

Abstract
Transcription factors with aberrant activity in disease are promising yet untested targets for therapeutic development, particularly in oncology. Directly inhibiting or activating the function of a transcription factor requires specific disruption or recruitment of protein-protein or protein-DNA interactions. The discovery or design of small molecules that specifically modulate these interactions has thus far proven to be a significant challenge and the protein class is often perceived to be ‘undruggable.’ This review will summarize recent progress in the development of small-molecule probes of transcription factors and provide evidence to challenge the notion that this important protein class is chemically intractable.
Keywords: transcription factor, undruggable, nuclear hormone receptor, latent cytoplasmic transcription factor, resident nuclear factor, translocation, fusion protein
Introduction
Cells use transcription factors to regulate specific gene expression patterns in response to developmental and environmental cues. Transcription factors form specialized multiprotein complexes in a combinatorial fashion to regulate expression at the promoters of genes involved in cellular responses [1]. Transcription factors that become overactive in cancers are promising therapeutic targets as they mediate the disproportionate transcription of genes whose products are required for tumor growth and metastasis. In 2002, James Darnell of Rockefeller University made a highly publicized call for a new transcription factor-based focus in medicine, especially with respect to cancer [2]. He argued that selected transcription factors having increased activity in a large percentage of cancers may serve as the most direct and promising targets for therapeutic development as they are less numerous than upstream signaling enzymes and reside at a focal point in deregulated pathways. Darnell noted the dearth of small molecules that directly modulate transcription factors other than nuclear receptors, which bind to endogenous small-molecule ligands and selected xenobiotics. He acknowledged the challenges associated with developing direct small-molecule modulators of transcription factors. Unlike enzymes, directly modulating the function of a transcription factor requires specific disruption or recruitment of DNA-protein or protein-protein interactions. The discovery or design of small molecules that specifically modulate these interactions has historically proven to be a challenge and the protein class is often perceived to be recalcitrant or ‘undruggable.’ Darnell challenged chemical biologists in his closing statement “Finally, a query might be offered: what is the benefit to medicine in all of the twenty-first century promise of proteomics if we cannot selectively inhibit protein-protein interactions?” Darnell’s challenge is especially significant considering that the human genome encodes 2,000–3,000 transcription factors and this class may represent more than 10% of all genes [3,4]. Genome-wide association and linkage studies are generating a growing list of disease-relevant transcription factors for which relatively little is known about structure or function. Notable examples include THAP1, a regulator of cell proliferation associated with early-onset torsion dystonia [5] and BCL11A, a stage-specific regulator of fetal hemoglobin and emerging target in β-hemoglobin disorders [6]. In the absence of small-molecule probes, several other approaches have been used to modulate transcription factors including antisense oligonucleotides [7], transcription factor decoy oligonucleotides [8], poylamides [9], RNAi approaches [9] and stapled peptides [10]. Whether motivated to develop therapeutics or to develop probes of function, chemical biologists have responded to Darnell’s challenge by rallying around this important and challenging protein class. This review will highlight recent developments involving small molecules that directly modulate selected members of the transcription factor classes Darnell noted as attractive anti-cancer targets- nuclear receptors, resident nuclear factors, latent cytoplasmic transcription factors, and fusion proteins arising from chromosomal translocations [2, 11]. The review will emphasize research published from 2008 onward. The reader is referred to several earlier review articles that provide a comprehensive summary of small molecules modulators of transcription described before 2008 [12–14].
Nuclear Receptors
The human genome contains 48 members of the nuclear receptor (NR) superfamily of transcription factors [15]. NRs regulate gene expression upon binding to a small-molecule ligand such as a vitamin or hormone that modulates interactions with coactivators or corepressors by inducing a conformational change. Drugs that modulate selected NRs have been used clinically for many years and include agonists, antagonists, inverse agonists, and selective receptor modulators. Despite being druggable, challenges and opportunities associated with developing novel small-molecule modulators of NRs remain. First, a number of NRs, called orphan receptors, have no assigned endogenous ligand partner. Several orphan receptors are associated with human disease and considerable effort has been placed on identifying both endogenous ligands and novel synthetic ligands for these NRs [16]. For example, nuclear receptor HNF4α, a regulator of hepatic lipid metabolism implicated in diabetes and atherosclerosis, was recently ‘deorphanized’ when linoleic acid (Figure 1a) was identified as an endogenous ligand using an affinity isolation/mass spectrometry approach [17•]. Salbert and coworkers identified novel direct nitronapthofuran activators of HNF4α using a yeast one-hybrid screen [18•]. The nitronapthofurans bound directly to the ligand binding domain of HNF4α and Compound 5 (Figure 1a) enhanced HNF4α-mediated transcription in HepG2C3A cells whereas lineolic acid does not. Early efforts to develop novel NR modulators focused on the development of ligands that targeted the ligand binding domain and altered receptor conformation such that transactivation was dissociated from transrepression activity [19]. The dissociated ligands typically had an improved therapeutic index relative to classical NR agonists as they were capable of inducing transrepression with little transactivating activity and they were useful tools in studying the role of NRs in inflammation. More recently, several groups have developed direct inhibitors of interactions between NRs and coactivator proteins (CBIs). Guy and colleagues developed β-aminoketones that disrupt steroid receptor coactivator SRC2 binding to the thyroid receptor (TR) with submicromolar IC50s (Figure 1b) [20,21]. Several compounds inhibit binding of estrogen receptor (ER) to SRCs and estrogen-induced transcriptional activity with single-digit micromolar IC50 values, including amphipathic benzenes, guanylhydrazones, and compounds containing a pyrimidine core [22–24]. Katzenellenbogen and coworkers further explored the pyrimidine core using a structure-based peptidomimetic approach to develop several CBIs that selectively target the androgen receptor (AR) interaction with SRC over the ER/SRC interaction [25••]. Small molecules capable of inhibiting AR transcriptional activity and cell proliferation by blocking AR-SRC interactions involving agonist-occupied receptors may be used to treat androgen-independent prostate cancer or patients that have developed resistance to anti-androgen therapies [25••,26]. New approaches to NRs, particularly those involving small molecules that target interactions with other proteins rather than the ligand-binding pocket of the receptor, may lead to novel therapeutics for diseases involving NR malfunctions.
