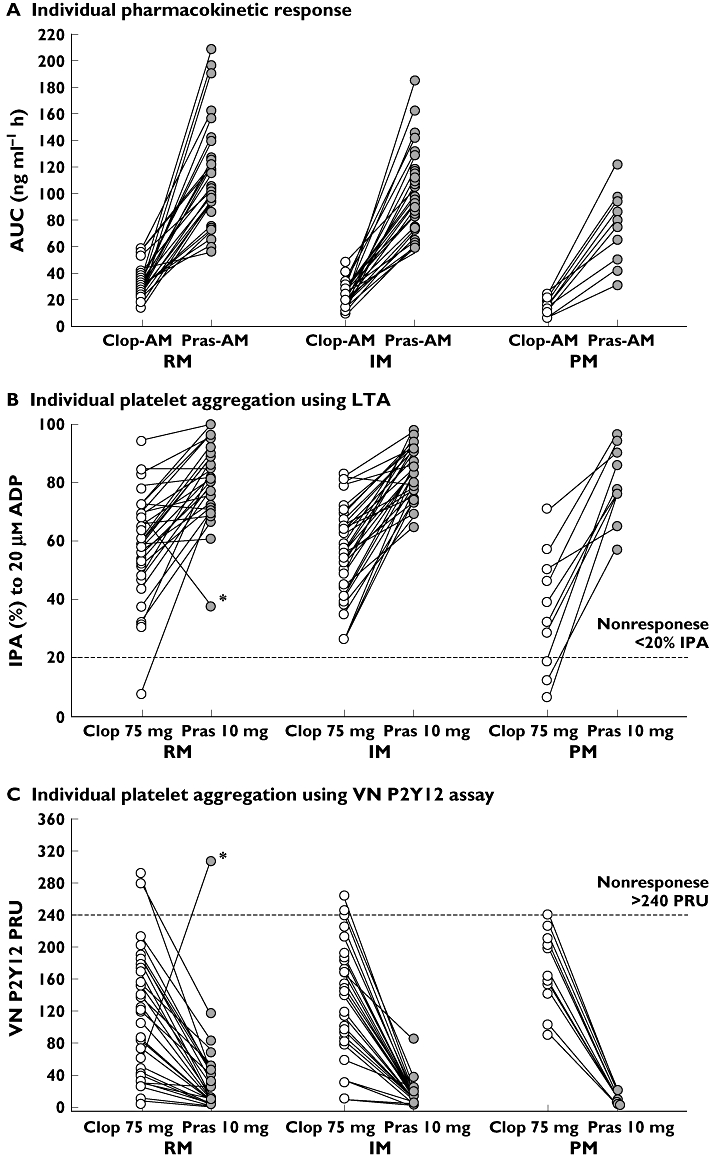
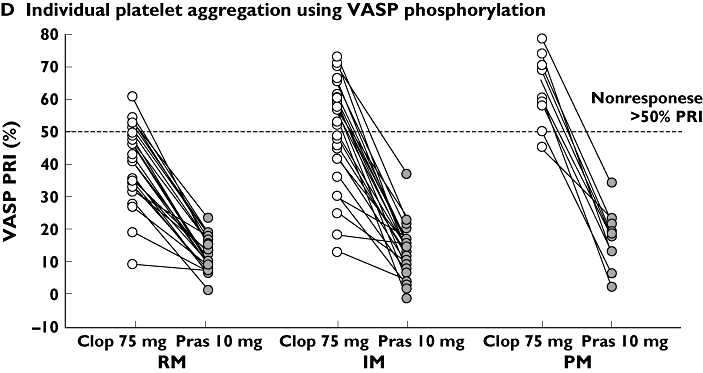
Pharmacokinetic and pharmacodynamic responses to clopidogrel and prasugrel in individual subjects on day 10 following daily doses of 10 mg prasugrel and 75 mg clopidogrel stratified by predicted CYP2C19 phenotype. Panel A shows active metabolite exposure, which is consistently higher for prasugrel than for clopidogrel across all 3 groups. Inhibition of platelet aggregation as measured by LTA (B), VN P2Y12 assay (C), and VASP phosphorylation (D) is also shown. The asterisked RM subject in panels B and C is subject 169, who was very likely noncompliant during prasugrel treatment. See text (Pharmacodynamic results) for details. AUC, area under the plasma concentration–time curve; ADP, adenosine diphosphate; Clop-AM, clopidogrel's active metabolite; CYP, cytochrome P450; RM, rapid metabolizer; IM, intermediate metabolizer; IPA, inhibition of platelet aggregation; LTA, light transmittance aggregometry; PM, poor metabolizer; Pras-AM, prasugrel's active metabolite; PRI, platelet reactivity index; PRU, P2Y12 reactivity units; VASP, vasodilator-stimulated phosphoprotein; VN, Accumetrics VerifyNow™
Among prasugrel-treated subjects, only subject 169 (*1/*1 genotype) met the criteria for nonresponse based on PRU. This subject showed a lower IPA at 24 h after the day 10 prasugrel dose compared with day 10 clopidogrel dose. This subject had no measurable Pras-AM concentrations on day 10, which indicated noncompliance on that day, at a minimum. However, in the absence of direct proof of sustained noncompliance, the subject was included in the PD analyses with the exception of PRI, as data assessed by VASP phosphorylation were not available for this subject.
Adverse events
Daily 10 mg doses of prasugrel were safe and well tolerated by healthy Chinese subjects when administered daily for 10 days. Most adverse events were mild in severity and no serious or drug-related severe adverse events were reported. The most frequently reported adverse events were mild bleeding-related events, specifically post procedural bleeding and contusion (i.e. bruising), with a higher incidence following prasugrel (72%) compared with clopidogrel (58%). For both treatments, the majority of bleeding-related adverse events were mild in severity.
