

The prevalence of cutaneous lesions in genetically confirmed adenomatous polyposis coli (APC) mutation carriers was prospectively determined and their potential usefulness in the identification of familial adenomatous polyposis (FAP) patients was assessed. FAP-associated skin lesions were not found to be useful as markers for FAP in clinical practice. However, the study showed that the common congenital hypertrophy of the retinal pigment epithelium–associated region should be extended to APC codons 148-2043.
Keywords: Familial adenomatous polyposis, Cutaneous marker, Extracolonic manifestations, Lipoma
Abstract
Background and Aims.
Benign skin tumors such as lipomas, fibromas, and epidermal cysts are among the extracolonic manifestations of familial adenomatous polyposis (FAP). Readily detectable by inspection, they could serve as presymptomatic diagnostic markers to identify FAP patients. We therefore prospectively determined the prevalence of cutaneous lesions in genetically confirmed adenomatous polyposis coli (APC) mutation carriers and assessed their potential usefulness in the identification of FAP patients.
Methods.
Whole-skin examination was performed in 56 adult APC mutation carriers, compared with a control group (n = 116). In addition, FAP patients were investigated for the presence of congenital hypertrophy of the retinal pigment epithelium (CHRPE), an established clinical marker for FAP, and a detailed review of medical records was performed.
Results.
Nearly half of all FAP patients (48.2%) had at least one FAP-associated skin lesion, compared with one third (34.5%) of controls. Only multiple lipomas and combined skin lesions were significantly more prevalent in APC mutation carriers. CHRPE was observed in 22 (43.1%) of 51 FAP patients, including 14 (37.8%) of 37 individuals with APC mutations outside the CHRPE-associated region between codons 311 and 1465.
Conclusions.
Despite a significantly higher prevalence of multiple lipomas, occurring at younger age, and combined skin lesions in APC mutation carriers, the low diagnostic sensitivity of FAP-associated skin lesions precludes their use as markers for FAP in clinical practice. Based on our findings, the common CHRPE-associated region should be extended to APC codons 148-2043.
Introduction
Familial adenomatous polyposis (FAP) (Online Mendelian Inheritance in Man identifier, OMIM #175100) is an autosomal dominant colorectal cancer predisposition syndrome caused by germline mutations in the adenomatous polyposis coli (APC) gene, which is located on chromosome 5q21 and acts as a tumor suppressor [1–3]. Typically, the disease is characterized by the development of hundreds to thousands of colorectal adenomatous polyps that, if not surgically removed, inevitably progress to colorectal cancer, usually before the age of 40 years [4, 5]. Early medical surveillance of APC mutation carriers coupled with individual surgical interventions has been shown to successfully prevent colorectal cancer [6].
The APC gene has an 8,538-bp open reading frame, with the most common variant spanning 15 exons and encoding a multifunctional protein consisting of 2,843 amino acids. The APC protein is part of the Wnt signaling pathway. The tumor suppressor function of APC relies on its capacity to downregulate intracellular β-catenin levels. Mutant APC lacking this ability leads to β-catenin accumulation that translocates into the nucleus and interacts with members of the T-cell factor/lymphoid enhancement factors family of transcriptional activators involved in cell proliferation and differentiation. In addition, APC participates in several other cellular processes, including cell adhesion, cell cycle regulation, migration, apoptosis, signal transduction, spindle assembly, chromosome segregation, and neuronal differentiation [7].
With regard to the colonic phenotype, a classical and an attenuated variant of FAP have been described. In contrast to the classical form, whereby patients develop ≥100 colorectal polyps, attenuated FAP (AFAP) is characterized by the occurrence of <100 adenomas and, consequently, later onset of polyposis as well as colorectal cancer [8, 9]. An association between AFAP and germline mutations at the 5′ (codons 1–157, 312–412) and 3′ (codons 1596–2843) ends of the APC gene has been described [10].
Extracolonic manifestations of FAP include potentially life-threatening manifestations, such as desmoid tumors, small intestinal adenomas, and carcinomas as well as other, more benign disease symptoms such as osteomas, dental anomalies, various skin lesions, and congenital hypertrophy of the retinal pigment epithelium (CHRPE). CHRPEs constitute pigmented fundus lesions with sharply demarcated borders, which are present at birth. They are more frequent in FAP patients than in the general population and typically occur multiply and/or bilaterally [11–13]. Thus far, FAP-associated CHRPE lesions have only been described in patients with APC germline mutations between codons 311 and 1465 [14].