Figure 1.
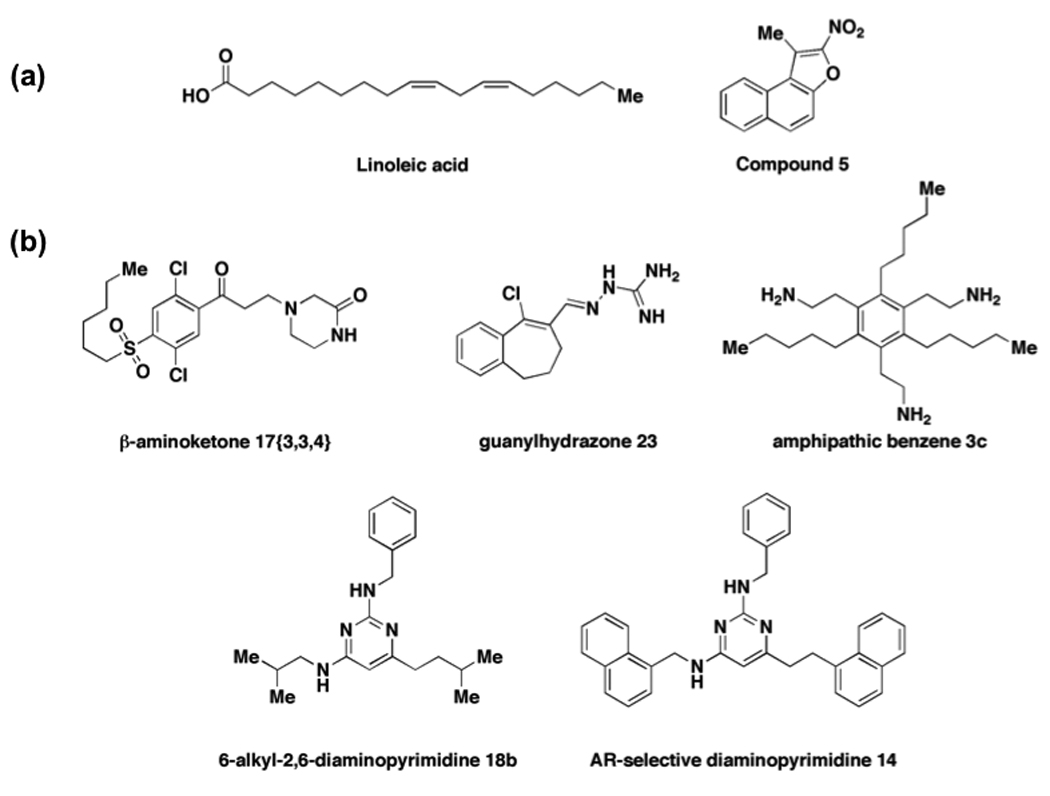
Novel modulators of nuclear receptors. (a) Recently discovered ligands for HNFα include endogenous linoleic acid and synthetic nitronapthofuran compound 5. (b) Coactivator binding inhibitors that target TR-SRC (β-aminoketone 17{3,3,4}), ER-SRC (guanylhydrazone 23, amphipathic benzene 3c, 6-alkyldiaminopyrimidine 18b), and AR-SRC interactions (AR-selective diaminopyrimidine 14).
Resident Nuclear Factors
Resident nuclear factors (RNFs) enter the nucleus upon synthesis where they bind to DNA constitutively and are activated by serine/threonine phosphorylation as the terminal step in signaling cascades [11]. Oncogenic proteins affect serine kinase cascades that terminate in phosphorylation of RNFs, leading to altered transcriptional patterns and cellular phenotypes [2, 11]. RNFs stimulate transcription by associating with a variety of transcription factors and coactivators in multiprotein complexes. Hundreds of proteins fall into this class and most genes are regulated in part by one or more of these transcription factors [11]. Several RNFs are aberrantly overexpressed in cancers including the prolific oncogene c-Myc [2, 14, 27]. Historically, efforts have focused on understanding the pathways that modulate the function of these factors and targeting the signaling enzymes was considered to be a path forward for therapeutic development. Small molecules that restore overactive transcription factors to normal levels of transcription may provide a novel approach toward treating selected cancers. Unlike NRs, these factors are considered to be ‘undruggable’ as they lack ligand binding domains or intrinsic enzymatic activities. Potential strategies for modulation include perturbing DNA-binding capacity or protein-protein interactions with other transcription factors or cofactors. Designing such molecules has proven to be a challenge as many of the protein domains involved in DNA recognition or protein recognition are intrinsically disordered in the absence of interacting partners [14,28]. Increasingly, interactions involving intrinsically disordered (ID) proteins are considered accessible with small molecules [29–31••]. Identifying small molecules that prevent disorder-to-order transitions associated with binding to a partner protein or DNA may trap selected proteins, including many transcription factors, in an inactive state and may represent a general strategy for therapeutic development for ID proteins.
Several RNFs have been successfully modulated directly with small molecules [12–14]. A number of direct inhibitors of c-Myc have been developed in the last decade and have been the subject of a previous review by Berg [14]. One of the first success stories involves the peptide mimetic IIA6B17 (Figure 2a), developed by Vogt and coworkers, that inhibits formation of the c-Myc/Max heterodimer [32] and transcription mediated by c-Myc in a reporter gene assay with an IC50 of 28 µM [33]. These studies paved the way for the design or discovery of new antagonists of c-Myc/Max [31••–43] or c-Myc/Max-DNA interactions [44,45] with comparable or improved potencies in binding assays and cellular studies involving transcription or transformation (Figure 2a). In one example, Prochownik and coworkers executed a high-throughput yeast-two hybrid screen to identify seven compounds that modestly inhibited the c-Myc/Max interaction and HL60 cellular proliferation [38]. They performed subsequent computational studies to prepare improved derivatives of the thioxothiazolidinone 10058-F4 (IC50 = 49 µM) [39,40,42]. Representative derivatives include 28RH-NCN-1 and #764 with IC50s in the HL60 proliferation assay of 29 µM and 4.6 µM respectively [39]. Metallo and coworkers performed NMR studies to identify the residues of Myc that were directly involved in binding to the original seven compounds and identified three distinct binding sites for the molecules in the bHLHZip domain [31••,43]. Importantly, they demonstrated that the compounds can bind independently and simultaneously at all three sites, inducing only local changes in conformation and preserving the overall disorder of c-Myc. These compounds inhibit heterodimer formation with Max as they trap c-Myc in an ID state.
