

Abstract
RNA 3′-phosphate cyclase (RtcA) synthesizes RNA 2′,3′ cyclic phosphate ends via three steps: reaction with ATP to form a covalent RtcA-(histidinyl-Nϵ)-AMP intermediate; transfer of adenylate to an RNA 3′-phosphate to form RNA(3′)pp(5′)A; and attack of the vicinal O2′ on the 3′-phosphorus to form a 2′,3′ cyclic phosphate and release AMP. Here we report the crystal structures of RtcA•ATP, RtcA•ATP•Mn2+, and RtcA•ATP•Co2+ substrate complexes and an RtcA•AMP product complex. Together with the structures of RtcA apoenzyme and the covalent RtcA–AMP intermediate, they illuminate the mechanism of nucleotidyl transfer, especially the stereochemical transitions at the AMP phosphate, the critical role of the metal in orienting the PPi leaving group of ATP during step 1, and the protein conformational switches that accompany substrate binding and product release. The octahedral metal complex of RtcA•ATP•Mn2+ includes nonbridging oxygens from each of the ATP phosphates, two waters, and Glu14 as the sole RtcA component. Whereas the RtcA adenylylation step is metal-catalyzed, the subsequent steps in the cyclization pathway are metal-independent.
Keywords: covalent catalysis, RNA modification
The RNA 3′-terminal phosphate cyclase (Rtc) enzymes that catalyze cyclization of an RNA 3′-phosphomonoester comprise an enzyme family with members distributed widely among bacterial, archaeal, and eukaryal taxa (1). Cyclization occurs via a series of three nucleotidyl transfer reactions (2, 3) (Fig. 1A). In the first step, Rtc reacts with ATP and a divalent cation to form a covalent Rtc–AMP intermediate and liberate PPi. The AMP is linked via a P–N bond to an invariant histidine side chain of Rtc (4). In the second step, the adenylate is transferred from Rtc–AMP to the RNA 3′-phosphate terminus to form an activated phosphoanhydride intermediate, RNA(3′)pp(5′)A. In the third step, the terminal ribose 2′-OH attacks the 3′-phosphate of RNA(3′)pp(5′)A to generate an RNA 2′,3′ cyclic phosphate product and release AMP. Rtc enzymes are strongly selective for ATP as the nucleotide substrate; dATP is ineffective (1, 3, 5, 6).
Fig. 1.

RNA 3′-phosphate cyclase: reaction pathway, tertiary structure, and ATP-binding modes. (A) The three chemical steps of the Rtc reaction pathway are shown: (1) Rtc adenylylation; (2) RNA adenylylation; and (3) cyclization. (B) The tertiary structure of E. coli RtcA (from pdb 3KGD) is shown as a ribbon trace with the four component domain modules colored as follows: N-terminal domain 1 in magenta (aa 4–85), domain 2 in yellow (aa 86–177), domain 3 in blue (aa 186–277), and domain 4 in green (aa 178–185 plus 278–339). Domains 1, 2, and 4 comprise a single globular unit with pseudo-threefold symmetry, into which domain 3 is inserted. (C and D) Stereo views of electron density maps of the active sites of the RtcA•ATP binary complex (C) and the RtcA•ATP•Mn2+ ternary complex (D). The gray meshes denotes 2Fo-Fc density of the refined models, contoured at 1.3σ. Amino acids and ATP are shown as stick models with beige and gray carbons, respectively. Waters are depicted as red spheres. The red mesh in (C) is an omit map (Fo-Fc) of the ATP density contoured at 3.5σ. The cyan mesh in (D) is the anomalous difference density for the manganese ion (cyan sphere) contoured at 8σ. Contacts to the ligands in the octahedral metal coordination complex are denoted by dashed lines in (D).
Crystal structures of the Escherichia coli cyclase RtcA highlighted a distinctive fold composed of four tandem modules, each comprising a four-stranded β sheet overlying two α helices (7, 8) (Fig. 1B). Domains 1, 2, and 4 are homologous to one other and arranged in a radial pattern with pseudo-threefold symmetry. Domain 3, which has a different fold, is inserted en bloc between the β1 strand and α1 helix of domain 4. The essential His309 nucleophile is located within domain 4. The crystal structure of the covalent RtcA–AMP intermediate (8) revealed how the adenylate is linked via a phosphoamide bond to the His309 Nϵ atom and provided insights to substrate specificity, whereby: (i) a network of hydrogen bonds by Asp287 and Gln288 to the ribose O2′ and O3′ accounts for the stringent ribonucleotide preference; and (ii) the adenine base is sandwiched in a hydrophobic pocket between Tyr284 and Pro131 and the preference for adenine is enforced by Phe135, which packs against the purine C2 edge.