The triad of polyposis, osteomas, and soft tissue tumors (epithelial cysts, fibromas, desmoid tumors) was initially described as Gardner syndrome [15]. Since the identification of APC gene mutations, Gardner syndrome is commonly considered a clinical variant of FAP [16, 17].
Skin manifestations observed in FAP patients include lipomas, fibromas, epithelial cysts, desmoid tumors, and, more rarely, leiomyomas, neurofibromas, and pilomatricomas [18, 19]. Because they are readily detectable by inspection, skin lesions could serve as diagnostic markers and help to identify, in particular, those FAP patients with de novo APC mutations (up to 25% of all patients). The aim of this study was to systematically and prospectively assess the prevalence of FAP-associated cutaneous lesions and their usefulness as marker lesions in the diagnosis of FAP.
Methods
Patients
From 18 unrelated families, 56 adult FAP patients with confirmed pathogenic germline mutations in the APC gene were enrolled in this study, which was performed from September 2008 to August 2010. Twenty-eight patients were members of the same family (family 1981) with an APC germline mutation in exon 15n (c.5942delA, p.Asn1981fsX62). The remaining patients (n = 28) from 17 unrelated families had germline mutations within exon 4 to 15u (Table 1). The control group consisted of 116 healthy probands who had neither anamnestic evidence for FAP nor a personal history of colorectal cancer. The study was approved by the Ethics Committee of Basel, Switzerland (Basler Studie über familiäre Tumorkrankheiten EK258/05 and EK15/08).
Table 1.
Overview of APC germline mutations present in 56 FAP patients from 18 unrelated families

aBased on the oldest patient examined.
bParents with normal colonoscopy or died at old age, mutational status unknown.
cConfirmed de novo mutation.
Abbreviations: APC, adenomatous polyposis coli; FAP, familial adenomatous polyposis.
Clinical Assessment
A whole-body skin examination was performed with particular attention to FAP-associated cutaneous lesions like lipomas, fibromas, epithelial cysts, and trichilemmal cysts. With regard to lipomas, only superficial (s.c.) lesions were included, because detection of deep lipomas requires further diagnostic procedures like computed tomography, magnetic resonance imaging, and/or biopsy and these only account for a minor fraction (∼1%) of all lipomas [20]. All patients were examined by a dermatologist as well as an ophthalmologist. In addition, a personal medical history was taken and clinicopathological reports were reviewed.
CHRPE status could be assessed by indirect ophthalmoscopy (20 diopter lens) without pupil dilation in 51 of 56 FAP patients. Five patients could not be investigated because of cataract (n = 3) or refusal (n = 2). The Berk classification divides CHRPE lesions into four groups [12, 21–24]. Type A lesions are oval, pigmented, and surrounded by a depigmented halo. They are specific for FAP, whereas type B lesions also occur in the general population. Type B lesions are small, round, and pigmented, and in FAP patients typically occur bilaterally and/or multiply (three or more) (Fig. 1). Type C lesions are round, large, and pigmented, and type D lesions are round, large, atrophic lesions with or without depigmented halo. A lesion is classified as large if it is greater than half a disc diameter.
Figure 1.
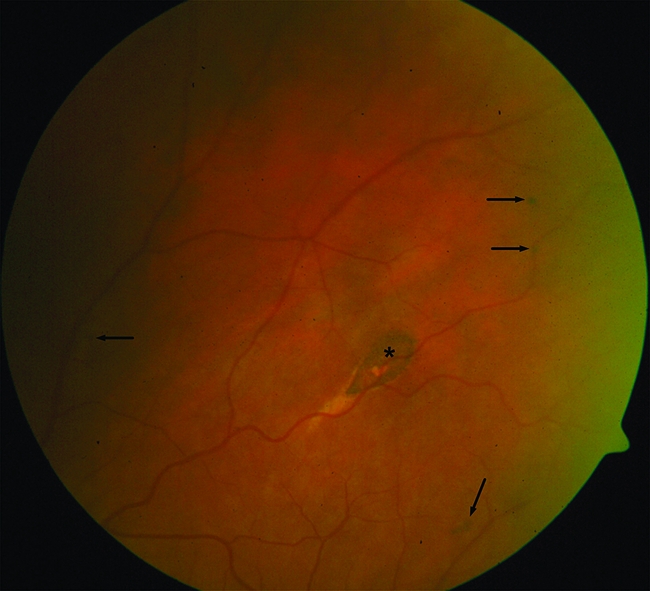
Fundoscopic image of the right eye of patient 44–2008 displaying multiple type B congenital hypertrophy of the retinal pigment epithelium lesions (arrows). A scar is marked by an asterisk.