Figure 2.
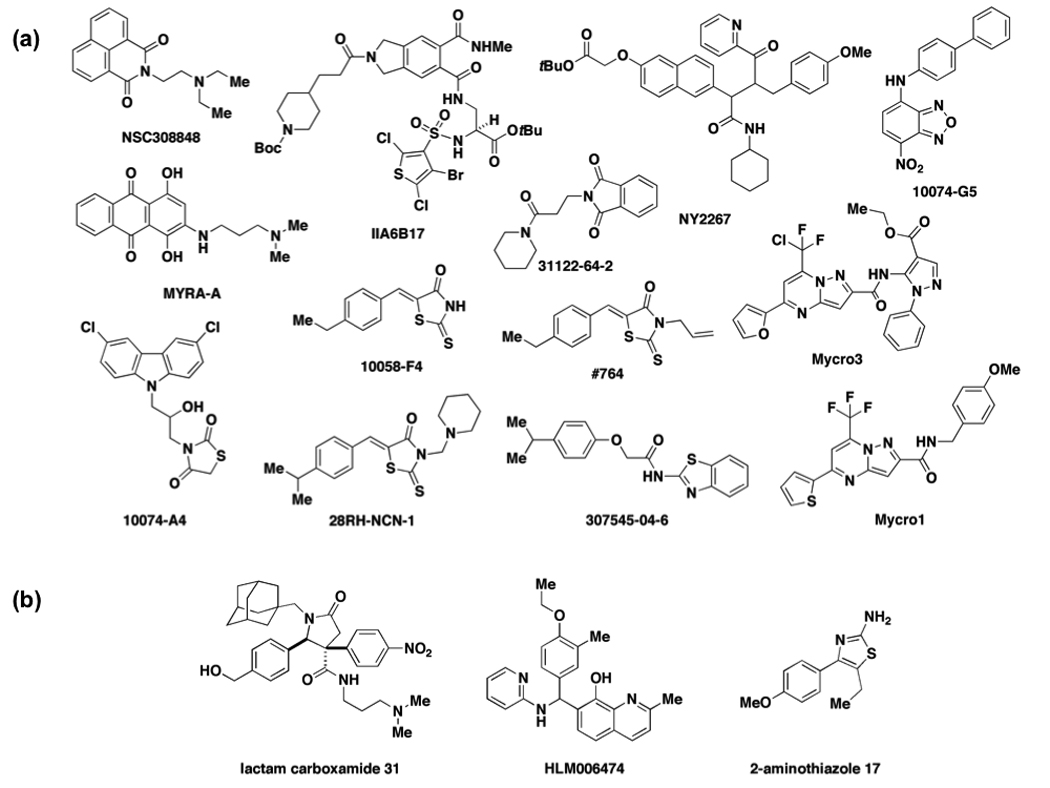
Small-molecule modulators of resident nuclear factors. (a) Selected small-molecule inhibitors of c-Myc function, including inhibitors of c-Myc/Max heterodimer formation and c-Myc/Max DNA binding. (b) Small-molecule modulators of HOXA13 (lactam carboxamide 31), E2F (HLM006474), and CBFB-RUNX2 (2-aminothiazole 17).
As reviewed previously [12–14], small molecules modulate a variety of other RNFs including CREB binding protein (CBP) [46–50] and C/EBPα [50]. These small molecules include compounds that inhibit transcription as well as compounds that mimic the functions of transcriptional activation domains [50]. More recently, Shaw and coworkers described a stereochemically complex lactam carboxamide (Figure 2b) prepared through diversity-oriented synthesis that inhibits binding of HOXA13 to DNA with an IC50 of 6.5 µM and inhibits HOXA13-mediated repression of transcription in a reporter-gene assay [51]. In another example, Bushweller and colleagues used a combined virtual screening/FRET approach to identify 2-aminothiazole compounds that inhibit the interaction between CBFβ and Runx1 with IC50 values below 10 µM [52]. 2-aminothiazole 17 (Figure 2b) exhibited a dose-dependent reduction in proliferation of ME-1 leukemia cells expressing the CBFβ-SMMHC transcription factor translocation fusion required for leukemogenesis. Cress and coworkers also used a virtual screening approach to identify putative small-molecule binders to the E2F4/DP2 heterodimer combined with EMSA assays to identify HLM006474 (Figure 2b) as an inhibitor of intracellular binding of E2F4 to DNA with an IC50 of 29.8 µM [53••]. The compound was shown to increase apoptosis as well as inhibit the proliferation and subsequent invasion of A375 melanocytes into an underlying dermal substrate. Although the compound is not as potent as some of the other inhibitors of transcription factors described in this review, the results provide a proof of principle for E2F blockers as anti-proliferative agents. More work is required to develop small-molecule probes for all members of the class but these early examples demonstrate that RNFs are chemically tractable.
Latent Cytoplasmic Factors
Latent cytoplasmic transcription factors (LCTFs) reside in the cytoplasm in an inactive form until activation is triggered by a cell surface receptor-ligand interaction [11]. A wide variety of mechanisms exist for activating these factors, including phosphorylation by serine or tyrosine kinases at the cell surface (e.g. SMADs, STATs), proteolytic regulation in combination with post-translational modification events (e.g. HIFs, NF-κB, Notch, β-catenin), and secondary messenger signaling combined with phosphorylation events (e.g. NFAT). Upon activation, these transcription factors translocate into the nucleus where they interact with other transcription factors, including RNFs, to regulate transcription.
