

Abstract
Studies of liquid water in its supercooled region have helped us better understand the structure and behavior of water. Bulk water freezes at its homogeneous nucleation temperature (approximately 235 K), but protein hydration water avoids this crystallization because each water molecule binds to a protein. Here, we study the dynamics of the hydrogen bond (HB) network of a percolating layer of water molecules and compare the measurements of a hydrated globular protein with the results of a coarse-grained model that successfully reproduces the properties of hydration water. Using dielectric spectroscopy, we measure the temperature dependence of the relaxation time of proton charge fluctuations. These fluctuations are associated with the dynamics of the HB network of water molecules adsorbed on the protein surface. Using Monte Carlo simulations and mean-field calculations, we study the dynamics and thermodynamics of the model. Both experimental and model analyses are consistent with the interesting possibility of two dynamic crossovers, (i) at approximately 252 K and (ii) at approximately 181 K. Because the experiments agree with the model, we can relate the two crossovers to the presence at ambient pressure of two specific heat maxima. The first is caused by fluctuations in the HB formation, and the second, at a lower temperature, is due to the cooperative reordering of the HB network.
Keywords: hydrated proteins, model calculations, dielectric relaxation, water dynamics, water specific heat
Recent experiments have studied water in the first hydration shell of globular proteins (1–5). Unlike bulk water, this water does not freeze until the temperature T is well below 235 K (6), a property that may be essential to biological functioning (7). Although quasi-elastic neutron scattering investigations (1) and molecular dynamics simulations (8, 9) support the presence of a dynamic crossover at approximately 220 K, other experiments and simulations do not (2–4, 10). It has been demonstrated that the suggested crossover could be related to the anomalous behavior of water, but that it is independent of any possible liquid–liquid critical point at finite T (11).
Here, we show by experiments, simulations, and model calculations that the dynamic properties of the hydrogen bond (HB) network at the protein–water interface exhibit not one, but two dynamic crossovers in the one-phase region at low pressure. We show how the two crossovers are related to the thermodynamics of water. We investigate the dielectric relaxation time of water protons, due to charged defects—such as H3O+—moving with a diffusive or hopping mechanism along the HB network (6, 12). These measurements are a sensitive probe for HB breaking and formation (13). We perform dielectric relaxation experiments on lysozyme powder with hydration level h = 0.30 g H2O/g dry protein, over a broad frequency (10-2 s-1–108 s-1) and temperature range (150 K ≤ T ≤ 300 K). The experimental setup and the data analysis (14–18) are described in Methods and SI Text.
In the dielectric spectrum for lysozyme powder at 215 K (Fig. 1), we identify (i) a low-frequency tail, (ii) a relatively broad process at intermediate frequencies, and (iii) a small high-frequency relaxation. The low-frequency tail (i) is due to electrode polarization, to interfacial dispersion (also known as the Maxwell–Wagner effect), and to sample conductivity (see Methods). The high-frequency process (iii) has a relaxation time with a T dependence and absolute values identical to recent dielectric measurements on the same protein with the same water content h (3, 4). This process is labeled “main” in the literature cited above, and we will be using this terminology (see Methods and SI Text). This relaxation is undetectable at hydration levels below h ∼ 0.3 g H2O/g dry protein, and we assign it to a local relaxation of protein hydration water (3), although its assignment and temperature dependence are controversial (19, 20).
Fig. 1.

Dielectric relaxation data for lysozyme powder at 215 K and hydration level h = 0.30 g H2O/g dry protein. (A) ϵ′ (◯) and (B) ϵ″ (▵) are real and imaginary components, respectively, of the complex permittivity 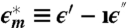 , and ω is the angular frequency. Solid lines through symbols result from the fitting procedure in the complex plane (14–18), described in Methods and SI Text. As shown in B for ϵ″,
, and ω is the angular frequency. Solid lines through symbols result from the fitting procedure in the complex plane (14–18), described in Methods and SI Text. As shown in B for ϵ″, 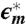 is resolved into (i) a low-ω tail (omitted for clarity), (ii) a broad process at intermediate ω deconvoluted into two relaxations (continuous and dotted lines), and (iii) a high-ω relaxation (dashed line).
is resolved into (i) a low-ω tail (omitted for clarity), (ii) a broad process at intermediate ω deconvoluted into two relaxations (continuous and dotted lines), and (iii) a high-ω relaxation (dashed line).
The broad relaxation process (ii), whose width at half maximum is about 3.5 frequency decades, can be resolved into two contributions (14), relatively close in frequency and largely overlapping but with a markedly different T dependence (Fig. 2). This decomposition has not been discussed in previous work (3, 4, 7), but a quantitative test for the presence of two relaxation processes is described in detail in ref. 14 (see Methods and SI Text). The quality of the two-relaxation fit, the T dependence, the h dependence, and the shape of each relaxation strongly suggest that two separate relaxations are present (see SI Text). Here, we label these processes “side-chain” relaxation and “proton” relaxation (Fig. 1). This labeling is based on previous studies of the T dependence and h dependence of the dielectric response of the same protein and of a similar globular protein (myoglobin). In particular, the side-chain process has a relaxation time in which the T dependence and absolute values agree with those measured by others for hydrated lysozyme powders (3) and hydrated myoglobin (7) (see Methods and SI Text). The side-chain relaxation has a symmetric shape over the entire temperature range investigated (see Fig. 3), in agreement with previous findings (3). This observation provides additional support for distinguishing the side-chain relaxation from the more asymmetric proton relaxation. Moreover, the wide temperature and frequency ranges investigated allow us to follow these two processes carefully and to identify them even when they are largely overlapping. The fitting parameters for each process, such as its relaxation time and shape parameters, change gradually with temperature, and this allows us to accurately distinguish between side-chain relaxation and proton relaxation at all temperatures.
