

Abstract
Radioiodinated photoactivatable photoprobes can provide valuable insights regarding protein structure. Previous work in our laboratory showed that the cocaine derivative and photoprobe 3-[125I]iodo-4-azidococaine ([125I]IACoc) binds to the sigma-1 receptor with 2–3 orders of magnitude higher affinity than cocaine [Kahoun, J. R. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 1393–1397]. Using this photoprobe, we demonstrated the insertion site for [125I]IACoc to be Asp188 [Chen, Y. (2007) Biochemistry 46, 3532–3542], which resides in the proposed steroid binding domain-like II (SBDLII) region (amino acids 176–194) [Pal, A. (2007) Mol. Pharmacol. 72, 921–933]. An additional photoprobe based on the sigma-1 receptor ligand fenpropimorph, 1-N-(2-3-[125I]iodophenyl)propane ([125I]IAF), was found to label a peptide in both the SBDLII and steroid binding domain-like I (SBDLI) (amino acids 91–109) [Pal, A. (2007) Mol. Pharmacol. 72, 921–933]. In this report, we describe two novel strategically positioned carrier-free, radioiodinated photoaffinity labels specifically designed to probe the putative “nitrogen interacting region” of sigma-1 receptor ligands. These two novel photoprobes are (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-[125I]iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-N-IACoc) and N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]IAC44). In addition to reporting their binding affinities to the sigma-1 and sigma-2 receptors, we show that both photoaffinity labels specifically and covalently derivatized the pure guinea pig sigma-1 receptor (26.1 kDa) [Ramachandran, S. (2007) Protein Expression Purif. 51, 283–292]. Cleavage of the photolabeled sigma-1 receptor using Endo Lys C and cyanogen bromide (CNBr) revealed that the [125I]-N-IACoc label was located primarily in the N-terminus and SBDLI-containing peptides of the sigma-1 receptor, while [125I]IAC44 was found in peptide fragments consistent with labeling of both SBDLI and SBDLII.
Once classified to be subtypes of opioid receptors and phencyclidine (PCP)1 receptors (5), sigma receptors are now considered to be unique nonopioid and non-PCP binding sites present in the central nervous system and peripheral organs which are distinct from other known neurotransmitter or hormone receptors (6). Currently, two subtypes of sigma receptors have been identified based on their pharmacology, function, and molecular weight: the sigma-1 receptor and the sigma-2 receptor. Although the sigma-2 receptor has yet to be cloned, the 223 amino acid sigma-1 receptor was first cloned from guinea pig liver in 1996 (7) and subsequently from other sources including human placental choriocarcinoma cells (8), human brain (9), rat brain (10, 11), and mouse brain (12). The sigma-1 receptor shares 90% identity and 95% similarity across species, and the guinea pig receptor shares 30% identity and 67% similarity with a yeast sterol C8-C7 isomerase (ERG2), which is involved in postsqualene sterol synthesis (13). Despite its amino acid sequence similarities with ERG2, however, the sigma-1 receptor does not have sterol isomerase activity (7), nor can it rescue sterol isomerase-deficient yeast (13). In addition, the sigma-1 receptor remarkably shares no sequence homology with any known mammalian proteins, including the mammalian C8-C7 sterol isomerase.
Recently, Hayashi and Su (14) discovered that the sigma-1 receptor has properties of a ligand- and Ca2+-regulated chaperone in forming a complex with BiP/GRP78 at the ER/mitochondrial interface in Chinese hamster ovary (CHO) cells. Their data, consistent with the previously reported progrowth and antiapoptotic nature of the sigma-1 receptor (15,16), support the conclusion that when sigma-1 receptors dissociate from the BiP complex in response to ligand binding or Ca2+ reduction, the result is an extended Ca2+ signaling into mitochondria through inositol 1,4,5-triphosphate receptors (IP3Rs) leading to apoptosis (14).
Sigma-1 receptor binding studies have revealed high affinities for a variety of naturally occurring compounds such as steroids and neuropeptides. However, the endogenous ligands for the sigma-1 receptor are as yet unknown. Previous pharmacological studies have indicated that this receptor binds to a wide range of compounds including opiates, antipsychotics, antidepressants, antihistamines, PCP-like compounds, beta-adrenergic receptor ligands, serotonergic compounds, cocaine, and cocaine analogues. In order to study the binding site of the sigma-1 receptor, we have developed a series of high-affinity radioiodinated ligands as photoprobes. Photoaffinity labeling approaches have been used extensively in various laboratories, including our own, to identify the binding sites of the β-adrenergic receptor, monoamine transporters, G-proteins, G-protein-linked effectors, and the sigma-1 receptor (1, 2, 17–28). Previous work from our laboratory demonstrated that a cocaine-based radioiodinated photoaffinity label [125I]-3-iodo-4-azidococaine ([125I]IACoc) (Figure 1) with the photoactive azide on the benzoyl moiety is a high-affinity ligand for the sigma-1 receptor (1), which specifically identified the 26 kDa sigma-1 receptor in brain homogenates, PC12 cells, and membranes derived from several organs (1, 18). We also discovered that [125I]IACoc covalently inserted at Asp188, which is located in the steroid binding domain-like II (SBDLII) region within the C-terminal third hydrophobic region of the sigma-1 receptor (2). Further examination of the binding site(s) using the high-affinity sigma-1 photoprobe 1-N-(2-3-[125I]iodophenyl)propane ([125I]IAF) (Figure 1) showed specific derivatization of the steroid binding domain-like I (SBDLI) in addition to the steroid binding domain-like II (SBDLII). This has led to the conclusion that SBDLI (amino acids 91–109) and SBDLII (amino acids 176–194) comprise part, if not all, of the main sigma-1 receptor binding site (3).
Figure 1.
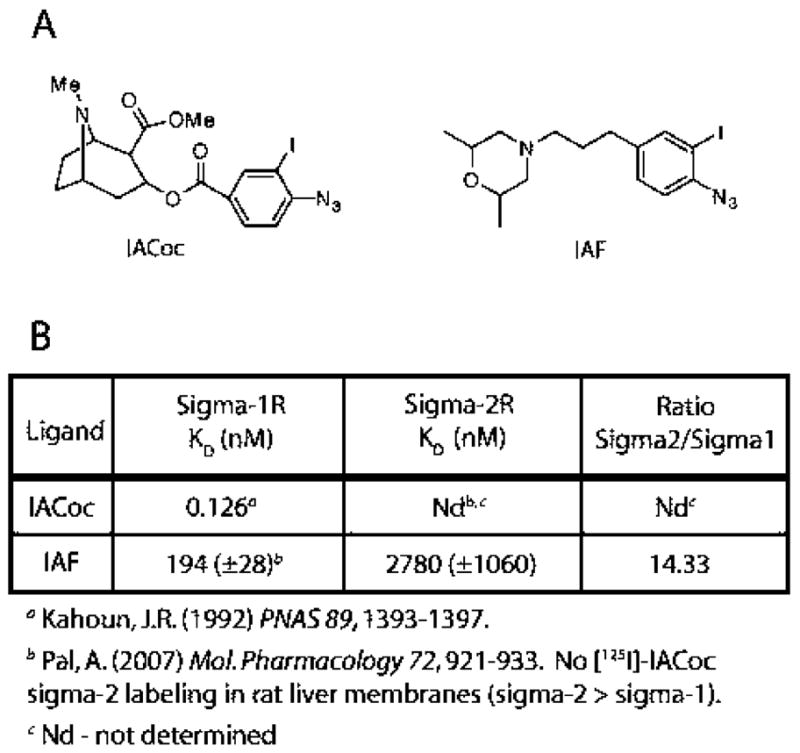
(A) Chemical structures of 3-iodo-4-azidococaine (IA-Coc) and 1-N-(2′,6′-dimethylmorpholino)-3-(4-azido-3-iodophenyl)propane (IAF). (B) Reported sigma-1 receptor KD values are shown for IACoc. As referenced, [125I]IACoc did not label the sigma-2 receptor in rat liver membranes where the amount of sigma-2 receptor is at least four times greater than the sigma-1 receptor. Sigma-1 and sigma-2 receptor KD values for IAF binding are also depicted.
With the aim of probing the “nitrogen interacting region” of sigma ligands that bind to the sigma-1 receptor we have synthesized two novel carrier-free, radioiodinated photoaffinity labels, (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-[125I]iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-N-IACoc) and N-propyl-N-(4-azido-3-[125I]iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]IAC44), by strategic placement of the radioactive iodine and a photoactivatable aryl azide on the nitrogen. Following specific binding site covalent derivatization and subsequent Endo Lys C and CNBr digestions to cleave the photolabeled pure protein, the sigma-1 receptor binding site peptides specifically derivatized by [125I]-N-IACoc and [125I]IAC44 were identified.
MATERIALS AND METHODS
Chemistry
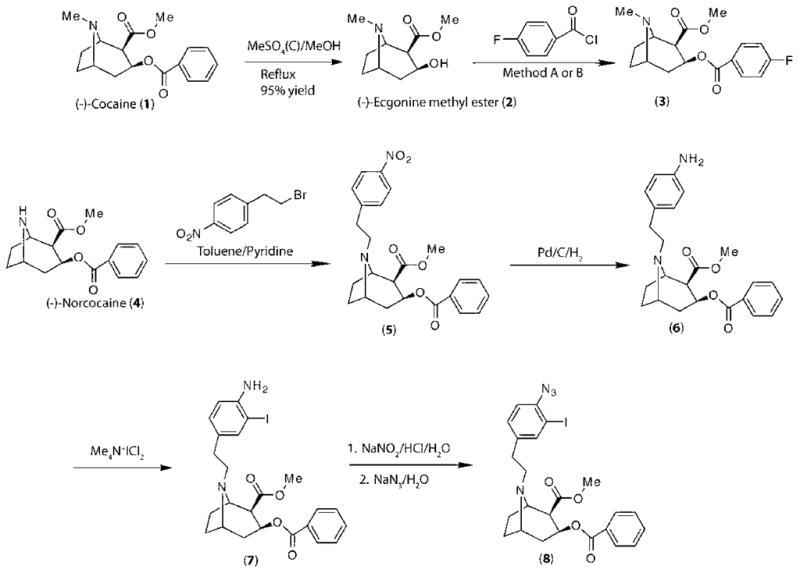
Yields refer to isolated pure products after column chromatography. The products were characterized by comparison of their spectral (IR, 1H and 13C NMR, CHN, and mass spectroscopy analysis) and physical data with those of authentic samples. All 1H NMR spectra were recorded at 300 MHz in CDCl3 relative to TMS (0.00 ppm), and IR spectra were recorded on a Shimadzu 435 IR spectrometer. Melting points were determined with a Thomas-Hoover capillary melting point apparatus and are reported uncorrected. Chemicals were obtained from Aldrich Chemical Co. and utilized without further purification. Elemental analysis was performed at the Research Institute of Petroleum Industry, Tehran, IR, Iran. The synthesis of compounds 1–8 is outlined in Scheme 1, and the synthesis of compounds 9–15 is outlined in Scheme 2.