As reviewed previously [14,54], a number of direct small-molecule inhibitors have been developed for LCTFs such as the STATs [54–64] and HIF-1 [65–72]. Small molecules have been identified that inhibit STAT3 by directly blocking binding to DNA, including the natural product galiellalactone [54] and the platinum (IV) complex IS3 295 [56] (Figure 3a). Several small molecules also inhibit the function of the STAT3 SH2 domain, involved in STAT activation and dimerization (Figure 3a) [57–62]. Structure-based virtual screening approaches lead to the discovery of STA-21 as an inhibitor of STAT3 DNA-binding [57,58] as well as S3I-201 and SF-1-066 as inhibitors of STAT3 dimerization [60], each compound affecting STAT3 function in cells. Berg and coworkers identified Stattic, an inhibitor of STAT3 activation and dimerization, through a high-throughput fluorescence polarization-based screen of more than 17,000 compounds [60]. The compound inhibits STAT3 dimerization in vitro with an apparent IC50 of 5.1 µM and with selectivity over other STAT family members. Stattic also inhibits translocation of STAT3 into the nucleus of HepG2 liver carcinoma cells and induces apoptosis of STAT3-dependent cancer cell lines. Berg and coworkers extended this screening approach to identify chromone-based inhibitors of STAT5b such as nicotinoyl hydrazone 1 (Figure 3a) [64].
Figure 3.
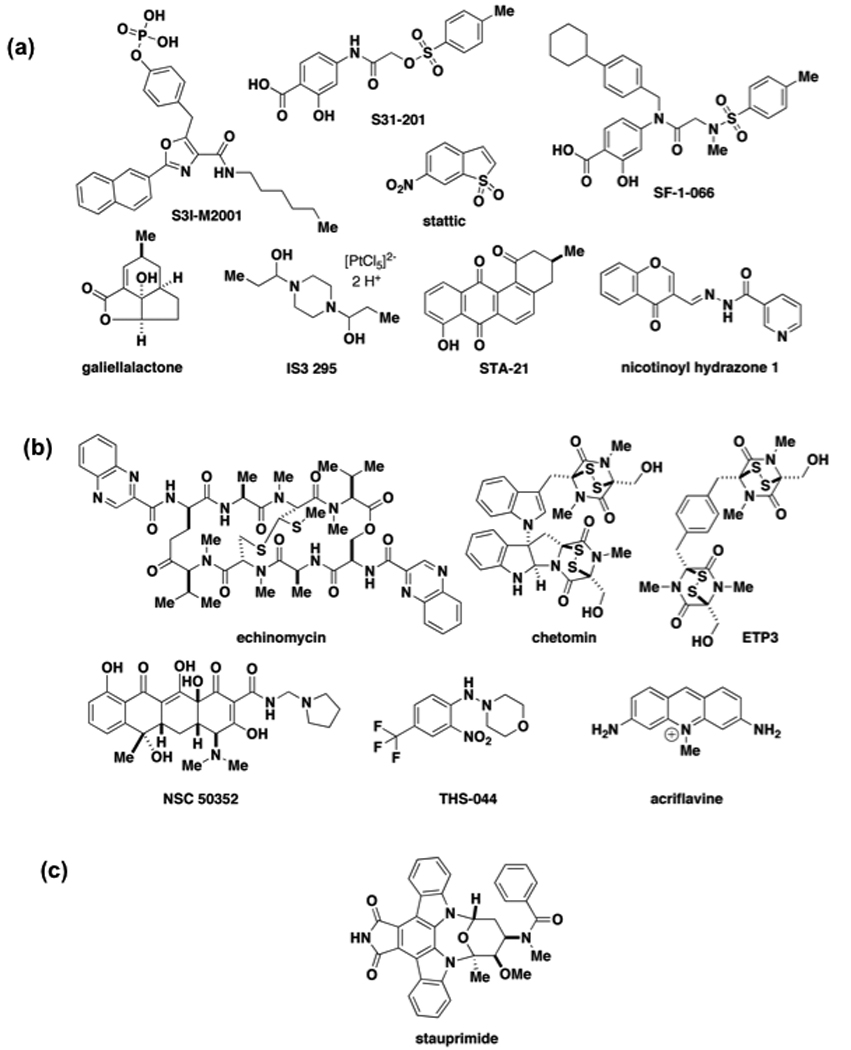
Representative small-molecule modulators of latent cytoplasmic transcription factors. (a) Direct STAT modulators (b) Direct modulators of HIF-1 or p300/CBP (c) Modulator of NME2.
Like the STATs, several compounds have been discovered that target binding of HIF-1 to DNA or HIF-1 dimerization through PAS domains (Figure 3b) [65–72]. The compounds include the natural product echinomycin [66], a potent yet nonspecific intercalator that blocks HIF-1 DNA binding and transcription, and NSC 50253 [67], a modest inhibitor of PAS-A domain interactions involving HIF-1α and HIF-1β. Semenza and coworkers recently reported that acriflavine, a topical antiseptic and dye, binds directly to HIF-1 and HIF-2, inhibits HIF-1 dimerization and transcriptional activity with IC50s around 1 µM, and inhibits tumor xenograft growth [68]. Gardner and colleagues recently reported a crystal structure of the HIF2α PAS-B domain with THS-044, a novel ligand with a KD of 2 µM that was identified using an NMR-based ligand binding assay. The crystal structure reveals a preformed solvent accessible cavity [69] and the same group published a separate study describing a binding-competent open protein conformation [70••]. Although the functional consequences of the ligand in cells is not known, the model may assist in virtual screens or rational design of HIF2 modulators. The epidithiodiketopiperazine (ETP) chetomin [71] was previously reported to inhibit the interaction of HIF-1α with p300/CBP although it was not clear whether the compound targeted the LCTF or the coactivator. In a recent report, Olenyuk and coworkers prepared a dimeric ETP antagonist of the HIF-1α-p300/CBP interaction called ETP3 [72•]. They demonstrated that both chetomin and ETP3 bind to the RNF p300 with submicromolar affinity and induce unfolding of p300 at 10 µM. Chetomin and ETP3 disrupt the HIF-1α-p300/CBP interaction with IC50s of 0.54 µM and 1.5 µM respectively and inhibit HIF-1a inducible promoter activity at submicromolar concentrations [72•].