Neither of the available RtcA structures contains a metal ion, which leaves open the question of how, and at which steps, the metal contributes to catalysis of cyclization. The apoRtcA and RtcA–AMP crystal structures contained a citrate anion and two sulfate anions, respectively, docked in the N-terminal domain module via electrostatic and hydrogen bonding interactions with conserved amino acid side chains. It was speculated that the citrate and sulfates might plausibly mimic either the 3′-terminal and penultimate phosphates of RNA or the pyrophosphate product of the RtcA adenylylation reaction (8, 9).
The unique 3′ end-modification pathway of the Rtc enzymes and the prominence of cyclic ends in RNA repair reactions (10–13) prompted us to extend the structural and mechanistic framework described above, by capturing additional crystal structures of RtcA at discrete steps along the cyclization reaction pathway. Here we present the structures of RtcA•ATP complexes, with and without a divalent cation cofactor, and an RtcA•AMP product complex, which provide unique insights to the mechanism and stereochemistry of nucleotidyl transfer, the contributions of metal-assisted catalysis exclusively to the RtcA adenylylation step, and the protein conformational switches that accompany substrate binding and product release.
Results and Discussion
Crystallization and Structure Determination of an RtcA•ATP Complex.
RtcA purified from E. coli comprises a mixture of apoRtcA and RtcA–AMP. RtcA crystallizes readily, either as a Cys308 disulfide-bridged apoRtcA homodimer (7) or as an RtcA–AMP monomer (8). We initially attempted to obtain RtcA•ATP crystals by: (i) soaking preformed apoRtcA crystals in a solution containing ATP; or (ii) regenerating ATP in situ by soaking preformed RtcA–AMP crystals in a solution containing magnesium and pyrophosphate. Neither maneuver was successful. We reasoned that the “closed” active site conformation characteristic of the Cys308-crosslinked apoRtcA homodimer (7, 8) might sterically hinder ATP binding to the preformed crystals. In an effort to bias the crystallization outcome, we mutated Cys308 to serine to avoid disulfide crosslinking and also mutated the His309 nucleophile to asparagine, which precludes covalent RtcA adenylylation. We then modified the RtcA purification procedure such that the Ni-agarose eluate fraction of His10RtcA was adjusted immediately to 20 mM ATP, and we included 10 mM ATP in all subsequent chromatography buffers. RtcA•ATP crystals in space group P21212 and one RtcA protomer in the asymmetric unit were grown by vapor diffusion against a precipitant solution containing ammonium sulfate and PEG5000-MME. The structure was determined by molecular replacement using as a search model the RtcA–AMP structure [from Protein Data Bank (PDB) ID code 3KGD] from which AMP and His309 had been subtracted. The final refined RtcA•ATP structure at 1.58 Å resolution had R/Rfree values of 0.200/0.253 and excellent geometry (Table S1).
The omit difference density maps revealed the presence of ATP in the active site (Fig. 1C). The adenosine nucleoside is in anti- conformation with the adenine base sandwiched between Tyr284, with which it forms a π stack, and Pro131, with which it makes van der Waals contacts. Multiple hydrogen bonds engage the ATP ribose O2′ and O3′ atoms (Fig. 2A). Asp287 accepts bidentate hydrogen bonds to its carboxylate oxygens from the ribose 2′-OH and 3′-OH groups. The ribose 2′-OH receives a hydrogen bond from Gln288 Nϵ atom. The ATP α-phosphate is coordinated only by Ser129 Oγ; the Asn309 side chain (in lieu of the native His309 nucleophile) is oriented toward the α-phosphate.
Fig. 2.

Atomic contacts to ATP and divalent cation in the RtcA active site. Stereo views of the active site of the RtcA•ATP complex (A) and the RtcA•ATP•Mn2+ complex (B). The main-chain cartoon traces are colored by domain as in Fig. 1 B. Amino acids and ATP are shown as stick models with beige and gray carbons, respectively. Waters are depicted as red spheres and Mn2+ as a cyan sphere. Atomic contacts are indicated by dashed lines.
The new RtcA•ATP structure reveals an extensive network of electrostatic and hydrogen-bonding contacts between RtcA domain 1 and the ATP β and γ phosphates (Fig. 1C and 2A). The γ-phosphate is coordinated by the Arg21, Arg40, Arg43, Gln51, and His52 side chains and by the main-chain amide of Gly17. The β-phosphate is engaged, directly or via water, by the Arg43 side chain and the Gly16 and Gly17 main-chain amides. The trio of ATP-binding arginines is essential for RNA cyclase activity (9). The essential Glu14 side chain is oriented away from the ATP phosphates, presumably because of electrostatic repulsion.
