

Abstract
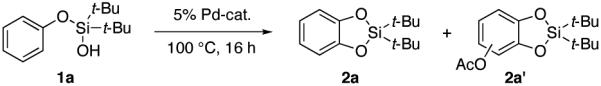
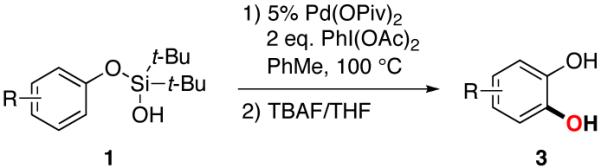
A silanol-directed, Pd-catalyzed C–H oxygenation of phenols into catechols is presented. This method is highly site selective and general, as it allows for oxygenation of not only electron-neutral- but also electron poor phenols. This method operates via a silanol-directed acetoxylation, followed by a subsequent acid-catalyzed cyclization reaction into a cyclic silicon-protected catechol. A routine desilylation of the silacyle with TBAF uncovers the catechol product.
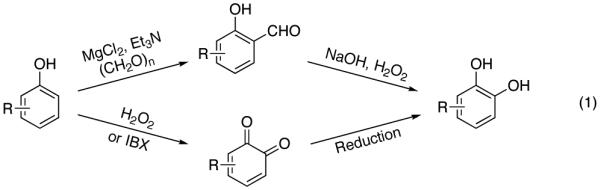
Catechols are widely present in natural products and extensively used in nearly every sector of chemical industries.1 They are common structural motifs found in many bioactive molecules and drugs (Figure 1).1a Due to the regiospecific nature of biotransformations, synthesis of substituted catechols is prevailed by a fermentation of phenols.2 In addition, a few synthetic procedures exist for transformation of substituted phenols into catechols.3 One practical procedure involves ortho-formylation of phenols followed by a subsequent Dakin oxidation (eq 1, top).3a However, this process suffers from low selectivity, particularly for meta-substituted phenols.4 Another method employs oxidation of phenols to o-quinones and a subsequent reduction of the latter into catechols (eq 1, bottom). The industrial version of this method employing H2O2 oxidation usually provides a mixture of catechol and para-hydroquinone,1a,c while the method using 2-iodoxybenzoic acid (IBX) as an oxidant is restricted to electron-rich substrates only.3b An improved version of the latter method somewhat expands the scope of phenols used, however provides lower regioselectivity of oxidation.3c Thus, the development of efficient, general, and selective methods for conversion of phenols into catechols is warranted. Herein, we wish to report a novel approach towards catechols from phenols via the Pd-catalyzed silanol-directed ortho C–H oxygenation (eq 2), a process featuring high site selectivity and a broad functional group tolerance.
Figure 1.
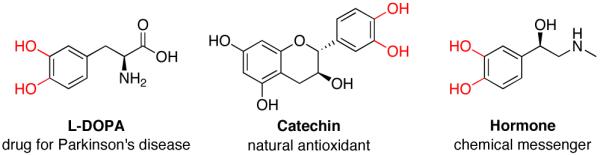
Catechol-containing natural products and pharmaceuticals.
 |
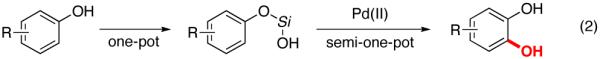 |
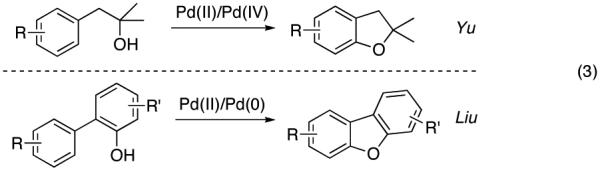
Transition metal-catalyzed directed C–H5 oxygenation of arenes has emerged as one of the most powerful tools for synthesis of phenol derivatives.6 Recently, Yu7 and Liu8 disclosed an intramolecular hydroxyl group-directed Pd-catalyzed oxygenation of arenes proceeding via a C–H activation/C–O cyclization protocol (eq 3). On the other hand, our group has recently introduced the silanol as a traceless directing group for the Pd-catalyzed ortho-alkenylation of phenols.9,10 Considering the similarity of the OH functionality in alcohols and in silanols, we hypothesized that phenoxy silanol 1 could also undergo the Pd-catalyzed C–H activation/C–O cyclization reaction into silacycle 2. The latter, upon subsequent desilylation, would furnish catechol 3.
