

Summary
Chemosensing in nature relies on biomolecular switches, biomolecules that undergo binding-induced changes in conformation or oligomerization to transduce chemical information into specific biochemical outputs. Motivated by the impressive performance of these natural “biosensors,” which support continuous, real-time detection in highly complex environments, significant efforts have gone into the adaptation of such switches into artificial chemical sensors. Ongoing advances in the fields of protein and nucleic acid engineering (e.g., computational protein design, directed evolution, selection strategies and labeling chemistries) have greatly enhanced our ability to design new structure-switching sensors. Coupled with the development of advanced optical read-out mechanisms, including genetically encoded fluorophores, and electrochemical read-outs supporting detection directly in highly complex sample matrices, switch-based sensors have already seen deployment in applications ranging from real time, in vivo imaging to the continuous monitoring of drugs in blood.
Keywords: nanosensors, nanomachines, structure-switching sensors, molecular switch, allosteric biosensors, synthetic biology, allosteric enzymes, allosteric ribozymes, aptasensors, switch-based sensors, conformational-switching, ligand sensing, signalling aptamer, molecular beacons, calcium sensor, point-of-care
Introduction
The impressive specificity, affinity and versatility of biomolecular recognition have motivated decades of research on the development of sensors based on this effect. A significant hurdle in the development of these technologies, however, is that most biomolecules do not respond in any easily measurable way upon binding their target ligands. Antibodies, for example, do not change their shape or emit electrons or photons upon binding their target antigens. Due to this, existing bio-analytical approaches, including ELISAs, western blots and PCR, are typically multistep, washing- and reagent-intensive processes. As such these approaches are ill suited for use outside the laboratory, and impossible to deploy in real-time or in situ applications. In order to overcome this limitation, a number of sensors have been developed that detect binding in real time by monitoring a change in mass, charge or optical properties that occurs when the target binds a biomolecule-coated surface (e.g., surface plasmon resonance, field-effect transistor, quartz crystal microbalance and microcantilevers). Unfortunately, however, these approaches also suffer from a serious drawback: because they detect adsorption to the sensor head rather than a specific binding per se, they cannot distinguish between the binding of the correct, authentic target and the non-specific binding of contaminants. They thus fail when challenged with realistically complex samples, such as blood serum [1]. In short, the goal of reagentless, real-time sensors that can be deployed directly in complex samples remains largely unmet [1].
Nature has, long ago, solved the problem of real time molecular sensing in complex environments; lessons learned from these natural, protein- and nucleic acid-based “biosensors” could therefore assist in the development of improved sensing technologies. Tellingly, these naturally occurring sensors do not sense their targets via binding-induced changes in adsorbed mass or charge. They instead respond to their targets by undergoing specific, binding-induced changes in conformation or oligomerization state [2]. These switching events, in turn, trigger specific output signals, such as the opening of an ion channel or the activation of an enzyme. Inspired by the speed, specificity and versatility of these naturally occurring sensors, recent years have seen significant efforts to fabricate artificial biosensors based on this principle. Here we review recent efforts to develop such structure-switching sensors.
The many advantages of biomolecular switches
As noted above, biomolecular switches are proteins or nucleic acids that reversibly shift between two or more conformations (or conformational ensembles) in response to the binding of a specific target ligand. Several attributes render such switches well suited for adaptation into artificial sensors. First, binding-specific conformational changes offer a robust means of transducing a binding event into an output signal that is not easily mimicked by non-specific effects. That is, because structure switching is induced by the formation of many weak, non-covalent bonds (e.g., hydrogen bonding, hydrophobic effect, van der Waals forces) it is generally specific to a given ligand-biomolecule interface and thus largely or entirely insensitive to the presence of other molecules present in even highly complex environments (Fig. 1). Second, switching, and thus signal transduction, is rapid, reversible, and reagent-free, allowing these nano-scale switches to support continuous, real-time detection even inside living cells (Fig. 1a). Third, biomolecular switches are versatile; as discussed at length below, switching can be coupled to a number of specific optical, electrochemical and biochemical outputs (Fig. 2) and can be engineered into a wide range of biomolecules characterized by an equally wide range of binding specificities (Fig. 3). Finally, the conformational equilibria of biomolecular switches are quantitatively related to both target concentration and to the switch’s underlying thermodynamics. This renders switch-based sensors quantitative and provides a means by which their dynamic ranges can be rationally optimized without altering their binding specificity (Fig. 4). Given these attributes, it is perhaps not surprising that a large number of structure-switching biosensors have been reported in recent years. Here we review the mechanisms by which biomolecular switches have been coupled to optical, electronic and biochemical “read-outs,” current approaches to the design of structure-switching sensors and, finally, methods by which the thermodynamics of such switches can be modified in order to optimize their signaling.
Figure 1.

Real-time molecular detection in complex samples using structure-switching sensors. (a) Tsien and co-workers have developed a genetically encoded, calcium-sensitive switch by fusing calmodulin (a calcium binding protein), M13 (a calmodulin binding polypeptide) and two fluorescent protein reporter domains. As shown (lower left), they have employed this sensor to monitor calcium ion dynamics at the plasma membrane of hippocampal neurons following a simulation with 100 mM histamine [3]. (b). We have demonstrated an electrochemical, aptamer-based switch that folds upon cocaine binding. Affixed via one terminus to an electrode and modified at the other with a redox-active methylene blue molecule, the binding-induced folding of this aptamer leads to a large increase in current, supporting the real-time detection of micromolar cocaine in blood serum as it flows through the depicted, sub-microliter micro-fluidic device [4*]. Figures adapted with permission.
Figure 2.