Structure of an RtcA•ATP•Mn2+ Complex.
A preformed RtcA•ATP crystal that had been soaked in precipitant solution containing 20 mM MnCl2 for 2 h prior to cryoprotection diffracted to 1.85 Å resolution and was virtually isomorphous to the original RtcA•ATP lattice. The refined structural model at 1.85 Å resolution had R/Rfree values of 0.185/0.251 (Table S1). The electron density maps revealed uptake of a single manganese ion in the RtcA active site (colored cyan in Figs. 1D and 2B), defined by the anomalous scattering peak overlying the modeled manganese atom (Fig. 1D). Whereas the position and atomic contacts of the adenosine nucleoside moiety of ATP were unchanged in RtcA•ATP•Mn2+ versus RtcA•ATP (Fig. 2), the binding of the metal elicited significant changes in the positions and atomic contacts of the ATP phosphates that provide key insights to catalysis of adenylate transfer.
The manganese atom is at the center of an octahedral coordination complex that includes three nonbridging phosphate oxygens of ATP (one each from the α, β, and γ phosphates), two waters, and the Oϵ2 atom of the Glu14 side chain (Fig. 1D). Glu14 Oϵ1 coordinates one of the waters in the metal complex. Binding of the metal changes the conformation of the triphosphate moiety from a catalytically unproductive mode seen in the RtcA•ATP active site (Figs. 1C and 2A), in which the Pα-O-Pβ linkage of the PPi leaving group is nearly orthogonal to the attacking nucleophile (aa 309), to a catalytically productive mode in which the Pα-O-Pβ linkage is nearly apical to the 309 side chain (Fig. 1D and 2B). This transition is easily appreciated when the RtcA•ATP and RtcA•ATP•Mn2+ structures are superimposed in Fig. 3B, where the arrow indicates the projected orientation of the His309 side chain. In addition to its clear role in ensuring proper orientation of the PPi leaving group, the metal ion might also stabilize the putative pentacoordinate phosphorane transition state via its atomic contact to the nonbridging oxygen of the α phosphate (Fig. 2B).
Fig. 3.

Snapshots of the RtcA active site at sequential stages along the reaction pathway. Pairwise superpositions of the active sites of apoRtcA and RtcA•ATP (A), RtcA•ATP and RtcA•ATP•Mn2+ (B), RtcA•ATP•Mn2+ and RtcA–AMP ( C), and RtcA–AMP and RtcA•AMP (D). In each image the fold of RtcA in the antecedent state along the reaction pathway is colored gray and its amino acids and nucleotide are rendered with gray carbons. The fold of RtcA in the subsequent state along the reaction pathway is colored by domain and its amino acids and nucleotide are rendered with beige carbons. Mn2+ is depicted as a cyan sphere in (B) and (C).
The atomic contacts of the ATP phosphates are extensively reorganized in response to metal binding. For example, the position of the Pγ atom shifts by 2.2 Å in transition from RtcA•ATP to RtcA•ATP•Mn2+, which results in severance of the direct contacts of the γ-phosphate with Arg21, Gln51, and His52 and their replacement by water-mediated interactions (Fig. 2 A and B). Simultaneously, the γ-phosphate in the metal-bound RtcA gains a pair of bidentate electrostatic interactions with Arg40 and Arg43 (Fig. 2B). The Pβ atom moves 2.8 Å in transition from RtcA•ATP to RtcA•ATP•Mn2+, during which the β-phosphate contacts to Arg43 are lost while new contacts to Mn2+ and Ser129 Oγ are acquired (Fig. 2A and B). The Pα atom moves 1.8 Å, surrenders its contact with Ser129, and gains contacts to Arg21 and Mn2+ (Fig. 2A and B). In addition, the ATP phosphates in RtcA•ATP•Mn2+ receive direct and water-mediated hydrogen bonds from the main-chain amides of Gly16, Gly17, and Gln18 that comprise a P-loop-like element within domain 1 of RtcA. We infer that the active site configuration in the RtcA•ATP•Mn2+ structure (especially the interactions of domain 1 with the triphosphate) reflects that of the Michaelis complex for step 1 catalysis. This view is consistent with available mutational data showing that Glu14, Arg21, Arg40, Arg43, Asp287, and Gln288 (or their equivalents in human Rtc1) are essential for cyclization and Rtc adenylylation (8, 9).