 |
Accordingly, phenoxy silanol 1a was tested in this oxygenation process. The reaction of 1a under the Pd-catalyzed cyclization conditions developed by Yu7 provided 43% GC yield of silacycle 2a along with 3% of an over-oxidized byproduct 2a’ (Table 1, entry 1). Gratifyingly, a better yield of 2a was obtained in the absence of base11 (entry 2). Pd(OPiv)2 was found to be superior among different palladium sources tested (entries 2-4). It was found that during the reaction, toluene was partially oxidized into isomeric tolyl acetates. To avoid that, fluorinated solvents were tested. However, their employment was not beneficial (entries 5-6). Employment of larger amounts of PhI(OAc)2 improved the yield to 58% (79% bsrm, entry 7). Further increase of the oxidant resulted in no improvement (entry 8). Expectedly, there was no reaction without palladium catalyst (entry 9).
Table 1.
Screening of Reaction Conditions for C–O Cyclization
 | |||||
|---|---|---|---|---|---|
| entry | Pd-cat. | oxidant | solvent | yield, %a |
|
| 2a | 2a’ | ||||
| 1b | Pd(OPiv)2 | PhI(OAc)2 (1.5 eq) | PhMe | 43 (74) | 2 |
| 2 | Pd(OPiv)2 | PhI(OAc)2 (1.5 eq) | PhMe | 50 (65) | 3 |
| 3 | Pd(OAc)2 | PhI(OAc)2 (1.5 eq) | PhMe | 40 (67) | 3 |
| 4 | Pd(OTf)2 | PhI(OAc)2 (1.5 eq) | PhMe | 40 (78) | 2 |
| 5 | Pd(OPiv)2 | PhI(OAc)2 (1.5 eq) | C6F6 | 37 (45) | 3 |
| 6 | Pd(OPiv)2 | PhI(OAc)2 (1.5 eq) | PhCF3 | 47 (53) | 14 |
| 7 | Pd(OPiv)2 | PhI(OAc)2 (2.0 eq) | PhMe | 58 (79) | 6 |
| 8 | Pd(OPiv)2 | PhI(OAc)2 (3.0 eq) | PhMe | 47 (52) | 6 |
| 9 | none | PhI(OAc)2 (1.5 eq) | PhMe | 0 | 0 |
GC yields against tetradecane as internal standard, brsm yields in the parentheses (based on recovered starting material).
Li2CO3 (1 equiv) was added.
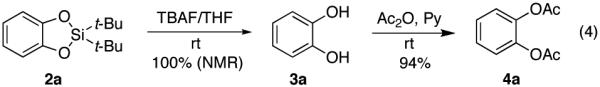
A routine desilylation of 2a with TBAF quantitatively released catechol 3a (eq 4). To ease separation, catechol 3a was efficiently converted into its bis-acetate derivative 4a.
 |
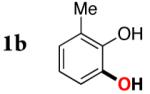
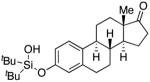
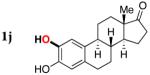
Next, the scope of the combined semi-one-pot cyclization/desilylation procedure from silanols 1 to catechols 3 was investigated (Table 2). It was found that substrates with electron donating groups typically reacted faster, providing good to excellent yields of the catechols (3b-h). Remarkably, in contrast to the previous catechol syntheses,3 this transformation demonstrated excellent site selectivity, directing the newly installed hydroxyl group to the sterically less hindered C–H site. Of note, estrone was highly efficiently and selectively converted into 2-hydroxyestrone (3j), an important intermediate of the estrone metabolism in human body.13
Table 2.
