

Abstract
Proteins evolve at very different rates and, most notably, at rates inversely proportional to the level at which they are produced. The relative frequency of highly expressed proteins in the proteome, and thus their impact on the cell budget, increases steeply with growth rate. The maximal growth rate is a key life-history trait reflecting trade-offs between rapid growth and other fitness components. We show that the maximal growth rate is weakly affected by genetic drift. The negative correlation between protein expression levels and evolutionary rate and the positive correlation between expression levels of highly expressed proteins and growth rates, suggest that investment in growth affects the evolutionary rate of proteins, especially the highly expressed ones. Accordingly, analysis of 61 families of orthologs in 74 proteobacteria shows that differences in evolutionary rates between lowly and highly expressed proteins depend on maximal growth rates. Analyses of complexes with key roles in bacterial growth and strikingly different expression levels, the ribosome and the replisome, confirm these patterns and suggest that the growth-related sequence conservation is associated with protein synthesis. Maximal growth rates also shape protein evolution in the other bacterial clades. Long-branch attractions associated with this effect might explain why clades with persistent history of slow growth are attracted to the root when the tree of prokaryotes is inferred using highly, but not lowly, expressed proteins. These results indicate that reconstruction of deep phylogenies can be strongly affected by maximal growth rates, and highlight the importance of life-history traits and their physiological consequences for protein evolution.
Keywords: evolutionary rate heterogeneity, microbial growth, tree of life
Protein families evolve at very diverse rates because of different specific structural and functional constraints and to the costs of protein production (1, 2). The evolutionary rates of proteins are inversely proportional to their expression level in all investigated clades, from bacteria to mammals (3). The distribution of the concentration of proteins in bacterial cells follows approximately a log-normal distribution, where most proteins are present at low concentrations and a small percentage of highly expressed proteins (HEP) account for the majority of the proteome (4, 5). Hence, the large variance in protein abundance is partly responsible for the variation in protein conservation. There is some controversy as to whether the cost of proteins is dominated by the cost of producing them [i.e., transcription and translation (6)] or by the cost of the product (7). In any case, the cost is expected to scale with the expression level. Along these lines, three hypotheses have been put forward to explain the dependency of evolutionary rates on expression levels (2). First, selection for translational-associated codon usage bias is stronger in highly expressed genes (8). Hence, nonsynonymous substitutions from an optimal codon toward a suboptimal codon are more deleterious in these genes, effectively decreasing their evolutionary rates (9). Second, purifying selection at the amino acid level is expected to be stronger in HEP because compensation of lost activity by gene overexpression requires a larger share of the cell budget (10). Third, substitutions increasing the frequency of mistranslation and protein misfolding are expected to be more deleterious in HEP because of the associated higher production cost and because abundant misfolded proteins can be toxic for the cell (7, 11).
In microorganisms, protein expression levels are intimately linked to the cell's growth rate: as bacteria grow faster, protein expression increases and HEP become even more abundant. When the generation time of Escherichia coli decreases from 100 to 20 min, the total cellular protein content increases almost fivefold and the fraction of ribosomal proteins in the proteome increases from 9% to 21% (12). This increased expression during periods of fast growth is costly. As a case in point, protein production represents the largest fraction of the ATP budget of fast growing E. coli cells (6, 13). Bacterial species growing slowly even under optimal growth conditions also exhibit high expression of the same housekeeping genes. Ribosomal proteins and RNA polymerase are among the most abundant proteins in the slow-growing Synechococcus elongatus (cyanobacteria) (14), Mycoplasma pneumoniae (tenericutes) (15), and Leptospira interrogans (spirochaetes) (4). Ribosomal proteins are 8–12% of the proteome of M. pneumoniae and L. interrogans cells growing at nearly optimal rates (>6 h per generation). This percentage is close to the value observed in slow-growing E. coli cells (<9% at 100 min per generation) (12). In summary, periods of fast growth require heavy investment in protein expression, especially in HEP, and the magnitude of this investment relative to the cell budget increases with growth rate.
