

Abstract
We develop and validate the XYGJ-OS functional, based on the adiabatic connection formalism and Görling-Levy perturbation theory to second order and using the opposite-spin (OS) ansatz combined with locality of electron correlation. XYGJ-OS with local implementation scales as N3 with an overall accuracy of 1.28 kcal/mol for thermochemistry, bond dissociation energies, reaction barrier heights, and nonbonded interactions, comparable to that of 1.06 kcal/mol for the accurate coupled-cluster based G3 method (scales as N7) and much better than many popular density functional theory methods: B3LYP (4.98), PBE0 (4.36), and PBE (12.10).
Keywords: ACM, DHDF, GGA, LDA , MAD
Obtaining chemical accuracy (∼1 kcal/mol) to quantify key chemical quantities (e.g., heats of formation, bond dissociation energies, and reaction barrier heights) using quantum mechanics (QM) has been a major focus in the development of the theory. This has led to, for example, the Gn method (1, 2) that approaches this chemical accuracy. Because G3 is a coupled-cluster based method, it scales on the order of N7, where N measures the system size, limiting to fairly small species for routine use.
The desire to predict unique physiochemical phenomena (e.g., solvation, catalysis, self-assembly, and drug design) in practical (large) systems has brought about a second major focus of theoretical development, leading, for example, to divide-and-conquer formulations to attain more efficient scaling (3), but with much lower accuracy than Gn.
Density functional theory (DFT) in the framework of Kohn-Sham (KS) scheme (4, 5) provides a “shortcut” to the many-body problem. Many density functional approximations (DFAs), provide typical scaling of N3 ∼ N4, while yielding significantly more accurate results than Hartree-Fock (HF) theory, the lowest level wave function based method with similar scaling, but they still lead to significant errors for some systems. For example, current DFAs lead to a poor description of London dispersion (van der Waals attraction), which is essential to predict the packing of molecules into solids, and the binding of drug molecules to proteins. These DFAs are also poor in predicting the magnitude of reaction barriers.
In this article, we develop and present a unique functional, XYGJ-OS, that provides a good combination of high accuracy and speed. XYGJ-OS involves a doubly hybrid density functional (DHDF), containing both a nonlocal orbital-dependent component in the exchange term (HF-like exchange), and also information about the unoccupied KS orbitals in the electron correlation part (PT2, perturbation theory up to second order) using the opposite-spin (OS) ansatz to include the locality of electron correlation. XYGJ-OS provides accuracy comparable to that of Gn for the test datasets and speed with N2 ∼ N3 for the local implementation. Hence XYGJ-OS is both accurate and fast.
Theory
The Holy Grail in KS-DFT is to find the exact, yet unknown, exchange-correlation functional Exc[ρ] using density ρ as the basic variable (4, 5). In practice, an approximate Exc must be adopted, which is often partitioned into the exchange and correlation parts
| [1] |
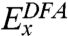 has been extended to include a portion of nonlocal
has been extended to include a portion of nonlocal 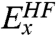 , where the superscript “HF” emphasizes that the exchange part has the same form as in Hartree-Fock theory (6). The exchange-correlation potential υxc in the nth cycle of the self-consistent-field (SCF) process to solve the KS equation is obtained as a functional of ρ of the previous cycle.
, where the superscript “HF” emphasizes that the exchange part has the same form as in Hartree-Fock theory (6). The exchange-correlation potential υxc in the nth cycle of the self-consistent-field (SCF) process to solve the KS equation is obtained as a functional of ρ of the previous cycle.
On the other hand, Görling and Levy (GL) (7, 8) argued that the same KS scheme should work as well in terms of KS orbitals φi and eigenvalues εi. GL proposed a formally exact KS scheme based on perturbation theory, where Exc was expressed as:
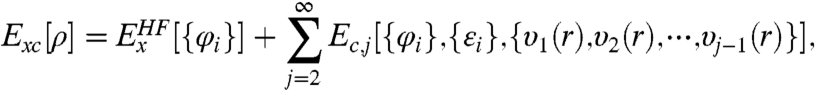 |
[2] |
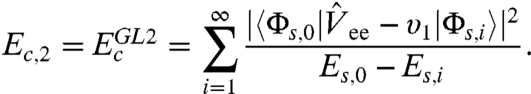 |
[3] |
Here 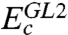 stands for the GL perturbation theory up to the second order, {Φs,0,Es,0} and {Φs,i,Es,i} are the wave function and energy for the ground state and the ith excited state of an N-electron KS system, respectively;
stands for the GL perturbation theory up to the second order, {Φs,0,Es,0} and {Φs,i,Es,i} are the wave function and energy for the ground state and the ith excited state of an N-electron KS system, respectively; 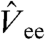 is the operator of electron-electron repulsion, and the υ1 potential may be determined from the “exchange-only” KS equation (8). With knowledge of the potentials υj(r), Eq. 2 gives the formally exact exchange-correlation energy as functional of the KS orbitals φi and eigenvalues εi, with which the approximate
is the operator of electron-electron repulsion, and the υ1 potential may be determined from the “exchange-only” KS equation (8). With knowledge of the potentials υj(r), Eq. 2 gives the formally exact exchange-correlation energy as functional of the KS orbitals φi and eigenvalues εi, with which the approximate 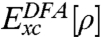 defined in [1] can be compared. However, in practice, such a procedure is difficult to apply to higher than second order due to the unfavorable scaling with the system size, and in many cases perturbation theory is nonconvergent. Thus the scheme must be simplified to make it applicable for including higher-order contributions.