Statistical Analysis
Only clinical findings present at the time of examination were used for statistical analysis. Anamnestically reported, previously removed lesions were not considered. Statistical comparison of patients' features, encompassing phenotypic characteristics (gender, age at diagnosis, etc.) and mutational status, was done using the χ2 and Fisher's exact tests for categorical variables or Student's t-test for continuous variables, with all probabilities reported as two-tailed p-values, considering p < .05 to be statistically significant. A putative age dependency of cutaneous lesions was analyzed using a simple linear regression model (StatView, version 4.57; Abacus Concepts Inc., Berkeley, CA).
Results
A complete skin examination was performed on 56 genetically confirmed FAP patients (male:female, 29:27; median age, 42.0 years; interquartile range [IQR], 28.0 years) including 28 patients from one large kindred (ID 1981; male:female, 10:18; median age, 45.5 years; IQR, 28.0 years).
Twenty-seven (48.2%) FAP patients presented with FAP-associated cutaneous lesions at the time of the examination (Table 2). Lipomas accounted for the majority of skin lesions (n = 15, 55.5%), followed by fibromas (n = 13, 48.1%), and epidermal cysts (n = 7, 25.9%). No pilomatricomas or leiomyomas were observed. Lipomas were predominantly located on the extremities (59.1%), fibromas were primarily located on the neck (41.7%), and epidermal cysts were primarily located on the upper back (44.4%). Additional, non–FAP-associated skin lesions included two fibrolipomatous hamartomas [25] as well as seven ganglion cysts, with six of these occurring in FAP kindred 1981. Considering members of this family only, a similar prevalence of FAP-associated cutaneous lesions was observed (16 of 28, 57.1%), with lipomas being the most common finding (n = 9, 56.3%), followed by fibromas (n = 7, 43.8%) and epidermal cysts (n = 5, 31.3%) (Fig. 2). The sites of the lesions were similar to those of the overall FAP group (data not shown).
Table 2.
Prevalence of FAP-specific skin lesions in FAP patients and control group

Abbreviations: CI, confidence interval; FAP, familial adenomatous polyposis; NS, not significant.
Figure 2.

Extract from familial adenomatous polyposis kindred 1981 depicting intrafamilial variability with regard to the occurrence of skin lesions and congenital hypertrophy of the retinal pigment epithelium (CHRPE). Symbol description: upper right quadrant, lipoma; lower right quadrant, fibroma; lower left quadrant, epidermal cyst; upper left quadrant, FAP-associated CHRPE. Superscript +, adenomatous polyposis coli mutation carrier; superscript −, noncarrier. Polyposis status is not depicted.
In parallel, a consecutive series of 116 individuals, all without anamnestic evidence for FAP and without a personal history of colorectal cancer, had a complete skin examination (male:female, 36:80; median age, 49.0 years; IQR, 25.0 years). Compared with FAP patients, the control group was significantly older (on average, 5.7 years; p = .037) and consisted of more female probands (69.0% versus 48.2%; p = .009). In general, FAP-associated skin lesions were less frequent among controls (34.5% versus 48.2%; p = .965). Lesion-specific comparison demonstrated that only lipomas, in contrast to fibromas (17.2% versus 23.2%; p = .41) and epidermal cysts (9.5% versus 12.5%; p = .598), were statistically significantly less prevalent in the control group (10.3% versus 26.8%; p = .008) (Table 2).
When subdivided according to the number of lesions, the difference observed between FAP patients and controls only remained significant for those with two or more (multiple) lesions at the time of the examination (n = 14, 25.0% versus n = 9, 7.8%; p = .003) (Table 2).
Among these, the occurrence of multiple lipomas alone as well as the lipoma-fibroma combination were significantly more frequently observed in FAP patients (p = .020 and p = .039, respectively).