Following administration of prasugrel, there appeared to be a higher incidence of drug-related contusion in subjects with lower body weights; contusion occurred in 17 of 34 subjects (50%) weighing <60 kg compared with 11 of 51 subjects (22%) with a body weight >60 kg. There was no apparent correlation between the incidence of bleeding-related events and phenotype for prasugrel treatment or for clopidogrel treatment. There was, however, a higher incidence of contusion in RMs compared with IMs and PMs following clopidogrel dosing; contusion was reported by 24% of RMs, 12% of IMs and 9% of PMs. There were no safety concerns in terms of clinical laboratory evaluations, vital signs, fundoscopy, and petechiae examinations following daily 10 mg doses of prasugrel or 75 mg doses of clopidogrel.
Discussion
Recent studies have indicated that the interpatient variability of response to clopidogrel is related to the presence of reduced function CYP2C19 alleles in poor responders and nonresponders, with a corresponding increased risk of adverse cardiovascular events and stent thrombosis [16, 19, 23, 38]. Further, the relative risk of having the CYP2C19*2 loss-of-function gene for patients who are heterozygous (IMs) vs. homozygous (PMs) has been a matter of some controversy. However, a meta-analysis of nine studies demonstrated that, compared with RMs, both groups are at increased risk of MACE when on clopidogrel treatment; the relative risk of MACE was 1.50 for IMs and 1.61 for PMs with a 2.51- and 4.78-fold increased risk of stent thrombosis, respectively [39]. These results suggest that clopidogrel-treated IMs and PMs are at greater risk for experiencing adverse cardiovascular events relative to clopidogrel-treated RMs.
It has also been proposed that patients who are non-responsive to the standard maintenance dose of clopidogrel may have higher IPA if treated with prasugrel as maintenance therapy [17]. Two trials have investigated the effects of switching from clopidogrel to prasugrel. In one, healthy subjects received clopidogrel 75 mg maintenance dose for 10 days and switched directly to either a prasugrel 60 mg loading dose or 10 mg maintenance dose for 10 days [40]. In another study, patients with ACS who had received a clopidogrel 900 mg loading dose were randomized to receive a prasugrel 10 mg or clopidogrel 150 mg maintenance dose for 14 days and then switched to the alternative treatment for 14 days [41]. Results of both studies demonstrated that switching from clopidogrel to prasugrel was well tolerated and resulted in improved platelet response [40, 41]. We undertook a prospectively designed, randomized, crossover study in which healthy Chinese subjects were stratified based on CYP2C19 genotype to specifically evaluate the effect of CYP2C19 genetic variation on the PK and PD of prasugrel and clopidogrel.