defined in [1] can be compared. However, in practice, such a procedure is difficult to apply to higher than second order due to the unfavorable scaling with the system size, and in many cases perturbation theory is nonconvergent. Thus the scheme must be simplified to make it applicable for including higher-order contributions.
The adiabatic connection method [(ACM), ref. 9, 10], which defines a family of partially interacting N-electron systems with a coupling constant for a fixed ρ, provides a powerful tool for developing and understanding Exc (6, 11–13). ACM suggests that Eq. 2 up to second order, while being more appropriate for coupling close to zero, is only exact if the potential energy of exchange-correlation depends linearly on the coupling strength, whereas [1] is more associated with full electron-electron coupling (13, also see SI Text for more discussion). To go beyond the linear approximation and to take advantages of both [1] and Eq. 2, we define an empirical DHDF that combines Eq. 2 (j = 2) with [1]:
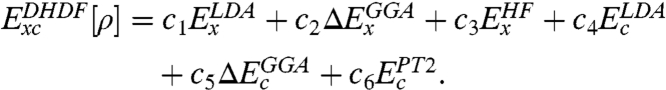 |
[4] |
Here 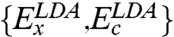 are the exchange and correlation components within the local density approximation (LDA), and
are the exchange and correlation components within the local density approximation (LDA), and 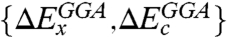 are the corresponding correction terms to LDA within the generalized gradient approximation (GGA). The meta-GGA functionals that include kinetic energy density or the Laplacian of density can also be used in place of GGAs. Such a combination suggests that only the energetically most important double excitation PT2 term in Eq. 2 need be calculated explicitly, the higher-order correlations are embedded into the parameterized terms such as
are the corresponding correction terms to LDA within the generalized gradient approximation (GGA). The meta-GGA functionals that include kinetic energy density or the Laplacian of density can also be used in place of GGAs. Such a combination suggests that only the energetically most important double excitation PT2 term in Eq. 2 need be calculated explicitly, the higher-order correlations are embedded into the parameterized terms such as 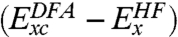 (12). Other examples of DHDFs include MC3BB (14), B2PLYP (15), B2GP-PLYP (16), and ωB97X-2 (17), which are derived and constructed differently (see below and SI Text for more discussion).
(12). Other examples of DHDFs include MC3BB (14), B2PLYP (15), B2GP-PLYP (16), and ωB97X-2 (17), which are derived and constructed differently (see below and SI Text for more discussion).
Several key issues distinguish our approach from GL perturbation theory (7, 8) and other DHDFs (14–23).
Instead of seeking for {φi,εi,υj} self-consistently and order by order via Eq. 2, we use a conventional DFA defined in [1] to generate {φi,εi} and to evaluate Exc defined in Eq. 4 in a post-SCF manner. In fact, the GL perturbation theory, Eq. 2, was built on KS orbitals with a local effective potential, while we have extended it to include hybrid functionals with generalized KS orbitals (12, 13).
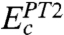 differs from
differs from 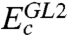 in that the singles contribution is not explicitly calculated, but we argue (12, 13) that it can be reasonably absorbed into the parameterization in Eq. 4. Higher-order contributions Ec,j (j > 2) are also implicitly included via the parameterization.
in that the singles contribution is not explicitly calculated, but we argue (12, 13) that it can be reasonably absorbed into the parameterization in Eq. 4. Higher-order contributions Ec,j (j > 2) are also implicitly included via the parameterization.Neglecting the nonlocal exchange-correlation effects
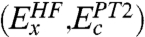 in Eq. 4 leads to a pure DFA, while neglecting the local exchange-correlation effects, Eq. 4 gives an approximation to GL2. Hence our DHDF may be regarded as an interpolation approach between a pure DFA and GL2, while both of them are in the framework of the KS scheme. As different orbitals are used to construct the PT2 term, neglecting the local exchange-correlation effects in other DHDFs (14–23) will bring back the conventional MP2, such that these DHDFs may be regarded as an interpolation approach between a DFA and MP2, the wave function based lowest level correlation method. These functionals go beyond the framework of the KS scheme (23).
in Eq. 4 leads to a pure DFA, while neglecting the local exchange-correlation effects, Eq. 4 gives an approximation to GL2. Hence our DHDF may be regarded as an interpolation approach between a pure DFA and GL2, while both of them are in the framework of the KS scheme. As different orbitals are used to construct the PT2 term, neglecting the local exchange-correlation effects in other DHDFs (14–23) will bring back the conventional MP2, such that these DHDFs may be regarded as an interpolation approach between a DFA and MP2, the wave function based lowest level correlation method. These functionals go beyond the framework of the KS scheme (23).