Scheme 1.
Chemical Synthesis of N-Substituted Cocaine Derivatives
Scheme 2.

Chemical Synthesis of N-Alkyl-N′-aralkyl Derivatives
Preparation of Methyl (1R-2-exo-3-exo)-8-Methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (Ecgonine Methyl Ester) (2)
(−)-Cocaine hydrochloride (10.0 g, 29 mmol) was dissolved in 200 mL of MeOH, combined with 20 mL of concentrated H2SO4, and refluxed overnight. The reaction mixture was allowed to cool to room temperature, and excess solvent was evaporated under reduced pressure. The residual yellow oil was taken up in a small amount of water and neutralized with saturated Na2CO3 solution at 0 °C. The product was extracted into 4 × 50 mL portions of Et2O. The combined Et2O extracts were dried with Na2SO4, filtered, and evaporated to dryness. The resulting yellow oil was used in the following reaction without further purification. If a pure product was desired, the crude product was readily purified by column chromatography (silica gel, toluene: Et2NH, 20:1), yield >95%. Yellow oil. 1H NMR: δ 4.80 (m, 1 H), 4.2 (m, 1 H), 4.08 (s, 1 H, OH), 3.80 (s, 3 H, OMe), 3.59 (m, 1 H), 2.90 (s, 3 H, NMe), 2.46–2.20 (m, 7 H). 13C NMR: 180.0, 68.2, 65.1, 61.8, 51.7, 50.2, 41.3, 35.7, 25.8, 25.5.
General Method for (−)-4-Fluorobenzoylecgonine Methyl Ester (3)
Method A
In a mortar, a mixture of (−)-ecgnonine methyl ester (free base) (1 mmol, 0.1 g), 4-fluorobenzoyl chloride (1.1 mmol, 0.17 g), and DABCO (1 mmol, 0.12 g) was ground with a pestle at room temperature for 5 min and then kept at room temperature for 20 min. The reaction mixture dissolved in 3 mL of MeOH and was purified by column chromatography (silica gel, toluene:Et2NH, 20:1) to afford pure product in >93% yield. Yellow oil. 1H NMR: δ 8.1 (m, 2 H), 7.2 (m, 2 H), 4.6 (m, 1 H), 3.78 (s, 3 H, OMe), 2.9 (m, 1 H), 2.3 (m, 1 H), 2.29 (s, 3 H), 1.9–1.6 (m, 7 H). 13C NMR: 180.0, 170.0. 132.0, 128.0, 68.4, 58.2, 56.8, 54.9, 42.5, 37.8, 21.9, 18.2. Anal. Calcd for C17H20FNO4: C, 63.54; H, 6.27; N, 4.36%. Found: C, 63.55; H, 6.31; N, 4.43%.
Method B
To a stirring mixture of (−)-ecgonine methyl ester (free base) (1 mmol, 0.2 g) and 1 equiv of K2CO3 in toluene was added 4-fluorobenzoyl chloride (1.1 mmol, 0.17 g). The mixture was refluxed for 2 h and allowed to cool to room temperature. After filtration, the solvent was removed to give a yellow oil. The crude products were purified by column chromatography (silica gel, toluene: Et2NH, 20:1) to afford pure product in >86% yield.
3-(Benzoyloxy)-8-2-(4-nitrobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (5)
To a stirring mixture of (−)-norcocaine (free base) (1 mmol, 0.24 g) and 1 equiv of Et3N (1.1 mmol, 0.11 g) in Et2O was added 2-(4-nitrobenzene)-1-ethyl bromide in anhydrous ether (0.9 mmol, 0.21 g). The mixture was stirred at room temperature for 5 h. After filtration, solvent was removed to give a yellow oil. The crude products were then purified by column chromatography (silica gel, toluene:Et2NH, 20:1) to afford pure product in >86% yield. 1H NMR: δ 8.3 (d, J = 6.8 Hz, 2 H), 8.2 (d, J = 6.8 Hz, 2 H), 7.7–7.4 (m, 5 H), 5.3 (m, 1 H), 3.78 (s, 3 H, OMe), 3.5 (m, 1 H), 3.32 (m, 1 H), 2.89 (t, J = 7.6 Hz, 2 H), 2.29 (t, J = 7.6 Hz, 2 H), 2.14–1.2 (m, 6 H). 13C NMR: 180.0, 170.0. 145.0, 144.0, 135.0, 132.0, 128.0, 126.0, 68.4, 65.1, 60.8, 50.9, 50.2, 41.3, 35.7, 25.8, 25.5, 24.8. Anal. Calcd for C24H22N2O6: C, 66.35; H, 5.10; N, 6.44%. Found: C, 66.23; H, 5.24; N, 6.36%.
3-(Benzoyloxy)-8-2-(4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (6)
A mixture of 3-(benzoyloxy)-8-2-(4-nitrobenzene)-1-ethyl-8-azabicyclo[3.2.1]-octane-2-carboxylate (5) (0.5 mmol, 0.22 g) and 10 mg of Pd/C (10%) in methanol was reduced with H2 at normal pressure. The mixture was stirred at room temperature overnight. After filtration, the solvent was removed to give a yellow oil, and the crude products were purified by column chromatography (silica gel, toluene:Et2NH, 20:1) to afford pure product in quantitative yield. Yellow solid, mp 198–199 °C. 1H NMR: δ 7.76 (d, J = 8.4 Hz, 2 H), 7.7–7.4 (m, 5 H), 6.62 (d, J = 8.4 Hz, 2 H), 5.3 (m, 1 H), 4.1 (b, 2 H, NH2), 3.78 (s, 3 H, OMe), 3.5 (m, 1 H), 3.32 (m, 1 H), 2.89 (t, J = 7.6 Hz, 2 H), 2.29 (t, J = 7.6 Hz, 2 H), 2.46–1.20 (m, 6 H). 13C NMR: 176.0, 170.0, 150.0, 145.0, 144.0, 135.0, 132.0, 128.0, 126.0, 68.4, 65.1, 60.8, 50.9, 50.2, 41.3, 35.7, 25.8, 25.5, 24.8. Anal. Calcd for C24H24N2O4: C, 71.27; H, 5.98; N, 6.93%. Found: C, 71.19; H, 6.04; N, 6.87%.
3-(Benzoyloxy)-8-2-(3-iodo-4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (7)
A mixture of 3-(benzoyloxy)-8-2-(3-iodo-4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (0.5 mmol, 0.2 g) and tetramethylammonium dichloroiodate (0.5 mmol, 0.14 g) (29) in a mortar was ground with a pestle to produce a homogeneous powder and left at room temperature until TLC (toluene:Et2NH, 20:1) showed complete disappearance of the starting material, 3-(benzoyloxy)-8-2-(3-iodo-4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (7). After adding 5 mL of sodium bisulfate (5%) to the brown solid, the reaction mixture was extracted with dichloromethane (3 × 5 mL). The combined extracts were dried with MgSO4, and evaporation of the solvent gave the corresponding iodo derivative (7). The product was purified by column chromatography (silica gel, toluene:Et2NH, 20: 1). Yellow solid, mp 176–178 °C, 86%. 1H NMR: δ 7.99–6.18 (m, 8 H), 4.68 (m, 1 H), 4.16 (br, 2 H, NH2), 3.74 (s, 3 H, OMe), 2.91 (m, 1 H), 2.87 (m, 1 H), 2.69 (t, J = 8.6 Hz, 2 H), 2.65 (t, J = 8.6 Hz, 2 H), 1.55 (m, 4 H). 13C NMR: δ 176.0, 167.1, 152.8, 137.6, 132.8, 131.9, 130.5, 129.7 (2 C), 128.4, 127.6, 116.8, 83.4, 63.6, 53.5, 51.2, 50.7, 50.6, 41.6, 35.2, 31.5, 25.6, 22.6. Anal. Calcd for C24H23IN2O4: C, 54.35; H, 4.73; N, 5.28%. Found: C, 54.26; H, 4.94; N, 5.21%.
3-(Benzoyloxy)-8-2-(3-iodo-4-azidobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (8)
To a cold mixture (0 °C) of 3-(benzoyloxy)-8-2-(3-iodo-4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (7) (0.1 mmol, 42.2 mg) in H2O (2 mL) and concentrated HCl (0.4 mL) in a round-bottomed flask was added an aqueous solution of NaNO2 (0.1 mmol, 21 g, in 0.5 mL of H2O) within 5 min. The reaction mixture was left to stir at room temperature for 30 min. Then, in darkness and at room temperature, an aqueous solution of NaN3 (0.36 mmol, 23 mg, in 0.5 mL of H2O) was added dropwise to the reaction mixture, which stirred for 30 min and was then extracted with EtOAc (3 × 3 mL). The combined EtOAc solution was dried with MgSO4, and the solvent was evaporated by rotary evaporator to afford an orange oil. The crude product was purified by column chromatography (silica gel, first toluene:Et2NH, 20: 1, and then toluene:Et2NH, 4:1) to give the product in 96%. Yellow solid, mp 187–189 °C. 1H NMR: δ 7.99–7.00 (m, 8 H), 4.68 (m, 1 H), 3.69 (s, 3 H, OMe), 2.91 (m, 1 H), 2.87 (m, 1 H), 2.69 (t, J = 8.6 Hz, 2 H), 2.65 (t, J = 8.6 Hz, 2 H), 2.24 (m, 1 H), 1.88–1.55 (m, 5 H). 13C NMR: δ 176.0, 167.1, 152.8, 137.6, 132.8, 131.9, 130.5, 129.7 (2 C), 128.4, 127.6, 116.8, 83.4, 63.6, 53.5, 51.2, 50.7, 50.6, 41.6, 35.2, 31.5, 25.6, 22.6. Anal. Calcd for C24H21IN4O4: C, 51.81; H, 3.80; N, 10.07%. Found: C, 51.76; H, 3.88; N, 9.98%.