Scope of Catechol Synthesis
 | ||||
|---|---|---|---|---|
| entry | silanol | catechol | yield, %a | |
| 1 |
|
|
3b | 81 |
| 2 |
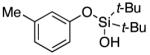
|
|
3c | 94 |
| 3 |
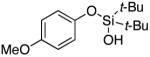
|

|
3d | 57b |
| 4 |
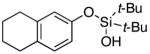
|
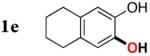
|
3e | 77 |
| 5 |
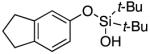
|
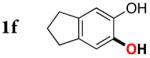
|
3f | 68c |
| 6 |
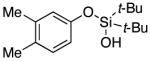
|
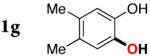
|
3g | 93 |
| 7 |
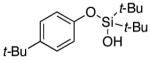
|
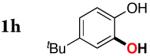
|
3h | 78 |
| 8 |
|
|
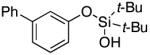
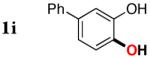
3i | 87 |
| 9 |
|
|
3j | 88 |
| 10 |
|
|
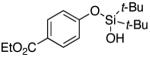
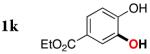
3k | 76c |
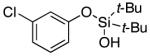
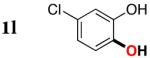
| 1l |
|
|
3l | 62b,d |
| 12 |
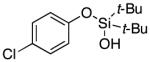
|
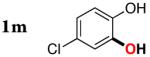
|
3m | 84b,d |
| 13 |
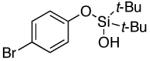
|
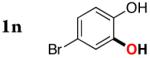
|
3n | 83b,d |
| 14 |
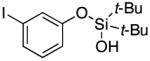
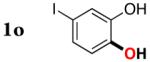
|
|
3o | 70b,d |
| 15 |
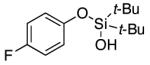
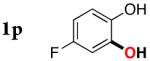
|
|
3p | 60b,d |
| 16 |
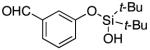
|
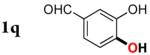
|
3q | 47b,d,e |
| 17 |
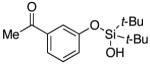
|
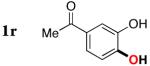
|
3r | 65b,d |
| 18 |
|
|
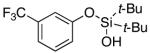
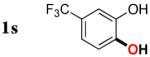
3s | 35b,d |
| 19 |
|
|
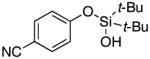
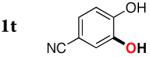
3t | 29b,d |
| 20 |
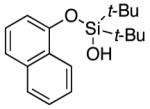
|
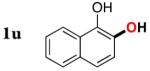
|
3u | 76b,d |
| 21 |
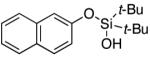
|
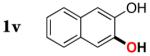
|
3v | 54b,d |
Isolated yields.
Isolated as bis-acetates by further treatment of the catechols with Ac2O and pyridine in the same pot.12
Major isomer is shown (34:1).
PhCF3 was used instead of PhMe, PhI(OAc)2 (1.5 equiv), 120 °C.
10 mol% Pd(OPiv)2 was used.