Assuming LDA and GGA in Eq. 4 are SVWN (24, 25) and BLYP (26, 27), respectively, we previously developed XYG3 (12), a DHDF which is remarkably accurate for a wide range of systems and important chemical properties (12, 13, 28–31). Nevertheless, the PT2 term in XYG3 and other DHDFs is evaluated in a way similar to MP2:
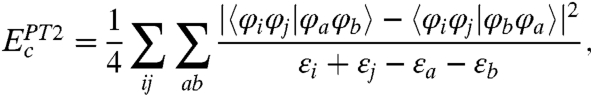 |
[5] |
where the subscripts (i, j) and (a, b) denote the occupied and unoccupied KS spin-orbitals, respectively, leading to a formal scaling as N5, as opposed to a formal scaling of N4 as in B3LYP (24–27, 32, 33). This unfavorable scaling raises an issue for the practicality to apply DHDFs to large systems.
Density fitting approximations have often been used in electronic structure theories to reduce computational expense. In the so-called “resolution-of-the-identity” RI-MP2 method (34, 35), the product of occupied and virtual orbitals (ia pair) is expanded with auxiliary functions, such that numerous 4-center 2-electron integrals based on molecular orbitals (MO) are replaced by fewer 3 and 2-center integrals with cheaper transformation from atomic orbitals (AO) to MO. RI-MP2 is about 5–20 times faster than conventional MP2 (36, 37). Nevertheless, RI-MP2 reduces only the prefactor, it does not change the scaling. Similarly, we have RI-XYG3, which retains the original accuracy but is faster for small systems with large basis sets.
Here we propose a unique OS ansatz for DHDF, that yields a balanced description of nonlocal correlation effects while considerably reducing computational time. Our OS ansatz is motivated by the observation that the most important electron correlation effects involve correlations of the OS electrons in the same orbital. The OS ansatz leads to N4 scaling (36, 37) [using auxiliary basis expansions (34, 35) and Laplace quadrature approximations (38)]. It should be emphasized that as the same-spin (SS) correlation is very important in accurate description of open-shell systems and magnetic properties, such contributions cannot be simply neglected. In XYGJ-OS, the SS correlation effects are included within the standard DFA.
In recognition of the “nearsightedness of electron correlation” as emphasized by Kohn (39), we then build upon XYGJ-OS to introduce the local approximation for the OS electron correlation by utilizing the sparsity of the RI expansion coefficients, integral matrices, and Laplace transform matrices. The local implementation of XYGJ-OS scales as N3 while retaining the accuracy of the original XYGJ-OS (see SI Text for more discussion).
Thus our proposed functional form (XYGJ-OS) is
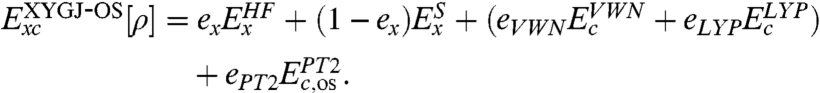 |
[6] |
In Eq. 6 we normalize the HF exchange and Slater exchange (24), while eliminating the 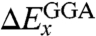 contribution. The correlation part consists of
contribution. The correlation part consists of 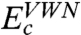 (25),
(25), 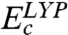 (27), and
(27), and 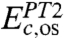 , where the first term includes both the SS and OS effects while the second and third terms include only OS components. Our concept is that the combination of VWN, LYP, and PT2-OS yields a balanced description of both local and nonlocal spin dependent correlation terms. To determine the optimal four parameters in Eq. 6, we use the experimental heats of formation (HOF) data for the G3/99 set of 223 molecules (1, 2) as the training set, leading to {ex,eVWN,eLYP,ePT2} = {0.7731,0.2309,0.2754,0.4364}.
, where the first term includes both the SS and OS effects while the second and third terms include only OS components. Our concept is that the combination of VWN, LYP, and PT2-OS yields a balanced description of both local and nonlocal spin dependent correlation terms. To determine the optimal four parameters in Eq. 6, we use the experimental heats of formation (HOF) data for the G3/99 set of 223 molecules (1, 2) as the training set, leading to {ex,eVWN,eLYP,ePT2} = {0.7731,0.2309,0.2754,0.4364}.