Few LCTFs beyond the STATs and HIF-1 have been directly modulated with small molecules. In one recent example, Schultz and colleagues describe a small molecule named stauprimide (Figure 3c) that increases the efficiency of directed embryonic stem cell (ESC) differentiation in conjunction with defined extracellular signaling cues [73••]. The compound, which resembles known kinase inhibitors staurosporine and UCN-01, was identified from a collection of 20,000 kinase-biased compounds through a high-content imaging screen for definitive endoderm. Stauprimide inhibited several kinases at 5 µM but it only inhibited Flt3 and MLK1 at 500 nM, a concentration close to the EC50 in the endoderm differentiation assay. Treatment with staurosporine, UCN-01, and Flt3 or MLK inhibitors in the ESC differentiation assays did not increase differentiation. The authors identified NME2, an LCTF that is highly expressed in ESCs and that regulates c-Myc expression, as a direct target of stauprimide using affinity-based target identification methods. shRNA knock down of NME2 increased differentiation efficiency to levels similar to those observed during stauprimide treatment. The authors demonstrated that stauprimide inhibits translocation of NME2 to the nucleus and represses c-Myc expression in ESCs. Targeting NME2 may provide an alternative strategy to restoring aberrantly overactive c-Myc to normal levels in selected cancers. This example illustrates an unbiased approach to discovering a small-molecule modulator of a transcription factor using a phenotypic assay.
Targeting oncogenic translocation fusion proteins
Fusion protein products arising from chromosomal translocations involving transcriptional regulators are attractive targets for therapeutic development. Translocations that give rise to gene fusions play an important role in tumorigenesis [74–76]. A recent review noted that at least 358 gene fusions involving 337 different genes are known and have been described in all main subtypes of human neoplasia, accounting for roughly 20% of human cancer morbidity [76]. More than 50% of acute myeloid leukemia (AML) cases are associated with non-random translocation events and the resulting fusion proteins often contain a transcription factor that retains the DNA-binding motif fused to an unrelated protein that interacts with co-repressor complexes [75]. AML1-ETO is an example of a common transcription factor translocation (TFT) that functions via repression of myeloid development genes and differentiation block [77]. Small molecules that selectively target oncogenic AML1-ETO over the normal full-length AML1 and ETO proteins may provide a novel therapeutic strategy for AML that may be explored in combination with approved drugs such as retinoic acid or vorinostat [75,77]. As with the other transcription factor classes described in this review, TFTs are considered to be ‘undruggable’ proteins and no selective therapeutics for important TFT targets such as AML1-ETO exist.
Toretsky and colleagues recently published a seminal proof of principle study involving the oncogenic fusion protein EWS-FLI1 that arises from the t(11;22) translocation characteristic of Ewing’s sarcoma family tumors (ESFTs) [78••]. The translocation fuses the amino portion of EWS to the carboxy portion of ets family DNA binding protein FLI1 (Figure 4a). Like several of the transcription factors previously discussed, EWS-FLI1 is an intrinsically disordered protein that engages in several protein-protein interactions as part of transcriptional complexes [29]. Toretsky and coworkers reasoned that the flexible TFT protein would have a greater potential for small-molecule binding due to higher induced-fit sampling probabilities and that the sites of interactions with other proteins may serve as binding sites for small molecules. They used a direct binding assay involving surface plasmon resonance to screen 3,000 small molecules leading to the discovery of NSC635473 (Figure 4b) as a direct binder to EWS-FLI1. The compound reduced direct binding of partner protein RNA helicase A (RHA) in vitro. An improved compound, YK-4-279 (Figure 4b), binds to EWS-FLI1 with KD of 9.48 µM and inhibits EWS-FLI1 binding to RHA in vitro and in ESFT cells at 10 µM. YK-4-279 also exhibits dose-dependent inhibition in a luciferase reporter assay, induces apoptosis in ESFT cells, and reduces the growth of ESFT orthotopic xenografts. Current efforts involve medicinal chemistry and pharmacology studies aimed at advancing an improved analog of YK-4-279 into clinical trials. This study is a pioneering example of targeting and modulating oncogenic TFTs in cells.
Figure 4.

Targeting the oncogenic EWS-FLI1 transcription factor translocation product with small molecules. (a) Schematic representation of the EWS-FLI1 fusion protein resulting from the t(22;11) translocation. The RNA binding domain (RBD), ETS DNA binding domains (DBD), and transactivation domains (TAD) are indicated. The fusion gene can vary depending on whether exons 5–9 or 6–9 of FLI1 are involved. (b) Small molecules that disrupt the EWS-FLI1 interaction with RNA helicase A.
Conclusions
Darnell challenged the medical and chemical biology communities to innovate in small-molecule discovery and development aimed at transcription factors, particularly those that become overactive in a variety of cancers [2]. Several examples of small molecules that directly bind and modulate function of transcription factors now exist, including compounds that modulate protein-protein interactions and protein-DNA interactions [12–14]. These compounds come from various sources and include small and flat compounds [18•,52] as well as large and stereochemically complex natural products [66,71] and products of diversity-oriented synthesis [51,79]. Progress has been made for most classes of recalcitrant transcription factors including RNFs, LCTFs, and oncogenic TFTs. Novel approaches to modulating druggable NRs using coactivator binding inhibitors have also been developed. It should be noted that few of the modulators of transcription factors developed to date achieve submicromolar potencies. Many of these early modulators stem from high-throughput screens or in silico screens and may be improved using additional knowledge gained from structure-activity relationship studies, structural biology efforts, and studies aimed at mechanism of action in cells. While these examples demonstrate that transcription factors are indeed chemically tractable, it is important to note that the chemical biology community has yet to develop general and systematic strategies for identifying modulators for any transcription factor of interest. Most of the approaches used to date required knowledge about structure or function in order to develop assays aimed at preventing or disrupting a specific interaction or to execute structure-based virtual screens. New technologies that can be applied to large sets of transcription factors in a general manner will expedite discovery and provide clues about specificity. Following a similar rationale to that provided by Toretsky and coworkers, Koehler et al. used high-throughput and direct binding assays involving small-molecule microarrays (SMMs) to identify a direct modulator of the yeast transcription factor Hap3p in cells known as haptamide [79]. Unbiased SMM binding assays involving purified full-length transcription factors can identify multiple types of probes in a single screen, including compounds that target DNA binding domains and ligand binding domains. Assay positives may inhibit any number of interactions with partner proteins or DNA either directly or through allosteric influence of protein conformation. The SMM approach offers unprecedented scope and throughput for this target class, allowing screens of various types of factors, including all of the classes described in this review, against a common collection of diverse compounds using essentially the same protocol [80]. Koehler and coworkers recently undertook a screen involving 100 structurally and functionally diverse transcription factors using SMMs containing roughly 20,000 small molecules. Details about this screen will be provided in a subsequent publication and we hope this effort increases the number of direct ligands to transcription factors dramatically. Through the efforts of many labs around the globe, the chemical biology community has already demonstrated that transcription factors are chemically tractable. By escalating this community effort, we may develop a small-molecule toolkit to comprehensively study transcriptional regulation and rise to Darnell’s challenge of developing therapeutics against overactive, cancer-specific transcription.