The maximal growth rate (or minimum generation time) achievable by an organism under optimal conditions is a key and costly life-history trait. The evolution of very high growth rates can be maladaptive because it leads to lower yield metabolism, it is associated with low affinity transporters, and renders bacteria more sensitive to stress, to starvation, and to predators (16). Fast-growers, bacteria that can grow fast under optimal growth conditions, are poorly adapted to the suboptimal conditions that are likely to predominate in nature (17). Slow-growers, bacteria growing slowly even under optimal conditions, are the most abundant bacterial species in the ocean and stabilized soil communities, which are the natural habitats containing the largest fraction of the planet's bacteria (18–20). Therefore, low minimal generation times do not necessarily implicate low average generation times in natural populations or very large bacterial populations. The trade-offs between high growth rates and other important traits imply that different species will select for different optimal minimal generation times. Thus, one should not equate slow growth with inefficient selection. Instead, growth-related trade-offs have led to the evolution of very diverse minimal generation times, from a few minutes to several days, and have shaped genome organization (21, 22).
In short, at higher growth rates bacteria synthesize more proteins and HEP account for a larger fraction of the cell proteome. Because HEP evolve slower (because they account for a larger fraction of protein expression), we propose that minimal generation times shape the variation in protein evolutionary rates. Highly abundant housekeeping proteins (e.g., ribosomal proteins) should thus evolve slower in fast-growing bacteria. On the other hand, lowly expressed proteins (LEP) account for a smaller fraction of the proteome in fast growers, and are thus expected to show slightly relaxed selection in fast-growing bacteria. However, this effect is expected to be weak because LEP are very numerous, each with small individual contributions to the cell proteome. Hence, purifying selection on LEP is most likely associated with function than with expression. In any case, our hypothesis predicts an increase in the difference of evolutionary rates between HEP and LEP with maximal growth rates. Additionally, housekeeping HEP are becoming the favorite markers of phylogenetic studies (23). Our hypothesis implicates that such markers can be affected by the way species evolve relative to growth-related life-history traits.
Results
Ubiquitous Highly Expressed Proteobacterial Proteins Evolve Slower.
Proteobacteria are the most sampled bacterial phylum and have very diverse minimal generation times (21). Therefore, we selected a set of proteobacteria with experimentally determined minimal generation times recovered from the primary literature (21) and inferred their phylogeny. We eliminated a few species that produced very short branches (to minimize topological uncertainty), or that were not mesophiles [to avoid temperature-adaptation biases (21, 24)]. We further eliminated species with long terminal branches because these branches are likely to include different historical periods of slow and fast growth. The precise threshold for a long terminal branch (>0.95 substitutions per site) was determined from the analysis of the phylogenetic inertia of minimal generation times (Materials and Methods and Fig. S1A). We identified the 61 families of orthologs shared by all remaining 74 species and used them to reconstruct the species tree. All of the nodes of this tree are very well supported (Fig. S2). Fast and slow growing bacteria are present in all major subdivisions of the proteobacteria (Fig. 1 and Fig. S2). Nevertheless, the tree shows very long branches for vertically inherited obligatory endomutualists, such as Buchnera, which are slow-growers with very small effective population sizes (25). We redid all of the analyses described in the next sections excluding these clades (Buchnera, Wigglesworthia, Blochmannia, Sodalis, and Wolbachia) and found similar qualitative results. We compared the species tree built with the 61 orthologs with a tree of 158 nearly ubiquitous homolog families from which horizontal gene transfers were expunged (26) (Materials and Methods and Fig. S2). The two trees are topologically identical, further suggesting that our tree accurately represents the evolutionary relations between the taxa and that it is not strongly affected by horizontal gene transfer.
Fig. 1.

Cladogram representation of the reference tree of the 74 proteobacteria with published minimal generation times (g). Branch lengths and bootstraps are provided in Fig. S2.