Results and Discussion
We emphasize that we use the fully optimized B3LYP orbitals to generate the density and to calculate each term in Eqs. 4 or 6 (12, 13, 29–31). But the choice of LDA = SVWN and GGA = BLYP in Eqs. 4 or 6, as well as using B3LYP orbitals as input, is not unique. We find that any conventional DFAs defined in [1] can also serve the same purpose, leading to similar performance, albeit with a reoptimized set of mixing parameters. In our method, we assume that our KS wave function is the zeroth-order wave function in the GL perturbation theory that gives the correct ground-state electron density. Instead, MC3BB is a multicoefficient method, which mixes the total energies from the conventional MP2 and a conventional DFA calculation. Hence there are two independent SCF calculations in the MC3BB type of DHDFs, which lead to two sets of different orbitals, yielding two different densities. In MC3BB, the SCF-HF orbitals are used for MP2 evaluation (14), while it is well known that HF wave function is the one-determinantal wave function which gives the lowest expectation value with the fully interacting Hamiltonian. It has been shown recently (23) that the B2PLYP family of functionals works in a way which is technically similar to MP2. These functionals adopt orbitals that minimize the one-determinantal wave function based on the so-called density-scaled one parameter hybrid approximation. Hence, the density from the B2PLYP type of functionals is by construction not meant to be the true density. In contrast to what was originally proposed (15), there is no singles’ contribution by construction in the B2PLYP family of functionals, and its theoretical basis is provided by the multideterminant extension of the Kohn-Sham scheme (23). On the other hand, the key idea of the XYG3 type of functionals is to combine the GL perturbation theory and the standard KS scheme in the framework of ACM. In XYG3, only the energetically most important double excitation PT2 term in Eq. 2 is calculated explicitly using orbitals generated from a conventional DFA in [1]. Our present OS ansatz further reduces the computational cost by only calculating the most important electron correlation effects contributed by the OS electrons in the same orbital. XYGJ-OS presents a unique combination of speed and accuracy. Hence, we propose that the current DHDFs shall be categorized into three types (13), as represented by MC3BB (14), B2PLYP (15), and XYG3 (12). The former two go beyond the KS scheme.
It is possible to carry out a SCF calculation for any orbital-dependent Exc using, for example, the direct optimization approach to compute the optimized effective potential (OEP) as proposed by Yang and coworkers (40). It was found that such a SCF procedure can often lead to unphysically unbound state with too low total energy mainly due to the near-degeneracy of the orbitals in the single excitation terms. This unbound issue can be largely remedied by calculating only the double excitation terms in the post-SCF manner (40). This procedure is indeed the calculation approach adopted by the XYG3 type of functionals. Furthermore, we note that extra terms will appear when formulating the analytical gradients for doing geometry optimization with our functionals, which, however, will not impose any practical difficulty in implementation as compared to other types of DHDFs.
We find that XYGJ-OS is remarkably accurate for a broad range of systems and important chemical properties (1, 2,41–45) other than HOF which are not used in fitting the parameters. Table 1 (more details are in SI Text) compares the overall performance of some representative DFT methods, showing that XYGJ-OS is the best or nearly best for essentially all properties, leading to chemical accuracy (1.28 kcal/mol) comparable to the G3 theory (which contains four empirical parameters involving the number of electron pairs) (1.06) and much better than MP2 (7.49 kcal/mol).
Table 1.