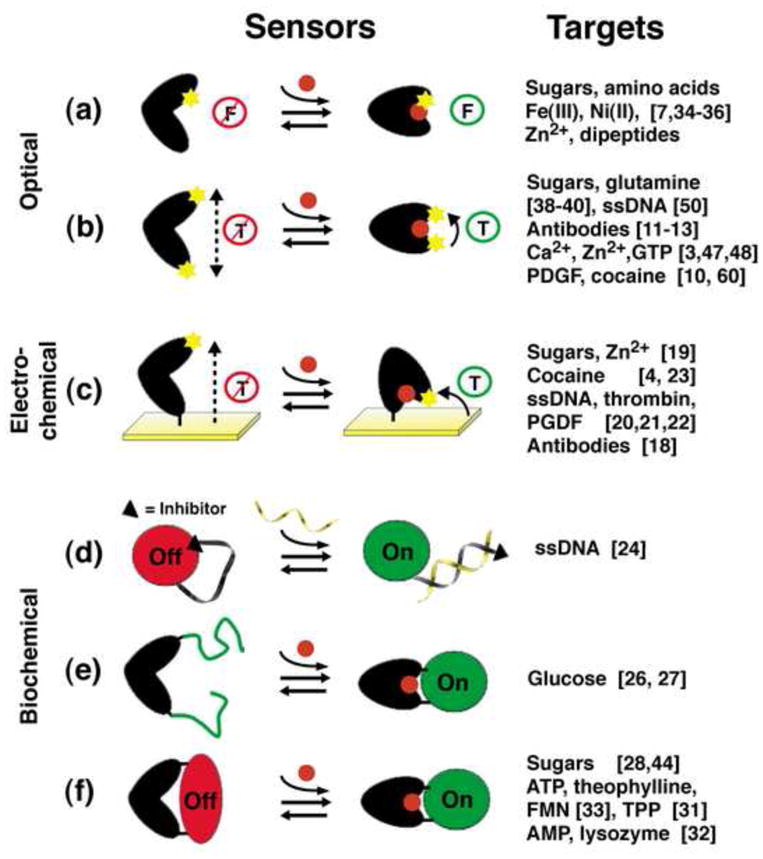
Biomolecular switches have been coupled to a number of optical, electrochemical and biochemical read-out mechanisms. (a) Environmentally-sensitive, single-fluorophore readouts; (b) Distance-dependent, dual-component optical readouts, such as FRET, electron-transfer-based quenching and excimer formation; (c) Distance-dependent electrochemical readouts; (d) Switch-linked segregation of a catalytic reporter domain and an inhibitor; (e) Switching-induced assembly of a functional reporter domain; (f) Switch-driven allosteric regulation of a reporter domain. “F” and “T” represent the fluorescence emission or the FRET and electron transfer efficiencies, respectively.
Figure 3.
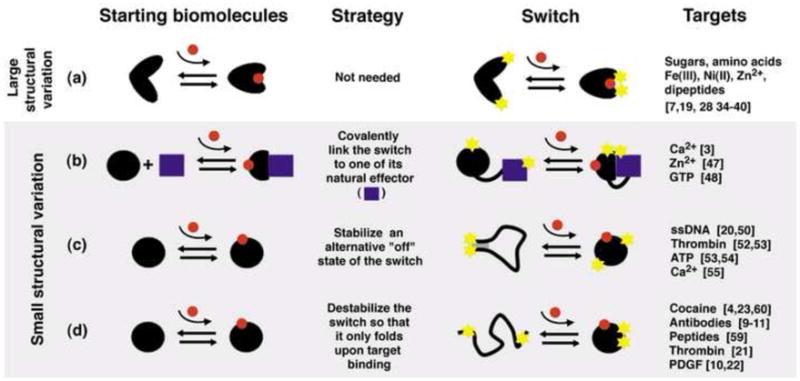
Strategies for the design of switches from a biomolecule that binds the desired molecular target. (a) If a naturally occurring switch exists that binds the target of interest it can readily be converted into a sensor. Alternatively, computational redesign or in vitro selections can be used to expand the range of targets recognized by such a switch, allowing the generation of sensors for novel targets. In a complementary set of approaches, a non-switching biomolecule that binds the target of interest can be re-engineered to undergo binding-induced switching via: (b) linking the recognition biomolecule to a naturally occurring effector; (c) stabilizing an alternative, nonbinding conformation of the recognition biomolecule; or, (d) destabilizing the recognition biomolecule so that it only folds upon target binding.
Figure 4.
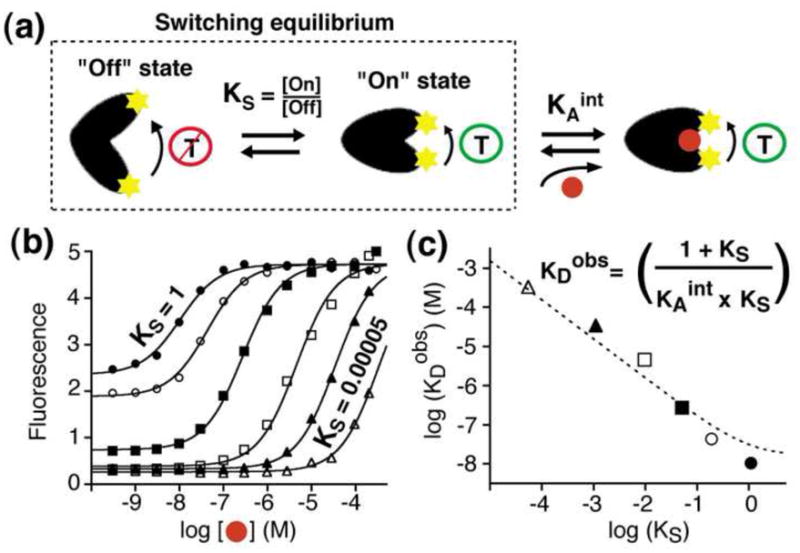
Tuning the dynamic range of a switch by optimizing its switching equilibrium, KS. (a) Switching is well described by a population-shift model in which a pre-existing equilibrium between a nonbinding “off” state and a binding-competent “on” state is pushed towards the latter by target binding [49**]. (b-c) Increasing the stability of the “off” state (reducing KS) proportionally decreases the switch’s affinity for its target (higher KDobs), providing a means of rationally “tuning” the dynamic range of a switch without altering its binding specificity. The lowest possible detection limits are generally achieved with equilibrium constants near unity (KS ≈ 1) [49**]; under these conditions many switches are poised to respond to the target (leading to high sensor gain) without a concomitantly grave reduction in affinity. Figure adapted with permission.