The 61 orthologs common to all 74 proteobacteria necessarily correspond to highly conserved proteins; otherwise homology would not be recognized at this large time scale. We therefore first checked that protein expression levels and evolutionary rates were negatively associated in this set. We quantified gene expression/protein abundance using mRNA, proteome and codon usage data (Materials and Methods). Qualitative results are similar for all types of data and we concentrate here on mRNA data (see SI Materials and Methods for the other analyses). We found a significant negative correlation between expression and evolutionary rates across the orthologs of E. coli and Salmonella enterica (ρmRNAindex = −0.68, P < 0.0001), whichever the type of expression data we used (Fig. S3 and Table S1). These results are in agreement with previous results on a much larger dataset of around 3,000 orthologs between these two species (10). At a larger scale, we find a negative association between the expression ranks of these genes in E. coli and the average substitution rates of the protein in the tree (ρmRNAindex = −0.32, P = 0.01) (Table S1). We conclude that the association of expression and evolutionary rates is common to proteobacteria and that the set of 61 families is representative of the association we wish to test.
HEP Evolve Even Slower in Fast-Growing Bacteria.
Because no calibration points (e.g., using fossil records) are available to estimate divergence times for bacteria, we cannot perform direct correlations between absolute estimations of evolutionary rates and life-history traits. Therefore, we focused on how differences of evolutionary rates between pairs of genes across proteobacteria were correlated with minimal generation times. These paired tests effectively control for genome-wide effects because genomic mutation rates, effective population sizes, and number of generations are similar to both genes in the pair. To exemplify our approach, we took a randomly chosen pair of differently expressed genes among the families of orthologs not involved in replication or translation (the replisome and the ribosome are analyzed in detail in the next section). The differences in evolutionary rates between one highly expressed member of the general secretory pathway (SecY, mRNAindex ∼37) and a lowly expressed protease (Lon, mRNAindex∼6) do covary in the expected way with minimal generation times [Spearman rank correlation: ρ = −0.51; phylogenetically independent contrasts (Materials and Methods) (PIC)-P value < 10−4) (Fig. 2A). Interestingly, although for fast-growers SecY is the most conserved protein, Lon is more conserved in slow growers. This example clearly supports the hypothesis of evolutionary rate heterogeneity driven by selection for fast growth.
Fig. 2.

Highly expressed proteins evolve slower in bacteria with smaller minimal generation times. (A) Association between minimal generation times and the differences in evolutionary rates between one highly expressed secretion-related protein (HEP) and a LEP (ρ = −0.51, P < 10−4, PIC-P value < 10−4, n = 74). (B) Under the null hypothesis (H0) the median (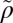 ) of the Spearman's rank correlation, coefficients should be close to zero. Under our alternative hypothesis (H1) the correlations between LEP and HEP should be predominantly negative. (C) Distribution of Spearman coefficients of all pairwise comparisons between proteins with a 10-fold difference in expressivity. (a) Median over the entire distribution (
) of the Spearman's rank correlation, coefficients should be close to zero. Under our alternative hypothesis (H1) the correlations between LEP and HEP should be predominantly negative. (C) Distribution of Spearman coefficients of all pairwise comparisons between proteins with a 10-fold difference in expressivity. (a) Median over the entire distribution (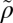 = −0.16, n = 215); (b and c) median of the pairs that show individually statistically significant correlations [P < 0.05 (light gray, n = 72) and P < 0.01 (dark gray, n = 44), respectively].
= −0.16, n = 215); (b and c) median of the pairs that show individually statistically significant correlations [P < 0.05 (light gray, n = 72) and P < 0.01 (dark gray, n = 44), respectively].