MAD, (in kcal/mol) for various benchmarks
| Methods |
HOF | IP | EA | PA | BDE | NHTBH | HTBH | NCIE | All | Time* |
|
| (223) |
(38) |
(25) |
(8) |
(92) |
(38) |
(38) |
(31) |
(493) |
C100H202 |
C100H100 |
|
| DFT methods | |||||||||||
| SVWN (LDA) | 130.88 | 15.14 | 17.30 | 5.68 | 18.14 | 12.53 | 17.95 | 3.29 | 67.28 | ||
| BLYP | 10.16 | 6.02 | 2.47 | 1.75 | 7.00 | 8.29 | 7.68 | 1.49 | 7.84 | ||
| PBE | 20.71 | 5.13 | 2.40 | 1.56 | 3.91 | 8.57 | 9.48 | 1.17 | 12.10 | ||
| TPSS | 5.01 | 5.36 | 2.41 | 1.66 | 5.88 | 9.04 | 8.26 | 1.14 | 5.33 | ||
| B3LYP | 6.08 | 3.74 | 2.45 | 1.40 | 5.51 | 4.84 | 4.26 | 0.98 | 4.98 | 2.8 | 12.3 |
| B3LYP-D3 | 4.15 | 3.77 | 2.47 | 1.18 | 4.29 | 5.17 | 4.97 | 0.64 | 3.93 | ||
| PBE0 | 5.64 | 3.84 | 2.97 | 1.25 | 3.67 | 3.56 | 4.38 | 0.71 | 4.36 | ||
| M06-2X | 2.26 | 2.72 | 2.37 | 1.94 | 1.40 | 1.26 | 1.25 | 0.28 | 1.86 | ||
| XYG3† | 1.81 | 1.31 | 1.84 | 1.61 | 1.57 | 1.29 | 0.75 | 0.32 | 1.51 | 200.0 | 81.4 |
| XYGJ-OS | 1.65 | 1.23 | 1.97 | 1.68 | 0.71 | 1.18 | 0.88 | 0.35 | 1.28 | 7.8 | 46.4 |
| MC3BB | 3.28 | 2.78 | 4.01 | 1.03 | 2.43 | 1.44 | 0.80 | 0.58 | 2.58 | ||
| B2PLYP‡ | 2.74 | 2.48 | 2.15 | 1.52 | 2.95 | 2.23 | 1.73 | 0.55 | 2.45 | ||
| B2PLYP-D‡ | 1.67 | 2.48 | 2.15 | 1.34 | 2.27 | 2.47 | 2.11 | 0.45 | 1.88 | ||
| ωB97X-2(LP)§ | 1.52 | 1.73 | 1.56 | 1.09 | 1.62 | 1.67 | 0.74 | 0.47 | 1.44 | ||
| Wavefunction based methods | |||||||||||
| HF | 213.42 | 23.19 | 26.46 | 3.09 | 32.70 | 9.08 | 13.51 | 2.37 | 107.71 | ||
| MP2 | 10.63 | 3.49 | 3.59 | 2.13 | 7.73 | 5.42 | 3.91 | 0.60 | 7.49 | ||
| G2 | 1.89 | 0.97 | 1.31 | 1.34 | 1.80 | 0.97 | 1.24 | 0.57 | 1.56 | ||
| G3 | 1.06 | 1.27 | 1.13 | 1.06 | 1.08 | 0.97 | 1.24 | 0.57 | 1.06 | ||
HOF is heat of formation (1, 2), IP is ionization potential (2), EA is electron affinity (2), PA is proton affinity (2), BDE is bond dissociation energy (40), NHTBH and HTBH are barrier heights for reactions (42, 43), NCIE is the binding in molecular clusters (42, 43). The basis sets used for final energetics are G3Large unless otherwise stated. See SI Text for computational details.
*The time is single CPU hours on 2.5 GHz Xeon for linear alkane C100H202 and the C100H100 diamond structure. The basis sets used are cc-pVDZ.
†Taken from ref. 12 with 6-311+G(3df,2p).
‡HOF are taken from ref. 22 with very large basis set of CQZV3P. BDEs are calculated using the corresponding HOF.
§Taken from ref. 17 or calculated with 6-311++G(3df,3pd). BSSE corrections are included for the NCIE set.
Heats of Formation.
The 223 molecule G3/99 set (1, 2) provides a test of accuracy for the main group covalent systems. XYGJ-OS gives mean absolute deviation (MAD) of 1.65 kcal/mol, lying between those of G2 (1.89) and G3 (1.06) theories. Note that the MADs for HOF listed in Table 1 associated with XYG3, B2PLYP, B2PLYP-D, and ωB97X-2(LP) are taken from the original refs. 12, 17, 22, which were obtained by using the way these functionals were parameterized. It was shown that HOF calculations with DHDFs are most prone to the basis set effects (29). The results with the G3Large basis set (1) used for optimization of the XYGJ-OS functional can be found in SI Text, where severe basis set dependences are clearly shown.
Charged Species.
Charged species are not included in our training set. The G2-1 set (2) for ionization potential (IP) contains 14 atoms and 24 molecules. XYGJ-OS gives a MAD of 1.23 kcal/mol, being one of the best DFT methods for calculating IP. Over the 25 cases in the G2-1 set for electron affinity (EA) (2), XYGJ-OS leads to MAD = 1.97 kcal/mol. Generally, increasing the size of the basis set will increase the accuracy for EA calculation with DHDFs. For the eight systems for proton affinity in the G2 and G3 sets (1), XYGJ-OS leads to MAD = 1.68 kcal/mol, comparable to the performance of conventional DFAs.
Bond Dissociation Enthalpy (BDE).
BDE is arguably the most important property for chemistry, carrying additional information other than HOFs (13). XYGJ-OS leads to MAD = 0.71 kcal/mol for the 92 reactions in the BDE92/07 set (40)! This accuracy surpasses the MAD of the most accurate ab initio methods, G2 (1.80) and G3 (1.09 kcal/mol). Indeed XYGJ-OS leads to half the MAD of XYG3 (1.57 kcal/mol), providing strong support for the OS ansatz and the local approximation.
Reaction Barrier Height (RBH).