Preparation of 3-(4-Nitrophenyl)propylbromine (10)
Two grams of P2O5/silica gel (65% w/w) (10 mmol) (30) and 3-phenylpropylbromine (9) (10 mmol, 1.98 g) was ground for 30 s, and then 5 mL of HNO3 (65%) was added. The mixture was ground with a mortar and pestle at room temperature until a deep yellow color appeared (2 min). When TLC (n-hexane:EtOAc, 90:10) showed complete disappearance of 3-phenylpropyl bromide (10 min), ether (100 mL) was added to the reaction mixture, and the solid was separated through a short pad of silica gel and washed with ether (3 × 20 mL). The filtrate was washed with 10% NaHCO3 (3 × 20 mL) and dried (MgSO4). The solvent was evaporated under reduced pressure, and the residue was purified by short column chromatography (n-hexane:EtOAc, 90:10). 3-(4-Nitrophenyl)propyl bromide was obtained (8.3 mmol, 2.02 g 83%) as a yellow oil. 1H NMR (CDCl3): δ 8.2 (d, J = 6.3 Hz), 7.38 (d, 2 H, 6.3), 3.3 (t, 2 H, J = 7.8 Hz), 2.55 (t, 2H, J = 7.8 Hz), 2.12 (m, 2 H). 13C NMR (CDCl3): δ 145.7, 144.9, 129.7, 123.5, 36.7, 34.7, 32.4. Anal. Calcd for C9H10BrNO2: C, 44.29; H, 4.13; N, 5.74%. Found: C, 44.50; H, 48.30; N, 5.80%.
Preparation of N-Propyl-N-(4-nitrophenylethyl)-3-(4-fluorophenyl)propionamide (11)
A mixture of DCC (1 mmol, 2.1 g) and 4-fluorophenylpropionic acid (1 mmol, 0.17 g) was ground with a mortar and pestle for 30 s, and then the amine (1 mmol) was added to the reaction mixture. The reaction was ground with a pestle until TLC showed no remaining acid (n-hexane:EtOAc, 75:25) (20 min). Then to the reaction mixture was added a mixture of ether (20 mL) and H2O (5 mL). The etheral layer was washed with saturated NaHCO3, dried (MgSO4), and evaporated by a rotary evaporator to give a residue, which was purified by column chromatography (silica gel, n-hexane:EtOAc, 75:25). Yield 0.29 g, 81%. Yellow oil. IR (KBr): 1657 cm−1. 1H NMR (CDCl3): δ 8.14 (d, 2 H, J = 6.8 Hz), 7.38 (d, 2 H, J = 6.8 Hz), 7.10 (m, 2 H), 6.92 (m, 2 H), 3.62 (t, 2 H, J = 7.2 Hz), 3.20 (m, 4 H), 2.83 (t, 2 H, J = 7.8 Hz), 2.61 (t, 2 H, J = 7.8 Hz), 1.69 (m, 2 H), 0.96 (t, 3 H, J = 7.8 Hz). 13C NMR (CDCl3): δ 173.6, 159.3, 146.7, 146.3, 135.8, 129.5, 128.8, 123.4, 115.4, 53.0, 40.2, 35.1, 34.2, 31.8, 22.4, 11.4. MS (CI) m/z 344 (100, M+). Anal. Calcd for C20H23FN2O3: C, 67.02; H, 6.47; N, 7.82%. Found: C, 67.10; H, 6.70; N, 7.90%.
Preparation of N-Propyl-N-(4-aminophenylethyl)-3-(4-fluorophenyl)propionamide (12)
A mixture of nitro compounds (11) (1 mmol) and 10 mg of Pd/C (10%) in methanol was reduced with H2 at normal pressure and then stirred at room temperature overnight. After filtration, the solvent was removed to give a yellow residue. The crude products were purified by column chromatography (silica gel, toluene: Et2NH, 20:1) to afford pure amine. Yield 0.31 g, 96%. Yellow oil. IR (KBr): 3258, 1657 cm−1. 1H NMR (CDCl3): 7.10 (m, 2 H), 6.92 (m, 2 H), δ 6.82 (d, 2 H, J = 6.8 Hz), 6.41 (d, 2 H, J = 6.8 Hz), 4.13 (s, 2 H, NH2), 3.63 (t, 2 H, J = 7.20 Hz), 3.20 (t, 2 H, J = 7.20 Hz), 2.83 (m, 4 H), 2.61 (t, 2H, J = 7.8 Hz), 1.69 (m, 2 H), 0.96 (t, 3 H, J = 7.8 Hz). 13C NMR (CDCl3): δ 173.6, 159.3, 143.0, 135.8, 130.2, 129.5, 115.0, 49.9, 35.6, 34.2, 31.8, 22.4, 11.4. MS (CI) m/z 328 (100, M+). Anal. Calcd for C20H25FN2O: C, 73.14; H, 7.67; N, 8.53%. Found: C, 73.10; H, 7.70; N, 8.50%.
Reduction of Amide 12 to N-Propyl-N-(4-aminophenylethyl)-3-(4-fluorophenyl)propylamine (13)
In a double-necked round-bottomed flask equipped with a septum and condenser, a solution of the amide (1 mmol) in anhydrous THF (5 mL) was added dropwise to a stirring solution of LiAlH4 (0.74 g, 2 mmol) in anhydrous THF (5 mL) under argon. TLC indicated the reaction to be almost complete after 15 min at room temperature. The reaction mixture was driven to completion by brief refluxing (15 min), and when it was cooled to room temperature, it was diluted with the addition of THF (5 mL). The excess LiAlH4 was destroyed by dropwise addition of water (1 mL), 15% aqueous NaOH (1 mL), and finally water (1 mL). The reaction mixture was stirred for 30 min at room temperature, and then the solids were removed by filtration. The filtrate was dried (MgSO4) and the solvent evaporated by a rotary evaporator to give pure product as a yellow oil in quantitative yield. IR (KBr): 3251 cm−1. 1H NMR (CDCl3): 7.10 (m, 2 H), 6.92 (m, 2 H), δ 6.81 (d, 2 H, J = 6.8 Hz), 6.34 (d, 2 H, J = 6.8 Hz), 4.12 (s, 2 H, NH2), 3.69–2.36 (m, 10 H), 1.72 (m, 2 H), 1.43 (m, 2H), 0.98 (t, 3 H, J = 7.8 Hz). 13C NMR (CDCl3): δ 159.4, 145.2, 134.4, 129.4, 128.3, 115.6, 114.8, 59.7, 56.1, 34.5, 32.1, 22.8, 11.7. MS (CI) m/z 314 (100, M+). Anal. Calcd for C20H27FN2: C, 76.39; H, 8.65; N, 8.91%. Found: C, 76.30; H, 8.80; N, 8.70%.
Iodination of Amine 13 to N-Propyl-N-(4-amino-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine (14)
A mixture of amine 13 (0.5 mmol) and tetramethylammonium dichloroiodate (0.5 mmol, 0.14 g) (29) was ground with a mortar and pestle to produce a homogeneous powder, and the mixture was left at room temperature until TLC (toluene: Et2NH, 20:1) showed complete disappearance of the amine. Five milliliters of sodium bisulfate (5%) was added to the brown solid, and the reaction mixture was extracted with dichloromethane (3 × 5 mL). The combined extracts were dried with MgSO4, and evaporation of the solvent gave the corresponding iodo derivative (14). The product was purified by column chromatography (silica gel, toluene:Et2NH, 20: 1). Oil, yield 81%. IR (KBr): 3258 cm−1. 1H NMR (CDCl3): δ 7.26–6.92 (m, 6 H), 6.18 (d, 1 H, J = 6.4 Hz), 4.12 (s, 2 H, NH2), 2.69–2.36 (m, 10 H), 1.72 (m, 2 H), 1.43 (m, 2H), 0.96 (t, 3 H, J = 7.8 Hz). 13C NMR (CDCl3): δ 159.4, 152.8, 137.6, 134.4, 131.8, 129.9, 127.6, 116.6, 115.6, 83.9, 56.5, 56.3, 34.6, 33.5, 32.1, 22.8, 11.7. MS (CI) m/z 440 (100, M+). Anal. Calcd for C20H26IFN2: C, 54.55; H, 5.95; N, 6.36%. Found: C, 54.40; H, 6.10; N, 6.30%.
Conversion of Amino Iodo Derivative 14 to N-Propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine (15)
To a cold mixture (0 °C) of 14 (0.1 mmol) in H2O (2 mL) was added concentrated HCl (0.4 mL) with an aqueous solution of NaNO2 (0.30 mmol, 21 mg, in 0.5 mL of H2O) in a round-bottomed flask within 5 min. The reaction mixture stirred at room temperature for 30 min. Then at room temperature and darkness an aqueous solution of NaN3 (0.36 mmol, 23 mg, in 0.5 mL of H2O) was added. The reaction mixture was stirred at room temperature for 30 min and then extracted with EtOAc (3 × 3 mL). The combined EtOAc solution was dried with MgSO4, and the solvent was evaporated with a rotary evaporator to afford an orange oil. The crude product was purified by column chromatography (silica gel, first toluene:Et2NH, 20:1, and then toluene:Et2NH, 4:1) to give the product as a yellow oil. Oil, yield 93%. 1H NMR (CDCl3): δ 7.50–6.92 (m, 7 H), 2.69–2.36 (m, 10 H), 1.72 (m, 2 H), 1.43 (m, 2H), 0.96 (t, 3 H, J = 7.8 Hz). 13C NMR (CDCl3): δ 159.4, 142.3, 137.4, 135.0, 129.9, 127.4, 116.6, 115.6, 97.0, 56.5, 56.3, 34.6, 33.5, 32.1, 22.8, 11.7. MS (CI) m/z 466 (100, M+). Anal. Calcd for C20H24IFN4: C, 55.51; H, 5.19; N, 12.01%. Found: C, 55.50; H, 5.30; N, 11.90%.