We then generalized the previous analysis by comparing all pairs of proteins among the 61 families in our analysis. In our model, the null hypothesis (H0) is that differences in evolutionary rates between pairs of HEP and LEP are independent of minimal generation times. Our alternative hypothesis (H1) is that HEP evolve slower in fast-growers. For each pair of genes we compute a correlation coefficient between the minimal generation time and the difference in evolutionary rate (as in the previous example for SecY and Lon). Under H0 the median of these correlations is centered on zero, whereas under H1 it is negative. Indeed, data analysis shows negative medians, rejecting H0 with great confidence in all cases: (i) When comparing pairs of proteins with at least 10- or 5-fold differences (to minimize the effect of noise in expression data) in the corresponding mRNA concentrations (Fig. 2C and Fig. S4B, respectively, both P < 10−4). (ii) When comparing all pairs of proteins without filtering for a minimal threshold difference (Fig. S4A). (iii) When using codon usage or proteomic data instead of mRNA concentration (Fig. S4A). A series of randomization tests to control for the effects of multiple comparisons and phylogenetic nonindependence confirmed the significant trend (Materials and Methods, PD-P value < 10−3). These results show that evolutionary rates and expression levels are associated with minimal generation times. In particular, they support the hypothesis that fast-growers exhibit stronger purifying selection on HEP.
Differences in the Evolution of the Replisome and the Ribosome.
We then restricted our analysis to the proteins families that are part of the replisome or of the ribosome (Table S2). These complexes have strikingly different expression levels (low and very high, respectively) and are involved in two essential processes in actively growing cells (replication and translation, respectively). Given their function, both complexes are expected to be under strong purifying selection in actively replicating bacteria. Because concatenates of the proteins involved in these complexes do not share a strictly identical evolutionary history, we initially restricted our analysis to the tree of 38 taxa that showed identical history for the ribosomal proteins, the replisome, the 16S rRNA, and the reference tree. As expected, the difference in terms of sequence conservation between ribosomal proteins and replisome proteins increases with growth rates, such that ribosomal proteins are relatively more conserved than replisome proteins in fast-growers compared with slow-growers (Fig. 3) [ρ = −0.61, P < 10−4, generalized estimating equations (GEE)-P value = 0.02]. Similar results were obtained using the full dataset of 74 taxa (Fig. S4C). Hence, these two essential protein complexes show the same association between minimal generation times and evolutionary rates as the individual proteins in our larger subset. Less expectedly, we find that the nucleotide substitution rates of the 16S rRNA do not follow the trend of protein substitution rates in ribosomal proteins (Fig. 3) (ρ = −0.61, P < 10−4, GEE-P value = 0.07). Instead, the rates are as affected as the replisome by minimal generation times. As a structural component of ribosomes, rRNA molecules are highly transcribed and even more so during fast growth (12). This finding suggests that the increased conservation associated to growth is much more important for protein coding genes than for RNA genes.
Fig. 3.

Ribosomal proteins evolve slower than the replisome proteins or the 16S rDNA with decreasing generation times.
Growth-Related Heterotachy and Phylogenetic Reconstruction.
Minimal generation times change quickly among proteobacteria (Fig. S1A). Among the other bacteria, some clades are essentially composed of either fast-growers (e.g., bacillales, clostridia) or slow-growers (e.g., spirochaetes, chlamydiae, cyanobacteria) (Fig. S1B). Our previous results suggest that HEP of clades experiencing such long-term consistent selection for fast growth should exhibit lower evolutionary rates and, conversely, those of clades of slow-growers should evolve faster. We reanalyzed two deep phylogenies of prokaryotes, one based on 31 HEP (27), and the other based on the abovementioned set of 158 proteins (158P, Materials and Methods), which is larger and includes LEP. Both phylogenies differ from the 16S rRNA tree and share 72 taxa with available minimal generation times. We observe an association between minimal generation times and root-to-tip distances in these trees (31 HEP tree: ρ = 0.32; 158P tree: ρ = 0.28; 16S tree: ρ = −0.02; n = 72). The highest value is obtained in the set with the largest fraction of HEP (31HEP). The lowest correlation is obtained for the 16S rRNA tree. The exclusion of proteobacteria from the dataset leads to even stronger trends, certainly because proteobacteria are more diverse in terms of minimal generation times (31HEP tree: ρ = 0.63; 158P tree: ρ = 0.39; 16S tree: ρ = 0.20; n = 39 excluding proteobacteria). Accordingly, the evolutionary rate differences in terminal branches between the protein trees and the 16S tree are more strongly correlated with minimum generation times in the tree based exclusively on HEP (16S tree vs. 31HEP tree: ρ = −0.37, P = 0.002) (Fig. S5) than in the other (16S tree vs. 158P tree: ρ = −0.26, P = 0.005; n = 72 taxa with congruent topology in the three trees). These results suggest that HEP evolve slower in fast-growers among all prokaryotes, not just in proteobacteria. They also indicate that trees built with HEP without accounting for heterotachy can be significantly affected by minimal generation times.