We use the NHTBH and HTBH datasets of Zhao and Truhlar as testing sets (42, 43) of RBHs. NHTBH contains six heavy-atom transfer reactions, eight nucleophilic substitution reactions, five unimolecular and association reactions, while HTBH comprises solely 19 hydrogen transfer reactions. XYGJ-OS leads to MAD = 1.03 kcal/mol for the two sets together, a significant improvement over B3LYP (4.55 kcal/mol) and MP2 (4.67 kcal/mol). Note that the dispersion corrected methods [e.g., B3LYP-D3 (46)] may deteriorate the performance for RBH calculations.
We test various methods in describing the whole H + CH4 → H2 + CH3 reaction path using the coupled-cluster method with single and double as well as perturbative triple excitations (CCSD)(T)/6-311++G(3df,2pd) data (43) as the reference. The results are depicted in Fig. 1A. XYGJ-OS results are nearly identical to the XYG3 and CCSD(T) results before the barrier. But XYGJ-OS overestimates the reaction endothermicity by 1.21 kcal/mol.
Fig. 1.

(A) Potential energy curves for H + CH4 → H2 + CH3 reaction along the reaction coordinate of [R(CH)-R(HH)] (in Å). (B) Intermolecular potentials for CH4-C6H6 complex. R is the distance from the carbon of CH4 to the ring center of C6H6.
Nonbonded Interaction (NBI).
We use the NCIE dataset of Zhao and Truhlar (42, 43) to test the XYGJ-OS performance for the description of NBIs. The NCIE set includes six hydrogen bond complexes, seven charge-transfer complexes, six dipole interaction complexes, seven weak interaction complexes, and five π-π stacking complexes. XYGJ-OS (MAD = 0.35 kcal/mol) does quite well for NBI, including the London dispersion dominant systems in the NCIE dataset. For the S22 set (47), which contains larger molecules (e.g., uracil dimer, adenine-thymine complexes) that are more biologically related, XYGJ-OS (MAD = 0.36 kcal/mol) leads to similar accuracy as the dispersion corrected methods (22, 46). On average, B3LYP-D3 improves B3LYP by 0.34 kcal/mol for the NCIE set. While significant improvements occur for the weak interaction complexes, and the π-π stacking complexes, B3LYP-D3 significantly overbinds the hydrogen bond complexes, and the charge-transfer complexes (see SI Text for more discussion).
We test various methods in describing the intermolecular potentials of the CH4-C6H6 complex calculated by various methods. The CCSD(T) results at the complete basis set (CBS) limit are used as reference (44). As shown in Fig. 1B, XYGJ-OS data are nearly on top of those of XYG3.
Scaling.
Equally important is that XYG3 and other DHDFs (15–19) scale formally with the fifth power of the size, while XYGJ-OS scales formally with the fourth power, and for the local implementation it scales formally with the third power. In contrast, the most accurate ab initio methods, CCSD(T), and G3, scale as the seventh power. Indeed, Table 1 shows that the total computational time for C100 chains and diamondoids with local XYGJ-OS is just 3 to 4 times that for B3LYP (a part of XYGJ-OS), with a scaling of N2.1for n-alkanes (See SI Text for more details).
Summary
We have developed and validated here a unique doubly hybrid density functional, XYGJ-OS, using Walter Kohn’s insight about “nearsightedness” of electron correlation by including explicitly only the correlation between electrons of OS and then only the parts that are in the same region of space. We show that XYGJ-OS achieves nearly chemical accuracy (1.28 kcal/mol) with computational costs scaling as N3. This unique combination of high accuracy and speed leads to a practical level of calculation while attaining chemical accuracy for large molecular systems.
Supplementary Material
Acknowledgments.
X.X. acknowledges the support of NSF of China (91027044, 21133004, 20923004), and the Ministry of Science and Technology of China (2007CB815206, 2011CB808505). Y.J. acknowledges the support of National Research Foundation (NRF) of Korea (2010-0023018, 2010-0029034, and 2010-0029728) by the Ministry of Education, Science and Technology (MEST) of Korea. W.A.G. acknowledges the support of USA National Science Foundation (NSF) (ECS-0609128, CTS-0608889), the Center for Catalytic Hydrocarbon Functionalization (Department of Energy, Basic Energy Sciences Award DE-SC0001298), and Office of Naval Research-Defense Advanced Research Projects Agency [ONR-DARPA_ (PROM N00014-06-1-0938 and N00014-05-1-0778)]. Y.J. and W.A.G. have also been supported by the World Class University (WCU) (NRF R-31-2008-000-10055-0) program funded by the Ministry of Education, Science and Technology.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115123108/-/DCSupplemental.