In the current study, PMs had lower exposure to both Pras-AM and Clop-AM when compared with their respective RMs. After adjusting for body weight as an intrinsic factor that is known to affect Pras-AM exposure, the AUC(0,tlast) for Pras-AM did not differ between IMs and RMs, but was 20% (90% CI 7%, 31%) lower in PMs than in RMs. In the sensitivity analysis that excluded data from one subject with atypically low exposure for both prasugrel and clopidogrel, the magnitude of the Pras-AM exposure difference between PMs and RMs was reduced to 13% (90% CI 0%, 25%). As with previous comparisons of exposure between Pras-AM and Clop-AM following 10 mg and 75 mg doses, respectively, AUC and Cmax estimates for Pras-AM were substantially higher overall than those for Clop-AM across all predicted phenotype groups (Table 2).
The PD response to prasugrel treatment was unaffected by CYP2C19 genetic variation based on IPA to 20 µm ADP as measured by LTA, the VN P2Y12 assay and VASP phosphorylation. Consistent with the higher exposure of Pras-AM, IPA was substantially higher during prasugrel treatment than during clopidogrel treatment for all three CYP2C19-predicted phenotype groups. Compared with prasugrel treated RMs, clopidogrel treated RMs had a 72 percentage point lower active metabolite exposure, translating to a 25 percentage point lower IPA. Previous studies showed that Pras-AM and Clop-AM are equipotent and that the PD response to these compounds correlates with the respective exposure to the active metabolite [1, 7–9, 12]. Since the exposure to Pras-AM is significantly greater than that for Clop-AM, the increase in IPA would be correspondingly greater after a prasugrel dose, and actually approaches the asymptote of the exposure–response curve [1]. Accordingly, while the exposure to Pras-AM was lower in CYP2C19 PMs, the impact on the resultant IPA, if any, was negligible, as small changes in exposure to Pras-AM did not affect the PD response to prasugrel.
One limitation of the current study is that the small number of PMs, which comprised only 11 (13%) of the 83 subjects who completed the study, may limit interpretation of results. For example, the atypically low AUC(0,tlast) for both Pras-AM and Clop-AM in one PM subject 105, had a disproportionately large influence on comparisons between CYP2C19-predicted phenotype groups, particularly for Pras-AM analyses. The low exposure for this subject was not associated with demographic characteristics, bioanalytical assay variability or improper sample handling. Thus, PK analyses are presented both with and without this subject's data for full disclosure (Table 2). It is noteworthy, however, that the 90% CI for the Clop-AM AUC(0,tlast) in the PM group did not include 1.0 for either analysis, suggesting a real difference in Clop-AM exposure for PMs relative to RMs and IMs.
The results of this study demonstrate that because of differences in the respective metabolic pathways leading to the formation of Pras-AM and Clop-AM and the resultant differences in exposure between the two drugs, the presence of reduced function CYP2C19 alleles does not affect the PD response to prasugrel, but significantly reduces the anti-platelet response to clopidogrel. This difference would also explain the greater IPA and fewer ischaemic events seen with prasugrel, as compared with clopidogrel, in the TRITON-TIMI 38 study [42]. These results are consistent with the findings of a study in which CYP3A was completely inhibited with ketoconazole in subjects treated with prasugrel or clopidogrel [43]. CYP3A is the predominant pathway for Pras-AM generation, yet potent CYP3A inhibition did not affect the extent of active metabolite generation as indicated by AUC. Given that ketoconazole is one of the most potent inhibitors of CYP3A, inhibitors or reduced function of other, less important CYPs would not be expected to affect the overall exposure or pharmacodynamic response to prasugrel maintenance or loading doses.