Variations in evolutionary rates across lineages create phylogenetic reconstruction artifacts where long branches cluster together and closer to distant outgroups, regardless of their true phylogenetic relationship (28). To test the hypothesis that minimal generation times may lead to such long-branch attraction artifacts, we split the 158P dataset into HEP and LEP subsets (Materials and Methods and Table S2) and reconstructed trees without removing any type of incongruence (unprocessed reference trees, Materials and Methods). This method was done to avoid spurious removal of incongruence because of variable evolutionary rates across lineages, which is what we wish to identify. First, we confirmed the negative association between minimal generation times and evolutionary rate differences between the LEP and HEP reconstructions for 316 prokaryotes, of which 154 have experimentally characterized minimal generation times (ρ = −0.33, P < 0.0001, n = 133 excluding archaea) (Fig. 4A). To study systematic topological divergences between HEP and LEP reconstructions, we then ran 500 jackknives on the HEP and LEP markers. This process allowed us to produce similar concatenate sizes (from HEP: 8,131 sites; LEP: 4,469 sites, to an average of around 4,500 sites). It also allowed testing the effect of randomizing the contribution of markers within the sets. Only between-clade rearrangements were permitted during the reconstruction, to focus on the comparison of clades branching deeper in the HEP and LEP jackknife trees (i.e., closer to the archaeal outgroup). We observed very clear topological differences between HEP and LEP jackknife reconstructions (Fig. 4B and Table S3): clades with an overrepresentation of slower growing organisms consistently branch closer to archaea in HEP-trees but not in LEP-trees. These differences fit the observation that HEP evolve slower in fast growers and suggest that this effect can lead to long-branch attraction artifacts.
Fig. 4.

Minimum generation times impact deep-phylogenetic reconstructions. (A) Association between minimal generation times and the differences in evolutionary rates between deep phylogenies reconstructed with two subsets of the 158P homolog families: 57 HEP and 22 LEP in 133 bacteria and an outgroup of 21 archaea. (ρ = −0.33, P < 10−4, n = 133). (B) Clades branching closest to the archaeal outgroup in deep-phylogenetic reconstructions based on highly (HEP) and lowly expressed (LEP) markers. Reconstructed trees (%) where the clade's ancestor is the node closest to the outgroup (dark color), or is part of several ancestor nodes equidistant to the root (light color), based on 500 HEP-jackknives and 500 LEP-jackknives. Clades are ordered according to the inferred ancestral minimal generation time (details in Table S3).
Discussion
The association between expression levels and evolutionary rates of proteins has been shown in a series of model organisms (3, 10). Here, we provide evidence that it extends to the full clade of proteobacteria. We then use this dataset to test the effect of selection for rapid growth on HEP evolutionary rates. We provide evidence that HEP of bacteria with low minimal generation times are more conserved than their orthologs in slow-growers. We interpret this observation as the result of more intense purifying selection in these proteins because they represent a higher fraction of protein production under faster growth. One could argue that some LEP might not be under strong selection under optimal growth conditions (e.g., repair genes). We therefore compared the ribosome and the replisome. Both of these essential protein complexes have strikingly different expression levels, but are expected to be, on functional terms, under strong selection in fast-growing bacteria. We still found the same significant difference between their patterns of sequence conservation in relation to minimal generation time. Interestingly, we found higher growth-associated deceleration of evolutionary rates in the protein than in the RNA component of ribosomes (16S rRNA), which correlates with minimal generation times like the replisome. This result fits all three previously proposed hypotheses for the association between expression levels and evolutionary rates (see introductory paragraphs): (i) proteins are much more expensive than RNA, both in terms of process and product; (ii) errors in proteins can be induced by transcription and translation; and (iii) ribosomal proteins show strong codon usage bias. These results suggest that protein synthesis is at the basis of a significant fraction of the cost leading to strong conservation of HEP (7).