Fig. 2.

Temperature dependence of the characteristic times of the relaxation processes shown in Fig. 1: side-chain relaxation (◯) and proton relaxation (•) of the intermediate-ω relaxation (ii), and “main” relaxation (▴) of the high-ω relaxation (iii). Solid lines are Arrhenius fits.
Fig. 3.

Imaginary component ϵ″(ω) of the measured complex permittivity as a function of the angular frequency ω, at three temperatures, (A) T = 186 K, (B) T = 215 K, and (C) T = 245 K. Solid lines through the symbols are the result of the fitting procedure in the complex plane (see Methods and SI Text). Here, we show the results of the decomposition into two processes of the broad relaxation peak. Dashed lines represent the side-chain relaxation, and thick solid lines represents the proton relaxation. The fitting procedure yields a β1 ≈ 1 (see Eq. 6) for the side-chain relaxation over the entire temperature range investigated, resulting in a symmetric relaxation process. Conversely, the same procedure gives a β2 < 1 for the proton relaxation, resulting in a characteristic asymmetric shape. We omit for clarity the contributions due to sample conductivity and to electrode polarization at low frequency.
The proton relaxation that contributes to the broad peak in Fig. 1 is the focus of our work here, and it has been extensively studied (6, 14–18) at hydration h < 0.30 g H2O/g dry protein. At these hydration levels, we find that the side-chain relaxation is indeed quite small and that it rapidly slows with dehydration, in agreement with ref. 3. The measured dielectric spectra for lysozyme at h < 0.30 g H2O/g dry protein is thus dominated by the proton relaxation (6, 14–18). Its characteristic relaxation time and associated dc conductivity has been described in terms of percolation theory (6, 15). Its assignment to water-assisted proton displacements over the protein surface has been tested by measuring hydrogenated and deuterated samples (6, 16), and its quantum character has been checked experimentally using dielectric spectroscopy and deep inelastic neutron scattering (16, 21), and the results agree with theoretical models (22).
At hydration h < 0.30 g H2O/g dry protein, the proton relaxation process has a characteristic relaxation time whose temperature dependence is well described by the Vogel–Fulcher–Tamman (VFT) equation τ(T) = τ0 exp[BT/R(T - T0)], where τ0, BT, and T0 are fitting parameters and R is the gas constant. Instead, for the sample water content, h = 0.30 g H2O/g dry protein, the proton relaxation time τ at high T shows a VFT behavior, with a clear kink at T ≈ 252 K (Fig. 4A), where we find a crossover from one high-T VFT behavior to a second VFT behavior at a lower T. We associate the crossover at ≈252 K with a change in the diffusion regime of water protons, from subdiffusive at lower T to freely diffusive at higher T, which has been previously reported for a lysozyme sample at h = 0.26 g H2O/g dry protein and 260 K (15). Note that the VFT description at high temperature of the water proton relaxation time is characterized by the same T0 ∼ 200 K independent of the sample hydration (14–16). Moreover, τ would reach 100 s at approximately 220 K (Fig. 4A), which is often defined as the dielectric glass transition temperature. This value is in good agreement with the calorimetric glass transition temperature of the hydrated protein at the same hydration level (3, 4, 13, 15, 17, 18).
Fig. 4.

Two crossovers in the proton relaxation time τ of hydration water. We find a non-Arrhenius to non-Arrhenius crossover at T ≈ 252 K and a non-Arrhenius to Arrhenius crossover at T ≈ 181 K. (A) Experimental τ (•) vs. 1/T. Solid line is the VFT function with fitting parameters τ0 = 7.8 × 10-12 s, BT = 9.4 kJ/mol, T0 = 180 K. Dotted line is the VFT function with τ0 = 6.5 × 10-8 s, BT = 6.2 kJ/mol, T0 = 140 K. Dashed line is the Arrhenius function with fitting parameters τ0 = 1.1 × 10-7 s, A = 25.2 kJ/mol. The behaviors at high and low T intersect at about 232 K. Relative errors (not shown) on τ are about 0.2% for 1/T < 6 × 10-3 K-1 and are at least one order of magnitude larger for lower T (see SI Text for more details). (B) MC relaxation time τMC (•) vs. 1/T, for P = 0.1 MPa. Solid line is the VFT function with τ0 = 1.61 × 10-8 s, BT = 5.2 kJ/mol, T0 = 181.2 K. Dotted line is the VFT function with τ0 = 7.5 × 10-10 s, BT = 15.9 kJ/mol, T0 = 95.2 K. Dashed line is the Arrhenius function with τ0 = 3.3 × 10-4 s, A = 13.7 kJ/mol. See the text for the discussion about the quantitative differences between the numerical and the experimental results.
At T ≈ 181 K we observe a second crossover, as the T dependence of τ changes from the VFT above 181 K to Arrhenius τ(T) = τ0 exp(A/RT) below 181 K, where τ0 is a characteristic relaxation time and A a constant activation energy (Fig. 4A). To our knowledge, the crossover at approximately 181 K has not been previously reported. Here, we offer a possible interpretation based on the results of the model described below, and we attribute this crossover to a structural rearrangement of the HB network.
Neutron scattering experiments on the same hydrated protein have also revealed two dynamical transitions, one at 220 K and the other at 150 K (23). The transition at 220 K has been attributed to the rotational motion of interfacial water, and the one at 150 K has been attributed to proton dynamics on a very local scale (of the order of a few angstroms). The low temperature transition (150 K) has been attributed to a sudden increase in the configurational entropy of the system, linked to a significant change in the HB length (23). Deep inelastic neutron scattering experiments on the same hydrated protein do not support this claim because they do not find changes in the HB length or proton potential below 290 K (16, 21).