Since the existing synthetic methods are marginally efficient and/or selective for oxidation of electron deficient phenols,3 it was interesting to probe the generality of this new C–H functionlization method. Thus, oxygenation of para-ester-substituted substrate 1k gave 24% NMR yield of the cyclization product 2k. Switching to PhCF3 at elevated temperature (120 °C) dramatically improved the yield of 3k (entry 10). Moreover, phenols possessing F, Cl, Br, and I, reacted well under the modified conditions (entries 11-15). Substrates possessing aldehyde (1q) and ketone (1r) functionalities were smoothly oxidized into the corresponding catechols in moderate yields (entries 16-17). Phenols possessing CF3 and CN groups were less efficient providing 35% and 29% yields, respectively (entries 18-19). 1- and 2-Naphthol derivatives were also competent reactants in this oxygenation reaction (entries 20-21). Remarkably, the silanol-directed oxygenation reaction allows access to naphthalene-2,3-diol 3v from 2-naphthol derivative 1v (entry 21), demonstrating orthogonal site selectivity of this method to the existing techniques, which convert 2-naphthol into regioisomeric naphthalene-1,2-diol 3u.3c
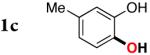
Interestingly, the GC/MS analyses of the oxygenation of 1c at the early stages of the reaction indicate formation of acetoxylated product 5c. This was further investigated by a careful monitoring of the reaction under the standard conditions. The reaction profile clearly shows the formation and decay of acetoxylated product 5c during the reaction course (Figure 2). Meanwhile, increasing amounts of the cyclization product 2c was also observed from the very beginning of the reaction. In order to understand how the acetoxylated product 5c is transformed into the silacyle 2c, several experiments with 5c, isolated from the reaction mixture, have been performed. Thus, simple heating of 5c in PhMe at 100 °C for 12 h gave no reaction. However, addition of 2 equiv HOAc led to a full conversion of 5c into 2c within 10 h. Expectedly, 5c was smoothly transformed into 2c under the standard reaction conditions. Based on these results, the transformation of the acetoxylated product 5c into the silacylcle 2c seems to be mediated by HOAc, which is generated during the reaction course (see Scheme 1).
Figure 2.
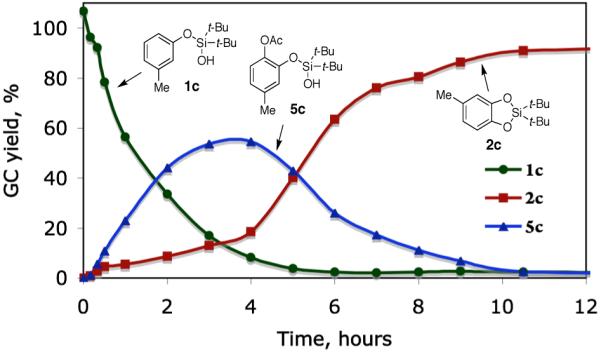
Reaction profile of PdII-catalyzed silanol-directed C–H acetoxylation and cyclization, picturing the formation and decline of acetoxylated product 5c as the intermediate in the reaction. Reaction conditions: 1c (0.2 mmol), Pd(OPiv)2 (0.01 mol), PhI(OAc)2 (0.4 mmol), PhMe (2 ml), 100 °C. The reaction was monitored by GC/MS with tetradecane as the internal standard
Scheme 1.
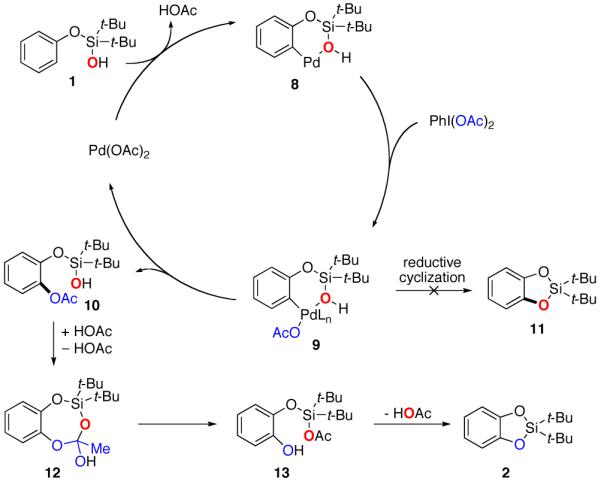
Plausible Reaction Pathway
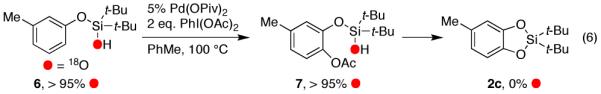
In order to verify whether silacyle 2c arises solely through a stepwise route involving acetoxylated product 5c or it also forms via a direct C–O reductive cyclization,7 the 18O-labeled silanol 6 was subjected to the standard reaction conditions (eq 6). It was found that 18O-labeled acetoxylated product 7 was formed and then gradually declined during the reaction producing the cyclized product 2c with no 18O label incorporated (eq 6). It deserves mentioning that throughout the reaction course the abundance of the 18O label in both the starting silanol 6 and the acetoxylated product 7 remained unchanged.