Radiosynthesis of (−)-Methyl 3-(Benzoyloxy)-8-2-(4-azido-3-[125I]iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-N-IACoc)
To 2.5 mCi of Na125I in 7 μL of 0.1 M NaOH was added 7 μL of 0.1 M HCl and 40 μL of 0.5 M sodium acetate buffer (pH 5.6). 3-(Benzoyloxy)-8-2-(4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (6) (10 μL of 2.5 mg/mL in 0.5 M sodium acetate buffer, pH 5.6) was added. Iodination was initiated by adding 30 μL of Chloramine-T (1 mg/mL in water) and continued for 15 min at room temperature. The reaction was terminated by the addition of 100 μL of Na2S2O5 (5 mg/mL in water). The pH of the reaction was adjusted to approximately 9 by adding 1 M NaOH. The reaction mixture was then extracted three times with 1 mL of ethyl acetate. The pooled extracts were evaporated under N2 to about 50 μL and streaked on a 0.25 mm thick silica gel (60/254) plate (10 × 20 cm), which was developed with a solvent system of toluene:diethylamine (20:1 v/v). 3-(Benzoyloxy)-8-2-(3-[125I]iodo-4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-7) was detected by autoradiography using Kodak X-Omat film (Rf = 0.78), and the corresponding silica gel was extracted three times with 1 mL of ethyl acetate and stored in −20 °C (yield 1.5 mCi, 30%).
The ethyl acetate extract of 3-(benzoyloxy)-8-2-(3-[125I]iodo-4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-7) was evaporated to dryness under a N2 stream. H2SO4 (50 μL of 3%) was added to the tube, vortexed, and kept on ice for approximately 30 min. To this reaction mixture, 10 μL of 1 M sodium nitrite was added and maintained on ice in the dark for 30 min. Sodium azide (50 μL of 1 M) was added, and the reaction was allowed to proceed on ice in the dark for 30 min. The reaction was terminated by addition of 0.5 mL of 10% sodium bicarbonate and then extracted three times with 1 mL of ethyl acetate. The extracts were pooled and back-extracted with 0.5 mL of water. The extract was then reduced to approximately 50 μL under a N2 stream and streaked on a 0.25 mm thick silica gel (60/254) plate (10 × 20 cm), which was developed in toluene:diethylamine (20:1). The major radioactive reaction product, 3-(benzoyloxy)-8-2-(3-[125I]iodo-4-azidobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-8), migrated with an Rf of 0.92. The material was detected by autoradiography, and the corresponding silica gel was extracted three times with ethyl acetate and stored at −20 °C overnight. The ethyl acetate extract was centrifuged the next day at 24000g for 10 min to remove dissolved silica (appeared as a solid white precipitate). The radioactive product comigrated with authentic nonradioactive 3-(benzoyloxy)-8-2-(3-[127I]iodo-4-azidobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([127I]-8) prepared as described above. The overall yield for the synthesis varied between 18% and 23%.
Radiosynthesis of N-Propyl-N-(4-azido-3-[125I]iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]IAC44)
To 5 mCi of Na125I in 14 μL of 0.1 M NaOH was added 14 μL of 0.1 M HCl and 50 μL of 0.5 M sodium acetate buffer (pH 5.6).N-Propyl-N-(4-aminophenylethyl)-3-(4-fluorophenyl)propylamine (13) (10 μL of 2.5 mg/mL in 0.5 M sodium acetate buffer, pH 5.6) was added. Iodination was initiated by adding 30 μL of Chloramine-T (1 mg/mL in water) and continued for 15 min at room temperature. The reaction was terminated by the addition of 100 μL of Na2S2O5 (5 mg/mL in water). The pH of the reaction was adjusted to approximately 9 by adding 20 μL of 1 M NaOH. The reaction mixture was then extracted three times with 1 mL of ethyl acetate. The pooled extracts were evaporated under N2 to about 50 μL and streaked on a 0.25 mm thick silica gel (60/254) plate (10 × 20 cm), which was developed with a solvent system of toluene:diethylamine (4:1 v/v). N-Propyl-N-(4-amino-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine (14) was detected by autoradiography using Kodak X-Omat film (Rf = 0.70), and the corresponding silica gel was extracted three times with 1 mL of ethyl acetate and stored in −20 °C (yield 1.5 mCi, 30%).
The ethyl acetate extract of N-propyl-N-(4-amino-3-[125I]iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]-14) was evaporated to dryness under a N2 stream. H2SO4 (50 μL of 3%) was added to the tube, vortexed, and kept on ice for approximately 15 min. To this reaction mixture, 10 μL of 1 M sodium nitrite was added and maintained on ice in the dark for 30 min. Sodium azide (50 μL of 1 M) was added, and the reaction was allowed to proceed on ice in the dark for 30 min. The reaction was terminated by addition of 0.5 mL of 10% sodium bicarbonate and then extracted three times with 1 mL of ethyl acetate. The extracts were pooled and back-extracted with 0.5 mL of water. The extract was then reduced to approximately 50 μL under a N2 stream and streaked on a 0.25 mm thick silica gel (60/254) plate (10 × 20 cm), which was developed in toluene:diethylamine (20:1 v/v). The major radioactive reaction product, N-propyl-N-(4-azido-3-[125I]iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]-15) migrated with an Rf of 0.91. The material was detected by autoradiography, and the corresponding silica gel was extracted three times with ethyl acetate and stored at −20 °C overnight. The ethyl acetate extract was centrifuged the next day at 24000g for 10 min to remove dissolved silica (appeared as a solid white precipitate). The radioactive product comigrated with authentic nonradioactive N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)-propylamine (15) prepared as described above. The overall yield for the synthesis of [125I]IAC44 varied between 18% and 20%.
Preparation of Rat Liver and Guinea Pig Liver Membranes
Rat liver and guinea pig liver membranes were prepared as previously reported (3). Once obtained from Pel-Freez (Rogers, AR), frozen rat livers or guinea pig livers (65 g) were minced and thawed in 100 mL of homogenization buffer (10 mM phosphate buffer, pH 7.4, containing 0.32 M sucrose, 1 M MgSO4, 0.5 M EGTA, 1 mM phenylmethanesulfonyl fluoride (PMSF), 10 μg/mL leupeptin, 1 μg/mL pepstatin A, 10 μg/mL p-toluenesulfonyl-L-arginine methyl ester (TAME)). They were then homogenized on ice with a Brinkman polytron homogenizer (setting 6, 4 bursts of 10 s each) followed by a glass homogenizer (Teflon pestle by 6 slow passes at 3000 rpm). The homogenized tissues were centrifuged at 17000g for 10 min, and the supernatants were recentrifuged at 100000g for 1 h. Microsomal pellets were subsequently resuspended in homogenization buffer, snap frozen with dry ice–ethanol, and stored at −80 °C at a final protein concentration of 20 mg/mL.
Protein Assays
Protein concentrations were determined using the microassay protocol (Bradford method) with the dye reagent from Bio-Rad (Bio-Rad Laboratories). Bovine serum albumin was used as the standard protein.
Preparation of Purified Sigma-1 Receptor
Pure guinea pig sigma-1 receptor was prepared as previously described in detail (4). To summarize, the maltose binding protein (MBP)–guinea pig sigma-1 receptor fusion protein with a factor Xa cleavage site linking the MBP and sigma-1 receptor and a six-histidine tag on the C-terminus of sigma-1 receptor was expressed in Escherichia coli.
The E. coli biomass was collected by centrifugation at 3000 rpm for 30 min, sonicated for 15 min at 4 °C, and extracted for 3 h with stirring at 4 °C in 20 mM Tris, pH 8.0, 0.2 M NaCl, 1 mM 2-mercaptoethanol, and 1 mM EDTA supplemented with a protease inhibitor cocktail and 1% Triton X-100 (w/v). The extract was centrifuged at 100000g for 1 h at 4 °C, and the supernatant containing the fusion protein was then purified on an amylose column and cleaved with factor Xa at room temperature for 36–48 h. The sigma-1 receptor with a six-histidine tag at the C-terminus was collected on a Ni2+ column and eluted using 0.25 M imidazole in wash buffer (50 mM sodium phosphate, pH 8.0, 0.3 M NaCl) containing 1% Triton X-100. The protein was then further purified by incubating with an agarose resin coupled with anti-maltose binding protein antibody (binding capacity of approximately 0.4 mg for MBP/mL) at 4 °C for 18–24 h. The mixture was centrifuged at 4000 rpm at room temperature, and the supernatant contained pure guinea pig sigma-1 receptor containing a six-histidine tag at the C-terminus.
Sigma Receptor Binding Assays
Competitive binding assays were performed to determine binding affinities of the compounds listed for the sigma-1 and sigma-2 receptors as previously described but with appropriate modifications (31,32). Sigma-1 assays were performed by first setting up reaction mixtures including 10 nM (+)-[3H]pentazocine, guinea pig liver homogenates, and serial concentrations of the compounds listed in Tables 1 and 2. The mixtures were incubated for 1 h at 30 °C, and the samples were filtered through 0.5% polyethylenimine (PEI) treated Whatman GF/B to measure their ability to displace the radioligands from the sigma-1 receptor. Haloperidol (5 μM) was used to determine non-specific binding. [3H]Pentazocine radioactivity on the filters was detected by liquid scintillation spectrometry using NEN formula 989 Ultima Gold (Perkin-Elmer) as scintillation cocktail. Assays for sigma-2 binding were performed using rat liver homogenate membranes and 3 nM [3H]DTG in the presence of 100 nM (+)-pentazocine. Values were fit to a nonlinear regression curve using graphing software (Graph-pad Prism), and reported dissociation equilibrium constants, KD, were calculated using the Cheng–Prusoff equation (33).
Table 1.
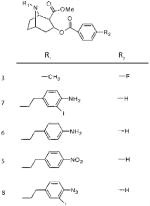
Sigma-1 Receptor Affinity (KD) and One-Site Linear Regression (R2) of N-Substituted Cocaine Derivatives. KD and R2 values of these compounds were calculated using GraphPad Prism software. (A) Competitive displacement curves against 10 nM (+)-[3H]pentazocine were generated, and data points were fit to a one-site nonlinear regression curve. R2 values are reported to examine the “goodness of fit”. Haloperidol (5 μM) was used to determine nonspecific binding. (B) Structures of the N-substituted cocaine derivatives used in competitive displacement assays
| A | |||
|---|---|---|---|
| Ligand | Sigma-1R KD (μM) (± SEM, n=3), R2 | Sigma-2R KD (μM) (± SEM, n=3), R2 | Ratio Sigma2/Sigma1 |
| 3 | 1300 (±401, 0.90) | a | Ndb |
| 7 | 63 (±21, 0.98) | >40000 | 682.5 |
| 6 | 30 (±29.2, 0.86) | 16.7 (±9.8, 0.91) | 0.557 |
| 5 | 18.7 (±8.1, 0.95) | 5350 | 286.1 |
| 8 | 10.97 (±3.2, 0.98) | N/a | Ndb |
| B |
|
Does not compete with [3H]-DTG
Nd - not determined
Table 2.