In order to study the microscopic mechanisms responsible for the dynamic crossovers observed at 252 K and 181 K, we use a coarse-grained model of a monolayer of water adsorbed on a generic inert substrate, which represents the low-hydrated protein powder. At this hydration level, the adsorption is such that water molecules are restricted to a surface geometry with a coordination number up to four (24). The temperature of their structural arrest is ≫250 K (25), hence they do not diffuse. Because water molecules do not crystallize (25), their positions and orientations fluctuate and they form and break HBs. These assumptions are based on the observation that, at the relatively low h value investigated, we have less than one monolayer of water molecules covering the protein surface, and the protein itself does not undergo any configurational transformation or large scale motion (6, 25). This model, originally proposed in ref. 26 and extensively studied, e.g., in refs. 11 and 27–32, reproduces the known properties of water at interfaces, including the shape of the locus of the temperatures of maximum density in the (T,P) plane, the anomalous behavior of thermodynamic response functions, the subdiffusive regime at low T for protein hydration water, and the occurrence of minima and maxima in diffusivity upon pressurization (33). It has also predicted behaviors for protein hydration water, successively verified by experiments (11, 34).
The model discretizes the coordinates of the water molecules in the monolayer into N cells, each containing one molecule, with a volume given by the inverse of the average water density and a height given by the monolayer thickness d. To take into account the change of configurational entropy upon HB formation, the model associates each molecule i with four variables σij = 1,2,…,q with the index j running among the four neighbor molecules and with a discrete number of states q describing the bonding state of the molecule i with the neighbor molecule j. The model chooses q by adopting the standard convention that 30° is the maximum deviation of a HB from a linear bond (i.e., q ≡ 180°/30° = 6). For every molecule there are q4 = 64 ≡ 1,296 total possible bonding states. The system is fully described by the average density V/N and the set of σij.
The model separates the interactions among molecules into three components. The first is the sum of all isotropic interactions (e.g., van der Waals) between molecules at a distance r ≡ (V/Nd)1/2, and is represented by a Lennard–Jones potential, U0(r) = ϵ[(r/r0)12 - (r/r0)6]. On the basis of previous experiments, the model uses attractive energy ϵ = 5.8 kJ/mol (35) and r0 ≡ (v0/d)1/2 = 2.9 Å (36).
The second component is the directional component of the HB interaction. Neighboring molecules i and j form a HB when their facing bonding variables are in the same state (i.e., σij = σji). When a bond is formed, there is a J = 2.9 kJ/mol decrease in local energy, and a vHB increase in local volume. Based on the change of density between tetrahedral ice Ih and the interpenetrating tetrahedral network in ice VI or ice VIII, the model uses vHB/v0 = 0.5. In real water the formation of HBs does not imply a larger separation between molecules but only a larger excluded volume per molecule. Therefore, in the model the increase vHB does not affect the U0(r) term but only the total enthalpy with a contribution -(J - PvHB)NHB, where P is pressure and NHB is the number of HBs in the system. NHB is a function of the configuration of variables σij
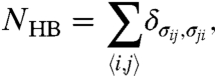 |
[1] |
where 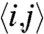 indicates nearest neighbors and δa,b = 1 if a = b or δa,b = 0 if a ≠ b.
indicates nearest neighbors and δa,b = 1 if a = b or δa,b = 0 if a ≠ b.
The final interaction is the cooperative (i.e., many-body) interaction among HBs, which gives rise to an O–O–O correlation (37) that drives the molecules toward a local ordered configuration. This is modeled using an interaction among the four σij belonging to the same molecule, driving them toward the same state. For each molecule, there is a local energy decrease of Jσ = 0.29 kJ/mol for each of the six possible pairs of σij in the same state.
The enthalpy of this model at pressure P is (26–28)
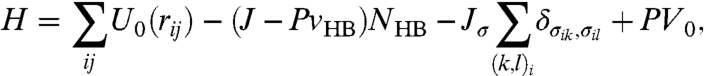 |
[2] |
where the first sum is over all pairs of molecules (i,j), the second sum is over all pairs of σik belonging to molecule i, and V0⩾Nv0 is the water volume apart from the contribution of the HBs.
We perform Monte Carlo (MC) simulations at constant N = 104, P and T, where V0 fluctuates and the configuration of variables σij changes, and therefore NHB also changes. The result is a variable total volume V ≡ V0 + NHBvHB. We explore the thermodynamics of the system in the P–T plane, confirming previous results (11, 26–31).
We next study the dynamic evolution of the system by adopting the single-spin flip Metropolis algorithm, which corresponds to Model A in the classification used by Hohenberg and Halperin (38). The most straightforward quantity to compare with the experiments considered here is the autocorrelation function
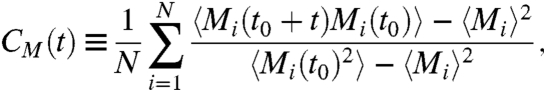 |
[3] |
where t is the time measured in MC steps, t0 is a time larger than the equilibration time of the system, and 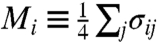 quantifies the order of the four bond indices σij of molecule i. Defining the MC relaxation time τMC from CM(τMC) = 1/e, we observe two crossovers in the computed τMC, from VFT to VFT at T ≈ 252 K (in ref. 11 this crossover was estimated as VFT to Arrhenius due to the limited resolution at low T), and from VFT to Arrhenius at T ≈ 181 K (Fig. 4B) (to compare with experiments, MC T is offset of 134.5 K, MC P is rescaled by 5 and offset of -1 × 10-4 GPa). Fig. 4 shows that the MC results agree well with the experimental data (with some difference at high T, which we will discuss below). The agreement suggests that the model is able to adequately describe the dynamics and connectivity of a real HB network and is thus a useful tool when investigating the thermodynamic origin of the crossovers.