References
- 1.Curtiss LA, Redfern PC, Raghavachari K, Pople JA. Gaussian-3X (G3X) theory: use of improved geometries, zero-point energies, and Hartree-Fock basis sets. J Chem Phys. 2001;114:108–117. [Google Scholar]
- 2.Curtiss LA, Redfern PC, Raghavachari K, Pople JA. Assessment of Gaussian-2 and density functional theories for the computation of ionization potentials and electron affinities. J Chem Phys. 1998;109:42–55. [Google Scholar]
- 3.Yang WT. Direct calculation of electron density in density-functional theory. Phys Rev Lett. 1991;66:1438–1441. doi: 10.1103/PhysRevLett.66.1438. [DOI] [PubMed] [Google Scholar]
- 4.Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev B. 1964;3:864–871. [Google Scholar]
- 5.Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev A. 1965;4:1133–1138. [Google Scholar]
- 6.Becke AD. A new mixing of Hartree-Fock and local density-functional theories. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 7.Görling A, Levy M. Correlation-energy functional and its high-density limit obtained from a coupling-constant perturbation expansion. Phys Rev B. 1993;47:13105–13113. doi: 10.1103/physrevb.47.13105. [DOI] [PubMed] [Google Scholar]
- 8.Görling A, Levy M. Exact Kohn-Sham scheme based on perturbation theory. Phys Rev A. 1994;50:196–204. doi: 10.1103/physreva.50.196. [DOI] [PubMed] [Google Scholar]
- 9.Langreth DC, Perdew JP. Exchange-correlation energy of a metallic surface: wave-vector analysis. Phys Rev B. 1977;15:2884–2902. [Google Scholar]
- 10.Gunnarsson O, Lundqvist B. Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism. Phys Rev B. 1976;13:4274–4298. [Google Scholar]
- 11.Mori-Sanchez P, Cohen AJ, Yang WT. Self-interaction-free exchange-correlation functional for thermochemistry and kinetics. J Chem Phys. 2006;124:1–4. doi: 10.1063/1.2179072. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Xu X, Goddard W A., III Doubly hybrid density functional for accurate description of nonbond interactions, thermochemistry, and thermochemical kinetics. Proc Natl Acad Sci USA. 2009;106:4963–4968. doi: 10.1073/pnas.0901093106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang IY, Xu X. Doubly hybrid density functional for accurate description of thermochemistry, thermochemical kinetics and nonbonded interactions. Int Rev Phys Chem. 2011;46:115–160. [Google Scholar]
- 14.Zhao Y, Lynch BJ, Truhlar DG. Doubly hybrid meta DFT: new multi-coefficient correlation and density functional methods for thermochemistry and thermochemical Kkinetics. J Phys Chem A. 2004;108:4786–4791. [Google Scholar]
- 15.Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J Chem Phys. 2006;124:1–16. doi: 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- 16.Karton A, Tarnopolsky A, Lamère J-F, Schatz GC, Martin JML. Highly accurate first-principles benchmark datasets for the parametrization and validation of density functional and other approximate methods Derivation of a robust, generally applicable, double-hybrid functional for thermochemistry and thermochemical kinetics. J Phys Chem A. 2008;112:12868–12886. doi: 10.1021/jp801805p. [DOI] [PubMed] [Google Scholar]
- 17.Chai JD, Head-Gordon M. Long-range corrected double-hybrid density functionals. J Chem Phys. 2009;131:1–13. doi: 10.1063/1.3244209. [DOI] [PubMed] [Google Scholar]
- 18.Sancho-García JC, Pérez-Jiménez AJ. Assessment of double-hybrid energy functionals for π-conjugated systems. J Chem Phys. 2009;131:1–11. doi: 10.1063/1.3212881. [DOI] [PubMed] [Google Scholar]
- 19.Graham DC, Menon AS, Goerigk L, Grimme S, Radom L. Optimization and basis-set dependence of a restricted-open-shell form of B2-PLYP double-hybrid density functional theory. J Phys Chem A. 2009;113:9861–9873. doi: 10.1021/jp9042864. [DOI] [PubMed] [Google Scholar]
- 20.Benighaus T, DiStasio RA, Lochan R, Chai JD, Head-Gordon M. Semiempirical double-hybrid density functional with improved description of long-range correlation. J Phys Chem A. 2008;112:2702–2712. doi: 10.1021/jp710439w. [DOI] [PubMed] [Google Scholar]
- 21.Schwabe T, Grimme S. Towards chemical accuracy for the thermodynamics of large molecules: new hybrid density functionals including non-local correlation effect. Phys Chem Chem Phys. 2006;8:4398–4401. doi: 10.1039/b608478h. [DOI] [PubMed] [Google Scholar]