Lipomas, fibromas, and epidermal cysts occurred regardless of type and site of the APC germline mutation and were equally frequent in men and women (data not shown). Furthermore, no association with type of colon polyposis was observed (classical, 64.7%; attenuated, 62.5%; p = .9).
Patients with FAP-associated skin lesions at the time of the examination were significantly older (median age, 53.0 years; IQR, 23.0 years) than those without lesions (median age, 35.5 years; IQR, 18.5 years; p = .0074), which was similar for the control group (median age, 54.0 years; IQR, 20.0 years versus median age, 46.0 years; IQR, 33.0 years; p = .032). Subdividing according to type of skin lesion, the observed age difference remained only significant for lipomas in both the FAP and control groups (FAP, 47.0 years versus 38.0 years; p = .01; controls, 62.0 years versus 48.0 years; p = .04). Split according to 10 year-age class, lipomas were, in general, more prevalent among FAP patients, in particular in the age 20–49 years group (p = 0.02) (Fig. 3).
Figure 3.

Lipoma frequency split according to 10-year age class.
Abbreviation: FAP, familial adenomatous polyposis.
Anamnestically, 16 (28.6%) FAP patients reported cutaneous lesions (seven lipomas, two fibromas, 10 epidermal cysts) that had been removed previously, in four (25.0%) of them before the age of 20 years.
To assess the usefulness of FAP-associated skin lesions in the identification of FAP patients, we determined their diagnostic accuracy (Table 2). In our study, the occurrence of FAP-related cutaneous lesions, overall, did not differ significantly between FAP patients and controls. Lipomas alone had a diagnostic sensitivity of 26.8% (confidence interval [CI], 16.2%–40.5%) and a specificity of 89.7% (CI, 82.8%–94.3%). If only patients with multiple lesions were considered, the sensitivity was lower, at 25.0% (CI, 14.8%–38.6%), with higher specificity of 92.2% (85.4%–96.2%). The specificity further improved when only multiple lipomas (96.6%; CI, 90.8%–98.9%) or a combination of lipomas and fibromas (99.1%; CI, 94.6%–99.9%) were taken into account, albeit with a concomitant lower sensitivity (14.3% and 7.1%, respectively).
Indirect ophthalmoscopic assessment identified FAP-associated CHRPE in 22 (43.1%) of 51 FAP patients examined (Table 3). The most frequent lesions were Berk type B—most were multiple (n = 19, 86.4%) and in one patient two lesions were bilateral (4.5%)—followed by type C and type B/C lesions (n = 1 each, 4.5%). No type A or D lesions were observed. Eight (57.1%) of 14 FAP patients harboring an APC germline mutation within the CHRPE-associated region (codons 311-1465) presented with CHRPE. In addition, these lesions were also observed in 14 (37.8%) of 37 FAP patients with an APC germline mutation either proximal to codon 311 or distal to codon 1465. Notably, CHRPE was present in 10 (35.7%) of 28 family members from kindred 1981, who all carry a 1-bp deletion (c.5942delA, p.Asn1981IlefsX62) in APC that leads to a premature stop at codon 2043. No statistically significant correlations were found among the presence of CHRPE, skin lesions, or the severity of polyposis. Overall, 35 (68.6%) FAP patients exhibited FAP-associated CHRPE and/or skin lesions; the presence of CHRPE and/or lipoma was observed in 30 (58.8%) patients.
Table 3.
Prevalence of FAP-associated CHRPE lesions split by colonic phenotype and FAP-specific skin lesions

a,bCHRPE could not be examined in threea and twob patients.
Abbreviaions: CHRPE, congenital hypertrophy of the retinal pigment epithelium; FAP, familial adenomatous polyposis.
Discussion
Up to 25% of all APC mutations are thought to have occurred de novo [5]. Because these FAP patients only come to medical attention at late disease stages, with 67% of them already presenting with colorectal cancer, they clearly would benefit from early diagnosis. Cutaneous lesions, in particular epidermal cysts, fibromas, and lipomas, have been frequently reported in FAP patients. Because they are readily detectable by inspection without the need for expensive diagnostic tools, they could serve as useful markers for an early diagnosis of FAP.