quantifies the order of the four bond indices σij of molecule i. Defining the MC relaxation time τMC from CM(τMC) = 1/e, we observe two crossovers in the computed τMC, from VFT to VFT at T ≈ 252 K (in ref. 11 this crossover was estimated as VFT to Arrhenius due to the limited resolution at low T), and from VFT to Arrhenius at T ≈ 181 K (Fig. 4B) (to compare with experiments, MC T is offset of 134.5 K, MC P is rescaled by 5 and offset of -1 × 10-4 GPa). Fig. 4 shows that the MC results agree well with the experimental data (with some difference at high T, which we will discuss below). The agreement suggests that the model is able to adequately describe the dynamics and connectivity of a real HB network and is thus a useful tool when investigating the thermodynamic origin of the crossovers.
Next, we discuss the thermodynamic interpretation of the crossover in the model. Ref. 11 shows by direct calculations of the model in Eq. 2 that a maximum in isobaric specific heat, CP(T) ≡ (∂H/∂T)P, implies a crossover in the temperature dependence of τ. This result is consistent with the Adam–Gibbs theory (39). In the present work we find the CP maximum observed in ref. 11, and also another maximum at a lower T, in a region not explored in ref. 11 (Fig. 5A). To understand the origin of the two CP maxima, we write the enthalpy as the sum of two terms H = HHB + HCoop, where 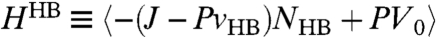 and HCoop ≡ H - HHB. We define the HB contribution to the specific heat
and HCoop ≡ H - HHB. We define the HB contribution to the specific heat 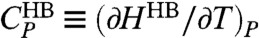 , and the cooperative contribution
, and the cooperative contribution 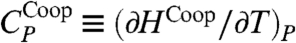 .
. 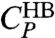 is responsible for the broad maximum at higher T (Fig. 5A). To show that
is responsible for the broad maximum at higher T (Fig. 5A). To show that 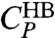 captures the enthalpy fluctuations due to the HB formation, we calculate the locus of maximum fluctuation of
captures the enthalpy fluctuations due to the HB formation, we calculate the locus of maximum fluctuation of 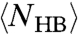 , related to the maximum of
, related to the maximum of 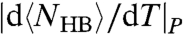 . The temperatures of these maxima coincide with the locus of maxima of
. The temperatures of these maxima coincide with the locus of maxima of 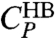 (Fig. 5B).
(Fig. 5B).
Fig. 5.

Two maxima in the specific heat for the model. (A) The MC isobaric specific heat CP (•), at P = 0.1 MPa has two maxima, decomposed into the components 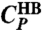 (□) and
(□) and 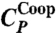 (⋄) described in the text. (B)
(⋄) described in the text. (B) 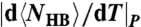 (□) and
(□) and 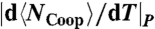 (⋄) show maxima where
(⋄) show maxima where 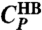 and
and 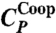 , respectively, have maxima. (C) MF calculations at P = 0 for the case with cooperative interaction (Jσ/ϵ = 0.05, solid line labeled as
, respectively, have maxima. (C) MF calculations at P = 0 for the case with cooperative interaction (Jσ/ϵ = 0.05, solid line labeled as 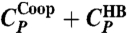 ) show two maxima for CP, whereas for the case without cooperative interaction (Jσ = 0, dashed line labeled as
) show two maxima for CP, whereas for the case without cooperative interaction (Jσ = 0, dashed line labeled as 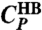 ) there is no low-T maximum that, hence, is due to the cooperativity.
) there is no low-T maximum that, hence, is due to the cooperativity.
The maximum of CP at lower T is given by the maximum of 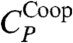 (Fig. 5A). To confirm that
(Fig. 5A). To confirm that 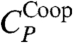 corresponds to the enthalpy fluctuations due to the cooperative Jσ term in Eq. 2, we calculate
corresponds to the enthalpy fluctuations due to the cooperative Jσ term in Eq. 2, we calculate 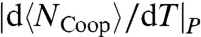 , where
, where 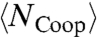 is the average number of molecules with perfect local order of their bond indices. We find that the locus of maxima of |dNCoop/dT|P overlaps with the locus of maxima of
is the average number of molecules with perfect local order of their bond indices. We find that the locus of maxima of |dNCoop/dT|P overlaps with the locus of maxima of 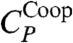 (Fig. 5B). The same qualitative behavior for CP is predicted from mean-field (MF) calculations (31) for the model (Fig. 5C).
(Fig. 5B). The same qualitative behavior for CP is predicted from mean-field (MF) calculations (31) for the model (Fig. 5C).