This work shows that intragenomic variation of evolutionary rates between essential housekeeping proteins depends on life-history traits shaping selection for maximal growth rates. It was known that within genomes HEP evolve slower. We show that HEP evolve even slower in the fastest-growing bacteria. Hence, natural selection for fast growth affects differentially the proteome. Our method is based on the comparative analysis of genes in the same genomes and should therefore control for genome-wide effects that are expected to affect the evolution of proteins, such as the number of generations or the mutation rate. We further removed nonmesophiles to control for the effect of temperature (24) and controlled for the low effective population size (Ne) lineages of endomutualists. Different Ne could alter the evolutionary patterns of different genes if Ne were strongly associated with minimal generation times. The tradeoff hypothesis suggests that low Ne does not necessarily cause high minimal generation times because fast growth is only one of a series of conflicting traits shaping the organisms fitness. To verify this assertion, we computed the effective population sizes of 38 species using published data on their genetic diversity (29) (SI Materials and Methods). These results show no significant correlation between Ne scaled by mutation rate (Ne.u) and minimal generation times (ρ = −0.042, P value = 0.8) (Fig. S6). This finding further suggests that minimal generation times result from adaptation by natural selection, not just from unequal role of genetic drift in different lineages. These findings also confirm that decreased evolutionary rates in HEP of fast-growers are not caused by preponderance of drift in the lineages of slow-growers.
Finally, our results suggest that consistent selection for fast or slow growth will be deeply imprinted in the evolutionary patterns of HEP. This suggestion is of practical importance because protein-based deep-phylogenies are done with HEP, exactly because they evolve slowly. Here, we show that the pace of evolution of these proteins is dependent on minimal generation times, possibly leading to systematic biases in phylogenetic reconstructions and inferences of divergence times. We cannot formally exclude horizontal transfer from the causes of the apparent long-branch attractions we observe. However, explaining the different placements of clades in the tree by horizontal gene transfer from or to archaea is not simple. First, many of the slow-growing basal clades have no known extremophiles, but extreme habitats are frequent among archaea. Instead, the former clades include many bacteria interacting with eukaryotic cells, which are very rare among archaea. Furthermore, such a scenario might require the transfer of a significant number of essential highly conserved genes between distant clades and should assume different directionality for transfers of HEP and LEP. As the incongruence of reconstructions using LEP and HEP is consistent with the rest of our analyses, we are inclined to interpret them as a sign that deep phylogenies can be strongly affected by minimal generation times. As a result, such reconstructions would gain by using diverse panels of incongruence-controlled protein markers (26), tackling heterogeneous evolutionary rates (30), and jointly modeling the evolution of the trait and the phylogeny as recently proposed for DNA sequences (31). Such developments are likely to provide more accurate reconstruction of the deep branches in the tree of life and will deepen our understanding of the coevolution of the proteome and the organism's life-history traits.
Materials and Methods
Analysis of Evolutionary Rates.
Genomes were retrieved from GenBank (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/). Orthologs were identified as bidirectional best hits, using end-gap free global alignment, between the proteome of E. coli as a pivot and each of the other proteomes. Hits with less than 40% sequence similarity or more than 20% difference in length were discarded. The 61 families of orthologs were checked for paralogs, classed in terms of function, aligned, and expunged of poorly aligned regions (SI Materials and Methods). Protein evolutionary rates were estimated by maximum likelihood using PAML (32), with the WAG+Γ(8) model and with the fixed topology given by the reference tree pruned to the 74 proteobacteria (see below). The evolutionary rate of a given ortholog in one taxon was taken as the substitution rate in the terminal branch. Trees and distances based on protein sets were obtained from concatenated superalignments. The evolutionary rates of 16S rRNA sequences were estimated using PAML, with the GTR + Γ(8) model and the same fixed topology.