The results also demonstrate that, compared with clopidogrel, prasugrel has greater exposure to the active metabolite and more consistent PD responses across all three CYP2C19-predicted phenotypes. This genetics study confirms observations from previous research that CYP2C19 phenotype plays an important role in the variability of response to clopidogrel, but has no impact on response to prasugrel.
Acknowledgments
This study was funded by Daiichi Sankyo Co., Ltd. and Eli Lilly and Company. The authors thank Mei Teng Loh (Eli Lilly) for support in protocol development and study implementation, Mary Pritchard (i3 Statprobe) for assistance in drafting the manuscript, Christopher Konkoy (Eli Lilly) for assistance in drafting the manuscript and project management, Angela Lorio (i3 Statprobe) for editorial and content review and Julie Sherman (Eli Lilly) for formatting and submitting the manuscript.
Competing Interests
RPK, KJW, LS, FN, JAJ, MH and DSS are all employees and shareholders of Eli Lilly and Company. SLC is a former employee of Eli Lilly and Company and received fees for consulting from Eli Lilly and Daichii Sankyo. NAF is a retired employee and shareholder of Eli Lilly and Company. JRW is an employee of Daiichi Sankyo, Inc. and a shareholder of Daiichi Sankyo Company, Ltd.
REFERENCES
- 1.Farid NA, Kurihara A, Wrighton SA. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans. J Clin Pharmacol. 2010;50:126–42. doi: 10.1177/0091270009343005. [DOI] [PubMed] [Google Scholar]
- 2.Rehmel JL, Eckstein JA, Farid NA, Heim JB, Kasper SC, Kurihara A, Wrighton SA, Ring BJ. Interactions of two major metabolites of prasugrel, a thienopyridine antiplatelet agent, with the cytochromes P450. Drug Metab Dispos. 2006;34:600–7. doi: 10.1124/dmd.105.007989. [DOI] [PubMed] [Google Scholar]
- 3.Kazui M, Nishiya Y, Ishizuka T, Hagihara K, Farid NA, Okazaki O, Ikeda T, Kurihara A. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38:92–9. doi: 10.1124/dmd.109.029132. [DOI] [PubMed] [Google Scholar]
- 4.Mega JL, Close SL, Wiviott SD, Shen L, Walker JR, Simon T, Antman EM, Braunwald E, Sabatine MS. Genetic variants in ABCB1, CYP2C19, and cardiovascular outcomes following treatment with clopidogrel and prasugrel. Lancet. 2010;376:1312–9. doi: 10.1016/S0140-6736(10)61273-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farid NA, Smith RL, Gillespie TA, Rash TJ, Blair PE, Kurihara A, Goldberg MJ. The disposition of prasugrel, a novel thienopyridine, in humans. Drug Metab Dispos. 2007;35:1096–104. doi: 10.1124/dmd.106.014522. [DOI] [PubMed] [Google Scholar]
- 6.Williams ET, Jones KO, Ponsler GD, Lowery SM, Perkins EJ, Wrighton SA, Ruterbories KJ, Kazui M, Farid NA. The biotransformation of prasugrel, a new thienopyridine prodrug, by the human carboxylesterases 1 and 2. Drug Metab Dispos. 2008;36:1227–32. doi: 10.1124/dmd.107.020248. [DOI] [PubMed] [Google Scholar]
- 7.Payne CD, Li YG, Small DS, Ernest CS, 2nd, Farid NA, Jakubowski JA, Brandt JT, Salazar DE, Winters KJ. Increased active metabolite formation explains the greater platelet inhibition with prasugrel compared to high-dose clopidogrel. J Cardiovasc Pharmacol. 2007;50:555–62. doi: 10.1097/FJC.0b013e3181492209. [DOI] [PubMed] [Google Scholar]
- 8.Sugidachi A, Ogawa T, Kurihara A, Hagihara K, Jakubowski JA, Hashimoto M, Niitsu Y, Asai F. The greater in vivo antiplatelet effects of prasugrel as compared to clopidogrel reflect more efficient generation of its active metabolite with similar antiplatelet activity to that of clopidogrel's active metabolite. J Thromb Haemost. 2007;5:1545–51. doi: 10.1111/j.1538-7836.2007.02598.x. [DOI] [PubMed] [Google Scholar]