To date, to the best of our knowledge, no prospective dermatological assessment of FAP-associated skin lesions in APC mutation carriers has ever been carried out. Thus far, published studies relied either on questionnaire-based surveys [26, 27], hospital- or registry-based records [10, 28–31], or both [32]. These methods of data collection, however, are susceptible to introduce reporting and/or ascertainment bias. Here, we conducted a systematic and prospective study performing whole-skin examination in 56 adult genetically confirmed APC mutation carriers and, in parallel, a control group of 116 individuals with neither anamnestic evidence for FAP nor a personal history of colorectal cancer.
Overall, FAP-associated skin lesions were more frequently observed in FAP patients than in controls (48.2% versus 34.5%), although this difference was not statistically significant (Table 2). Our observation that the prevalence of FAP-associated skin lesions increased with age in both the FAP and control groups is in accordance with previously reported data from the literature [33–35]. With regard to specific cutaneous lesions, lipomas were statistically significantly more prevalent in FAP patients (26.8% versus 10.3%). Lipomas are among the most common soft tissue tumors in the general population, and nearly one third of all benign tumors are lipomas [36]. Lipomas are benign lesions composed of mature adipocytes with a sharply demarcated border often defined by a capsule. Histologically, they are similar to normal adipose tissue, but cytogenetic abnormalities have been found in approximately 50%—60% of cutaneous lipomas [37]. Because most of them do not cause discomfort and thus do not come to medical attention, they often remain undetected and no reliable epidemiological data exist [38].
Subdivision into single and multiple lesions showed that the significant difference in lipoma prevalence applied solely to multiple lipomas (14.3% versus 3.4%) and to the lipoma-fibroma combination (7.1% versus 0.8%) (Table 2). Of all FAP patients with lipomas, 53.3% presented with multiple lipomas, compared with 33.3% of controls. Previous studies regarding the general population recorded multiple lipomas in approximately 6.9%–18.0% of patients with lipomas [39, 40]. We assume that the higher proportion of multiple lipomas in our control group is mainly a result of methodological reasons, such as patient recruitment and assessment (prospective versus registry based).
Interestingly, when we subdivided our study group according to 10-year age class, lipomas were consistently up to three times more frequent among FAP patients, with statistical significance, for the age range 20–49 years (Fig. 3). Although we found that lipomas occurred at a younger age and as multiple lesions in FAP patients, as expected by “Schnyder's rule,” they appear unsuitable as screening markers for the identification of an underlying FAP predisposition because of their low diagnostic sensitivity (7.1%–26.8%) [41].
Although this prospective study has its limitations with respect to the number of patients investigated, it clearly has the advantage that the data solely rely on thorough dermatological examination, thus minimizing detection as well as reporting bias. Obviously, prospective studies on larger cohorts of APC mutation carriers are needed to verify these findings, in particular with regard to the lipoma prevalence in the 20–49 years age group. In view of the fact that 25% of our FAP patients reported previously removed FAP-associated skin lesions before the age of 20 years, future studies could benefit from including adolescents in their assessment.
CHRPE is a well-known phenotypic marker of FAP, characterized by the congenital, age-independent occurrence of specific pigmented fundus lesions, with a reported prevalence of 70%–75% [42]. In our study, FAP-related CHRPE lesions could only be identified in 43.1% of APC mutation carriers. This may, in part, be a result of methodological reasons because fundoscopy was performed without pupil dilation, potentially resulting in an underestimation. Nevertheless, 57.1% of FAP patients with an APC germline mutation within the CHRPE-associated region (codons 311-1465) presented with such lesions, which is similar to previous reports [21]. Interestingly, CHRPE was also observed in 14 (37.8%) of 37 FAP patients harboring APC mutations outside the typical region. Thus, our findings challenge the prevailing view that the occurrence of CHRPE is restricted to APC codons 311-1465 [42]. Furthermore, we observed pronounced intrafamilial variability with, on average, 40%–50% of family members presenting with CHRPE, pointing to additional genetic modifiers (Fig. 2).
No significant correlations between the colonic phenotype and single or multiple FAP-associated cutaneous lesions or CHRPE were observed. In contrast to generally accepted genotype–phenotype correlations [42], great variability in the colonic phenotype was observed in kindred 1981, which harbors an APC germline mutation in a region commonly associated with attenuated FAP (p.Asn1981fsX62) [43]. As described before [30], the pronounced inter- and intrafamilial variability in families with 3′ APC mutations makes it difficult to predict the extent of colonic disease in the individual family member. With regard to skin manifestations, affected members of family 1981 showed approximately the same rate of FAP-associated cutaneous lesions as the remaining patients. In our set of FAP patients, we thus found no evidence for an interdependence between the type or site of the APC germline mutation and the occurrence of skin lesions.