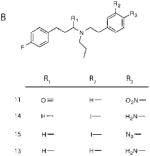
Sigma-1 Receptor Affinity (KD) and One-Site Linear Regression (R2) of N-Alkyl-N′-aralkyl Derivatives. KD and R2 values of these compounds were calculated using GraphPad Prism software. (A) Competitive displacement curves against 10 nM (+)-[3H]pentazocine were generated, and data points were fit to a one-site nonlinear regression curve. R2 values are reported to examine the “goodness of fit”. Haloperidol (5 μM) was used to determine nonspecific binding. (B) Structures of the N-alkyl-N′-aralkyl derivatives used in competitive displacement assays
| A | |||
|---|---|---|---|
| Ligand | Sigma-1R KD (μM) (± SEM, n=3), R2 | Sigma-2R KD (μM) (± SEM, n=3), R2 | Ratio Sigma2/Sigma1 |
| 11 | a | 12.45 (±8.1, 0.88) | Ndb |
| 14 | 28.9 (±8.4, 0.97) | 42.0 (±34, 0.86) | 0.689 |
| 15 | 7.24 (±2.03, 0.98) | 1.29 (±3.4, 0.96) | 0.178 |
| 13 | 2.59 (±0.63, 0.96) | 0.12 (±0.045, 0.91) | 0.046 |
| B |
|
Does not compete with [3H]-DTG
Nd - not determined
Photoaffinity Labeling of the Sigma-1 Receptor
Sigma-1 receptor photoaffinity labeling methods are similar to those previously described (3). In the presence or absence of 10 μM haloperidol, purified guinea pig sigma-1 receptor (200 μg/lane) was suspended to a final volume of 100 μL of incubation buffer (50 mM Tris-HCl, pH 7.4) and incubated at 37 °C for 10 min. The radioactive photoprobes, (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-[125I]iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-N-IACoc) or N-propyl-N-(4-azido-3-[125I]iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]IAC44), were then added to the reactions to a final concentration of 1 nM and incubated for 10 min at 37 °C. The membrane suspensions were diluted 10-fold with ice-cold incubation buffer containing 20 mM β-mercaptoethanol immediately prior to photolysis. The reactions were then exposed to a high-pressure AH-6 mercury lamp (10 cm distance) for 6 s in order to activate the photoaffinity label. After photolysis, the membrane suspensions were centrifuged at 100000g for 1 h at 4 °C and the pellets resuspended in 0.1 mL of incubation buffer. Proteins were separated by SDS–Tricine–PAGE and visualized by phosphorimaging (Molecular Dynamics).
Endo Lys C Digestion of the Photolabeled Sigma-1 Receptor
Endo Lys C cleavage of the sigma-1 receptor was carried out using previous methods as a guideline (3). After the samples of full-length photolabeled sigma-1 receptor were run on a 15% SDS–polyacrylamide gel and visualized by autoradiography, the autoradiogram was then used as a template. Using this template, specifically labeled and haloperidol-protected sigma-1 receptor bands were excised from the gel with a razor blade, which were minced and placed into 1 mL of water to elute overnight at 4 °C. More than 80% of photolabeled sigma-1 receptor was generally recovered in the aqueous supernatant as measured by radioactivity. The eluted materials were concentrated to 100 μL by lyophilization and incubated with 0.25 μg/tube of Endo Lys C (Promega, Madison, WI) overnight at room temperature. Finally, the enzymatic digestions were terminated by adding 5× SDS–PAGE sample buffer and separated by 16.5% Tricine–SDS–PAGE.
CNBr Digestion of the Endo Lys C Photolabeled Sigma-1 Receptor Peptides
Methods for CNBr cleavage were adapted from previously published techniques (3). After the Endo Lys C photolabeled peptides were separated by 16.5% Tricine–SDS–PAGE and visualized by wet gel (unstained) autoradiography, the autoradiogram was used as a template to excise the specifically labeled Endo Lys C peptides with a razor blade. The gel slices were minced, placed into 1 mL of water, and kept overnight at 4 °C. The eluted Endo Lys C peptides were then concentrated to 30 μL, and each was digested with 70 μL of CNBr (0.15 M in 70% formic acid solution) and for 15–18 h at room temperature with tumbling. More than 80% of the photolabeled sigma-1 receptor peptides were recovered in the aqueous supernatant (as measured by radioactivity). The reaction mixtures were then removed of their formic acid with the addition of 1 mL of water and lyophilization (3×). The samples were then separated by 16.5% Tricine–SDS–PAGE, and the radiolabeled peptides were identified by autoradiography.
RESULTS
Chemistry. (−)-Methyl 3-(Benzoyloxy)-8-2-(4-azido-3-iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([127I]-N-IACoc (8)) and Derivatives (3, 5–7)
The synthesis of the N-substituted cocaine derivatives (3–8) is outlined in Scheme 1. (−)-Ecgonine methyl ester (2) was obtained from (−)-cocaine (1) by refluxing (−)-cocaine in a mixture of MeOH and concentrated H2SO4 overnight and was obtained in 95% yield as a yellow oil. 4-Fluorobenzoyl chloride was reacted with (−)-ecgonine methyl ester under solvent-free conditions or in refluxing toluene conditions in the presence of DABCO or triethylamine to afford the (−)-4-fluorobenzoylecgonine methyl ester (3). (−)-Methyl 3-(benzoyloxy)-8-2-(4-nitrobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (5) was prepared by reaction of (−)-norcocaine (4) with 2-(4-nitrobenzene)-1-ethyl bromide in anhydrous ether and was reduced to the (−)-methyl 3-(benzoyloxy)-8-2-(4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (6) in methanol with Pd/C–H2 in 80% yield. Reaction of (−)-methyl 3-(benzoyloxy)-8-2-(4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (6) with Me4N+ICl2− under solid-state conditions afforded (−)-methyl 3-(benzoyloxy)-8-2-(4-amino-3-iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (7) in 85% yield after purification of the crude product by preparative thin-layer chromatography (TLC) (toluene:Et2NH, 4:1). The (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-iodoobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (8) was obtained from (−)-methyl 3-(benzoyloxy)-8-2-(4-amino-3-iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (7) with NaNO2 and then NaN3 in quantitative yield. Compounds 3 and 5–8 were deduced by 1H and 13C NMR and CHN analysis.
N-Propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine ([127I]IAC44 (15)) and Derivatives (11–14)
The synthesis of the N-alkyl-N′-aralkyl derivatives (11–15) is outlined in Scheme 2. 3-(4-Nitrophenyl)propylbromine (10) was obtained as a yellow oil (83% yield) from 3-phenylpropylbromine (9) by reacting under solvent-free conditions until TLC (n-hexane:EtOAc, 90:10) showed complete disappearance of 3-phenylpropylbromine. After reacting 10 with n-propylamine, the resulting amine product was added to a mixture of 4-fluorophenylpropionic acid and DCC to afford N-propyl-N-(4-nitrophenylethyl)-3-(4-fluorophenyl)propionamide (11). N-Propyl-N-(4-aminophenylethyl)-3-(4-fluorophenyl)propionamide (12) was prepared by reduction of N-propyl-N-(4-nitrophenylethyl)-3-(4-fluorophenyl)propionamide (11) in methanol with Pd/C–H2 in 96% yield. The reduction of N-propyl-N-(4-aminophenylethyl)-3-(4-fluorophenyl)propionamide (12) to the amine N-propyl-N-(4-aminophenylethyl)-3-(4-fluorophenyl)propionamine (13) occurred by adding a mixture of 12 and THF dropwise to a solution of LiAlH4 and THF under argon, brief refluxing, and dropwise addition of water, NaOH, and water to destroy excess LiAlH4. Reaction of N-propyl-N-(4-aminophenylethyl)-3-(4-fluorophenyl)propionamine (13) with Me4N+ICl2− under solid-state conditions afforded N-propyl-N-(4-amino-3-iodophenylethyl)-3-(4-fluorophenyl)propionamine (14) in 81% yield after purification of the crude product by column chromatography (silica gel, toluene:Et2NH, 20:1). The N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)propionamine (15) was obtained from N-propyl-N-(4-amino-3-iodophenylethyl)-3-(4-fluorophenyl)propionamine (14) with NaNO2 and then NaN3 in quantitative yield. Compounds 10–15 were deduced by 1H and 13C NMR and CHN analysis.
Binding of N-Substituted Cocaine Derivatives and N-Alkyl-N′-aralkyl Derivatives to Sigma-1 and Sigma-2 Receptors
Compounds were tested for their binding affinities to sigma-1 receptors in guinea pig liver homogenates and to sigma-2 receptors in rat liver homogenates. The binding affinities of these compounds were determined by competitive displacement of radioactive ligands with high affinity and selectivity for sigma-1 and sigma-2 receptors. [3H]-(+)-Pentazocine, for example, was used for competition at the sigma-1 receptor. Due to the absence of a selective sigma-2 receptor ligand, however, the sigma-1 and sigma-2 radioligand, [3H]di-tolylguanidine ([3H]DTG), was utilized in the presence of nonradioactive (+)-pentazocine to generate sigma-2 receptor competitive displacement curves. The results from the described competitive displacement assays for the N-substituted cocaine derivatives are summarized in Table 1. Binding affinity results for the N-alkyl-N′-aralkyl compounds are summarized in Table 2. With the exception of compounds 6, 11, 13, and 15, the relative affinities of these derivatives, while modest for the sigma-2 receptor, are higher for the sigma-1 receptor. The N-substituted cocaine derivative, which was used as a photoprobe (8), was found to have a sigma-1 receptor KD of 10.97 (±3.2) μM, while the N-alkyl-N′-aralkyl photoprobe (15) was found to have a sigma-1 receptor KD of 7.24 (±2.03) μM.