1- Coupling switching to a readily measurable output signal
The development of structure-switching sensors requires that the switch’s conformational state be linked to a readily detectable output. To date, structure-switching sensors have been described that respond to their targets via conformation-linked changes in fluorescence emission (for optical detection), electron transfer (for electrochemical detection) or biochemical (catalytic or binding) activity. Here we review these approaches.
The most widely employed method for linking structure switching to a specific output has been to use conformation-linked fluorescence quenching. This has been achieved via: 1) a change in the microenvironment around a single, structure-sensitive fluorophore (Fig. 2a); 2) a distance-dependent change in the Forster Resonance Energy Transfer (FRET) between a donor/acceptor fluorophore pair (Fig. 2b); or 3) a distance-dependent change in excimer formation or electron-transfer based fluorescence quenching (Fig. 2b). Fluorescent reporters utilized in such applications include a range of convenient, commercially available organic dyes as well as a number of brighter, if more complex, structures including dye-labeled microspheres, fluorescent dendrimers and polymers, narrow-bandwidth semiconductor nanocrystals (quantum dots), and, for in vivo applications, fluorescent proteins and fluorophore-binding polypeptides and nucleic acids (reviewed in [5, 6]).
Hellinga and coworkers have published an exhaustive study of the design of sensors employing single, environmentally sensitive fluorophores. They constructed more than 320 such sensors using 8 different fluorophores and 11 different proteins in the bacterial perisplasmic binding protein (bPBP) superfamily (Fig. 2a) [7]. Using the known or modeled structures of the bound and unbound states of their proteins, the authors placed reporting fluophores in positions thought likely to undergo significant environmental changes upon the binding-induced conformational change. By doing so they succeeded in generating efficient fluorescent sensors from all 11 proteins in this superfamily. A caveat of this approach, however, is that even when the structures of the two conformations of the switch are known (or can be accurately modeled), it is difficult to predict the detailed and complex interactions that occur between the fluorophore and the protein surface. Indeed, only 4% of their 320 variant sensors produced a substantive change in absolute fluorescence upon target binding [7].
Despite the inherently greater complexity associated with their design and/or fabrication, sensors employing dual optical reporters (Fig. 2b) offer important advantages over single-fluorophore sensors and have thus seen more widespread application [5]. For example, FRET-based sensors are ideally suited for in vivo imaging; by measuring the ratio of donor to acceptor emission FRET automatically corrects for variation in the concentration of the sensor within a cell [6]. Likewise, two-reporter fluorphore/quencher constructs often produce very “dark” non-signaling conformations. Because of this, binding can produce very large increases in fluorescence, rendering such sensors particularly sensitive [8].
While the 1/r6 donor-to-acceptor distance dependence of FRET supports robust signaling in many applications, the characteristic Förster radii (distance at which the energy transfer efficiency reaches 50%) of visible-light FRET pairs are large relative to the 1–2 nm conformational changes produced by many biomolecular switches. Recent years have thus seen the development of fluorescent read-outs that rely instead on electron transfer-based quenching [9], which falls off exponentially with distance, or excimer (excited state dimer) formation, which is still more strongly distance-dependent [10, 11]. The strong distance dependencies of these mechanisms ensure that conformational changes of even a few tenths of a nanometer can produce large, multifold changes in fluorescence intensity. To date these readout mechanisms have been employed to monitor a number of binding-induced conformational changes, including the folding of short polypeptide epitopes upon binding their target antibodies [11–13] and the folding of DNA aptamers upon binding to their target ligands [10].
While optical read-out mechanisms have proven well suited for applications such as in vivo imaging (Fig. 1a), fluorescent and strongly absorbing materials are common in clinical and environmental samples, reducing the utility of optical approaches in many applications. Electro-active contaminants, in contrast, are rare and thus electrochemical methods for monitoring biomolecular switching perform well even in highly heterogeneous samples (Fig. 1b). Switch-based electrochemical sensors are typically engineered by attaching an electroactive reporter (e.g., ferrocene or methylene blue) to one position on the biomolecule that, in turn, is fixed onto an electrode surface via a second, distal position. Binding-induced conformational changes will thus alter the redox current produced by the reporter, leading to a readily measurable electrochemical signal (Fig. 2c) [14]. To date, such structure-switching electrochemical sensors have been fabricated from a range of nucleic acid- [15–17], polypeptide- [18] and protein-based switches [19]. Sensors based on nucleic-acid switches have proven particularly amenable to this approach, with sensors directed against specific nucleic acid sequences [20], proteins (e.g., thrombin [21] and PDGF [22]) and small-molecule targets (e.g., cocaine [23]) having been described to date. In every case, these electrochemical sensors are rapid (responding in seconds to minutes), sensitive (detecting sub-picomolar to micromolar concentrations), and selective enough to support detection in complex samples (e.g., blood serum (Fig. 1b)). They are also operationally convenient: they are supported on micron-scale electrodes [4], enabling parallelizability, are low cost, and because the switch is covalently attached to the electrode, are typically reusable [15–17].