- 22.Schwabe T, Grimme S. Double-hybrid density functionals with long-range dispersion corrections: higher accuracy and extended applicability. Phys Chem Chem Phys. 2007;9:3397–3406. doi: 10.1039/b704725h. [DOI] [PubMed] [Google Scholar]
- 23.Sharkas K, Toulouse J, Savin A. Double-hybrid density-functional theory made rigorous. J Chem Phys. 2011;134:1–9. doi: 10.1063/1.3544215. [DOI] [PubMed] [Google Scholar]
- 24.Dirac PAM. Note on exchange phenomena in the Thomas atom. Math Proc Cambridge. 1930;26:376–385. [Google Scholar]
- 25.Vosko SH, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys. 1980;58:1200–1211. [Google Scholar]
- 26.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 27.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 28.Zhang IY, Luo Y, Xu X. XYG3s: speedup of the XYG3 fifth-rung density functional with scaling-all-correlation method. J Chem Phys. 2010;132:1–11. doi: 10.1063/1.3424845. [DOI] [PubMed] [Google Scholar]
- 29.Zhang IY, Luo Y, Xu X. Basis set dependence of the doubly hybrid XYG3 functional. J Chem Phys. 2010;133:1–12. doi: 10.1063/1.3488649. [DOI] [PubMed] [Google Scholar]
- 30.Zhang IY, Wu JM, Luo Y, Xu X. Trends in R-X bond dissociation energies (R• = Me, Et, i-Pr, t-Bu, X• = H, Me, Cl, OH) J Chem Theory Comput. 2010;6:1462–1469. doi: 10.1021/ct100010d. [DOI] [PubMed] [Google Scholar]
- 31.Zhang IY, Wu JM, Xu X. Extending the reliability and applicability of B3LYP. Chem Commun. 2010;46:3057–3070. doi: 10.1039/c000677g. [DOI] [PubMed] [Google Scholar]
- 32.Becke AD. Density-functional thermochemistry III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 33.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 34.Feyereisen M, Fitzgerald G, Komornicki A. Use of approximate integrals in ab initio theory. An application in MP2 energy calculations. Chem Phys Lett. 1993;208:359–363. [Google Scholar]
- 35.Weigend F, Haser M, Patzelt H, Ahlrichs R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem Phys Lett. 1998;294:143–152. [Google Scholar]
- 36.Jung YS, Lochan RC, Dutoi AD, Head-Gordon M. Scaled opposite-spin second order Møller-Plesset correlation energy: an economical electronic structure method. J Chem Phys. 2004;121:9793–9802. doi: 10.1063/1.1809602. [DOI] [PubMed] [Google Scholar]
- 37.Jung YS, Shao YH, Head-Gordon M. Fast evaluation of scaled opposite spin second-order Møller-Plesset correlation energies using auxiliary basis expansions and exploiting sparsity. J Comput Chem. 2007;28:1953–1964. doi: 10.1002/jcc.20590. [DOI] [PubMed] [Google Scholar]
- 38.Almlöf J. Elimination of energy denominators in Møller-Plesset perturbation theory by a Laplace transform approach. Chem Phys Lett. 1991;181:319–320. [Google Scholar]
- 39.Kohn W. Density functional and density matrix method scaling linearly with the number of atoms. Phys Rev Lett. 1996;76:3168–3171. doi: 10.1103/PhysRevLett.76.3168. [DOI] [PubMed] [Google Scholar]
- 40.Mori-Sánchez P, Wu Q, Yang WT. Orbital-dependent correlation energy in density-functional theory based on a second-order perturbation approach: success and failure. J Chem Phys. 2005;123:1–14. doi: 10.1063/1.1904584. [DOI] [PubMed] [Google Scholar]
- 41.Wu JM, Xu X. The X1 method for accurate and efficient prediction of heats of formation. J Chem Phys. 2007;127:1–8. doi: 10.1063/1.2800018. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Truhlar DG. Density functionals with broad applicability in chemistry. Theor Chem Acc. 2008;120:215–241. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, Truhlar DG. Design of density functionals that are broadly accurate for thermochemistry, thermochemical kinetics, and nonbonded interactions. J Phys Chem A. 2005;109:5656–5667. doi: 10.1021/jp050536c. [DOI] [PubMed] [Google Scholar]
- 44.Zhang LL, Lu YP, Lee S-Y, Zhang DH. A transition state wave packet study of the H + CH4 reaction. J Chem Phys. 2007;127:1–7. doi: 10.1063/1.2812553. [DOI] [PubMed] [Google Scholar]
- 45.Takatani T, Sherrill CD. Performance of spin-component-scaled Møller-Plesset theory (SCS-MP2) for potential energy curves of noncovalent interactions. Phys Chem Chem Phys. 2007;9:6106–6114. doi: 10.1039/b709669k. [DOI] [PubMed] [Google Scholar]
- 46.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132:1–19. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 47.Jurecka P, Sponer J, Cerny J, Hobza P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys Chem Chem Phys. 2006;8:1985–1993. doi: 10.1039/b600027d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.