Specific Photolabeling of the Pure Sigma-1 Receptor and Endo Lys C Digestion of [125I]-N-IACoc and [125I]IAC44 Specifically Photolabeled Sigma-1 Receptor
Figure 2 demonstrates specific labeling of full-length purified guinea pig sigma-1 receptor with (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-[125I]iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-N-IACoc) and N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]IAC44). The 26.1 kDa position of the gels, which showed specific labeling by both photo-probes, was excised and eluted with water. Generally, 80% of the 125I-iodinated photoprobe-labeled sigma-1 receptor was eluted. As predicted by theoretical cleavage at lysine 142 (Figure 3A), Endo Lys C digestion resulted in cleavage of the eluted [125I]iodine photolabeled sigma-1 receptor into two large fragments of 16.3 and 9.8 kDa (Figure 3B). As previously reported (2), [125I]IACoc specifically labeled the SBDLII domain (amino acids 170–195) at Asp188, which corresponds to the 9.8 kDa fragment (Figure 3A, lanes 5 and 6). This same SBDLII domain containing peptide (9.8 kDa) was also labeled by [125I]IAC44 (Figure 3B, lanes 1 and 2) in addition to the 16.3 kDa fragment, indicating simultaneous labeling of the region of the sigma-1 receptor, which includes SBDLI (amino acids 91–109). [125I]-N-IACoc, labeling, on the other hand, occurred mainly in the 16.3 kDa fragment, indicating that positioning of the radioactive photoactivatable moiety on the nitrogen of the tropane ring effectively engaged the SBDLI region (Figure 3B, lanes 3 and 4).
Figure 2.
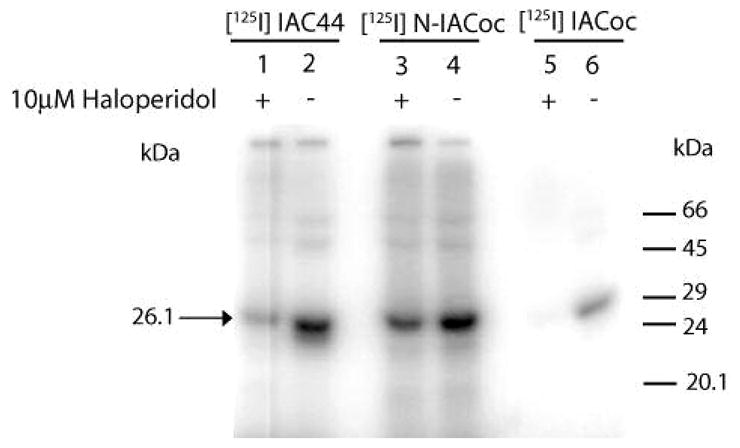
Autoradiogram of [125I]IAC44 (lanes 1 and 2), [125I]-N-IACoc (lanes 3 and 4), and [125I]IACoc (lanes 5 and 6) photolabeling of purified guinea pig sigma-1 receptor (26.1 kDa). Lanes 1, 3, and 5 show specificity of photolabeling with addition of 10 μM haloperidol. Lanes 2, 4, and 6 show photolabeling in the absence of 10 μM haloperidol.
Figure 3.
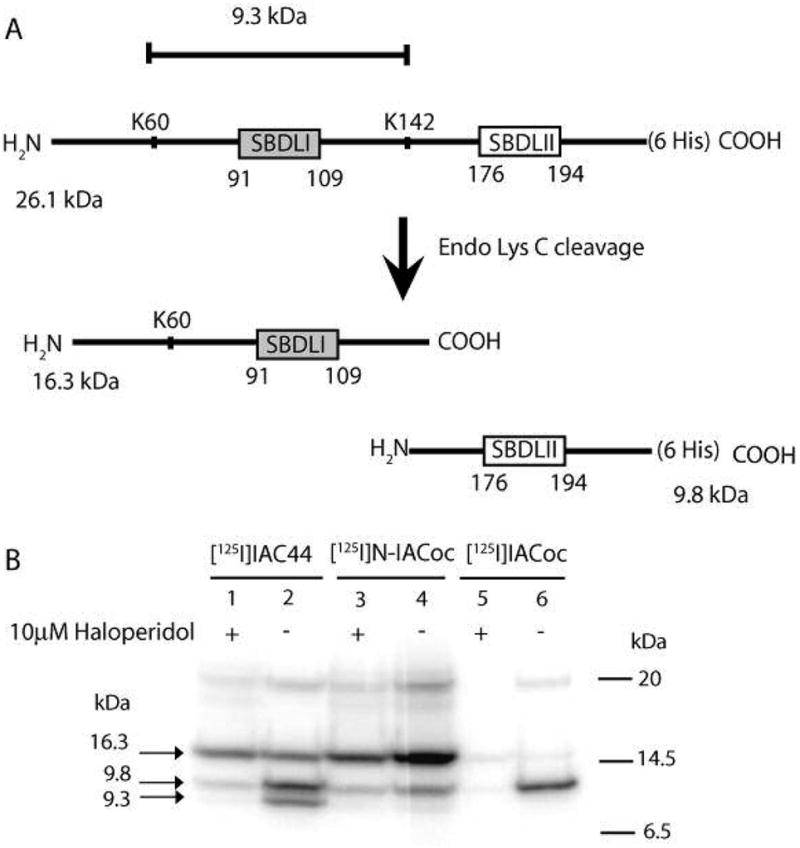
(A) Schematic diagram of Endo Lys C cleavage of the pure guinea pig sigma-1 receptor. (B) Autoradiogram of Endo Lys C cleaved [125I]IAC44 (lanes 1 and 2), [125I]-N-IACoc (lanes 3 and 4), and [125I]IACoc (lanes 5 and 6) photolabeled peptides. Specific labeling was determined by comparing sigma-1 receptor photolabeling in the absence (lanes 2, 4, and 6) and presence of 10 μM haloperidol (lanes 1, 3, and 5). Upon Endo Lys C cleavage, the [125I]IAC44 (lanes 1 and 2), [125I]-N-IACoc (lanes 3 and 4), and [125I]IACoc (lanes 5 and 6) photolabeled receptors resulted in two peptides with molecular masses of 16.3 and 9.8 kDa. The [125I]IAC44 (lanes 1 and 2) photolabeled receptor was also cleaved by Endo Lys C to produce a 9.3 kDa peptide. As expected, the specific radiolabel from [125I]IACoc was located on the SBDLII region (9.8 kDa) (lanes 5 and 6). The specific [125I]-N-IACoc labeled sigma-1 receptor (lanes 3 and 4) afforded radiolabel on the SBDLI (16.3 kDa) containing peptide. Alternatively, the specific [125I]I-AC44 labeled sigma-1 receptor (lanes 1 and 2) showed radiolabel on the SBDLI (16.3 kDa) containing peptide and the SBDLII (9.8 kDa) containing peptide. An additional specific radiolabeled band at 9.3 kDa (lane 2) was observed with [125I]IAF, consistent with the cleavage at position 60 by Endo Lys C, which contains the SBDLI domain.
An additional observation from the Endo Lys C cleavage experiments that was reproducibly observed was that the guinea pig sigma-1 receptor, which contains a lysine at position 60, was not cleaved by the enzyme as would be predicted. The reason for the lack of cleavage by Endo Lys C in guinea pig sigma-1 receptor at position 60 is not readily explained but may be due to a conformational constraint or otherwise lack of access to the proteolytic enzyme. As previously observed (3) covalent labeling by flexible photoprobes such as [125I]IAF (3) and [125I]IAC44 (Figure 3B, lanes 1 and 2) showed a band at 9.3 kDa, which does correspond to a cleavage at lysine 60.
Cyanogen Bromide (CNBr) Cleavage of the Endo Lys C-Derived 16.3 and 9.8 kDa Peptides from the Photolabeled Pure Guinea Pig Sigma-1 Receptor
In order to cleave with cyanogen bromide, the 16.3 and 9.8 kDa peptides from Endo Lys C cleavage were excised and eluted from the SDS gels with water. As with the Endo Lys C cleavage, approximately 80% of the 125I-iodinated photoprobe labeled peptides was eluted. As predicted by theoretical cleavage at methionines within the 16.3 and 9.8 kDa peptides (Figure 4A), CNBr digestion resulted in cleavage of the 16.3 kDa fragment into two fragments (10.5 and 5.5 kDa) as well as cleavage of the 9.8 kDa fragment into two fragments (3 and 6.8 kDa), the smaller of which could not be resolved on Tricine gels. Using the rigid tropane photoprobe [125I]-N-IACoc, haloperidol-protectable labeling was found on the N-terminal 10.5 kDa peptide and the SBDLI-containing 5.5 kDa peptide (Figure 4B, lanes 1 and 3). Nonspecific labeling was also found on the SBDLII-containing 6.8 kDa peptide (Figure 4B, lanes 2 and 4). [125I]IAC44 showed haloperidol-protectable labeling on all the fragments (10.5, 5.5, 6.8 kDa) (Figure 4B, lanes 5 and 6 compared with lanes 7 and 8), indicating access to both SBDLI and SBDLII.
Figure 4.

(A) Schematic diagram depicting CNBr cleavage of the 16.3 and 9.8 kDa peptides derived from Endo Lys C cleavage of pure guinea pig sigma-1 receptors. (B) Autoradiogram of CNBr-cleaved [125I]IAC44 (lanes 1–4) and [125I]-N-IACoc (lanes 5–8) photolabeled peptides from Endo Lys C cleavage (16.3 and 9.8 kDa). Specific labeling was determined by comparing sigma-1 receptor photolabeling in the absence (lanes 3, 4, 7, and 8) and presence of 10 μM haloperidol (lanes 1, 2, 5, and 6). Upon treatment with CNBr, the 16.3 kDa peptides resulted in two peptides with molecular masses of 10.5 and 5.5 kDa (lanes 1, 3, 5, and 7). Likewise, the 9.8 kDa Endo Lys C peptide resulted in a 6.8 kDa peptide after CNBr cleavage (lanes 2, 4, 6, and 8). When [125I]-N-IACoc was used as the photoprobe, the majority of haloperidol-protectable label was found on the 10.5 kDa fragment, with some protectable labeling of the 5.5 kDa peptide (lanes 1 and 3). Photolabeling of the 9.8 and 6.8 kDa bands (lanes 2 and 4) was not specific. When [125I]IAC44 was used as the photoprobe, haloperidol-protectable label was found on the 10.5, 5.5, and 6.8 kDa fragments (lanes 5–8).