In addition to optical and electrochemical outputs, other strategies have been described in which structure switching activates or inhibits the function of a second, reporting domain to produce a biochemical output. Advantages offered by these readouts include the opportunity to amplify the output signal via catalysis and the ability to encode such sensors genetically for in vivo sensing. An early example of such a sensor was created by Ghadiri and co-workers, who covalently linked a protease to its inhibitor via a single-stranded DNA linker. Hybridization of the DNA with its complement rigidifies this linker, removing the inhibitor from the enzyme and allowing it to proteolize -and thus activate- a fluorescent reporter (Fig. 2d) [24]. A second approach, inspired by protein-fragment complementation assays (a strategy used to monitor protein-protein interactions in vivo [25]), utilizes switching to assemble two fragments of a catalytic signaling domain (Fig. 2e). Examples include switches designed to activate the enzymes aequorin [26**] and luciferase [27], producing bioluminescence in response to target binding. Finally, switch controlled-enzymes and rybozymes have been constructed in which switching induces or releases mechanical tension in the reporting domain, thus modulating its activity (Fig. 2f) ([28], see, also, [29, 30] for recent reviews). Examples include sensors based on the switch-induced distortion of enzymes (e.g., β-galactosidase [28, 29]), ribozymes (e.g., the hammerhead ribozyme) [31]), DNAzymes (e.g., a peroxidase-mimicking DNAzyme [32]) and fluorophore-binding aptamers (e.g., the binding of malachite green dye [33]).
2- Sensors based on naturally occurring switches
Not surprisingly, the first reported examples of structure-switching sensors employed naturally occurring switches in the detection of their natural binding partners. Specifically, in the late 1990s Hellinga and co-workers converted maltose binding protein, a member of the Escherichia coli superfamily of periplasmic binding proteins, into a fluorescent sensor for maltose [34]. Conveniently, members of this superfamily undergo large hinge bending motion when their target binds to a cleft separating the protein’s two domains. In the intervening years, numerous other members of this superfamily have been employed in sensing, with target including maltose, glucose, ribose, arabinose, glutamine, glutamate, histidine, Fe(III), Ni(II), phosphate, sulfate and, finally, dipeptides (reviewed in [35, 36]). Several of these, including the maltose [37], glucose [38] and glutamate [39, 40*] sensors, have been adapted into expressible, genetically encoded sensors using fluorescent protein reporters and used to map target concentrations in vivo. The glucose and maltose binding proteins have also been adapted into electrochemical sensors, supporting the electronic monitoring of glucose in blood serum and maltose in beer [19]. Further highlighting the versatility switch-based readout mechanisms, maltose binding protein has also been coupled to an enzymatic readout via linkage to a β-galactosidase catalytic domain [28].
3- Designing switches for the detection of novel targets
Naturally occurring switches can only detect a limited number of targets (Fig. 3a). Fortunately, however, two approaches have been developed by which biomolecular switches can be engineered to respond to novel molecular targets. The first of these approaches starts with an existing, naturally occurring switch and redesigns or re-selects its binding site to support recognition of the desired new target. The second starts with an existing binding site (either naturally occurring or the product of in vitro selection) and re-engineers the host biomolecule so that it undergoes switching upon target binding.
Altering the specificity of an existing switch
Using the above-described maltose binding protein as a scaffold, Hellinga and coworkers pioneered the use of computational protein engineering to convert existing, naturally occurring switches into sensors for novel targets. Using this approach they have reported fluorescent sensors for a number of small molecule and inorganic ion targets, including Zn(II), trinitrotoluene (TNT), lactate, serotonine and pinacolyl methyl phosphonic acid (PMPA) (reviewed in [35]). We must note, however, that recent characterisation of the binding properties of the switches directed against serotonin, lactate, TNT and PMPA and related crystallographic studies of the serotonin sensor suggest that none of these redesigned sensors bind their ligands as anticipated; the computational redesign of binding sites appears to be far from a solved problem [41*]. Ongoing advances in this field [42], however, including the recent successful design of nonnatural enzymes [43], suggest that this approach may soon reach fruition. In the meantime, directed evolution provides an alternative means of generating new binding specificities. As an example, Ostermeier and coworkers have created a switch from maltose binding protein that reports via β-lactamase activity (see Fig. 2e), allowing them to use an ampicillin-survival assay to screen for sucrose-responsive switches from a library of 4 × 106 random switch variants [44]. Strategies for the in vitro selection of structure-switching aptamers have also been described. Nutiu and Li, for example, demonstrated a method by which specific, structure-switching aptamers can be eluted from a column when ligand-binding induces a conformational change that disrupts the hybridization of a portion of their sequence to a complementary sequence fixed to the column [45].
Engineering switching into an arbitrary biomolecule
Despite some successes, re-engineering a biomolecular switch so that it binds new ligands remains a complex and time consuming challenge. Fortunately, however, several strategies have been reported by which arbitrary biomolecules can be redesigned to undergo switching upon target binding (Fig. 3), an approach that allows bioengineers to exploit the binding specificities of biomolecules that lack structure-switching activity. These strategies include: 1) fusion of the recognition biomolecule to an effector that binds only its target-bound form; 2) engineering an alternative, nonbinding conformation of the recognition biomolecule in equilibrium with its target-binding conformation or 3) destabilizing the recognition biomolecule, thus creating a biomolecule that folds only upon binding its target ligand. As described below, each of these approaches have seen notable recent successes.
Many biomolecules bind a second, “effector” biomolecule after binding their target ligand (Fig. 3b, left). An example is the protein calmodulin, which, in its calcium-bound state, binds to many other proteins and polypeptides [46]. Tsien and coworkers have employed this effect to design a now “classic,” genetically encoded calcium sensor comprised of four components: calmodulin, a polypeptide named M13 that binds the calcium-bound form of calmodulin, and two fluoresent protein FRET reporters (Fig. 1a) [3]. Similar sensors have more recently been reported for the in vivo detection of Zn2+ [47] and GTPase activation (i.e., when guanosine triphosphate is bound to a specific GTPase) [48], further speaking to the utility of this approach.