 |
In light of these observations, a plausible reaction pathway for the Pd-catalyzed silanol-directed ortho C–H oxygenation is proposed (Scheme 1). First, Pd(OAc)2 (or palladium pivalate) reacts with silanol 1 producing palladacycle 8,14 in which silanol acts as a neutral directing group for palladium. 15 Next, PdII in palladacycle 8 is oxidized by PhI(OAc)2 to a higher oxidation state (PdIV or PdIII)16 to give intermediate 9. The direct C–O reductive cyclization from Pd to form 11 was ruled out by the 18O-labeling studies (vide supra). Instead, a reductive acetoxylation from 9 regenerates the PdII catalyst and produces the observed acetoxylated intermediate 10. The latter, presumably via an acid-catalyzed17 transesterification into 13 and a subsequent loss of the 18O labeled acetic acid produces cyclic silyl-proctected catechol 2.
In summary, we have developed a semi-one-pot Pd(II)-catalyzed silanol-directed C–H oxygenation of phenols into catechols. This new method operates via a consecutive C–H acetoxylation/acid-catalyzed transesterification/cyclization sequence. In a striking contrast to the known alcohol-7 and phenol-directed8 C–O cyclization methods, where the directing group serves as the oxygen source, in our oxygenation method the oxygen atom of the newly installed hydroxyl group is delivered by the oxidant. This new method allows for efficient and site selective construction of substituted catechols, including electron-defficient catechols, which are not easily accessible via existing synthetic approaches.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (GM-64444) for financial support of this work.
Footnotes
Supporting Information. Detailed experimental procedures and charcterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Fiegel H, Voges H-W, Hamamoto T, Umemura S, Iwata T, Miki H, Fujita Y, Buysch H-J, Garbe D, Paulus W. Phenol Derivatives in Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH; New York: 2002. [Google Scholar]; (b) Barner BA. “Catechol”. In: Paquette L, editor. Encyclopedia of Reagents for Organic Synthesis. John Wiley and Sons; New York: 2004. [Google Scholar]; (c) Karakhanov EA, Maximov AL, Kardasheva YS, Skorkin VA, Kardashev SV, Ivanova EA, Lurie-Luke E, Seeley JA, Cron SL. Ind. Eng. Chem. Res. 2010;49:4607. [Google Scholar]
- (2).(a) Draths KM, Frost JW. J. Am. Chem. Soc. 1991;113:9361. [Google Scholar]; (b) Bui VP, Hansen TV, Stenstrøm Y, Hudlicky T. Green Chem. 2000;2:263. [Google Scholar]; (c) Nolan LC, O’Connor KE. Biotechnol. Lett. 2008;30:1879. doi: 10.1007/s10529-008-9791-5. [DOI] [PubMed] [Google Scholar]; (d) Berberian V, Allen CCR, Sharma ND, Boyd DR, Hardacre C. Adv. Synth. Catal. 2007;349:727. [Google Scholar]; (e) Prakash D, Pandey J, Tiwary BN, Jain RK. BMC Biotechnology. 2010;10:49. doi: 10.1186/1472-6750-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Montersino S, Tischler D, Gassner GT, van Berkel WJH. Adv. Synth. Catal. 2011;353:2301. [Google Scholar]
- (3).(a) Hansen TV, Skattebøl L. Tetrahedron Lett. 2005;46:3357. [Google Scholar]; (b) Magdziak D, Rodriguez AA, Van De Water RW, Pettus TRR. Org. Lett. 2002;4:285. doi: 10.1021/ol017068j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pezzella A, Lista L, Napolitano A, d’Ischia M. Tetrahedron Lett. 2005;46:3541. [Google Scholar]
- (4).Hofsløkken NU, Skattebøl L. Acta Chem. Scand. 1999;53:258. [Google Scholar]
- (5).For general reviews on transition metal-catalyzed C–H activation of arenes, see: Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077. Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. Yu J-Q, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k. Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. Campeau L-C, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35. Ackermann L, Vicente R, Kapdi AR. Angew. Chem., Int. Ed. 2009;48:9792. doi: 10.1002/anie.200902996. Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058. McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447. doi: 10.1039/b805701j. Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890. doi: 10.1021/cr900206p. Ashenhurst JA. Chem. Soc. Rev. 2010;39:540. doi: 10.1039/b907809f. Satoh T, Miura M. Synthesis. 2010:3395. Sun C-L, Li B-J, Shi Z-J. Chem. Rev. 2011;111:1293. doi: 10.1021/cr100198w. Wencel-Delord J, Droge T, Liu F, Glorius F. Chem. Soc. Rev. 2011;40:4740. doi: 10.1039/c1cs15083a.