Acknowledgements
Dr. Arturo J. Vegas is thanked for critical review of this manuscript. Funding from the US National Cancer Institute’s Initiative for Chemical Genetics (N01-CO-12400) is gratefully acknowledged. The content of this publication does not necessarily reflect the views or policies of the US Department of Health and Human Service, nor does the mention of trade names, commercial products or organizations imply endorsement by the US government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of the review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Marr MT, 2nd, Isogai Y, Wright KJ, Tjian R. Coactivator cross-talk specifies transcriptional output. Genes Dev. 2006;20:1458–1469. doi: 10.1101/gad.1418806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 3.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA. Structure and evolution of transcriptional regulatory networks. Curr Opin Struct Biol. 2004;14:283–291. doi: 10.1016/j.sbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Erlich ME, Bressman SB, Ozelius LJ. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- 6.Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HKA, Hirschhorn JN, Cantor AB, Orkin SH. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 7.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 8.Mann MJ, Dzau VJ. Therapeutic applications of transcription factor decoy oligonucleotides. J Clin Invest. 2000;106:1071–1075. doi: 10.1172/JCI11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nickols NG, Jacobs CS, Farkas ME, Dervan PB. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem Biol. 2007;2:561–571. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brivanlou AH, Darnell JE., Jr Signal transduction and the control of gene expression. Science. 2002;295:813–818. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- 12.Majmudar CY, Mapp AK. Chemical approaches to transcriptional regulation. Curr Opin Chem Biol. 2005;9:467–474. doi: 10.1016/j.cbpa.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 13.Arndt H-D. Small molecule modulators of transcription. Angew Chem Int Ed Engl. 2006;45:4552–4560. doi: 10.1002/anie.200600285. [DOI] [PubMed] [Google Scholar]
- 14.Berg T. Inhibition of transcription factors with small organic molecules. Curr Opin Chem Biol. 2008;12:464–471. doi: 10.1016/j.cbpa.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Burch PE, Cooney AJ, Lanz RB, Pereira FA, Wu J, Gibbs RA, Weinstock G, Wheeler DA. Genomic analysis of the nuclear hormone receptor family: new insights into structure, regulation, and evolution from the rate genome. Genome Res. 2004;14:580–590. doi: 10.1101/gr.2160004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y. Orphan nuclear receptors in drug discovery. Drug Discov Today. 2007;12:440–445. doi: 10.1016/j.drudis.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuan X, Ta TC, Lin M, Evans JR, Dong Y, Bolotin E, Sherman MA, Forman BM, Sladek FM. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One. 2009;4:e5609. doi: 10.1371/journal.pone.0005609. • The authors describe a general strategy involving affinity isolation/mass spectrometry to identify endogenous ligands for orphan receptors and other lipid binding proteins under physiologically relevant conditions.
- 18. Le Guével R, Oger F, Lecorgne A, Dudasova Z, Chevance S, Bondon A, Barath P, Simonneaux G, Salbert G. Identification of small molecule regulators of the nuclear receptor HNF4α based on napthofuran scaffolds. Bioorg Med Chem. 2009;17:7021–7030. doi: 10.1016/j.bmc.2009.07.079. • The authors identified novel activators of the nuclear receptor HNFαincluding a compound that interacts directly with the ligand binding domain of the transcription factor.
- 19.Schäcke H, Schottelius A, Döcke W-D, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA. 2004;101:227–232. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnold LA, Estébanez-Perpiñá E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J Biol Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 21.Hwang JY, Arnold LA, Zhu F, Kosinski A, Mangano TJ, Setola V, Roth BL, Guy RK. Improvement of pharmacological properties of irreversible thyroid receptor coactivator binding inhibitors. J Med Chem. 2009;52:3892–3901. doi: 10.1021/jm9002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunther JR, Moore TW, Collins ML, Katzenellenbogen JA. Amphipathic benzenes are designed inhibitors of the estrogen receptor alpha/steroid receptor coactivator interaction. ACS Chem Biol. 2008;3:282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LaFrate AL, Gunther JR, Carlson KE, Katzenellenbogen JA. Synthesis and biological evaluation of guanylhydrazone coactivator binding inhibitors for the estrogen receptor. Bioorg Med Chem. 2008;16:10075–10084. doi: 10.1016/j.bmc.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parent AA, Gunther JR, Katzenellenbogen JA. Blocking estrogen signaling after the hormone: pyrimidine-core inhibitors of estrogen receptor-coactivator binding. J Med Chem. 2008;51:6512–6530. doi: 10.1021/jm800698b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gunther JR, Parent AA, Katzenellenbogen JA. Alternative inhibition of androgen receptor signaling: peptidomimetic pyrimidines as direct androgen receptor/coactivator disruptors. ACS Chem Biol. 2009;4:435–440. doi: 10.1021/cb900043e. •• The authors describe the development of compounds that selectively disrupt interactions between the androgen receptor and steroid receptor coactivator and that inhibit transcriptional activity in LNCaP androgen-sensitive human prostate adenocarcinoma cells.