Switching occurs when the ground state (most stable conformation) of a biomolecule differs from the conformation that binds its target ligand [49**]. Binding stabilizes the latter conformation, causing the population to shift to this state. A second approach by which non-switching biomolecules can be re-engineered to undergo a large scale conformational change upon target binding is thus to stabilize an alternative conformation of the biomolecule that does not bind the target. Molecular beacons, which were introduced by Kramer and coworkers in the late 1990s, provide an illustrative example [50]. Comprised of a reporter-modified DNA strand, molecular beacons switch between a nonbinding stem-loop conformation and a binding-competent linear conformation. Hybridization with a complementary target stabilizes the latter state, leading to a large increase in net fluorescence (for fluorophore/quencher-modified molecular beacons [51]), or redox current (for electrochemical, E-DNA sensors [15, 20]). Similar strategies have been used to create structure-switching aptamers, which in turn have been adapted to optical [52, 53] and electrochemical [16, 17] sensors for the detection of a wide range of analytes. We note, however, that most of the reported examples of this approach to switch design involve nucleic acid-based switches. This is because the rational design of an alternative fold is far easier for nucleic acids than for proteins [42]: the simple complementarity of base pairing renders the design of alternate nucleic acid conformations quite straightforward [54]. Nevertheless, Loh and co-workers have demonstrated a strategy for the design of novel protein-based switches [55**]. Their method, which they term “alternate frame folding,” involves duplication of a portion of a protein’s sequence, which introduces a second, low-energy “circularly permuted” conformation. The introduction of mutations that disrupt target binding for the lower energy of these two conformations thus links binding to a large shift in the conformational population of the switch. Using this approach, they have converted the protein calbindin D9k into a fluorescent sensor that responds robustly to calcium [55**].
Despite the success of Loh’s alternative frame folding approach, the design of stable, alternative conformations remains a challenge for protein-based sensors. Fortunately, a still easier strategy exists by which proteins and nucleic acids can be re-engineered to undergo binding-induced conformational changes. This strategy relies upon the principle that the folding of simple, single-domain biomolecules is a highly cooperative, largely two-state process (see, e.g. [56]). By introducing sufficiently destabilizing mutations (typically remote from the target binding site so as to ensure that specificity is retained) it is possible to push the folding equilibrium constant arbitrarily far towards the unfolded state (Fig. 3d). Such an unfolded biomolecule still samples its native, binding-competent conformation. In presence of its target, the unfolded-folded equilibrium thus shifts back towards the native configuration. This transduction mechanism, which is extensively employed in nature [57, 58], has seen use in the design of a number of artificial protein- [59] and nucleic acid-based [60] sensors to date, in both optical (reviewed in [13]) and electrochemical formats (e.g., Fig. 1b) [17, 18].
4- Optimizing performance of biomolecular switches
Biomolecular switching typically occurs via a population-shift mechanism in which the equilibrium between a nonbinding state and a binding-competent state is shifted when the target binds the latter (Fig. 4a) [49**]. The switching equilibrium constant, KS, thus significantly impacts sensor performance. Specifically, KS must favor the nonbinding state (KS<1) in order to generate a large population of switches that are poised to respond upon the introduction of target. Conversely, however, binding is coupled to the switching equilibrium (Fig. 4a), and thus KS also affects the switch’s affinity for its ligand [61]; as Ks becomes smaller, binding must overcome a more unfavorable switching equilibrium constant and thus affinity is reduced. For these reasons, care must be taken to tune KS so that it matches the specific application at hand. For example, KS can be tuned to optimize the detection limit of the sensor (i.e., for detecting the lowest possible concentration of target) or it can be altered so as to shift the dynamic range of a sensor so that its optimal sensitivity (change in signal/change in target concentration) is achieved at the relevant range of target concentrations.
As an exploration of the “population-shift” model of switching and to highlight the importance of KS in defining sensor performance, we recently designed a set of six switches differing only in KS (Fig. 4b) [49**]. Consistent with the population-shift model, the observed affinities of this set of related switches are inversely proportional to KS as it is varied over four orders of magnitude (Fig. 4c). The lowest detection limits are achieved at a KS of approximately unity (KS ≈1), conditions under which the sensor achieves reasonable gain (50% of the maximal possible signal change is captured) with only a two-fold reduction in affinity. The precise value of KS at which the lowest possible detection limit is achieved, however, will depend on the gain of the sensor (the signal change upon target binding) and on whether the detection limit is defined by its absolute signal change or the relative change in signal relative to any background.
The link between switching thermodynamics and switch affinity (Fig. 4b-c) provides a convenient route by which the, typically two-order of magnitude, dynamic range of structure-switching sensors can be tuned in order to suit specific applications. Historically this has been performed by reducing or increasing the sensor’s affinity via mutations in its binding interface. For example, using this approach, Hellinga and coworkers have pushed the dynamic range of the E. coli maltose and glucose binding proteins up by several orders of magnitude to allow these switches to better suit the measurement of maltose and glucose in beer (~ 100 mM) [19] and blood serum (~ 5 mM) [7], respectively. Unfortunately, however, this strategy can also alter the specificity of the switch. Optimization of KS, in contrast, provides a ready means of tuning the dynamic range of a switch via alterations at locations distal from the binding site that thus preserve specificity [61]. For example, by comparing the structures of the bound and unbound states of natural switches (Fig. 3a), substitutions distal to the binding site can be identified that specifically stabilize, or destabilize, the binding-competent state [61, 62]. Likewise, the KS of switches made via effector fusions (Fig. 3b) can be tuned without impacting specificity by altering the switch-effector interface [63], or by varying the size of the linker between the two [64]. Finally, switches created by stabilization of the unfolded state, or another nonbinding conformation (Fig. 3c,d), can be optimized by altering the stability of this latter conformation [49**, 55**]. Thus, this approach to tuning and optimizing the dynamic ranges of switch-based sensors appears quite general.