Conclusion
To the best of our knowledge, this study is the first to systematically and prospectively assess the type and prevalence of FAP-associated cutaneous lesions in a set of 56 adult APC mutation carriers. Thorough dermatological examination could identify these benign skin tumors in nearly half of all FAP patients. Despite a significantly higher prevalence of multiple lipomas and combined skin lesions in APC mutation carriers, their diagnostic sensitivity was too low to serve as reliable FAP-specific marker lesions in clinical practice. With regard to the occurrence of CHRPE, an established clinical marker for FAP, based on our inter- and intrafamilial observations, we propose to extend the CHRPE-associated region to codons 148-2043. Clearly, additional prospective studies on larger FAP collectives, preferably including adolescent APC mutation carriers, are needed to validate our findings.
Acknowledgments
We thank the patients and families who participated in this study, their respective doctors for contribution of clinical information, Marianne Haeusler for data administration, Hansjakob Mueller for patient recruitment support, and Josef Flammer for support in photographing the macula.
This work was supported by grants from the Krebsliga beider Basel and Oncosuisse.
Author Contributions
Conception/Design: Bettina Burger, Nadja Cattani, Swantje Trueb, Rosaria de Lorenzo, Mauro Albertini, Christoph Itin, Nathalie Schaub, Peter H. Itin, Karl Heinimann, Emanuele Bontognali
Provision of study material or patients: Bettina Burger, Rosaria de Lorenzo, Mauro Albertini, Christoph Itin, Nathalie Schaub, Peter H. Itin, Karl Heinimann, Emanuele Bontognali
Collection and/or assembly of data: Bettina Burger, Nadja Cattani, Swantje Trueb, Rosaria de Lorenzo, Mauro Albertini, Christoph Itin, Nathalie Schaub, Peter H. Itin, Karl Heinimann, Emanuele Bontognali
Data analysis and interpretation: Bettina Burger, Nadja Cattani, Peter H. Itin, Karl Heinimann
Manuscript writing: Bettina Burger, Nadja Cattani, Peter H. Itin, Karl Heinimann
Final approval of manuscript: Bettina Burger, Nadja Cattani, Swantje Trueb, Rosaria de Lorenzo, Mauro Albertini, Christoph Itin, Nathalie Schaub, Peter H. Itin, Karl Heinimann, Emanuele Bontognali
References
- 1.Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 2.Bodmer WF, Bailey CJ, Bodmer J, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987;328:614–616. doi: 10.1038/328614a0. [DOI] [PubMed] [Google Scholar]
- 3.Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 4.Bülow S. Clinical features in familial polyposis coli. Results of the Danish Polyposis Register. Dis Colon Rectum. 1986;29:102–107. doi: 10.1007/BF02555389. [DOI] [PubMed] [Google Scholar]
- 5.Bisgaard ML, Fenger K, Bülow S, et al. Familial adenomatous polyposis (FAP): Frequency, penetrance, and mutation rate. Hum Mutat. 1994;3:121–125. doi: 10.1002/humu.1380030206. [DOI] [PubMed] [Google Scholar]
- 6.Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP) Gut. 2008;57:704–713. doi: 10.1136/gut.2007.136127. [DOI] [PubMed] [Google Scholar]
- 7.Brocardo M, Henderson BR. APC shuttling to the membrane, nucleus and beyond. Trends Cell Biol. 2008;18:587–596. doi: 10.1016/j.tcb.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Davies DR, Armstrong JG, Thakker N, et al. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet. 1995;57:1151–1158. [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch HT, Smyrk T, McGinn T, et al. Attenuated familial adenomatous polyposis (AFAP). A phenotypically and genotypically distinctive variant of FAP. Cancer. 1995;76:2427–2433. doi: 10.1002/1097-0142(19951215)76:12<2427::aid-cncr2820761205>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 10.Nieuwenhuis MH, Bülow S, Björk J, et al. Genotype predicting phenotype in familial adenomatous polyposis: A practical application to the choice of surgery. Dis Colon Rectum. 2009;52:1259–1263. doi: 10.1007/DCR.0b013e3181a0d33b. [DOI] [PubMed] [Google Scholar]