- 26.Schulman IG, Heyman RA. The flip side: identifying small molecule regulators of nuclear receptors. Chem Biol. 2004;11:639–646. doi: 10.1016/j.chembiol.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 27.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 28.Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 29.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng Y, LeGall T, Oldfield CJ, Mueller JP, Van Y-Y, Romero P, Cortese MS, Uversky VN, Dunker AK. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 31. Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. 2009;131:7390–7401. doi: 10.1021/ja900616b. •• The authors used various biophysical methods to establish the presence of novel small-molecule binding sites on c-Myc. They also studied conformational changes of intrinsically disordered c-Myc upon binding to known inhibitors of Myc/Max heterodimer formation.
- 32.Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, Vogt PK. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci USA. 2002;99:3830–3835. doi: 10.1073/pnas.062036999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu X, Vogt PK, Boger DL, Lunec J. Disruption of the Myc transcriptional function by a small-molecule antagonist of MYC/MAX dimerization. Oncol Rep. 2008;19:825–830. [PubMed] [Google Scholar]
- 34.Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. A credit-card library approach for disrupting protein-protein interactions. Bioorg Med Chem. 2006;14:2660–2673. doi: 10.1016/j.bmc.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 35.Shi J, Stover JS, Whitby LR, Vogt PK, Boger DL. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg Med Chem Lett. 2009;19:6038–6041. doi: 10.1016/j.bmcl.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiessling A, Sperl B, Hollis A, Eick D, Berg T. Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem Biol. 2006;13:745–751. doi: 10.1016/j.chembiol.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 37.Kiessling A, Wiesinger R, Sperl B, Berg T. Selective inhibition of c-Myc/Max dimerization by a pyrazolo[1,5-a]pyrimidine. ChemMedChem. 2007;2:627–630. doi: 10.1002/cmdc.200600294. [DOI] [PubMed] [Google Scholar]
- 38.Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, Prochownik EV. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6:2399–2408. doi: 10.1158/1535-7163.MCT-07-0005. [DOI] [PubMed] [Google Scholar]
- 40.Mustata G, Follis AV, Hammoudeh DI, Metallo SJ, Wang H, Prochownik EV, Lazo JS, Bahar I. Discovery of novel myc-max heterodimer disruptors with a three-dimensional pharmacophore model. J Med Chem. 2009;52:1247–1250. doi: 10.1021/jm801278g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Follis AV, Hammoudeh DI, Daab AT, Metallo SJ. Small-molecule perturbation of competing interactions between c-Myc and Max. Bioorg Med Chem Lett. 2009;19:807–810. doi: 10.1016/j.bmcl.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 42.Guo J, Parise RA, Joseph E, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. Efficacy, pharmacokinetics, tissue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidene]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol. 2009;63:615–625. doi: 10.1007/s00280-008-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol. 2008;15:1149–1155. doi: 10.1016/j.chembiol.2008.09.011. •• The authors present data to suggest that previously identified Myc ligands induce a global conformational disordering thereby limiting its ability to interact with Max and form the highly ordered heterodimer
- 44.Mo H, Henriksson M. Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proc Natl Acad Sci USA. 2006;16:6344–6349. doi: 10.1073/pnas.0601418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mo H, Vita M, Crespin M, Henriksson M. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle. 2006;5:2191–2194. doi: 10.4161/cc.5.19.3320. [DOI] [PubMed] [Google Scholar]
- 46.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, Ha JR, Kahn M. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc Natl Acad Sci USA. 2004;101:12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M. Identification of small-molecule antagonists that inhibit an activator: coactivator interaction. Proc Natl Acad Sci USA. 2004;101:17622–17627. doi: 10.1073/pnas.0406374101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. A cell-permeable synthetic transcription factor mimic. Angew Chem Int Ed Engl. 2007;46:2865–2868. doi: 10.1002/anie.200604485. [DOI] [PubMed] [Google Scholar]
- 49.Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. Amphipathic small molecules mimic the binding mode and function of endogenous transcription factors. ACS Chem Biol. 2009;4:335–344. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rishi V, Potter T, Laudeman J, Reinhart R, Silvers T, Selby M, Stevenson T, Krosky P, Stephen AG, Acharya A, Moll J, Oh WJ, Scudiero D, Shoemaker RH, Vinson C. A high-throughput fluorescence anisotropy screen that identifies small molecule inhibitors of the DNA-binding of B-ZIP transcription factors. Anal Biochem. 2005;340:259–271. doi: 10.1016/j.ab.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 51.Ng PY, Tang Y, Knosp WM, Stadler HS, Shaw JT. Synthesis of diverse lactam carboxamides leading to the discovery of a new transcription-factor inhibitor. Angew Chem Int Ed Engl. 2007;46:5352–5355. doi: 10.1002/anie.200700762. [DOI] [PubMed] [Google Scholar]
- 52.Gorczynski MJ, Grembecka J, Zhou Y, Kong Y, Roudala L, Douvas MG, Newman M, Bielnicka I, Baber G, Corpora T, Shi J, Sridharan M, Lilien R, Donald BR, Speck NA, Brown ML, Bushweller JH. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFbeta. Chem Biol. 2007;14:1186–1197. doi: 10.1016/j.chembiol.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 53. Ma Y, Kurtyka CA, Boyapalle S, Sung SS, Lawrence H, Guida W, Cress WD. A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. 2008;68:6292–6299. doi: 10.1158/0008-5472.CAN-08-0121. •• The authors used the crystal structure of the E2F4/DP2 heterodimer bound to DNA to guide a virtual screen to identify small molecules that inhibit heterodimer binding to DNA. The screen yielded a compound that disrupts DNA-binding in EMSA assays and is a potent inhibitor of melanocyte proliferation and invasion in a three-dimensional culture model system.