- (6).For reviews on transition metal-catalyzed oxygenation of arenes, see: Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e. Sehnal P, Taylor RJK, Fairlamb IJS. Chem. Rev. 2010;110:824. doi: 10.1021/cr9003242. Muñiz K. Angew. Chem., Int. Ed. 2009;48:9412. doi: 10.1002/anie.200903671. Alonso DA, Nájera C, Pastor IM, Yus M. Chem. Eur. J. 2010;16:5274. doi: 10.1002/chem.201000470. Enthaler S, Company A. Chem. Soc. Rev. 2011;40:4912. doi: 10.1039/c1cs15085e. For recent representative examples, see: Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300. doi: 10.1021/ja031543m. Kalyani D, Sanford MS. Org. Lett. 2005;7:4149. doi: 10.1021/ol051486x. Kim SH, Lee HS, Kim SH, Kim JN. Tetrahedron Lett. 2008;49:5863. Chernyak N, Dudnik AS, Huang C, Gevorgyan VJ. Am. Chem. Soc. 2010;132:8270. doi: 10.1021/ja1033167. Desai LV, Malik HA, Sanford MS. Org. Lett. 2006;8:1141. doi: 10.1021/ol0530272. Desai LV, Stowers KJ, Sanford MS. J. Am. Chem. Soc. 2008;130:13285. doi: 10.1021/ja8045519. Wang G-W, Yuan T-T, Wu X-L. J. Org. Chem. 2008;73:4717. doi: 10.1021/jo8003088. Gou F-R, Wang X-C, Huo P-F, Bi H-P, Guan Z-H, Liang Y-M. Org. Lett. 2009;11:5726. doi: 10.1021/ol902497k. Zhang Y-H, Yu J-Q. J. Am. Chem. Soc. 2009;131:14654. doi: 10.1021/ja907198n. Zheng X, Song B, Xu B. Eur. J. Org. Chem. 2010;4376 Racowski JM, Dick AR, Sanford MS. J. Am. Chem. Soc. 2009;131:10974. doi: 10.1021/ja9014474. Wang G-W, Yuan T-T. J. Org. Chem. 2010;75:476. doi: 10.1021/jo902139b. Anand M, Sunoj RB. Org. Lett. 2011;13:4802. doi: 10.1021/ol201830r. Gu S, Chen W. J. Org. Chem. 2009;74:7203. doi: 10.1021/jo901316b. Richter H, Beckendorf S, Mancheno OG. Adv. Synth. Catal. 2011;353:295. Huang C, Chernyak N, Dudnik AS, Gevorgyan V. Adv. Synth. Catal. 2011;353:1285. doi: 10.1002/adsc.201000975.