The nonmonotonic behavior of 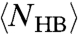 and
and 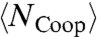 explains the two crossovers in the HB correlation time. At very low T, both experimental τ and simulation τMC have an Arrhenius behavior with constant activation energy A, 25.2 kJ/mol in the experiments and 13.7 kJ/mol in the model. The quantitative difference between the two arises from the choice of the parameters J and Jσ. In both experiments and model, A is consistent with the average energy
explains the two crossovers in the HB correlation time. At very low T, both experimental τ and simulation τMC have an Arrhenius behavior with constant activation energy A, 25.2 kJ/mol in the experiments and 13.7 kJ/mol in the model. The quantitative difference between the two arises from the choice of the parameters J and Jσ. In both experiments and model, A is consistent with the average energy 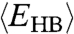 necessary to break a HB in a locally ordered environment. The relation
necessary to break a HB in a locally ordered environment. The relation 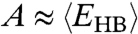 in both experiments and model suggests that the dynamics is dominated by the breaking and formation of a single HB at low T. This is well understood in the model where the energies
in both experiments and model suggests that the dynamics is dominated by the breaking and formation of a single HB at low T. This is well understood in the model where the energies 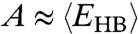 are both functions of
are both functions of 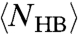 and
and 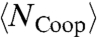 (11). Therefore, the saturation of the HB network (
(11). Therefore, the saturation of the HB network (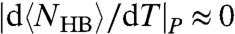 ) and its ordering (
) and its ordering (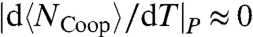 ) at low T imply constant A and an Arrhenius behavior for the HB correlation time.
) at low T imply constant A and an Arrhenius behavior for the HB correlation time.
At high T where CP is monotonic, 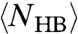 and
and 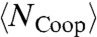 increase for decreasing T. Hence, the activation energy and
increase for decreasing T. Hence, the activation energy and 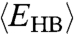 also increase, implying a non-Arrhenius behavior.
also increase, implying a non-Arrhenius behavior.
At intermediate T, between the two maxima of CP, the rate of change of 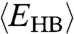 is proportional to the decreasing
is proportional to the decreasing 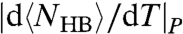 and the increasing
and the increasing 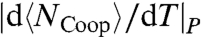 , giving rise to another non-Arrhenius behavior down to the temperature of the maximum
, giving rise to another non-Arrhenius behavior down to the temperature of the maximum 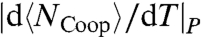 and the crossover to Arrhenius behavior (in the model there is no water diffusion, hence the low-T crossover is not due to a bulk glass transition as hypothesized for fully hydrated cases in ref. 40). The difficulty to separate the large lysozyme contribution and the low-h water contribution from the total experimental CP makes not possible a straightforward comparison of our CP calculations with experimental data (23, 25).
and the crossover to Arrhenius behavior (in the model there is no water diffusion, hence the low-T crossover is not due to a bulk glass transition as hypothesized for fully hydrated cases in ref. 40). The difficulty to separate the large lysozyme contribution and the low-h water contribution from the total experimental CP makes not possible a straightforward comparison of our CP calculations with experimental data (23, 25).
The relaxation time calculated for the model is characteristic to the breaking and forming of H bonds, which is analogous to the proton relaxation measured by dielectric spectroscopy. We find good qualitative agreement between τ and τMC, but at high T the crossover for τ is more pronounced than that for τMC (Fig. 4). This difference arises from two factors.
The experiments are carried out at constant h, corresponding to a decreasing effective P (possibly negative due to the surface adsorption) acting on water for decreasing T, while the MC results are at constant P = 0.1 MPa. Our MF calculations predict that CP displays two maxima along any path P(T) ≲ 0.1 MPa. Along a path such as in the experiments, in which P(T) decreases monotonically upon cooling,
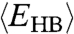 increases more rapidly by decreasing T, because
increases more rapidly by decreasing T, because 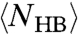 and
and 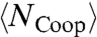 increase more rapidly when both P and T decrease (28, 29). This allows τMC to converge to the experimental τ at high T.
increase more rapidly when both P and T decrease (28, 29). This allows τMC to converge to the experimental τ at high T.The fluctuations in the HB network and distance between water oxygens, predicted by the model, could enhance the probability for a proton to be delocalized between two first-neighbor oxygens, inducing shorter proton relaxation times than those predicted on the base of classical simulations at high T. Experiments (16) show that this effect is maximum around 250 K, approximately where the model predicts the maximum fluctuation of the HB network and the experimental τ shows a stronger cusp than τMC.
To conclude, in dielectric spectroscopy experiments on hydrated lysozyme at low hydration level, we observe a relaxation mode associated to water protons, with two crossovers, one at approximately 252 K and another at approximately 181 K. At the same time, we find that a coarse-grained model of an adsorbed monolayer of water shows in simulations two crossovers for the HB dynamics. In the model these two crossovers can be fully understood as the effects of two structural changes of the HB network. These two structural reorganizations are marked by two maxima in CP, as well as in the compressibility KT and the isobaric thermal expansion coefficient αP (not shown here). The two structural changes are (i) at higher T, associated with the maximum fluctuations of the formation and breaking of the HBs, and (ii) at lower T, associated with the maximum fluctuation of the ordering of the local arrangement of the HBs. We argue that the model predictions provide an interpretation for our experimental findings.
Methods
Experimental Setup and Data Analysis.
We use an Alpha Analyzer dielectric apparatus (Novocontrol) to study crystallized and highly purified lysozyme powder from chicken egg white (Sigma-Aldrich), dialyzed and lyophilized to set its pH, then rehydrated (14). The capacitor containing the sample has blocking electrodes, coated by Teflon with thickness approximately 1/40th of the sample thickness. This choice of thickness eliminates the possibility of artifacts in our raw dielectric data, as discussed in a recent publication (41), and provides a “first check” of the reliability of the data analysis procedure adopted here.