Conclusion
Biomolecular switches offer a rapid and selective mean of transducing binding events into specific output signals in a single step and without the addition of exogeneous reagents. Combined with their nanoscale format and their ability to work reversibly and autonomously, structure-switching sensors are therefore well suited for the continuous, real-time monitoring of specific molecules even in environments as complex as the interiors of living cells, or blood serum (Fig. 1). The versatility of biomolecular switches –their ability to be coupled to a range of specific outputs (Fig. 2), the methods available to engineer switches with widely varying binding specificities (Fig. 3), and the ability to tune their dynamic range (Fig. 4)- further speaks to their utility. Due to these attributes, structure-switching sensors have already contributed significantly to biology (see, e.g., the Nobel prize lecture of Tsien [65]) (also Fig. 1a), and have begun to make inroads in the diagnosis of genetic and infectious diseases (e.g., molecular beacons [8]). Coupled with their ability to be readily adapted in a point-of care format (see, e.g., Fig. 1b, [4*]), we predict that structure-switching sensors are poised to drive numerous advances in clinical diagnostics [66], environmental monitoring and industrial process control.
Acknowledgments
The authors acknowledge Dr. F.-X. Campbell-Valois and members of the Plaxco group for critical reading of the manuscript. This work was supported by the NIH through grants R01EB007689 and R01EB002046 (to KWP). AVB is a Fond Québécois de la Recherche sur la Nature et les Technologies Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walt DR. Ubiquitous sensors: when will they be here? ACS Nano. 2009;3:2876–2880. doi: 10.1021/nn901295n. [DOI] [PubMed] [Google Scholar]
- 2.Gerstein M, Krebs W. A database of macromolecular motions. Nucleic Acids Res. 1998;26:4280–4290. doi: 10.1093/nar/26.18.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer AE, Tsien RY. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat Protoc. 2006;1:1057–1065. doi: 10.1038/nprot.2006.172. [DOI] [PubMed] [Google Scholar]
- *4.Swensen JS, Xiao Y, Ferguson BS, Lubin AA, Lai RY, Heeger AJ, Plaxco KW, Soh HT. Continuous, real-time monitoring of cocaine in undiluted blood serum via a microfluidic, electrochemical aptamer-based sensor. J Am Chem Soc. 2009;131:4262–4266. doi: 10.1021/ja806531z. The authors use an Electrochemical Aptamer-based sensor to monitor low micromolar concentrations of cocaine in real-time (approximately 1 min time resolution) in undiluted, umodified blood serum as it flows through a sub-microliter chamber. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sapsford KE, Berti L, Medintz IL. Materials for fluorescence resonance energy transfer analysis: beyond traditional donor-acceptor combinations. Angew Chem Int Ed Engl. 2006;45:4562–4589. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- 6.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 7.de Lorimier RM, Smith JJ, Dwyer MA, Looger LL, Sali KM, Paavola CD, Rizk SS, Sadigov S, Conrad DW, Loew L, et al. Construction of a fluorescent biosensor family. Protein Sci. 2002;11:2655–2675. doi: 10.1110/ps.021860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marras SA, Tyagi S, Kramer FR. Real-time assays with molecular beacons and other fluorescent nucleic acid hybridization probes. Clin Chim Acta. 2006;363:48–60. doi: 10.1016/j.cccn.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 9.Doose S, Neuweiler H, Sauer M. Fluorescence quenching by photoinduced electron transfer: a reporter for conformational dynamics of macromolecules. Chemphyschem. 2009;10:1389–1398. doi: 10.1002/cphc.200900238. [DOI] [PubMed] [Google Scholar]
- 10.Yang CJ, Jockusch S, Vicens M, Turro NJ, Tan W. Light-switching excimer probes for rapid protein monitoring in complex biological fluids. Proc Natl Acad Sci U S A. 2005;102:17278–17283. doi: 10.1073/pnas.0508821102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh KJ, Cash KJ, Plaxco KW. Excimer-based peptide beacons: a convenient experimental approach for monitoring polypeptide-protein and polypeptide-oligonucleotide interactions. J Am Chem Soc. 2006;128:14018–14019. doi: 10.1021/ja0651310. [DOI] [PubMed] [Google Scholar]
- 12.Oh KJ, Cash KJ, Hugenberg V, Plaxco KW. Peptide beacons: a new design for polypeptide-based optical biosensors. Bioconjug Chem. 2007;18:607–609. doi: 10.1021/bc060319u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh KJ, Cash KJ, Plaxco KW. Beyond molecular beacons: optical sensors based on the binding-induced folding of proteins and polypeptides. Chemistry. 2009;15:2244–2251. doi: 10.1002/chem.200701748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan C, Plaxco KW, Heeger AJ. Biosensors based on binding-modulated donor-acceptor distances. Trends Biotechnol. 2005;23:186–192. doi: 10.1016/j.tibtech.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Ricci F, Plaxco KW. E-DNA: a convenient, label-free method for the electrochemical detection of hybridization. Microchim Acta. 2008;163:149–155. [Google Scholar]
- 16.Xiao Y, Plaxco KW. Electrochemical aptamer sensors. In: Lu YaL Y, editor. Functional nucleic acids for sensing and other analytical applications. Springer; 2009. pp. 179–198. [Google Scholar]
- 17.Lubin AA, Plaxco KW. Folding-Based Electrochemical Biosensors: The Case for Responsive Nucleic Acid Architectures. Acc Chem Res. 2010 doi: 10.1021/ar900165x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerasimov JY, Lai RY. An electrochemical peptide-based biosensing platform for HIV detection. Chem Commun (Camb) 2010;46:395–397. doi: 10.1039/b919070h. [DOI] [PubMed] [Google Scholar]