DISCUSSION
Previously, our laboratory synthesized the cocaine photoaffinity label, [125I]-3-iodo-4-azidobenzoylmethylecgonine ([125I]IACoc) and characterized it as a high-affinity photo-affinity label for the sigma-1 receptor in rat liver microsomal membranes (1). Further work using [125I]IACoc showed that the insertion site of this photoprobe is at Asp188, which is located in the C-terminal hydrophobic region containing steroid binding domain-like II (SBDLII) (2). Utilizing [125I]IACoc and the more flexible 1-N-(2′,6′-dimethylmorpholino)-3-(4-azido-3-[125I]iodophenyl)propane ([125I]IAF) (3), we previously reported systematic cleavage of the photolabeled pure guinea pig sigma-1 receptor using Endo Lys C and CNBr to identify the sigma-1 receptor regions involved in covalent labeling by these photoprobes (3). While [125I]IACoc exclusively labeled the C-terminal hydrophobic region containing SBDLII and Asp188, [125I]IAF labeled the SBDLII region in addition to the SBDLI hydrophobic region (3). This led to the conclusion that steroid binding domain-like I (SBDLI) (amino acids 91–109) and steroid binding domain-like II (SBDLII) (amino acids 176–194) comprise part, if not all, of the main sigma-1 receptor binding site (3). Therefore, in an effort to probe the “nitrogen interacting regions” of sigma-1 receptor ligands, we designed two new novel N-substituted photoprobes. One of these was also derived from cocaine ([125I]-N-IACoc), but unlike [125I]IA-Coc, the radioactive, photoactivatable moiety extended from the nitrogen of cocaine (Scheme 1, compound 8). The [125I]IAC44 photoprobe is also N-substituted and provides a more flexible backbone (Scheme 2, compound 15).
The sigma-1 receptor affinities of the various N-substituted derivatives were determined by competitive displacement of [3H]-(+)-pentazocine, a sigma-1 specific ligand (Tables 1 and 2). To determine the affinity of the various derivatives for sigma-2 receptors, competitive displacement of [3H]DTG in the presence of nonradioactive (+)-pentazocine (Tables 1 and 2) was utilized (31, 32). Of the N-substituted cocaine derivatives, our results indicate that (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-iodoobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (8) possessed the highest affinity for the sigma-1 receptor with a 100-fold higher affinity than the compound closely resembling cocaine, compound 4)(Table 1). Interestingly, (−)-methyl 3-(benzoyloxy)-8-2-(4-aminobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (6) is the only compound of this group to have low specificity for the sigma-1 receptor since it was observed to have comparable affinity for the sigma-2 receptor. Our findings indicate that compounds 5 and 7 have a high degree of specificity for the sigma-1 receptor and that compound 4, the compound structurally closest to cocaine, essentially does not compete with [3H]DTG at the sigma-2 receptor.
Of the N-alkyl-N′-aralkyl compounds, compound 13 had the highest affinity to the sigma-1 receptor with a KD of 2.59 μM. Specific binding of compound 11 either could not be detected to bind to the sigma-1 receptor (KD > 0.1 mM) or possessed very low affinity, presumably because the amide group present in this compound traps the nitrogen’s lone pair, which is needed for optimal sigma receptor binding as previously reported by Ablordeppey et al. (34). Interestingly, the N-alkyl-N′-aralkyl compounds in this group had reasonable affinity for the sigma-2 receptor in which compounds 11, 15, and 13 slightly favor sigma-2 over sigma-1.
Since (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-iodoobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate (8) and N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)-propylamine (15) both had a sufficient binding affinity for the sigma-1 receptor (KD = 10.97 and 7.24 μM, respectively), they were synthesized as radioiodinated photoaffinity labels and tested. Using the full-length purified guinea pig sigma-1 receptor (Figure 2) compounds 8 and 15 as carrier-free radioactive photoprobes ([125I]-N-IACoc and [125I]IAC44, respectively) showed specific haloperidol-protectable covalent labeling (5 μM).
As illustrated in Figure 3A, Endo Lys C cleavage of the full-length 26.1 kDa pure guinea pig sigma-1 receptor at lysine 142 yielded two large fragments, 16.3 and 9.8 kDa. For reasons that are currently unclear, Endo Lys C cleavage at lysine 60 does not occur readily but can at times be detected. The resultant 9.3 kDa fragment is contained within the larger 16.3 kDa fragment and contains the hydrophobic region, SBDLI (amino acids 91–109) (Figure 3A). The 9.8 kDa fragment, on the other hand, contains SBDLII (amino acids 176–194) (Figure 3A). As previously observed, [125I]IACoc (Figure 3B, lanes 5 and 6) showed one haloperidol-protectable 9.8 kDa fragment which contains both SBDLII and Asp188, the insertion site for [125I]IACoc (2). [125I]-N-IACoc, however, mainly photolabeled the 16.3 kDa fragment, which contains SBDLI (Figure 3B, lanes 3 and 4). The significance of this result is based on the fact that the cocaine photoprobes are rigid in structure due to their tropane ring but differ only in the placement of their radioactive azido moiety. It is reasonable, therefore, to conclude that the cocaine tropane ring is positioned in the binding site in such a way that it is flanked by SBDLI and SBDLII with the benzoyl moiety of cocaine interacting with SBDLII and the bridging tropane nitrogen interacting primarily with SBDLI.
Compared to the rigid tropane-based cocaine photoprobes, [125I]IAC44, on the other hand, is relatively flexible and specifically photolabeled the 9.8 kDa peptide (Figure 3B, lanes 2 and 3). As previously observed with [125I]IAF (3), however, Lys60 cleavage by Endo Lys C produced the 9.3 kDa peptide. This result indicates that the haloperidol-protectable labeling by [125I]IAC44 occurred simultaneously in the regions containing SBDLI and SBDLII, presumably due to the innate flexibility of the probe.
Figure 4A illustrates that additional cleavage of the 16.3 kDa peptide with CNBr resulted in a 10.5 kDa fragment and a 5.5 kDa, the latter of which contains the SBDLI region. Peptides resulting from cleavage at methionines 90 and 93, however, are too small to be detected. The 9.8 kDa peptide, on the other hand, contains a single methionine (Met170) which resulted in CNBr cleavage to produce a 6.8 kDa peptide which contains the SBDLII and a smaller 3 kDa fragment which was too small to resolve by the SDS–PAGE system. Surprisingly, the majority of specific [125I]-N-IACoc photolabeling occurred in the 10.5 kDa fragment (Figure 4B, lanes 1 and 3), which contains neither the putative SBDLI or SBDLII regions. The 10.5 kDa fragment contains the N-terminus and the first hydrophobic transmembrane domain (Figure 5). To a lesser extent, specific [125I]-N-IACoc photolabeling also occurred at the 5.5 kDa SBDLI-containing fragment, but there was no haloperidol-protectable labeling of the 6.8 kDa fragment (Figure 4B, compare lane 1 with 3 and 2 with 4). In contrast, specific haloperidol-protectable [125I]IAC44 photolabeling occurred in the 10.5 and 5.5 kDa fragments (Figure 4B, compare lanes 5 and 7). Unlike [125I]-N-IACoc, however, [125I]IAC44 specifically photolabeled the 6.8 kDa peptide as well (Figure 4B, compare lanes 6 and 8), indicating a likely shared role for the SBDLI and SBDLII regions in the binding site.
Figure 5.

Steroid binding domain-like I (SBDLI) and steroid binding domain-like II (SBDLII) regions of the sigma-1 receptor. Hydrophobic regions of the protein were predicted by the TMPred program (http://www.ch.embnet.org/software/TMPRED_form.html), and the PONDR program (http://www.pondr.com/background.html) was used to predict protein areas of order and disorder. Three highly ordered hydrophobic regions are shown, the latter two of which contain the highly conserved SBDLI and SBDLII regions, respectively. A model of the sigma-1 receptor illustrates the hypothesized spatial arrangement of the receptor binding site (highlighted) with cylinders depicting the hydrophobic regions as putative α-helices.
Previous work in our laboratory has shown that the rigid photoprobe [125I]IACoc labels the SBDLII-containing fragments following CNBr cleavage (3), which is expected since Asp188 is the insertion site for this photoaffinity label (2). The more flexible photoprobe [125I]IAF, however, reacted with residues in the 5.5 kDa, SBDLI-containing peptide in addition to the 6.8 kDa fragment, indicating that this flexible molecule has the ability to bind both the SBDLI and SBDLII regions (3). Support for a single binding site was further indicated by the R (2) values from competitive displacement curves that fit best to a one-site nonlinear regression curve (Tables 1 and 2). The data presented here, using a rigid and a flexible N-substituted photoprobe, support a model for the sigma-1 receptor binding site (Figure 5) in which the N-terminal region containing the first hydrophobic transmembrane domain is also part of the binding site. Site-directed mutagenesis studies have also indicated that amino acid substitutions at residues 28 and 13, which are located in TM1, as well as residue 86 in SBDLI, result in 60% of total haloperidol binding to the sigma-1 receptor (35).
In summary the sigma-1 receptor ligand binding site, as evidenced by the analysis of the specifically photolabeled peptides reported here, is composed of residues within the SBDLI region (amino acids 91–109) which surround the critical nitrogen atom found in all sigma-1 receptor ligands. In addition, the binding site is composed of the SBDLII region (amino acids 176–194), which surrounds the phenyl ring also found in all sigma-1 receptor ligands. It is further likely that the first transmembrane segment additionally supports ligand binding via a close interaction with SBDLI and/or SBDLII. The interaction of sigma-1 receptor ligands and/or endogenous ligands may be further regulated by cholesterol, which has been demonstrated to bind to two amino acid segments, 161 to 180 and 191 to 210, which flank the SBDLII region (36). Binding to cholesterol in this C-terminal region was further demonstrated to be reduced by the sigma-1 ligand, SKF-10047 (36). Taken together, the N-substituted photoprobes reported herein identify an intramolecular juxtaposition of the TM1/SBDLI region of the sigma-1 receptor to the SBDLII region to create a ligand binding site.
Acknowledgments
We acknowledge valuable discussion during these experiments with Subramaniam Ramachandran.
Footnotes
Supported by National Institutes of Health Grant R01 MH065503 to A.E.R. D.F. was supported in part by the MCP training grant from the National Institutes of Health (T32 GM08688) and by the National Institutes of Health under the Ruth L. Kirschstein National Research Service Award (F31 DA022932) from the National Institute on Drug Abuse.
Abbreviations: IACoc, 3-iodo-4-azidococaine; IAF, 1-N-(2′,6′-dimethylmorpholino)-3-(4-azido-3-iodophenyl)propane; N-IACoc, (−)methyl 3-(benzoyloxy)-8-2-(4-azido-3-iodobenzene)-1-ethyl-8-aza-bicyclo[3.2.1]octane-2-carboxylate; IAC44, N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine; SBDLI, sterol binding domain-like I; SBDLII, sterol binding domain-like II; CNBr, cyanogen bromide; PCP, phencyclidine; TMD, transmembrane domain; ERG2, yeast sterol C8-C7 isomerase; DTG, ditolylguanidine.