- 9.Brandt JT, Payne CD, Wiviott SD, Weerakkody G, Farid NA, Small DS, Jakubowski JA, Naganuma H, Winters KJ. A comparison of prasugrel and clopidogrel loading doses on platelet function: magnitude of platelet inhibition is related to active metabolite formation. Am Heart J. 2007;153:66. doi: 10.1016/j.ahj.2006.10.010. e9–16. [DOI] [PubMed] [Google Scholar]
- 10.Combescure C, Fontana P, Mallouk N, Berdague P, Labruyere C, Barazer I, Gris JC, Laporte S, Fabbro-Peray P, Reny JL, CLOpidogrel and Vascular ISchemic Events Meta-analysis Study Group Clinical implications of clopidogrel non-response in cardiovascular patients: a systematic review and meta-analysis. J Thromb Haemost. 2010;8:923–33. doi: 10.1111/j.1538-7836.2010.03809.x. [DOI] [PubMed] [Google Scholar]
- 11.Hulot JS, Bura A, Villard E, Azizi M, Remones V, Goyenvalle C, Aiach M, Lechat P, Gaussem P. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108:2244–7. doi: 10.1182/blood-2006-04-013052. [DOI] [PubMed] [Google Scholar]
- 12.Brandt JT, Close SL, Iturria SJ, Payne CD, Farid NA, Ernest CS, 2nd, Lachno DR, Salazar D, Winters KJ. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J Thromb Haemost. 2007;5:2429–36. doi: 10.1111/j.1538-7836.2007.02775.x. [DOI] [PubMed] [Google Scholar]
- 13.Fontana P, Hulot JS, De Moerloose P, Gaussem P. Influence of CYP2C19 and CYP3A4 gene polymorphisms on clopidogrel responsiveness in healthy subjects. J Thromb Haemost. 2007;5:2153–5. doi: 10.1111/j.1538-7836.2007.02722.x. [DOI] [PubMed] [Google Scholar]
- 14.Kim KA, Park PW, Hong SJ, Park JY. The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: a possible mechanism for clopidogrel resistance. Clin Pharmacol Ther. 2008;84:236–42. doi: 10.1038/clpt.2008.20. [DOI] [PubMed] [Google Scholar]
- 15.Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, Walker JR, Antman EM, Macias WL, Braunwald E, Sabatine MS. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360:354–62. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- 16.Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, Walker JR, Antman EM, Macias WL, Braunwald E, Sabatine MS. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation. 2009;119:2553–60. doi: 10.1161/CIRCULATIONAHA.109.851949. [DOI] [PubMed] [Google Scholar]
- 17.Giusti B, Gori AM, Marcucci R, Saracini C, Sestini I, Paniccia R, Buonamici P, Antoniucci D, Abbate R, Gensini GF. Relation of cytochrome P450 2C19 loss-of-function polymorphism to occurrence of drug-eluting coronary stent thrombosis. Am J Cardiol. 2009;103:806–11. doi: 10.1016/j.amjcard.2008.11.048. [DOI] [PubMed] [Google Scholar]
- 18.Varenhorst C, James S, Erlinge D, Brandt JT, Braun OO, Man M, Siegbahn A, Walker J, Wallentin L, Winters KJ, Close SL. Genetic variation of CYP2C19 affects both pharmacokinetic and pharmacodynamic responses to clopidogrel but not prasugrel in aspirin-treated patients with coronary artery disease. Eur Heart J. 2009;30:1744–52. doi: 10.1093/eurheartj/ehp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, Méneveau N, Steg PG, Ferrières J, Danchin N, Becquemont L. French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360:363–75. doi: 10.1056/NEJMoa0808227. [DOI] [PubMed] [Google Scholar]
- 20.Paré G, Mehta SR, Yusuf S, Anand SS, Connolly SJ, Hirsh J, Simonsen K, Bhatt DL, Fox KAA, Eikelboom JW. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med. 2010;363:1704–14. doi: 10.1056/NEJMoa1008410. [DOI] [PubMed] [Google Scholar]
- 21.Sibbing D, Koch W, Gebhard D, Schuster T, Braun S, Stegherr J, Morath T, Schomig A, von Beckerath N, Kastrati A. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation. 2010;121:512–8. doi: 10.1161/CIRCULATIONAHA.109.885194. [DOI] [PubMed] [Google Scholar]