Our measured complex admittance Ym(ω) is directly related to the complex permittivity 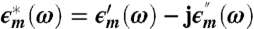 given that
given that
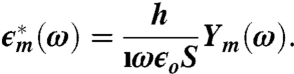 |
[4] |
Here, 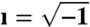 ; ϵo is the permittivity of free space; and S and h are, respectively, the electrode surface area and gap thickness. We extract the true sample permittivity from the measured frequency response by performing a complex function fit procedure that takes into account electrode polarization and capacity—that can be represented by a constant-phase-angle (CPA) element—interfacial dispersion, also known as the Maxwell–Wagner effect, along with relaxation processes due to the sample itself. Details about the deconvolution of the data are given in the SI Text.
; ϵo is the permittivity of free space; and S and h are, respectively, the electrode surface area and gap thickness. We extract the true sample permittivity from the measured frequency response by performing a complex function fit procedure that takes into account electrode polarization and capacity—that can be represented by a constant-phase-angle (CPA) element—interfacial dispersion, also known as the Maxwell–Wagner effect, along with relaxation processes due to the sample itself. Details about the deconvolution of the data are given in the SI Text.
We write the measured complex admittance 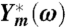 as
as
| [5] |
where A and d characterize the ω-dependent fractal polarization due to the blocking electrode, as in ref. 42, and
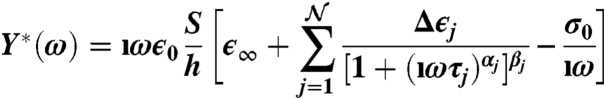 |
[6] |
is the admittance of the sample itself—expressed as a conductivity term plus a combination of Havriliak–Negami functions. Here, σ0 is the sample conductivity; ϵ∞ is the high-frequency limit of the permittivity; 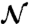 is the number of relaxation processes (i.e.,
is the number of relaxation processes (i.e., 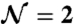 for the broad peak in Fig. 3); Δϵj and τj are the dielectric strength and the relaxation time for the jth contribution, respectively; and αj and βj characterize the shape of the relaxation time distribution function.
for the broad peak in Fig. 3); Δϵj and τj are the dielectric strength and the relaxation time for the jth contribution, respectively; and αj and βj characterize the shape of the relaxation time distribution function.
The presence of blocking electrodes eliminates the dc conductivity across the bulk sample, but not the sample conductivity term Eq. 6, related to local displacement of protons along the protein surface (15). This sample “local” conductivity has the same temperature dependence as that measured in ref. 2. Because the experimental setup in ref. 2 does not use blocking electrodes, this observation provides a “second check” for the reliability of our data analysis. The check provides also a verification of our data decomposition, because in ref. 2 a different deconvolution of the data is used, but the results for the same relaxation mode are the same. See SI Text, section I, for a “third check” of our data analysis.
Quantitative Analysis of the Shape of the Relaxation Time Distribution Function.
Raicu (43) has proposed a phenomenological “universal dielectric response” function able to describe a single Debye-like relaxation, such as the Havriliak-Negami relaxation, combined with interfacial dispersion and electrode polarization. An important result of this work is that a distribution function for the relaxation times in the frequency domain can be directly calculated using parameters appearing in the universal response. We adapt an algorithm (44) to obtain, from raw 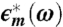 data, a distribution function in the frequency domain, by means of an inverse Laplace transform with no a priori assumption on the kind and number of relaxation processes This approach has proven to be a reliable tool to obtain a distribution function of relaxation times (45, 46). Figure 2 of ref. 14 compares the two distribution functions, one based on a single relaxation, the other with no assumption on the number of relaxation processes, for the dataset of hydrated lysozyme powder at 270.4 K and h = 0.26 g H2O/g dry protein. The analysis shows that the distribution function derived for a single relaxation process plus the effect of electrode polarization and interfacial dispersion does not totally account for the distribution calculated with no a priori assumptions. This implies that one or more additional relaxation processes are required to describe the raw dielectric data. We found that an additional relaxation is sufficient to completely account for the calculated distribution function. The addition of a third relaxation term results in unphysical negative Δϵi and therefore is not considered. We assume that a relatively small increase of water content from 0.26 to 0.3 g H2O/g dry protein does not change the results of the approach described above.
data, a distribution function in the frequency domain, by means of an inverse Laplace transform with no a priori assumption on the kind and number of relaxation processes This approach has proven to be a reliable tool to obtain a distribution function of relaxation times (45, 46). Figure 2 of ref. 14 compares the two distribution functions, one based on a single relaxation, the other with no assumption on the number of relaxation processes, for the dataset of hydrated lysozyme powder at 270.4 K and h = 0.26 g H2O/g dry protein. The analysis shows that the distribution function derived for a single relaxation process plus the effect of electrode polarization and interfacial dispersion does not totally account for the distribution calculated with no a priori assumptions. This implies that one or more additional relaxation processes are required to describe the raw dielectric data. We found that an additional relaxation is sufficient to completely account for the calculated distribution function. The addition of a third relaxation term results in unphysical negative Δϵi and therefore is not considered. We assume that a relatively small increase of water content from 0.26 to 0.3 g H2O/g dry protein does not change the results of the approach described above.
Simulations.
We simulate N = 10,000 water molecules in the NPT ensemble. To equilibrate such a large system at approximately 180 K for 100 s is a task that cannot be accomplished with molecular dynamics simulations of any detailed model of water. To overcome this problem, we (i) adopt a coarse-grained model, as described in the main text, and (ii) use MC simulations. Depending on whether we want to study thermodynamic quantities or dynamic quantities, we implement two different MC techniques (see SI Text, section II).
Supplementary Material
Acknowledgments.
We thank F. Caupin, H. Frauenfelder F. Mallamace, S. Sastry, and E. G. Strekalova for discussions and National Science Foundation Grants CHE0616489, CHE0911389, CHE0908218, and CHE0404673, and Ministerio de Ciencia e Innovación–Fondo Europeo de Desarrollo Regional (Spain) Grant FIS2009-10210 for support.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1104299108/-/DCSupplemental.