We also determined the 2.1 Å structure of an RtcA•ATP•Co2+ complex formed by soaking preformed RtcA•ATP crystals in a precipitant solution containing 20 mM CoCl2 (Table S1). The presence of a cobalt ion in the RtcA active site was verified by the anomalous scattering peak overlying the modeled metal atom (Fig. S1A). The octahedral Co2+ coordination complex, the conformation of ATP, and the atomic contacts in the active site of RtcA•ATP•Co2+ (Fig. S1B) were nearly identical to those seen in RtcA•ATP•Mn2+.
Structure of an RtcA•AMP Complex.
In an effort to capture a structure of RtcA mimicking the state immediately following the step 1 adenylylation reaction, we tried to crystallize an RtcA•AMP•Mn2+•PPi complex. To obtain AMP-bound RtcA, we modified the purification procedure for RtcA-(C308S-H309N) as follows: (i) the Ni-agarose eluate fraction of RtcA was adjusted immediately to 20 mM AMP; and (ii) 10 mM AMP was present in all subsequent chromatography buffers. The purified RtcA•AMP protein was adjusted to 30 mM manganese and 15 mM pyrophosphate and incubated for 1 h before mixing with precipitants. Crystals that grew by vapor diffusion against a solution containing ammonium sulfate and PEG5000-MME were in space group P21 (a different lattice than that seen for RtcA•ATP) and contained two RtcA protomers in the asymmetric unit. The RtcA•AMP structure was determined by molecular replacement and refined at 1.9 Å resolution with R/Rfree values of 0.180/0.233 (Table S1). The active site electron density confirmed the presence of AMP (Fig. S2), but there was no evidence of manganese or PPi in the active site. The position and atomic contacts of the adenosine nucleoside moiety in RtcA•AMP were similar to those of the covalent RtcA–AMP intermediate (Fig. 4D) and of the RtcA•ATP and RtcA•ATP•metal complexes. No direct contacts between RtcA and the AMP phosphate were apparent in the RtcA•AMP complex.
Fig. 4.

Protein conformational changes accompanying substrate binding and product release. (A) Stereo view of a superposition of apoRtcA, (colored gray) and RtcA•ATP (colored by domain) highlighting inward movements of domain 1 (denoted by arrows) in response to ATP binding. (B) Differences in main-chain Cα positions (Å) of apoRtcA and RtcA•ATP (Upper) and RtcA•ATP and RtcA•ATP•Mn2+ (Lower) are plotted for each amino acid in the primary structure. (C) Stereo view of a superposition of RtcA•ATP•Mn2+ (colored gray) and RtcA–AMP (colored by domain) highlighting outward movements of domain 1 (denoted by arrows) accompanying release of PPi and Mn2+. (D) Differences in main-chain Cα positions (Å) of RtcA•ATP•Mn2+ and RtcA–AMP (Upper) and RtcA–AMP and RtcA•AMP (Lower) are plotted for each amino acid in the primary structure.
Consistent with the absence of a metal, the Glu14 side chain moved away from the AMP phosphate, compared to its location in RtcA•ATP•Mn2+. The RtcA•AMP active site contains a sulfate anion bound to domain 1 via a bidentate salt bridge to Arg43, a hydrogen bond from the main-chain amide of Glu14, and a water-mediated contact to Arg40 (Fig. S2). Because the third step in the cyclization reaction liberates AMP as a reaction product, we regard the RtcA•AMP structure as a mimetic of the final postcyclization product complex.
Comparison of RtcA Structures Yields Insights to the Stereochemistry of Adenylate Transfer.
The ensemble of RtcA structures now captures multiple states along the reaction pathway, which are superimposed in pairwise sequence in Fig. 3. The comparison of apoRtcA and RtcA•ATP (Fig. 3A) underscores the need for active site remodeling prior to ATP binding, insofar as the positions and orientations of many of the amino acid side chains that bind ATP in the RtcA•ATP complex differ substantially from their conformations in the apoRtcA structure. In particular, Tyr284, which stacks on the ATP adenine, adopts a different rotamer in apoRtcA so that the phenol ring overlaps the ATP adenosine (Fig. 3A). Pro131 in apoRtcA also intrudes on the space occupied by adenine in RtcA•ATP. Consequently, the adenosine-binding site is effectively occluded in the apoenzyme. The superposition indicates that the “closed” adenosine pocket seen in apoRtcA transits to a more “open” conformation by reorientation of Tyr284 and outward movement of the domain 2 loop that includes Pro131. As noted above, ATP per se binds to the RtcA active site in a catalytically unproductive conformation that is transformed into a productive one by manganese-induced reorganization of the PPi leaving group to an apical position for in-line attack by His309 on the α-phosphate (Fig. 3B). The superposition of the RtcA•ATP•Mn2+ and RtcA–AMP structures in Fig. 3C highlights the stereochemical inversion of the α-phosphate during the adenylyltransferase reaction, consistent with a single-step in-line mechanism. By contrast, the superposition of the RtcA–AMP intermediate and the RtcA•AMP product structures in Fig. 3D shows that the configurations of the AMP phosphates are identical, which is in keeping with the occurrence of two nucleotidyl transfer steps and an RNA3′ppA intermediate (2) in the transition between these two structures, each of which is predicted to entail inversion of stereochemistry at the AMP phosphate, with the net result being retention of phosphate configuration, as observed.