- 19.Benson DE, Conrad DW, de Lorimier RM, Trammell SA, Hellinga HW. Design of bioelectronic interfaces by exploiting hinge-bending motions in proteins. Science. 2001;293:1641–1644. doi: 10.1126/science.1062461. [DOI] [PubMed] [Google Scholar]
- 20.Fan C, Plaxco KW, Heeger AJ. Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of DNA. Proc Natl Acad Sci U S A. 2003;100:9134–9137. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Y, Lubin AA, Heeger AJ, Plaxco KW. Label-free electronic detection of thrombin in blood serum by using an aptamer-based sensor. Angew Chem Int Ed Engl. 2005;44:5456–5459. doi: 10.1002/anie.200500989. [DOI] [PubMed] [Google Scholar]
- 22.Lai RY, Plaxco KW, Heeger AJ. Aptamer-based electrochemical detection of picomolar platelet-derived growth factor directly in blood serum. Anal Chem. 2007;79:229–233. doi: 10.1021/ac061592s. [DOI] [PubMed] [Google Scholar]
- 23.Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW. An electronic, aptamer-based small-molecule sensor for the rapid, label-free detection of cocaine in adulterated samples and biological fluids. J Am Chem Soc. 2006;128:3138–3139. doi: 10.1021/ja056957p. [DOI] [PubMed] [Google Scholar]
- 24.Saghatelian A, Guckian KM, Thayer DA, Ghadiri MR. DNA detection and signal amplification via an engineered allosteric enzyme. J Am Chem Soc. 2003;125:344–345. doi: 10.1021/ja027885u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6:569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- **26.Teasley Hamorsky K, Ensor CM, Wei Y, Daunert S. A bioluminescent molecular switch for glucose. Angew Chem Int Ed Engl. 2008;47:3718–3721. doi: 10.1002/anie.200704440. The authors create a glucose-responsive bioluminescent molecular by inserting glucose binding protein within the structure of the photoprotein aequorin. Binding-induced switching thus brings the two segments of aequorin together, activating bioluminescence and signaling the presence of the target. This strategy highlights a general approach by which switching can be linked to biochemical activity. [DOI] [PubMed] [Google Scholar]
- 27.Taneoka A, Sakaguchi-Mikami A, Yamazaki T, Tsugawa W, Sode K. The construction of a glucose-sensing luciferase. Biosens Bioelectron. 2009;25 :76–81. doi: 10.1016/j.bios.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Guntas G, Ostermeier M. Creation of an allosteric enzyme by domain insertion. J Mol Biol. 2004;336:263–273. doi: 10.1016/j.jmb.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Ostermeier M. Designing switchable enzymes. Curr Opin Struct Biol. 2009;19 :442–448. doi: 10.1016/j.sbi.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zocchi G. Controlling proteins through molecular springs. Annu Rev Biophys. 2009;38:75–88. doi: 10.1146/annurev.biophys.050708.133637. [DOI] [PubMed] [Google Scholar]
- 31.Wieland M, Benz A, Klauser B, Hartig JS. Artificial ribozyme switches containing natural riboswitch aptamer domains. Angew Chem Int Ed Engl. 2009;48:2715–2718. doi: 10.1002/anie.200805311. [DOI] [PubMed] [Google Scholar]
- 32.Teller C, Shimron S, Willner I. Aptamer-DNAzyme hairpins for amplified biosensing. Anal Chem. 2009;81:9114–9119. doi: 10.1021/ac901773b. [DOI] [PubMed] [Google Scholar]
- 33.Stojanovic MN, Kolpashchikov DM. Modular aptameric sensors. J Am Chem Soc. 2004;126:9266–9270. doi: 10.1021/ja032013t. [DOI] [PubMed] [Google Scholar]
- 34.Marvin JS, Corcoran EE, Hattangadi NA, Zhang JV, Gere SA, Hellinga HW. The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors. Proc Natl Acad Sci U S A. 1997;94:4366–4371. doi: 10.1073/pnas.94.9.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dwyer MA, Hellinga HW. Periplasmic binding proteins: a versatile superfamily for protein engineering. Curr Opin Struct Biol. 2004;14:495–504. doi: 10.1016/j.sbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Medintz IL, Deschamps JR. Maltose-binding protein: a versatile platform for prototyping biosensing. Curr Opin Biotechnol. 2006;17:17–27. doi: 10.1016/j.copbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 37.Fehr M, Frommer WB, Lalonde S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc Natl Acad Sci U S A. 2002;99 :9846–9851. doi: 10.1073/pnas.142089199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deuschle K, Chaudhuri B, Okumoto S, Lager I, Lalonde S, Frommer WB. Rapid metabolism of glucose detected with FRET glucose nanosensors in epidermal cells and intact roots of Arabidopsis RNA-silencing mutants. Plant Cell. 2006;18:2314–2325. doi: 10.1105/tpc.106.044073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okumoto S, Looger LL, Micheva KD, Reimer RJ, Smith SJ, Frommer WB. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proc Natl Acad Sci U S A. 2005;102:8740–8745. doi: 10.1073/pnas.0503274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *40.Hires SA, Zhu Y, Tsien RY. Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc Natl Acad Sci U S A. 2008;105:4411–4416. doi: 10.1073/pnas.0712008105. The authors describe a genetically encoded, structure-switching glutamate sensor that signals via FRET between two fluorescent proteins. Using this sensor the authors quantitatively measured the time course of synaptic glutamate release, spillover, and reuptake in cultured hippocampal neurons with 10-ms temporal and spatial resolution (femtoliter) good enough to resolve individual spines on the surface of neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *41.Schreier B, Stumpp C, Wiesner S, Hocker B. Computational design of ligand binding is not a solved problem. Proc Natl Acad Sci U S A. 2009;106:18491–18496. doi: 10.1073/pnas.0907950106. The structural re-analysis of five previously published high-affinity rationally designed switches in the periplasmic binding protein family highlights the difficulty of this approach. The authors find that that binding cannot be detected for any of the tested designed receptors. This work also highlights the critical need for rigorous controls when validating new sensors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ambroggio XI, Kuhlman B. Design of protein conformational switches. Curr Opin Struct Biol. 2006;16:525–530. doi: 10.1016/j.sbi.