Expression Data.
There is no available expression data for most species of proteobacteria. However, proteomic studies have shown that the relative abundance of orthologous proteins is conserved among distant species (15). Such conservation is observable even among orthologs of microbes and humans (33). Therefore, we made the necessary simplifying assumption that under exponential growth the ranking of expression levels of the 61 essential ubiquitous proteins in E. coli is representative of those of other proteobacteria. We used several expressivity indexes based on experimental data (mRNA and protein concentration) or predictions (codon usage bias) for E. coli (SI Materials and Methods). All these sources of expression data provided qualitatively similar results in all analysis. From the 158 families of homologs used for the reference tree, we separated 57 HEP and 22 LEP. Genes were ordered by their levels of expressivity according to the mRNA index of E. coli and the codon usage of Bacillus subtilis (for which no equivalent mRNA dataset was available). The sets of HEP/LEP were obtained from the intersection of the top/bottom 10%/50% of proteins in the E. coli and B. subtilis ranked lists of expression levels. Qualitatively similar results were found using E. coli's codon usage bias and when we used the union (instead of the intersection) of the lists of the two genomes.
Phylogenetic Analyses.
The reference tree included 316 bacteria and archaea. This tree was based on a previously identified reference set of 158 homolog families obtained from HOGENOM4, where incongruence because of horizontal gene transfer was removed using Prunier (26). The original 158 alignments without removal of predicted horizontal transfer events were used to build the unprocessed reference trees of 57 HEP (8,131 sites with 8% of invariant sites) and 22 LEP proteins (4,469 sites with 10% of invariant sites). We performed 500 jackknives on each set of homolog families (HEP and LEP) to obtain alignments of similar size (3,000 ± 250 sites). We then counted the number of trees where each of the 14 bacterial monophyletic clades branched closest, in number of nodes, to the archaeal outgroup. This was done separately for the HEP and LEP datasets. The topology within clades was fixed to the topology of the reference tree.
Controls for phylogenetic nonindependence were done using the R package ape (34). PIC/GEE-P value refers to the P value after correction using phylogenetically independent contrasts (PIC) or generalized estimating equations (GEE). Both methods provided similar results. PD-P value refers to the P value of the test on the median of the distribution of Spearman correlations using phylogenetically independent contrasts and controlling for multiple comparisons (SI Materials and Methods).
Supplementary Material
Acknowledgments
We thank Simonetta Gribaldo, Nicolas Lartillot, and the expert reviewers for comments and criticisms; and Vincent Daubin, Manolo Gouy, and Eric Tannier for agreeing to share the reference tree. This work was funded by the Centre National de la Recherche Scientifique and the Institut Pasteur.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110972108/-/DCSupplemental.