References
- 1.Chen S-H, et al. Observation of fragile-to-strong dynamic crossover in protein hydration water. Proc Natl Acad Sci USA. 2006;103:9012–9016. doi: 10.1073/pnas.0602474103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawlus S, Khodadadi S, Sokolov AP. Conductivity in hydrated proteins: No signs of the fragile-to-strong crossover. Phys Rev Lett. 2008;100:108103. doi: 10.1103/PhysRevLett.100.108103. [DOI] [PubMed] [Google Scholar]
- 3.Khodadadi S, Pawlus S, Sokolov AP. Influence of hydration on protein dynamics: Combining dielectric and neutron scattering spectroscopy data. J Phys Chem B. 2008;112:14273–14280. doi: 10.1021/jp8059807. [DOI] [PubMed] [Google Scholar]
- 4.Khodadadi S, et al. The origin of the dynamic transition in proteins. J Chem Phys. 2008;128:195106. doi: 10.1063/1.2927871. [DOI] [PubMed] [Google Scholar]
- 5.Vogel M. Origins of apparent fragile-to-strong transitions of protein hydration waters. Phys Rev Lett. 2008;101:225701. doi: 10.1103/PhysRevLett.101.225701. [DOI] [PubMed] [Google Scholar]
- 6.Rupley JA, Careri G. Protein hydration and function. Adv Protein Chem. 1991;41:37–172. doi: 10.1016/s0065-3233(08)60197-7. [DOI] [PubMed] [Google Scholar]
- 7.Frauenfelder H, et al. A unified model of protein dynamics. Proc Natl Acad Sci USA. 2009;106:5129–5134. doi: 10.1073/pnas.0900336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar P, et al. Glass transition in biomolecules and the liquid-liquid critical point of water. Phys Rev Lett. 2006;97:177802. doi: 10.1103/PhysRevLett.97.177802. [DOI] [PubMed] [Google Scholar]
- 9.Lagi M, et al. The low-temperature dynamic crossover phenomenon in protein hydration water: Simulations vs experiments. J Phys Chem B. 2008;112:1571–1575. doi: 10.1021/jp710714j. [DOI] [PubMed] [Google Scholar]
- 10.Vogel M. Temperature-dependent mechanisms for the dynamics of protein-hydration waters: A molecular dynamics simulation study. J Phys Chem B. 2009;113:9386–9392. doi: 10.1021/jp901531a. [DOI] [PubMed] [Google Scholar]
- 11.Kumar P, Franzese G, Stanley HE. Predictions of dynamic behavior under pressure for two scenarios to explain water anomalies. Phys Rev Lett. 2008;100:105701. doi: 10.1103/PhysRevLett.100.105701. [DOI] [PubMed] [Google Scholar]
- 12.Capaccioli S, Lucchesi M, Rolla PA, Ruggeri G. Dielectric response analysis of a conducting polymer dominated by the hopping charge transport. J Phys Condens Matter. 1998;10:5595–5617. [Google Scholar]
- 13.Peyrard M. Glass transition in protein hydration water. Phys Rev E Stat Nonlin Soft Matter Phys. 2001;64:011109. doi: 10.1103/PhysRevE.64.011109. [DOI] [PubMed] [Google Scholar]
- 14.Bruni F, Pagnotta SE. Dielectric investigation of the temperature dependence of the dynamics of a hydrated protein. Phys Chem Chem Phys. 2004;6:1912–1919. [Google Scholar]
- 15.Pagnotta SE, Gargana R, Bruni F, Bocedi A. Glassy behavior of a percolative water-protein system. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;71:031506. doi: 10.1103/PhysRevE.71.031506. [DOI] [PubMed] [Google Scholar]
- 16.Pagnotta SE, Bruni F, Senesi R, Pietropaolo A. Quantum behavior of water protons in protein hydration shell. Biophys J. 2009;96:1939–1943. doi: 10.1016/j.bpj.2008.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pizzitutti F, Bruni F. Glassy dynamics and enzymatic activity of lysozyme. Phys Rev E Stat Nonlin Soft Matter Phys. 2001;64:052905. doi: 10.1103/PhysRevE.64.052905. [DOI] [PubMed] [Google Scholar]
- 18.Pizzitutti F, Bruni F. Electrode and interfacial polarization in broadband dielectric spectroscopy measurements. Rev Sci Instrum. 2001;72:2502–2504. [Google Scholar]
- 19.Khodadadi S, Curtis JE, Sokolov AP. Nanosecond relaxation dynamics of hydrated proteins: Water versus protein contributions. J Phys Chem B. 2011;115:6222–6226. doi: 10.1021/jp1122213. [DOI] [PubMed] [Google Scholar]
- 20.Bruni F, Mancinelli R, Ricci MA. Multiple relaxation processes versus the fragile-to-strong transition in confined water. Phys Chem Chem Phys. 2011;13:19773–19779. doi: 10.1039/c1cp22029b. [DOI] [PubMed] [Google Scholar]
- 21.Senesi R, Pietropaolo A, Bocedi A, Pagnotta SE, Bruni F. Proton momentum distribution in a protein hydration shell. Phys Rev Lett. 2007;98:138102. doi: 10.1103/PhysRevLett.98.138102. [DOI] [PubMed] [Google Scholar]
- 22.Tuckerman ME, Marx D. Heavy-atom skeleton quantization and proton tunneling in “intermediate-barrier” hydrogen bonds. Phys Rev Lett. 2001;86:4946–4949. doi: 10.1103/PhysRevLett.86.4946. [DOI] [PubMed] [Google Scholar]