- 54.Berg T. Signal transducers and activators of transcription as targets for small organic molecules. ChemBioChem. 2008;9:2039–2044. doi: 10.1002/cbic.200800274. [DOI] [PubMed] [Google Scholar]
- 55.Hellsten R, Johansson M, Dahlman A, Dizeyi N, Sterner O, Bjartell A. Galiellalactone is a novel therapeutic candidate against hormone-refractory prostate cancer expressing activated Stat3. Prostate. 2008;68:269–280. doi: 10.1002/pros.20699. [DOI] [PubMed] [Google Scholar]
- 56.Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J Biol Chem. 2005;280:32979–32988. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- 57.Song H, Wang R, Wang S, Lin J. A low molecular weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA. 2005;13:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhasin D, Cisek K, Pandharkar T, Regan N, Li C, Pandit B, Lin J, Li P-K. Design, synthesis, and studies of small molecule STAT3 inhibitors. Bioorg Med Chem Lett. 2008;18:391–395. doi: 10.1016/j.bmcl.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 59.Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg Med Chem Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]
- 60.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor that modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 62.Schust J, Sperl B, Hollis A, Mayer TY, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 63.Fletcher S, Singh J, Zhang X, Yue P, Page BD, Sharmeen S, Shahani VM, Zhao W, Schimmer AD, Turkson J, Gunnin PT. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities. Chembiochem. 2009;10:1959–1964. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muller J, Sperl B, Reindl W, Kiessling A, Berg T. Discovery of chromone-based inhibitors of the transcription factor STAT5. ChemBioChem. 2008;9:723–727. doi: 10.1002/cbic.200700701. [DOI] [PubMed] [Google Scholar]
- 65.Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med. 2009;13:2780–2786. doi: 10.1111/j.1582-4934.2009.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kong D, Park EJ, Stephen AG, Calvani M, Cardellina JH, Monks A, Fisher RJ, Shoemaker RH, Melillo G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005;65:9047–9055. doi: 10.1158/0008-5472.CAN-05-1235. [DOI] [PubMed] [Google Scholar]
- 67.Park EJ, Kong D, Fisher R, Cardellina J, Shoemaker RH, Melillo G. Targeting the PAS-A domain of HIF-1alpha for development of small molecule inhibitors of HIF-1. Cell Cycle. 2006;5:1847–1853. doi: 10.4161/cc.5.16.3019. [DOI] [PubMed] [Google Scholar]
- 68.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA. 2009;106:17910–17915. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH. Artificial ligand binding within the HIF2alpa PAS-B domain of the HIF2 transcription factor. Proc Natl Acad Sci USA. 2009;106:450–455. doi: 10.1073/pnas.0808092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Key J, Scheuermann TH, Anderson OC, Daggett V, Gardner KH. Principles of ligand binding within a completely buried cavity in the HIF2alpha PAS-B. J Am Chem Soc. 2009;131:17647–17654. doi: 10.1021/ja9073062. •• The authors use structural and biophysical methods as well as molecular dynamics simulations to characterize binding of small molecules to a solvent inaccessible internal cavity of HIF2alpha PAS-B. The authors propose that a conformational equilibrium exists between a highly populated ligand-inaccessible ground state and a binding-competent state characterized by increased dynamics and altered structure.
- 71.Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 72. Block KM, Wang H, Szabó LZ, Polaske NW, Henchey LK, Dubey R, Kushal S, László CF, Makhoul J, Song Z, Meuillet EJ, Olenyuk BZ. Direct inhibition of hypoxia-inducible transcription factor complex with designed dimeric epidithiodiketopiperazine. J Am Chem Soc. 2009;131:18078–18088. doi: 10.1021/ja807601b. • The authors rationally design, synthesize and evaluate a simplified ETP inhibitors of the interaction between HIF-1α and the p300/CBP coactivator derived from the natural product chetomin that displays comparable activity and reduced toxicity.
- 73. Zhu S, Wurdak H, Wang J, Lyssiotas CA, Peters EC, Cho CY, Wu X, Schultz PG. A small molecule primes embryonic stem cells for differentiation. Cell Stem Cell. 2009;4:416–426. doi: 10.1016/j.stem.2009.04.001. •• The authors used high-content screening followed by affinity-based methods to identify a small molecule that increases the efficiency of differentiation of embryonic stem cells and interacts directly with the transcription factor NME2.
- 74.Rowley JD. The critical role of chromosome translocations in human leukemias. Annu Rev Genet. 1998;32:495–519. doi: 10.1146/annurev.genet.32.1.495. [DOI] [PubMed] [Google Scholar]
- 75.Gery S, Koeffler HP. Transcription factors in hematopoietic malignancies. Curr Opin Gen Dev. 2007;17:78–83. doi: 10.1016/j.gde.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 76.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 77.Fazi F, Zardo G, Gelmetti V, Travaglini L, Ciolfi A, Di Croce L, Rosa A, Bozzoni I, Grignani F, Lo-Coco F, Pelicci PG, Nervi C. Heterochromatic gene repression of the retinoic acid pathway in acute myeloid leukemia. Blood. 2007;109:4432–4440. doi: 10.1182/blood-2006-09-045781. [DOI] [PubMed] [Google Scholar]
- 78. Erzikan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou T-H, Dakshanamurthy S, Brown ML, Üren A, Toretsky JA. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med. 2009;15:750–756. doi: 10.1038/nm.1983. •• This study describes the use of direct binding assays to identify small molecules that target an oncogenic translocation fusion protein involving a transcription factor. The authors identified a small molecule that specifically blocks an interaction between the oncogenic transcription factor translocation product and a normal cellular binding partner that is required for oncogenic activity. This study is a proof of principle for targeting oncogenic transcription factors that result from chromosomal translocations.
- 79.Koehler AN, Shamji AF, Schreiber SL. Discovery of an inhibitor of a transcription factor using small molecule microarrays and diversity-oriented synthesis. J Am Chem Soc. 2003;125:8420–8421. doi: 10.1021/ja0352698. [DOI] [PubMed] [Google Scholar]
- 80.Vegas AJ, Fuller JH, Koehler AN. Small-molecule microarrays as tools in ligand discovery. Chem Soc Rev. 2008;37:1385–1394. doi: 10.1039/b703568n. [DOI] [PMC free article] [PubMed] [Google Scholar]