- 11.Caspari R, Olschwang S, Friedl W, et al. Familial adenomatous polyposis: Desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet. 1995;4:337–340. doi: 10.1093/hmg/4.3.337. [DOI] [PubMed] [Google Scholar]
- 12.Olschwang S, Tiret A, Laurent-Puig P, et al. Restriction of ocular fundus lesions to a specific subgroup of APC mutations in adenomatous polyposis coli patients. Cell. 1993;75:959–968. doi: 10.1016/0092-8674(93)90539-3. [DOI] [PubMed] [Google Scholar]
- 13.Coleman P, Barnard NA. Congenital hypertrophy of the retinal pigment epithelium: Prevalence and ocular features in the optometric population. Ophthalmic Physiol Opt. 2007;27:547–555. doi: 10.1111/j.1475-1313.2007.00513.x. [DOI] [PubMed] [Google Scholar]
- 14.Enomoto M, Konishi M, Iwama T, et al. The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. Jpn J Clin Oncol. 2000;30:82–88. doi: 10.1093/jjco/hyd017. [DOI] [PubMed] [Google Scholar]
- 15.Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet. 1953;5:139–147. [PMC free article] [PubMed] [Google Scholar]
- 16.Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- 17.Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101:385–398. doi: 10.1111/j.1572-0241.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 18.Ascari-Raccagni A, Baldari U, Righini MG. Cutaneous symptoms of Gardner's syndrome. J Eur Acad Dermatol Venereol. 1999;12:80–81. doi: 10.1111/j.1468-3083.1999.tb00824.x. [DOI] [PubMed] [Google Scholar]
- 19.Pujol RM, Casanova JM, Egido R, et al. Multiple familial pilomatricomas: A cutaneous marker for Gardner syndrome? Pediatr Dermatol. 1995;12:331–335. doi: 10.1111/j.1525-1470.1995.tb00195.x. [DOI] [PubMed] [Google Scholar]
- 20.Bancroft LW, Kransdorf MJ, Peterson JJ, et al. Benign fatty tumors: Classification, clinical course, imaging appearance, and treatment. Skeletal Radiol. 2006;35:719–733. doi: 10.1007/s00256-006-0189-y. [DOI] [PubMed] [Google Scholar]
- 21.Chen CS, Phillips KD, Grist S, et al. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam Cancer. 2006;5:397–404. doi: 10.1007/s10689-006-0011-y. [DOI] [PubMed] [Google Scholar]
- 22.Tiret A, Parc C. Fundus lesions of adenomatous polyposis. Curr Opin Ophthalmol. 1999;10:168–172. doi: 10.1097/00055735-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Berk T, Cohen Z, McLeod RS, et al. Congenital hypertrophy of the retinal pigment epithelium as a marker for familial adenomatous polyposis. Dis Colon Rectum. 1988;31:253–257. doi: 10.1007/BF02554355. [DOI] [PubMed] [Google Scholar]
- 24.Morton DG, Gibson J, Macdonald F, et al. Role of congenital hypertrophy of the retinal pigment epithelium in the predictive diagnosis of familial adenomatous polyposis. Br J Surg. 1992;79:689–693. doi: 10.1002/bjs.1800790733. [DOI] [PubMed] [Google Scholar]
- 25.Itin PH, Heinimann K, Attenhofer M, et al. Precalcaneal congenital fibrolipomatous hamartomas: Is there a pathogenetic relationship with Gardner syndrome? Eur J Dermatol. 2010;20:246–247. doi: 10.1684/ejd.2010.0888. [DOI] [PubMed] [Google Scholar]
- 26.Walon C, Kartheuser A, Michils G, et al. Novel germline mutations in the APC gene and their phenotypic spectrum in familial adenomatous polyposis kindreds. Hum Genet. 1997;100:601–605. doi: 10.1007/s004390050560. [DOI] [PubMed] [Google Scholar]
- 27.Bisgaard ML, Bülow S. Familial adenomatous polyposis (FAP): Genotype correlation to FAP phenotype with osteomas and sebaceous cysts. Am J Med Genet A. 2006;140:200–204. doi: 10.1002/ajmg.a.31010. [DOI] [PubMed] [Google Scholar]