- 23.Zanotti J-M, Gibrat G, Bellissent-Funel M-C. Hydration water rotational motion as a source of configurational entropy driving protein dynamics. Crossovers at 150 and 220 K. Phys Chem Chem Phys. 2008;10:4865–4870. doi: 10.1039/b808217k. [DOI] [PubMed] [Google Scholar]
- 24.Merzel F, Smith JC. Is the first hydration shell of lysozyme of higher density than bulk water? Proc Natl Acad Sci USA. 2002;99:5378–5383. doi: 10.1073/pnas.082335099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazaki Y, Matsuo T, Suga H. Low-temperature heat capacity and glassy behavior of lysozyme crystal. J Phys Chem B. 2000;104:8044–8052. [Google Scholar]
- 26.Franzese G, Stanley HE. Liquid-liquid critical point in a Hamiltonian model for water: Analytic solution. J Phys Condens Matter. 2002;14:2201–2209. [Google Scholar]
- 27.Franzese G, Marqués MI, Stanley HE. Intramolecular coupling as a mechanism for a liquid-liquid phase transition. Phys Rev E Stat Nonlin Soft Matter Phys. 2003;67:011103. doi: 10.1103/PhysRevE.67.011103. [DOI] [PubMed] [Google Scholar]
- 28.Franzese G, Stanley HE. The Widom line of supercooled water. J Phys Condens Matter. 2007;19:205126. [Google Scholar]
- 29.Kumar P, Franzese G, Stanley HE. Dynamics and thermodynamics of water. J Phys Condens Matter. 2008;20:244114. [Google Scholar]
- 30.Mazza MG, Stokely K, Strekalova EG, Stanley HE, Franzese G. Cluster Monte Carlo and numerical mean field analysis for the water liquid-liquid phase transition. Comput Phys Commun. 2009;180:497–502. [Google Scholar]
- 31.Stokely K, Mazza MG, Stanley HE, Franzese G. Effect of hydrogen bond cooperativity on the behavior of water. Proc Nat Acad Sci USA. 2010;107:1301–1306. doi: 10.1073/pnas.0912756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strekalova EG, Mazza MG, Stanley HE, Franzese G. Large decrease of fluctuations for supercooled water in hydrophobic nanoconfinement. Phys Rev Lett. 2011;106:145701. doi: 10.1103/PhysRevLett.106.145701. [DOI] [PubMed] [Google Scholar]
- 33.de los Santos F, Franzese G. Understanding diffusion and density anomaly in a coarse-grained model for water confined between hydrophobic walls. J Phys Chem B. 2011 doi: 10.1021/jp206197t. 10.1021/jp206197t. [DOI] [PubMed] [Google Scholar]
- 34.Franzese G, et al. Pressure effects in supercooled water: Comparison between a 2D model of water and experiments for surface water on a protein. J Phys Condens Matter. 2008;20:494210. [Google Scholar]
- 35.Henry M. Nonempirical quantification of molecular interactions in supramolecular assemblies. Chemphyschem. 2002;3:561–569. doi: 10.1002/1439-7641(20020715)3:7<561::AID-CPHC561>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 36.Narten AH, Danford MD, Levy HA. X-ray diffraction study of liquid water in the temperature range 4–200 °C. Discuss Faraday Soc. 1967;43:97–107. [Google Scholar]
- 37.Ricci MA, Bruni F, Giuliani A. “Similarities” between confined and supercooled water. Faraday Discuss. 2009;141:347–358. doi: 10.1039/b805706k. [DOI] [PubMed] [Google Scholar]
- 38.Hohenberg PC, Halperin BI. Theory of dynamic critical phenomena. Rev Mod Phys. 1977;49:435–479. [Google Scholar]
- 39.Adam G, Gibbs JH. On the temperature dependence of cooperative relaxation properties in glass-forming liquids. J Chem Phys. 1965;43:139–139. [Google Scholar]
- 40.Ngai KL, Capaccioli S, Shinyashiki N. The protein “glass” transition and the role of the solvent. J Phys Chem B. 2008;112:3826–3832. doi: 10.1021/jp710462e. [DOI] [PubMed] [Google Scholar]
- 41.Richert R. Insulated electrodes for eliminating conductivity in dielectric relaxation experiments. Eur Phys J B. 2009;68:197–200. [Google Scholar]
- 42.Feldman Y, Nigmatullin R, Polygalov E, Texter J. Fractal-polarization correction in time domain dielectric spectroscopy. Phys Rev E Stat Nonlin Soft Matter Phys. 1998;58:7561–7565. [Google Scholar]
- 43.Raicu V. Dielectric dispersion of biological matter: Model combining Debye-type and “universal” responses. Phys Rev E Stat Nonlin Soft Matter Phys. 1999;60:4677–4680. doi: 10.1103/physreve.60.4677. [DOI] [PubMed] [Google Scholar]
- 44.Provencher SW. A constrained regularization method for inverting data represented by linear algebraic or integral equations. Comput Phys Commun. 1982;27:213–227. [Google Scholar]
- 45.Alvarez F, Alegria A, Colmenero J. Relationship between the time-domain Kohlrausch-Williams-Watts and frequency-domain Havriliak-Negami relaxation functions. Phys Rev B Condens Matter. 1991;44:7306–7312. doi: 10.1103/physrevb.44.7306. [DOI] [PubMed] [Google Scholar]
- 46.Banys J, et al. Broadband dielectric spectroscopy of water confined in MCM-41 molecular sieve material. Ferroelectrics. 2005;318:201–207. doi: 10.1109/tuffc.2006.178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.