References
- 1.Kahoun JR, Ruoho AE. (125I)iodoazidococaine, a photoaffinity label for the haloperidol-sensitive sigma receptor. Proc Natl Acad Sci USA. 1992;89:1393–1397. doi: 10.1073/pnas.89.4.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Hajipour AR, Sievert MK, Arbabian M, Ruoho AE. Characterization of the cocaine binding site on the sigma-1 receptor. Biochemistry. 2007;46:3532–3542. doi: 10.1021/bi061727o. [DOI] [PubMed] [Google Scholar]
- 3.Pal A, Hajipour AR, Fontanilla D, Ramachandran S, Chu U, Mavlyutov T, Ruoho AE. Identification of regions of the sigma-1 receptor ligand binding site using a novel photoprobe. Mol Pharmacol. 2007;72:921–933. doi: 10.1124/mol.107.038307. [DOI] [PubMed] [Google Scholar]
- 4.Ramachandran S, Lu H, Prabhu U, Ruoho AE. Purification and characterization of the guinea pig sigma-1 receptor functionally expressed in Escherichia coli. Protein Expression Purif. 2007;51:283–292. doi: 10.1016/j.pep.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- 6.Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio JM, Rothman RB, Su TP, Tam SW, Taylor DP. A proposal for the classification of sigma binding sites. Trends Pharmacol Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 7.Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci USA. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kekuda R, Prasad PD, Fei YJ, Leibach FH, Ganapathy V. Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1) Biochem Biophys Res Commun. 1996;229:553–558. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- 9.Prasad PD, Li HW, Fei YJ, Ganapathy ME, Fujita T, Plumley LH, Yang-Feng TL, Leibach FH, Ganapathy V. Exon-intron structure, analysis of promoter region, and chromosomal localization of the human type 1 sigma receptor gene. J Neurochem. 1998;70:443–451. doi: 10.1046/j.1471-4159.1998.70020443.x. [DOI] [PubMed] [Google Scholar]
- 10.Mei J, Pasternak GW. Molecular cloning and pharmacological characterization of the rat sigma1 receptor. Biochem Pharmacol. 2001;62:349–355. doi: 10.1016/s0006-2952(01)00666-9. [DOI] [PubMed] [Google Scholar]
- 11.Seth P, Fei YJ, Li HW, Huang W, Leibach FH, Ganapathy V. Cloning and functional characterization of a sigma receptor from rat brain. J Neurochem. 1998;70:922–931. doi: 10.1046/j.1471-4159.1998.70030922.x. [DOI] [PubMed] [Google Scholar]
- 12.Pan YX, Mei J, Xu J, Wan BL, Zuckerman A, Pasternak GW. Cloning and characterization of a mouse sigma1 receptor. J Neurochem. 1998;70:2279–2285. doi: 10.1046/j.1471-4159.1998.70062279.x. [DOI] [PubMed] [Google Scholar]
- 13.Moebius FF, Reiter RJ, Hanner M, Glossmann H. High affinity of sigma 1-binding sites for sterol isomerization inhibitors: evidence for a pharmacological relationship with the yeast sterol C8-C7 isomerase. Br J Pharmacol. 1997;121:1–6. doi: 10.1038/sj.bjp.0701079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 15.Vagnerova K, Hurn PD, Bhardwaj A, Kirsch JR. Sigma 1 receptor agonists act as neuroprotective drugs through inhibition of inducible nitric oxide synthase. Anesth Analg. 2006;103:430–434. doi: 10.1213/01.ane.0000226133.85114.91. [DOI] [PubMed] [Google Scholar]
- 16.Marrazzo A, Caraci F, Salinaro ET, Su TP, Copani A, Ronsisvalle G. Neuroprotective effects of sigma-1 receptor agonists against beta-amyloid-induced toxicity. Neuroreport. 2005;16:1223–1226. doi: 10.1097/00001756-200508010-00018. [DOI] [PubMed] [Google Scholar]
- 17.Gopalakrishnan A, Sievert M, Ruoho AE. Identification of the substrate binding region of vesicular monoamine transporter-2 (VMAT-2) using iodoaminoflisopolol as a novel photoprobe. Mol Pharmacol. 2007;72:1567–1575. doi: 10.1124/mol.107.034439. [DOI] [PubMed] [Google Scholar]
- 18.Wilke RA, Mehta RP, Lupardus PJ, Chen Y, Ruoho AE, Jackson MB. Sigma receptor photolabeling and sigma receptor-mediated modulation of potassium channels in tumor cells. J Biol Chem. 1999;274:18387–18392. doi: 10.1074/jbc.274.26.18387. [DOI] [PubMed] [Google Scholar]
- 19.Kavanaugh MP, Tester BC, Scherz MW, Keana JF, Weber E. Identification of the binding subunit of the sigma-type opiate receptor by photoaffinity labeling with 1-(4-azido-2-methyl[6-3H]phenyl)-3-(2-methyl[4,6-3H]phenyl)guanidine. Proc Natl Acad Sci USA. 1988;85:2844–2848. doi: 10.1073/pnas.85.8.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moebius FF, Burrows GG, Hanner M, Schmid E, Striessnig J, Glossmann H. Identification of a 27-kDa high affinity phenylalkylamine-binding polypeptide as the sigma 1 binding site by photoaffinity labeling and ligand-directed antibodies. Mol Pharmacol. 1993;44:966–971. [PubMed] [Google Scholar]
- 21.Dhanasekaran N, Wessling-Resnick M, Kelleher DJ, Johnson GL, Ruoho AE. Mapping of the carboxyl terminus within the tertiary structure of transducin’s alpha subunit using the heterobifunctional cross-linking reagent, 125I-N-(3-iodo-4-azidophenylpropionamido)-S-(2-thiopyridyl) cysteine. J Biol Chem. 1988;263:17942–17950. [PubMed] [Google Scholar]
- 22.Hockerman GH, Girvin ME, Malbon CC, Ruoho AE. Antagonist conformations with the beta(2)-adrenergic receptor ligand binding pocket. Mol Pharmacol. 1996;49:1021–1032. [PubMed] [Google Scholar]
- 23.Rong Y, Arbabian M, Thiriot DS, Seibold A, Clark RB, Ruoho AE. Probing the salmeterol binding site on the beta 2-adrenergic receptor using a novel photoaffinity ligand, [125I]iodoazidosalmeterol. Biochemistry. 1999;38:11278–11286. doi: 10.1021/bi9910676. [DOI] [PubMed] [Google Scholar]
- 24.Sievert MK, Ruoho AE. Peptide mapping of the [125I]Iodoazidoketanserin and [125I]2-N-[(3′-iodo-4′-azidophenyl) propionyl]tetrabenazine binding sites for the synaptic vesicle monoamine transporter. J Biol Chem. 1997;272:26049–26055. doi: 10.1074/jbc.272.41.26049. [DOI] [PubMed] [Google Scholar]
- 25.Vaillancourt RR, Dhanasekaran N, Ruoho AE. Synthesis and use of radioactive photoactivatable NAD+ derivatives as probes for G-protein structure. Methods Enzymol. 1994;237:70–99. doi: 10.1016/s0076-6879(94)37054-0. [DOI] [PubMed] [Google Scholar]
- 26.Vaillancourt RR, Dhanasekaran N, Johnson GL, Ruoho AE. 2-Azido-[32P]NAD+, a photoactivatable probe for G-protein structure: evidence for holotransducin oligomers in which the ADP-ribosylated carboxyl terminus of alpha interacts with both alpha and gamma subunits. Proc Natl Acad Sci USA. 1990;87:3645–3649. doi: 10.1073/pnas.87.10.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, Ruoho AE. A high-affinity fluorenone-based beta 2-adrenergic receptor antagonist with a photoactivatable pharmacophore. Biochemistry. 2000;39:13044–13052. doi: 10.1021/bi001342k. [DOI] [PubMed] [Google Scholar]
- 28.Wu Z, Thiriot DS, Ruoho AE. Tyr199 in transmembrane domain 5 of the beta2-adrenergic receptor interacts directly with the pharmacophore of a unique fluorenone-based antagonist. Biochem J. 2001;354:485–491. doi: 10.1042/0264-6021:3540485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hajipour AR, Arbabian M, Ruoho AE. Tetramethylammonium dichloroiodate: An efficient and environmentally friendly iodination reagent for iodination of aromatic compounds under mild and solvent-free conditions. J Org Chem. 2002;67:8622–8624. doi: 10.1021/jo0264628. [DOI] [PubMed] [Google Scholar]
- 30.Hajipour AR, Kooshki B, Ruoho AE. Nitric acid in the presence of supported P2O5 on silica gel: an efficient and novel reagent for oxidation of sulfides to the corresponding sulfoxides. Tetrahedron Lett. 2005;46:5503–5506. [Google Scholar]
- 31.Matsumoto RR, Bowen WD, Tom MA, Vo VN, Truong DD, De Costa BR. Characterization of two novel sigma receptor ligands: antidystonic effects in rats suggest sigma receptor antagonism. Eur J Pharmacol. 1995;280:301–310. doi: 10.1016/0014-2999(95)00208-3. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen EC, McCracken KA, Liu Y, Pouw B, Matsumoto RR. Involvement of sigma (sigma) receptors in the acute actions of methamphetamine: receptor binding and behavioral studies. Neuropharmacology. 2005;49:638–645. doi: 10.1016/j.neuropharm.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 33.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 34.Ablordeppey SY, Fischer JB, Glennon RA. Is a nitrogen atom an important pharmacophoric element in sigma ligand binding? Bioorg Med Chem. 2000;8:2105–2111. doi: 10.1016/s0968-0896(00)00148-6. [DOI] [PubMed] [Google Scholar]
- 35.Ganapathy ME, Prasad PD, Huang W, Seth P, Leibach FH, Ganapathy V. Molecular and ligand-binding characterization of the sigma-receptor in the Jurkat human T lymphocyte cell line. J Pharmacol Exp Ther. 1999;289:251–260. [PubMed] [Google Scholar]
- 36.Palmer CP, Mahen R, Schnell E, Djamgoz MB, Aydar E. Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 2007;67:11166–11175. doi: 10.1158/0008-5472.CAN-07-1771. [DOI] [PubMed] [Google Scholar]