- 28.Moisio AL, Järvinen H, Peltomäki P. Genetic and clinical characterisation of familial adenomatous polyposis: A population based study. Gut. 2002;50:845–850. doi: 10.1136/gut.50.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinimann K, Müllhaupt B, Weber W, et al. Phenotypic differences in familial adenomatous polyposis based on APC gene mutation status. Gut. 1998;43:675–679. doi: 10.1136/gut.43.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brensinger JD, Laken SJ, Luce MC, et al. Variable phenotype of familial adenomatous polyposis in pedigrees with 3′ mutation in the APC gene. Gut. 1998;43:548–552. doi: 10.1136/gut.43.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giardiello FM, Krush AJ, Petersen GM, et al. Phenotypic variability of familial adenomatous polyposis in 11 unrelated families with identical APC gene mutation. Gastroenterology. 1994;106:1542–1547. doi: 10.1016/0016-5085(94)90408-1. [DOI] [PubMed] [Google Scholar]
- 32.Bertario L, Russo A, Sala P, et al. Genotype and phenotype factors as determinants of desmoid tumors in patients with familial adenomatous polyposis. Int J Cancer. 2001;95:102–107. doi: 10.1002/1097-0215(20010320)95:2<102::aid-ijc1018>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Leppard B, Bussey HJ. Epidermoid cysts, polyposis coli and Gardner's syndrome. Br J Surg. 1975;62:387–393. doi: 10.1002/bjs.1800620515. [DOI] [PubMed] [Google Scholar]
- 34.Kransdorf MJ. Benign soft-tissue tumors in a large referral population: Distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164:395–402. doi: 10.2214/ajr.164.2.7839977. [DOI] [PubMed] [Google Scholar]
- 35.Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52–57. doi: 10.1016/j.joms.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 36.Brenn T. Neoplasm of subcutaneous fat. In: Wolff K, Goldsmith LA, Katz SI, et al., editors. Fitzpatrick's Dermatology in General Medicine. Seventh Edition. New York: McGraw-Hill Professional; 2007. p. 1190. [Google Scholar]
- 37.Dei Tos AP, Dal Cin P. The role of cytogenetics in the classification of soft tissue tumours. Virchows Arch. 1997;431:83–94. doi: 10.1007/s004280050073. [DOI] [PubMed] [Google Scholar]
- 38.Weiss SW, Goldblum JR. Enzinger and Weiss's Soft Tissue Tumors. Fourth Edition. St. Louis, MO: Mosby; 2001. p. 1632. [Google Scholar]
- 39.Myhre-Jensen O. A consecutive 7-year series of 1331 benign soft tissue tumours. Clinicopathologic data. Comparison with sarcomas. Acta Orthop Scand. 1981;52:287–293. doi: 10.3109/17453678109050105. [DOI] [PubMed] [Google Scholar]
- 40.Rydholm A, Berg NO. Size, site and clinical incidence of lipoma. Factors in the differential diagnosis of lipoma and sarcoma. Acta Orthop Scand. 1983;54:929–934. doi: 10.3109/17453678308992936. [DOI] [PubMed] [Google Scholar]
- 41.Jung EG. [“Schnyder's rule” of heritability of skin tumors and its extension to cases of segmental presentation] Hautarzt. 1989;40:761–762. [PubMed] [Google Scholar]
- 42.Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit Rev Oncol Hematol. 2007;61:153–161. doi: 10.1016/j.critrevonc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Plasilova M, Russell AM, Wanner A, et al. Exclusion of an extracolonic disease modifier locus on chromosome 1p33–36 in a large Swiss familial adenomatous polyposis kindred. Eur J Hum Genet. 2004;12:365–371. doi: 10.1038/sj.ejhg.5201157. [DOI] [PubMed] [Google Scholar]
- 44.Scott RJ, van der Luijt R, Spycher M, et al. Novel germline APC gene mutation in a large familial adenomatous polyposis kindred displaying variable phenotypes. Gut. 1995;36:731–736. doi: 10.1136/gut.36.5.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sieber OM, Lamlum H, Crabtree MD, et al. Whole-gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or “multiple” colorectal adenomas. Proc Natl Acad Sci U S A. 2002;99:2954–2958. doi: 10.1073/pnas.042699199. [DOI] [PMC free article] [PubMed] [Google Scholar]