- (7).Wang X, Lu Y, Dai H-X, Yu J-Q. J. Am. Chem. Soc. 2010;132:12203. doi: 10.1021/ja105366u. [DOI] [PubMed] [Google Scholar]
- (8).Xiao B, Gong T-J, Liu Z-J, Liu J-H, Luo D-F, Xu J, Liu L. J. Am. Chem. Soc. 2011;133:9250. doi: 10.1021/ja203335u. [DOI] [PubMed] [Google Scholar]
- (9).Huang C, Chattopadhyay B, Gevorgyan V. J. Am. Chem. Soc. 2011;133:12406. doi: 10.1021/ja204924j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For o-arylation of phenols, see: Bedford RB, Coles SJ, Hursthouse MB, Limmert ME. Angew. Chem., Int. Ed. 2003;42:112. doi: 10.1002/anie.200390037. Bedford RB, Limmert ME. J. Org. Chem. 2003;68:8669. doi: 10.1021/jo030157k. Oi S, Watanabe S-I, Fukita S, Inoue Y. Tetrahedron Lett. 2003;44:8665. Bajracharya GB, Daugulis O. Org. Lett. 2008;10:4625. doi: 10.1021/ol801897m. For o-alkylation of phenols, see: Lewis LN, Smith JF. J. Am. Chem. Soc. 1986;108:2728. Dorta R, Tongi A. Chem. Commun. 2003;760 doi: 10.1039/b211672c. Carrión MC, Cole-Hamilton DJ. Chem. Commun. 2006;4527 doi: 10.1039/b610038d. Lewis JC, Wu J, Bergman RG, Ellman JA. Organometallics. 2005;24:5737. For o-borylation of phenols, see: Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2008;130:7534. doi: 10.1021/ja8015878.
- (11).It was shown by Yu group that the alcohol-directed C–O cyclization may proceed without the base, albeit with lower efficiency (ref 7).
- (12).See Supporting Information for details.
- (13).(a) Fishman J, Cox RR, Gallagher TF. Arch. Biochem. 1960;90:318. doi: 10.1016/0003-9861(60)90586-5. [DOI] [PubMed] [Google Scholar]; (b) Kraychy S, Gallagher TF. J. Am. Chem. Soc. 1957;79:754. [PubMed] [Google Scholar]
- (14).The observed intramolecular kinetic isotope effect (kH/kD = 2.6) is in the range for oxygenation reactions proceeding via analogous palladacycles, see: Stowers KJ, Sanford MS. Org. Lett. 2009;11:4584. doi: 10.1021/ol901820w. (b) See also refs 6a,i,k.
-
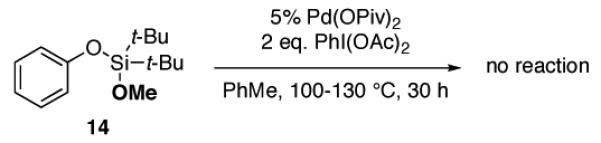
(15).Notably, the methyl-protected silanol 14 had no reactivity under the standard reaction conditions, suggesting the hydroxyl group of silanol being crucial for this transformation. Likewise, Liu8 reported that OH group of a phenol is required for the formation of hydrogen bonding with the acetate/pivalate group of the Pd(II) catalyst, which is necessary for a successful C–O cyclization.
- (16).For involvement of PdIV intermediates in C–H functionalization reactions, see: Yoneyama T, Crabtree RH. J. Mol. Catal. A: Chem. 1996;108:35. Engle KM, Mei T-S, Wang X, Yu J-Q. Angew. Chem., Int. Ed. 2011;50:1478. doi: 10.1002/anie.201005142. (c) See also refs 5d, 6a,b,f,h, 7. For involvement of bimetallic PdIII species, see: Powers DC, Geibel MAL, Klein JEMN, Ritter T. J. Am. Chem. Soc. 2009;131:17050. doi: 10.1021/ja906935c. Powers DC, Ritter T. Nat. Chem. 2009;1:302. doi: 10.1038/nchem.246.
- (17).This transesterification process could also take place in the presence of a base. Indeed, the cyclization completed within 10 min upon treatment of 5c with NaOtBu in MeOH at room temperature.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.