- 22.Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther. 2006;79:103–13. doi: 10.1016/j.clpt.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Sofi F, Giusti B, Marcucci R, Gori AM, Abbate R, Gensini GF. Cytochrome P450 2C19*2 polymorphism and cardiovascular recurrences in patients taking clopidogrel: a meta-analysis. Pharmacogenomics J. 2011;11:199–206. doi: 10.1038/tpj.2010.21. [DOI] [PubMed] [Google Scholar]
- 24.Bristol-Myers Squibb/Sanofi Pharmaceuticals Partnership [Internet] Plavix® (clopidogrel bisulfate) tablets: highlights of prescribing information. Available at http://products.sanofi-aventis.us/PLAVIX/PLAVIX.html (last accessed 2 September 2010.
- 25.Myrand SP, Sekiguchi K, Man MZ, Lin X, Tzeng RY, Teng CH, Hee B, Garrett M, Kikkawa H, Lin CY, Eddy SM, Dostalik J, Mount J, Azuma J, Fujio Y, Jang IJ, Shin SG, Bleavins MR, Williams JA, Paulauskis JD, Wilner KD. Pharmacokinetics/genotype associations for major cytochrome P450 enzymes in native and first- and third-generation Japanese populations: comparison with Korean, Chinese, and Caucasian populations. Clin Pharmacol Ther. 2008;84:347–61. doi: 10.1038/sj.clpt.6100482. [DOI] [PubMed] [Google Scholar]
- 26.Man M, Farmen M, Dumaual C, Teng CH, Moser B, Irie S, Noh GJ, Njau R, Close S, Wise S, Hockett R. Genetic variation in metabolizing enzyme and transporter genes: comprehensive assessment in 3 major East Asian subpopulations with comparison to Caucasians and Africans. J Clin Pharmacol. 2010;50:929–40. doi: 10.1177/0091270009355161. [DOI] [PubMed] [Google Scholar]
- 27.Farid NA, McIntosh M, Garofolo F, Wong E, Shwajch A, Kennedy M, Young M, Sarkar P, Kawabata K, Takahashi M, Pang H. Determination of the active and inactive metabolites of prasugrel in human plasma by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2007;21:169–79. doi: 10.1002/rcm.2813. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi M, Pang H, Kawabata K, Farid NA, Kurihara A. Quantitative determination of clopidogrel active metabolite in human plasma by LC-MS/MS. J Pharm Biomed Anal. 2008;48:1219–24. doi: 10.1016/j.jpba.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 29.Weerakkody GJ, Brandt JT, Payne CD, Jakubowski JA, Naganuma H, Winters KJ. Clopidogrel poor responders: an objective definition based on Bayesian classification. Platelets. 2007;18:428–35. doi: 10.1080/09537100701206790. [DOI] [PubMed] [Google Scholar]
- 30.Patti G, Nusca A, Mangiocapra F, Gatto L, D'Ambrosio A, Di Sciascio G. Point-of-care measurement of clopidogrel responsiveness predicts clinical outcome in patients undergoing percutaneous coronary intervention: results of the ARMYDA-PRO (Antiplatelet therapy for Reduction of Myocardial Damage during Angioplasty-Platelet Reactivity Predicts Outcome) study. J Am Coll Cardiol. 2008;52:1128–33. doi: 10.1016/j.jacc.2008.06.038. [DOI] [PubMed] [Google Scholar]
- 31.Bonello L, Paganelli F, Arpin-Bornet M, Auquier P, Sampol J, Dignat-George F, Barragan P, Camoin-Jau L. Vasodilator-stimulated phosphoprotein phosphorylation analysis prior to percutaneous coronary intervention for exclusion of postprocedural major adverse cardiovascular events. J Thromb Haemost. 2007;5:1630–36. doi: 10.1111/j.1538-7836.2007.02609.x. [DOI] [PubMed] [Google Scholar]
- 32.Dumaual C, Miao X, Daly TM, Bruckner C, Njau R, Fu DJ, Close-Kirkwood S, Bauer N, Watanabe N, Hardenbol P, Hockett RD. Comprehensive assessment of metabolic enzyme and transporter genes using the Affymetrix Targeted Genotyping System. Pharmacogenomics. 2007;8:293–305. doi: 10.2217/14622416.8.3.293. [DOI] [PubMed] [Google Scholar]
- 33.Human Cytochrome P450 (CYP) Allele Nomenclature Committee [Internet] Available at http://www.cypalleles.ki.se/ (last accessed 2 September 2010.