RtcA Domain Movements Accompany Substrate Binding and Product Release.
Whereas the β-sheet scaffolds of the four domains of RtcA are virtually identical in apoRtcA, RtcA•ATP, RtcA•ATP•Mn2+, RtcA–AMP, and RtcA•AMP, the pairwise comparisons of sequential states along the reaction pathway illuminates substantial movements of α-helices and connecting loops (reflected in Cα position changes; plotted in Fig. 4B and D) that correlate with ATP binding (Fig. 4A) and with formation of the covalent RtcA–AMP adduct and release of Mn2+ and PPi (Fig. 4C).
Focusing in Fig. 4A on the transition from apoRtcA (colored gray) to RtcA•ATP (colored by domain), we see that a 3.5 Å movement of a domain 2 loop (aa 129–131) served, together with side chain conformational changes (discussed above), to open up an otherwise obstructed adenosine-binding pocket. Most notable was that domain 1 also experienced a significant movement (highlighted by arrows in Fig. 4A) that brought the N-terminal segments of the two α-helices and the β1-α1 and β2-α2 loops closer to domains 2 and 4, and thereby established the extensive network of atomic contacts between domain 1 and the ATP β and γ phosphates seen in the RtcA•ATP structure. In effect, the binding of ATP triggers closure of domain 1 around the ATP phosphates. By contrast, the uptake of manganese by the bound ATP results in minimal protein main-chain movements, as reflected in the relatively flat topography of the Cα shift plot for the transition from RtcA•ATP to RtcA•ATP•Mn2+ (Fig. 4B, Lower). As discussed above, the primary consequences of metal binding are changes in the positions and contacts of the ATP phosphates and the reorientation of the Glu14 side chain.
The second instance of significant domain 1 movement occurred in the transition from RtcA•ATP•Mn2+ (colored gray in Fig. 4C) to RtcA–AMP (colored by domain in Fig. 4C), whereby the N-terminal segments of the two α-helices and the β1-α1 and β2-α2 loops retreat away from the bound AMP and domains 2 and 4 (see the arrows in Fig. 4C). In particular, the β1-α1 loop, which includes the metal ligand Glu14 and the P-loop like main-chain amides that coordinate the ATP phosphates, undergoes a significant conformational rearrangement (Fig. 4C) and approximately a 6 Å shift (Fig. 4D, Upper) when the α-β phosphoanhydride bond of ATP is severed. Thus, RtcA reverts to an open domain conformation after step 1 chemistry and release of the PPi leaving group (along with manganese). We presume that an open conformation facilitates access of the RNA 3′-phosphate substrate to the active site, in preparation for catalysis of step 2 of the cyclase pathway. Note that there are minimal positional differences between the RtcA–AMP and RtcA•AMP structures (Fig. 4D, Lower), suggesting that RtcA domain opening also accompanies release of the cyclized RNA product during the third step of the pathway.
RNA Cyclization by RtcA–AMP is Metal-Independent.
The RtcA crystal structures implicate an intact ATP triphosphate moiety and Glu14 as the critical factors in the binding of the metal to form the step 1 Michaelis complex, consistent with the requirement for a metal (either Mg2+ or Mn2+ in the case of RtcA) and Glu14 for RNA cyclization and step 1 enzyme adenylylation (5, 9). However, it is not known whether the second and third chemical steps of the cyclization pathway are also metal-dependent. Indeed, our findings that Mn2+ is not assimilated in the active site of RtcA•AMP crystals grown in the presence of Mn2+ and PPi, and that metals are not taken up when preformed RtcA–AMP crystals are soaked in buffers containing 10 mM Mg2+ or Mn2+ (not shown), raise the prospect that metal-assisted catalysis might be specific to the first step. To address this issue, we assayed RNA cyclization by preformed RtcA–AMP in reaction mixtures lacking exogenous ATP, either in the presence of Mn2+ or in the absence of added metal in reaction mixtures containing EDTA. The RNA 3′-phosphate substrate was a 20-mer oligonucleotide labeled with 32P at the penultimate phosphate (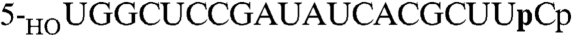 ). The reaction products were digested with RNase T1 prior to their analysis by PAGE. RNase T1 incised the RNAp substrate and cyclized RNA > p product 3′ of the most distal guanosine to yield 32P-labeled tetranucleotides—
). The reaction products were digested with RNase T1 prior to their analysis by PAGE. RNase T1 incised the RNAp substrate and cyclized RNA > p product 3′ of the most distal guanosine to yield 32P-labeled tetranucleotides—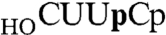 or
or 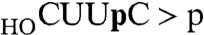 , respectively—with distinct electrophoretic mobilities (Fig. 5, Upper). In the absence of ATP, the cyclization reaction was limited to one round of step 2 + 3 catalysis by the preformed RtcA–AMP in the enzyme preparation. When RtcA was added in 10-fold excess over the RNAp substrate (representing a ∼4-fold excess of RtcA–AMP over substrate), we observed nearly complete conversion of RNAp to the cyclized product (Fig. 5, Upper). The salient findings were that the completeness of cyclization was unaffected by the omission of a divalent cation and inclusion of up to 20 mM EDTA in the reaction mixtures (Fig. 5, Upper).