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 43.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, 3rd, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guntas G, Mansell TJ, Kim JR, Ostermeier M. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc Natl Acad Sci U S A. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nutiu R, Li Y. In vitro selection of structure-switching signaling aptamers. Angew Chem Int Ed Engl. 2005;44:1061–1065. doi: 10.1002/anie.200461848. [DOI] [PubMed] [Google Scholar]
- 46.Chin D, Means AR. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 47.Vinkenborg JL, Nicolson TJ, Bellomo EA, Koay MS, Rutter GA, Merkx M. Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat Methods. 2009;6:737–740. doi: 10.1038/nmeth.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aoki K, Matsuda M. Visualization of small GTPase activity with fluorescence resonance energy transfer-based biosensors. Nat Protoc. 2009;4:1623–1631. doi: 10.1038/nprot.2009.175. [DOI] [PubMed] [Google Scholar]
- **49.Vallee-Belisle A, Ricci F, Plaxco KW. Thermodynamic basis for the optimization of binding-induced biomolecular switches and structure-switching biosensors. Proc Natl Acad Sci U S A. 2009;106:13802–13807. doi: 10.1073/pnas.0904005106. While a number of previous authors have discussed the link between the switching equilibrium constant and switch performance qualitatively, here we derive the quantitative relationship underlying the population-shift model, show that it holds experimentally and use it to rationally “tune” the dynamic range of a switch without altering its binding specificity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 51.Marras SA, Kramer FR, Tyagi S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002;30:e122. doi: 10.1093/nar/gnf121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hamaguchi N, Ellington A, Stanton M. Aptamer beacons for the direct detection of proteins. Anal Biochem. 2001;294:126–131. doi: 10.1006/abio.2001.5169. [DOI] [PubMed] [Google Scholar]
- 53.Tang Z, Mallikaratchy P, Yang R, Kim Y, Zhu Z, Wang H, Tan W. Aptamer switch probe based on intramolecular displacement. J Am Chem Soc. 2008;130:11268–11269. doi: 10.1021/ja804119s. [DOI] [PubMed] [Google Scholar]
- 54.White RJ, Rowe AA, Plaxco KW. Re-engineering aptamers to support reagentless, self-reporting electrochemical sensors. Analyst. 2010;135:589–594. doi: 10.1039/b921253a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **55.Stratton MM, Mitrea DM, Loh SN. A Ca2+-sensing molecular switch based on alternate frame protein folding. ACS Chem Biol. 2008;3:723–732. doi: 10.1021/cb800177f. The authors present a general approach, alternate frame folding, by which to engineer binding-induced structure-switching into a normally non-responsive protein. Their method involves duplication of a portion of a protein’s sequence, which introduces a second, low-energy “circularly permuted” conformation that competes with the native fold. Using this approach they have converted the protein calbindin D9k into a fluorescent sensor that responds robustly to calcium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fersht AR. Structure and mechanism in protein science : a guide to enzyme catalysis and protein folding. New-York: Freeman, W.H; 1999. [Google Scholar]
- 57.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 58.Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 59.Kohn JE, Plaxco KW. Engineering a signal transduction mechanism for protein-based biosensors. Proc Natl Acad Sci U S A. 2005;102:10841–10845. doi: 10.1073/pnas.0503055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stojanovic MN, de Prada P, Landry DW. Aptamer-based folding fluorescent sensor for cocaine. J Am Chem Soc. 2001;123:4928–4931. doi: 10.1021/ja0038171. [DOI] [PubMed] [Google Scholar]
- 61.Marvin JS, Hellinga HW. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat Struct Biol. 2001;8:795–798. doi: 10.1038/nsb0901-795. [DOI] [PubMed] [Google Scholar]
- 62.Mizoue LS, Chazin WJ. Engineering and design of ligand-induced conformational change in proteins. Curr Opin Struct Biol. 2002;12:459–463. doi: 10.1016/s0959-440x(02)00348-2. [DOI] [PubMed] [Google Scholar]
- 63.Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, Tsien RY. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol. 2006;13:521–530. doi: 10.1016/j.chembiol.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 64.van Dongen EM, Evers TH, Dekkers LM, Meijer EW, Klomp LW, Merkx M. Variation of linker length in ratiometric fluorescent sensor proteins allows rational tuning of Zn(II) affinity in the picomolar to femtomolar range. J Am Chem Soc. 2007;129:3494–3495. doi: 10.1021/ja069105d. [DOI] [PubMed] [Google Scholar]
- 65.Tsien RY. Constructing and exploiting the fluorescent protein paintbox (Nobel Lecture) Angew Chem Int Ed Engl. 2009;48:5612–5626. doi: 10.1002/anie.200901916. [DOI] [PubMed] [Google Scholar]
- 66.Giljohann DA, Mirkin CA. Drivers of biodiagnostic development. Nature. 2009;462:461–464. doi: 10.1038/nature08605. [DOI] [PMC free article] [PubMed] [Google Scholar]