References
- 1.Pál C, Papp B, Lercher MJ. An integrated view of protein evolution. Nat Rev Genet. 2006;7:337–348. doi: 10.1038/nrg1838. [DOI] [PubMed] [Google Scholar]
- 2.Rocha EPC. The quest for the universals of protein evolution. Trends Genet. 2006;22:412–416. doi: 10.1016/j.tig.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malmström J, et al. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature. 2009;460:762–765. doi: 10.1038/nature08184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taniguchi Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoebel DM, Dean AM, Dykhuizen DE. The cost of expression of Escherichia coli lac operon proteins is in the process, not in the products. Genetics. 2008;178:1653–1660. doi: 10.1534/genetics.107.085399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiler-Samerotte KA, et al. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akashi H. Synonymous codon usage in Drosophila melanogaster: Natural selection and translational accuracy. Genetics. 1994;136:927–935. doi: 10.1093/genetics/136.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rocha EPC, Danchin A. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol. 2004;21:108–116. doi: 10.1093/molbev/msh004. [DOI] [PubMed] [Google Scholar]
- 11.Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci USA. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bremer H, Dennis PP. Modulation of chemical composition and other parameters of the cell by growth rate. In: Neidhart FC, et al., editors. Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. Washington, DC: ASM Press; 1996. pp. 1553–1569. [Google Scholar]
- 13.Russell JB, Cook GM. Energetics of bacterial growth: Balance of anabolic and catabolic reactions. Microbiol Rev. 1995;59:48–62. doi: 10.1128/mr.59.1.48-62.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vijayan V, Jain IH, O'Shea EK. A high resolution map of a cyanobacterial transcriptome. Genome Biol. 2011;12:R47. doi: 10.1186/gb-2011-12-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maier T, et al. Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol Syst Biol. 2011;7:511. doi: 10.1038/msb.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gudelj I, et al. An integrative approach to understanding microbial diversity: from intracellular mechanisms to community structure. Ecol Lett. 2010;13:1073–1084. doi: 10.1111/j.1461-0248.2010.01507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch AL. Oligotrophs versus copiotrophs. Bioessays. 2001;23:657–661. doi: 10.1002/bies.1091. [DOI] [PubMed] [Google Scholar]
- 18.Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: The unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garbeva P, van Veen JA, van Elsas JD. Microbial diversity in soil: Selection microbial populations by plant and soil type and implications for disease suppressiveness. Annu Rev Phytopathol. 2004;42:243–270. doi: 10.1146/annurev.phyto.42.012604.135455. [DOI] [PubMed] [Google Scholar]
- 20.Scanlan DJ, et al. Ecological genomics of marine picocyanobacteria. Microbiol Mol Biol Rev. 2009;73:249–299. doi: 10.1128/MMBR.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vieira-Silva S, Rocha EPC. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 2010;6:e1000808. doi: 10.1371/journal.pgen.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rocha EPC. Codon usage bias from tRNA's point of view: Redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14:2279–2286. doi: 10.1101/gr.2896904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gribaldo S, Philippe H. Ancient phylogenetic relationships. Theor Popul Biol. 2002;61:391–408. doi: 10.1006/tpbi.2002.1593. [DOI] [PubMed] [Google Scholar]
- 24.Groussin M, Gouy M. Adaptation to environmental temperature is a major determinant of molecular evolutionary rates in archaea. Mol Biol Evol. 2011;28:2661–2674. doi: 10.1093/molbev/msr098. [DOI] [PubMed] [Google Scholar]
- 25.Moran NA. Accelerated evolution and Muller's rachet in endosymbiotic bacteria. Proc Natl Acad Sci USA. 1996;93:2873–2878. doi: 10.1073/pnas.93.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abby SS, Tannier E, Gouy M, Daubin V. Detecting lateral gene transfers by statistical reconciliation of phylogenetic forests. BMC Bioinformatics. 2010;11:324. doi: 10.1186/1471-2105-11-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciccarelli FD, et al. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 28.Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst Biol. 1978;27:401–410. [Google Scholar]
- 29.Lynch M. The origins of eukaryotic gene structure. Mol Biol Evol. 2006;23:450–468. doi: 10.1093/molbev/msj050. [DOI] [PubMed] [Google Scholar]
- 30.Philippe H, Zhou Y, Brinkmann H, Rodrigue N, Delsuc F. Heterotachy and long-branch attraction in phylogenetics. BMC Evol Biol. 2005;5:50. doi: 10.1186/1471-2148-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lartillot N, Poujol R. A phylogenetic model for investigating correlated evolution of substitution rates and continuous phenotypic characters. Mol Biol Evol. 2011;28:729–744. doi: 10.1093/molbev/msq244. [DOI] [PubMed] [Google Scholar]
- 32.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 33.Weiss M, Schrimpf S, Hengartner MO, Lercher MJ, von Mering C. Shotgun proteomics data from multiple organisms reveals remarkable quantitative conservation of the eukaryotic core proteome. Proteomics. 2010;10:1297–1306. doi: 10.1002/pmic.200900414. [DOI] [PubMed] [Google Scholar]
- 34.Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.