, respectively—with distinct electrophoretic mobilities (Fig. 5, Upper). In the absence of ATP, the cyclization reaction was limited to one round of step 2 + 3 catalysis by the preformed RtcA–AMP in the enzyme preparation. When RtcA was added in 10-fold excess over the RNAp substrate (representing a ∼4-fold excess of RtcA–AMP over substrate), we observed nearly complete conversion of RNAp to the cyclized product (Fig. 5, Upper). The salient findings were that the completeness of cyclization was unaffected by the omission of a divalent cation and inclusion of up to 20 mM EDTA in the reaction mixtures (Fig. 5, Upper).
Fig. 5.

RNA cyclization by RtcA–AMP is metal-independent. (Upper) Cyclase reaction mixtures (10 μL) containing 50 mM Tris-HCl (pH 7.4), 2 mM DTT, 200 fmol of 32P-labeled 20-mer RNA substrate (prepared as described in SI Materials and Methods and depicted with the radiolabeled phosphate denoted by •), and RtcA, ATP, MnCl2 and/or EDTA as specified were incubated at 37 °C for 30 min. The samples were then digested for 15 min at 37 °C with 1,000 U RNase T1 (Fermentas). The samples were mixed with 10 μL of 90% formamide, 0.01% bromophenol blue/xylene cyanol, 5 mM EDTA and then resolved by electrophoresis (at 50 W constant power) through a 40-cm 20% polyacrylamide gel containing 8 M urea in 45 mM Tris-borate, 1.2 mM EDTA. The 32P-labeled RNAs were visualized by autoradiography of the gel. The positions of the substrate and product T1 fragments are indicated on the left. (Lower) Cyclase reaction mixtures (10 μL) containing 50 mM Tris-HCl (pH 7.4), 2 mM DTT, 200 fmol of 32P-labeled 20-mer RNA substrate, either 5 mM MnCl2 or 10 mM EDTA, and RtcA as specified were incubated at 37 °C for 30 min. Following RNase T1 digestion, the products were resolved by denaturing PAGE. The extents of conversion of the 3′-phosphate substrate into 2′,3′-cyclic phosphate product were quantified by scanning the gel with a Fuji Film BAS-2500 imager and are plotted as a function of input RtcA. Each datum is the average of three independent RtcA titration experiments ± SEM.
The extents of single-turnover RNA cyclization as a function of input RtcA in the presence of Mn2+ versus no metal and 20 mM EDTA are plotted in Fig. 5, Lower. The titration curves were effectively identical; from the slopes of the curves, we calculated that preformed RtcA–AMP comprised ∼25% of the RtcA molecules in the enzyme preparation. These experiments demonstrate that the second and third steps of the RtcA cyclization pathway are metal-independent.
Materials and Methods
Crystallization and Data Collection.