- 34.Special report: genotyping for cytochrome P450 polymorphisms to determine drug-metaboliser status. Technol Eval Cent Asses Program Exec Summ. 2004;19:1–2. [PubMed] [Google Scholar]
- 35.Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211–21. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- 36.Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76:391–6. [PubMed] [Google Scholar]
- 37.Small DS, Li YG, Ernest CS, 2nd, April JH, Farid NA, Payne CD, Winters KJ, Rohatagi S, Ni L. Integrated analysis of pharmacokinetic data across multiple clinical pharmacology studies of prasugrel, a new thienopyridine antiplatelet agent. J Clin Pharmacol. 2011;51:321–32. doi: 10.1177/0091270010367429. [DOI] [PubMed] [Google Scholar]
- 38.Giusti B, Gori AM, Marcucci R, Saracini C, Vestrini A, Abbate R. Drug and medical device interactions: stent thrombosis and personalizing clopidogrel therapy. Curr Pharmacogenomics Pers Med. 2010;8:124–38. [Google Scholar]
- 39.Mega JL, Simon T, Anderson JL, Bliden K, Collet JP, Danchin N, Giusti B, Gurbel P, Horne BD, Kastrati A, Montalescot G, Neumann FJ, Shen L, Sibbing D, Steg PG, Trenk D, Wiviott SD, Sabatine MS. CYP2C19 genetic variants and clinical outcomes with clopidogrel: a collaborative meta-analysis. Circulation. 2009;120:S598–9. [Google Scholar]
- 40.Payne CD, Li YG, Brandt JT, Jakubowski JA, Small DS, Farid NA, Salazar DE, Winters KJ. Switching directly to prasugrel from clopidogrel results in greater inhibition of platelet aggregation in aspirin-treated subjects. Platelets. 2008;19:275–81. doi: 10.1080/09537100801891640. [DOI] [PubMed] [Google Scholar]
- 41.Montalescot G, Sideris G, Cohen R, Meuleman C, Bal dit Sollier C, Barthelemy O, Henry P, Lim P, Beygui F, Collet JP, Marshall D, Luo J, Petitjean H, Drouet L. Prasugrel compared with high-dose clopidogrel in acute coronary syndrome. The randomised, double-blind ACAPULCO study. Thromb Haemost. 2010;103:213–23. doi: 10.1160/TH09-07-0482. [DOI] [PubMed] [Google Scholar]
- 42.Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM. TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 43.Farid NA, Payne CD, Winters KJ, Ernest CS, 2nd, Brandt JT, Darstein C, Jakubowski JA, Salazar DE. Cytochrome P450 3A inhibition by ketoconazole affects prasugrel and clopidogrel pharmacokinetics and pharmacodynamics differently. Clin Pharmacol Ther. 2007;81:735–41. doi: 10.1038/sj.clpt.6100139. [DOI] [PubMed] [Google Scholar]