Crystals of RtcA•ATP and Rtc•AMP were grown by the hanging drop vapor diffusion method at room temperature. To obtain ATP-bound RtcA crystals, 2 μL of the RtcA protein sample (4.9 mg/mL, in buffer D with 10 mM ATP; purified as described in SI Materials and Methods) was mixed with an equal volume of the reservoir buffer containing 100 mM 2-(4-morpholino)-ethane sulfonic acid (MES), pH 6.5, 150 mM ammonium sulfate, and 28% (w/v) polyethyleneglycol-(5000)-monomethyl ether (PEG5000-MME). Crystals grew to their full size (approximately 300 × 100 × 50 μm) in 2 d. Single crystals were harvested, cryoprotected by suspension in 100 mM MES, pH 6.5, 10 mM ATP, 200 mM ammonium sulfate, 35% PEG5000-MME, 10% glycerol and then flash-frozen in liquid nitrogen. To obtain RtcA•ATP•Mn2+ and RtcA•ATP•Co2+ crystals, single preformed crystals of RtcA•ATP were soaked for 2 h in reservoir buffer containing 10 mM ATP and 20 mM MnCl2 or CoCl2. The soaked crystals were then harvested and cryoprotected by suspension in 100 mM MES, pH 6.5, 10 mM ATP, 200 mM ammonium sulfate, 35% PEG5000-MME, 10% glycerol, and 20 mM MnCl2 or CoCl2. To obtain the AMP-bound RtcA crystals, the protein sample (4.7 mg/mL, in buffer D with 10 mM AMP; purified as described in SI Materials and Methods) was adjusted to 15 mM sodium pyrophosphate (pH 6.5) and 30 mM MnCl2, then incubated for 1 h at 4 °C prior to mixing with an equal volume (2 μL) of reservoir buffer containing 100 mM MES (pH 6.5), 250 mM ammonium sulfate, and 23% (w/v) PEG5000 MME. Crystals grew to their full size (approximately 400 × 200 × 75 μm) in 3 d. Single crystals were harvested, cryoprotected by suspension in 100 mM MES, pH 6.5, 10 mM AMP, 30 mM MnCl2, 15 mM sodium pyrophosphate, 200 mM ammonium sulfate, 35% PEG5000-MME, 10% glycerol and then flash-frozen in liquid nitrogen.
X-ray diffraction was performed at the National Synchrotron Light Source (Brookhaven, NY) beamline X25 equipped with a Pilatus 6 M detector. Diffraction data for single RtcA•ATP and RtcA•ATP•Mn2+ crystals were collected at a wavelength of 0.9499 Å in 370 0.5° rotation increments in a shutterless mode with 0.5 s exposures per frame. Data for a single RtcA•ATP•Co2+ crystal were collected at a wavelength of 1.6051 Å (the energy of the K-edge adsorption for cobalt) in 730 0.5° rotation increments with 1 s exposures per frame. Data for a single RtcA•AMP crystal were collected at a wavelength of 1.282 Å in 370 0.5° rotation increments with 1 s exposures per frame. All data were processed using MOSFLM and SCALA (14). The diffraction statistics are compiled in Table S1.
Structure Determination and Refinement.
RtcA•ATP crystallized in orthorhombic space group P21212 (a = 69.2 Å, b = 82.7 Å, c = 52.0 Å; α = β = γ = 90°) with one monomer in the asymmetric unit. Phases for the RtcA•ATP structure were obtained via molecular replacement (MR) in PHENIX using the A protomer of the adenylylated RtcA structure (Protein Data Bank code 3KGD) as a search model, after removing the covalent adenylate and the His309 side chain. Because the RtcA•ATP•Mn2+, RtcA•ATP•Co2+, and RtcA•ATP crystals were isomorphous, the MR solution for RtcA•ATP was used as a starting model for the PHENIX.AUTOBUILD procedure for all three structures (15). The RtcA•AMP complex crystallized in monoclinic space group P21 (a = 52.2 Å, b = 79.8 Å, c = 85.7 Å; α = γ = 90°, β = 103.65°) with two monomers in the asymmetric unit. Phases for RtcA•AMP were obtained via a similar MR approach as above, looking for two copies of the search model. This MR solution yielded a starting model for a PHENIX.AUTOBUILD procedure. Following the AUTOBUILD procedure, all structures were iteratively rebuilt manually in COOT (16) and refined using standard procedures in PHENIX (15).
For the RtcA•ATP and RtcA•AMP complexes, initial rounds of refinement excluded nucleotide and solvent. Difference maps from these refinements revealed unambiguous density for all atoms of ATP and AMP, respectively (as depicted by the red mesh contours in Fig. 1C and Fig. S2, respectively). ATP or AMP was fit into the Fo-Fc difference density map and refined using standard stereochemistry with no bond angle restraints.
For the RtcA•ATP•Mn2+ and RtcA•ATP•Co2+ complexes, anomalous difference Fourier maps were calculated using model phases. For the Mn2+ complex, the anomalous map revealed a single peak at the active site corresponding to an octahedrally coordinated metal ion. The anomalous difference Fourier map for RtcA•ATP•Co2+ had three distinct Co2+ peaks, only one of which was at the active site in an octahedral coordination complex identical to that seen for Mn2+. The active sites of the RtcA•ATP•Mn2+ and RtcA•ATP•Co2+ were modeled by placing the nucleotide in the Fo-Fc density and coordinating metal at the position indicated by peak density in the anomalous difference Fourier maps. No Fo-Fc difference density for the pyrophosphate or anomalous difference Fourier peak for Mn2+ was observed in the RtcA•AMP model.
All the models were completed by adding heteroatoms and alternate amino acid conformations as warranted by the difference density maps and then processed further in PHENIX. The refinement and model statistics along with the model contents for all the different complexes are compiled in Table S1. Each modeled RtcA protomer comprised a continuous polypeptide from Met4 to Glu339 (the C-terminal residue).
Supplementary Material
Acknowledgments.
The work was supported by National Institutes of Health Grant GM46330. S.S. is an American Cancer Society Research Professor.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The coordinates for the refined models of RtcA•ATP, RtcA•ATP•Mn2+, RtcA•ATP•Co2+, and RtcA•AMP have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank, www.rcsb.org (PDB ID codes 3TUT, 3TUX, 3TW3, and 3TV1).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115560108/-/DCSupplemental.
References
- 1.Genschik P, Billy E, Swianiewicz M, Filipowicz W. The human RNA 3′-terminal phosphate cyclase is a member of a new family of protein conserved in eukarya, bacteria and archaea. EMBO J. 1997;16:2955–2967. doi: 10.1093/emboj/16.10.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filipowicz W, Strugala K, Konarska M, Shatkin AJ. Cyclization of RNA 3′-terminal phosphate by cyclase from HeLa cells proceeds via formation of N(3′)pp(5′)A activated intermediate. Proc Natl Acad Sci USA. 1985;82:1316–1320. doi: 10.1073/pnas.82.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reinberg D, Arenas J, Hurwitz J. The enzymatic conversion of 3′-phosphate terminated RNA chains to 2′,3′-cyclic phosphate derivatives. J Biol Chem. 1985;260:6068–6097. [PubMed] [Google Scholar]
- 4.Billy E, Hess D, Hofsteenge J, Filipowicz W. Characterization of the adenylation site in the RNA 3′-terminal phosphate cyclase from Escherichia coli. J Biol Chem. 1999;274:34955–34960. doi: 10.1074/jbc.274.49.34955. [DOI] [PubMed] [Google Scholar]
- 5.Genschik P, Drabikowski K, Filipowicz W. Characterization of the Escherichia coli RNA 3′-terminal phosphate cyclase and its σ54-regulated operon. J Biol Chem. 1998;273:25516–25526. doi: 10.1074/jbc.273.39.25516. [DOI] [PubMed] [Google Scholar]
- 6.Vicente O, Filipowicz W. Purification of RNA 3′-terminal phosphate cyclase from HeLa cells: Covalent modification of the enzyme with different nucleotides. Eur J Biochem. 1988;176:431–439. doi: 10.1111/j.1432-1033.1988.tb14300.x. [DOI] [PubMed] [Google Scholar]
- 7.Palm GJ, Billy E, Filipowicz W, Wlodawer A. Crystal structure of RNA 3′-terminal phosphate cyclase, a ubiquitous enzyme with unusual topology. Structure. 2000;8:13–23. doi: 10.1016/s0969-2126(00)00076-9. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka N, Smith P, Shuman S. Structure of the RNA 3′-phosphate cyclase-adenylate intermediate illuminates nucleotide specificity and covalent nucleotidyl transfer. Structure. 2010;18:449–457. doi: 10.1016/j.str.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka N, Shuman S. Structure-activity relationships in human RNA 3′-phosphate cyclase. RNA. 2009;15:1865–1874. doi: 10.1261/rna.1771509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konarska M, Filipowicz W, Gross HJ. RNA ligation via 2′-phosphomonoester, 3′,5′-phosphodiester linkage: requirement of 2′,3′-cyclic phosphate termini and involvement of a 5′-hydroxyl polynucleotide kinase. Proc Natl Acad Sci USA. 1982;79:1474–1478. doi: 10.1073/pnas.79.5.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greer CL, Peebles CL, Gegenheimer P, Abelson J. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell. 1983;32:537–546. doi: 10.1016/0092-8674(83)90473-7. [DOI] [PubMed] [Google Scholar]
- 12.Amitsur M, Levitz R, Kaufman G. Bacteriophage T4 anticodon nuclease, polynucleotide kinase, and RNA ligase reprocess the host lysine tRNA. EMBO J. 1987;6:2499–2503. doi: 10.1002/j.1460-2075.1987.tb02532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka N, Shuman S. RtcB is the RNA ligase component of an Escherichia coli RNA repair operon. J Biol Chem. 2011;286:7727–7731. doi: 10.1074/jbc.C111.219022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 15.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 16.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.